

Abstract
Chlamydia are obligate intracellular bacteria that replicate in a vacuole inside a host cell. Chlamydial infection has been shown to protect the host cell against apoptotic stimuli. This is likely important for the ability of Chlamydia to reproduce in human cells. Here we show that resistance to apoptosis is conveyed by the destruction of the proapoptotic BH3-only proteins Bim/Bod, Puma, and Bad during infection. Apoptotic stimuli were blocked upstream of the mitochondrial activation of Bax/Bak. During infection with both species, Chlamydia trachomatis and Chlamydia pneumoniae, Bim protein gradually disappeared without noticeable changes in Bim mRNA. The disappearance was blocked by inhibitors of the proteasome. Infected cells retained sensitivity to Bim expressed by transfection, indicating functional relevance of the Bim disappearance. Fusion to Bim targeted the green fluorescent protein for destruction during infection. Analysis of truncation mutants showed that a short region of Bim containing the BH3 domain was sufficient for destruction during chlamydial infection. Like Bim, Puma and Bad proteins disappeared during infection. These results reveal a novel way by which microbes can interfere with the host cell's apoptotic machinery, and provide a molecular explanation of the cellular resistance to apoptosis during infection with Chlamydia.
Keywords: bacteria, pathogens, cell death, proteasome, mitochondria
Introduction
Infections with bacteria of the genus Chlamydia are a common cause of human disease. Among these, Chlamydia trachomatis is of great importance as the cause of eye infections and sexually transmitted diseases (1), whereas Chlamydia pneumoniae is a common agent of respiratory infections (2). Chlamydia are obligate intracellular bacteria and replicate within a cytosolic vacuole in eukaryotic cells. Although contained within an occluded vacuole, Chlamydia can impact on host cell function in various ways.
Cell death by apoptosis is the result of the activation of an intracellular signal transduction pathway. It is becoming increasingly clear that apoptosis plays an important role in the defense against pathogens (3, 4) on the level of both the reaction of an individual host cell to an invading microorganism and the reacting immune system. For instance, in viral infections apoptosis is likely to act as a cellular defense mechanism. This is suggested by the finding that many viruses carry genes whose products inhibit apoptosis (5, 6). A number of bacteria have been found to induce apoptosis in the host's cells (for instance Legionella and Shigella; references 7 and 8). By contrast, a number of earlier studies have found that Chlamydia can decrease sensitivity of an infected cell against apoptotic stimuli (9–12).
The molecular function of the apoptotic pathway has been worked out in some detail. The final events of apoptosis are executed by the caspase proteases (13). In most circumstances, caspase activation requires the release of cytochrome c from the mitochondria into the cytosol (14). This release is governed by the Bcl-2 family of proteins. Bcl-2 proteins can be structurally and functionally divided into the following three groups: inhibitors of apoptosis (Bcl-2, Bcl-x, and others), effectors of cytochrome c release (Bax, Bak, and Bok), and triggers of apoptosis (BH3-only proteins). According to a plausible model, a stimulus to apoptosis activates one or several BH3-only proteins (nine are known at present; reference 15) that in turn activates Bax/Bak by an unknown mechanism. Active Bax/Bak then effect the release of cytochrome c. Bcl-2 blocks apoptosis by sequestering active BH3-only proteins (15).
Earlier studies have begun to map the apoptosis-inhibitory activity from Chlamydia within the apoptotic apparatus. Chlamydia efficiently block the release of cytochrome c from mitochondria upon the induction of apoptosis by external stimuli (9), whereas a death receptor signal that induces apoptosis independently of mitochondria is not inhibited (16). Here, we describe a molecular characterization of the apoptosis-inhibitory activity of Chlamydia in human host cells. We first focused on the BH3-only protein Bim, in part because Bim is bound to the microtubuli cytoskeleton, which is substantially reorganized during chlamydial infection (17). We found evidence that Bim is targeted for proteasomal destruction during chlamydial infection and extend this observation to the BH3-only proteins Puma and Bad. Because infected cells were not protected against active Bim or Puma, the disappearance of these proteins likely is the reason for the resistance of infected cells against apoptosis.
Materials and Methods
Cell Lines, Bacterial Organisms, and Infection.
The human laryngeal carcinoma cell line Hep2, the human cervical adenocarcinoma cell line HeLa, the human T lymphocyte cell line Jurkat, the pro-myeloblastic/myelocytic cell line HL60, and the human breast cancer cell line MCF-7 were obtained from the American Type Culture Collection (ATCC). The cell line HeLa Trex, which stably expresses the tetracycline repressor, was purchased from Invitrogen. All cells were cultured in either DMEM or RPMI 1640 complemented with 10% FCS. The mycoplasma-free strains C. pneumoniae strain CM-1 (VR-1360) and C. trachomatis strain L2 were obtained from ATCC. Chlamydia were grown in Hep2 cells and purified as described previously (11, 16). Human cells were infected with C. trachomatis or C. pneumoniae at a multiplicity of infection (MOI) of 3 unless otherwise mentioned (11, 16). Infection was checked routinely and was found to be >95% in the experiments shown. Legionella pneumophila 2064 was obtained from P. Hoffmann (Dalhousie University, Halifax, Canada; reference 18) and grown on BCYE plates. For infection of host cells with Legionella, a bacterial suspension in cell culture medium (McFarland 2 tubidity standard) was prepared and incubated with the cells for 8 h.
Induction of Apoptosis.
Host cells (3 × 105/well in 6-well plates seeded the day before) were infected or not and subjected to UV irradiation. Cells were washed with PBS and then exposed to UV light (1,000 J/m2) in a transilluminator box (cells were ∼80% confluent at this point; Stratagene). Medium was added and cells were analyzed at the time points given.
Assay for Nuclear Apoptosis and Caspase Activity.
Apoptosis was detected as described previously (11, 16). In brief, for detection of nuclear apoptosis, cells were stained with 20 μM Hoechst 33258 (Sigma-Aldrich) for 30 min and nuclear morphology was assessed under a fluorescence microscope. 300 nuclei per sample were scored. For detection of caspase 3–like activity, cells were lysed in NP-40 lysis buffer (11, 16), triplicates of aliquots were added to a peptide containing 10 μM of a caspase 3 recognition sequence (DEVD-AMC) in assay buffer containing BSA, Hepes, and fluorimetric substrate. Free AMC was measured after 1 h of incubation at 37°C and values are presented as arbitrary relative fluorescence units (mean ± the SD of triplicate reactions).
Microscopy.
Hep2 cells were grown on glass coverslips, infected with C. trachomatis or left uninfected, and some samples were treated with UV light for apoptosis induction as described above. Cells were fixed with 2% formalin for 30 min and consecutively stained with mouse anti–cytochrome c mAb (Becton Dickinson), FITC- or Cy3-labeled anti–mouse antiserum, and Alexa Fluor 546–labeled mouse antichlamydial LPS antibody (Progen) in PBS containing 1% FCS and 1% saponin. For detection of active Bax, cells were stained with anti-active Bax mAb (6A7; Upstate Biotechnology; reference 19) and Cy3-labeled anti–rabbit antiserum (Dianova) followed by staining with MitoTracker Green FM (Molecular Probes) or antichlamydial LPS antibody. Pictures were obtained with a laser scanning microscope (Carl Zeiss MicroImaging, Inc.). For detection of apoptosis in infected and transfected cells, HeLa cells were infected and transfected as described above. Cells were fixed and stained as described above (antichlamydia and Hoechst). Pictures were taken under a Zeiss microscope and analyzed with Zeiss AxioVision software. Brightness of whole pictures was adjusted electronically.
Determination of Bim mRNA by Microarrays.
HeLa cells were infected with C. trachomatis or C. pneumoniae. mRNA was harvested at various time points as indicated (see Results) and analyzed by a human micro array (U95A; Affymetrix, Inc.) as described recently in detail (20). Bim is represented on the chip by the sequence code 31611_s_at and the GenBank accession no. AF032457.
Detection of Bax and Bak by Flow Cytometry.
HeLa cells were infected with C. trachomatis or not and apoptosis was induced as described above. For some experiments, apoptosis was induced by transfection with expression vectors for enhanced GFP (EGFP) and BimS 10 h after infection. Cells were harvested by trypsinization and stained for active Bax or Bak. Anti-active Bak (amino acids 1–24) was purchased from Oncogene Research Products. Flow cytometry was performed with a FACSCalibur (Becton Dickinson). At least 105 cells per sample were recorded.
Transfection of HeLa Cells.
HeLa cells were plated in 12-well plates (2 × 105 per well) and infected with C. trachomatis as indicated. At the time points shown in the figure legends, cells were transfected in triplicate with 3 μg of control vector pEF, pEF-BimS expression vector, or Puma expression vector (provided by D. Huang, The Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia) together with 0.5 μg CMV-LacZ using ethylene imine polymer solution (Fluka; reference 21). After 3 h, medium was changed and 12–20 h later cells were stained for β-galactosidase activity. Blue cells were viewed under a microscope and scored alive or dead using morphological criteria (22). For inducible expression, BimS and Puma were cloned by PCR into the vector pCDNA4/TO. When transfected into cells that stably express the tetracycline repressor, the expression can be induced by removal of the suppressor upon the addition of tetracycline. The cell line HeLa Trex was transfected with these constructs or the empty vector, infected, and after 24 h the expression of BimS or Puma was induced by the addition of tetracycline. 20 h later, cells were scored as described above.
Western Blot Analysis.
For Western blot analysis, various cell types were either infected with C. trachomatis, C. pneumoniae, or L. pneumophila 2064, or left uninfected and harvested at the indicated time points. In some experiments, cells were treated with 10 μg/ml rifampin or 500 nM proteasome inhibitor MG132 (Calbiochem). For SDS extracts, 4 × 105 cells were directly lysed in SDS-containing loading buffer. For detergent extracts, 5 × 105 cells were lysed by incubation in 50 μl Triton buffer (1% Triton X-100, 0.05 M Pipes-NaOH, 0.05 M Hepes, pH 7.0, 2 mM MgCl2, 1 mM EDTA, 10 mM DTT, and protease inhibitors; Roche) for 30 min on ice. After centrifugation at 2,000 g at 4°C for 10 min, loading buffer was added and lysates were run on a 12% polyacrylamide gel. Proteins were transferred to nitrocellulose membranes, and membranes were probed with antibodies specific for Bax, Bak (Upstate Biotechnology), Bim (polyclonal from rabbit; Sigma-Aldrich), chlamydial Hsp60 (Affinity BioReagents, Inc.), GST (monoclonal from mouse; provided by H. Flaswinkel, GSF, Neuherberg, Germany), Puma NH2 terminus (rabbit; Sigma-Aldrich), tubulin (Sigma-Aldrich), Bad (Cell Signaling Technology), or GFP (CLONTECH Laboratories, Inc.). Proteins were visualized using peroxidase-conjugated secondary antibodies and a chemiluminescence detection system (PerkinElmer).
Measurement of Proteasomal Activity and In Vitro Bim Degradation.
Hep2 cells were infected with C. trachomatis or left uninfected. After 24 h of infection, cytosolic extracts were prepared as described previously (11). In brief, cells were resuspended in a cytoplasmic extraction buffer (1 mM Na-EGTA, 1 mM Na-EDTA, 1.5 mM MgCl2, 10 mM KCl, 20 mM Hepes-KOH, pH 7.5) containing a proteinase inhibitor mix and 1 mM DTT. After incubation for 1 h on ice, cells were disrupted by passages through a 22-gauge needle. For measurement of proteasomal activity, reactions were set up to contain 400 μg of cytosolic extract in a total of 200 μl of assay buffer (20 mM Tris/HCl, pH 7.5, and 50 mM NaCl) containing a final concentration of 10 μM Suc-LLVY-AMC fluorimetric substrate (proteasome substrate; Bachem). Free AMC was measured at 390 nm (excitation) and 440 nm (emission) every 5 min over 35 min.
GST-BimEL (provided by D. Huang) was expressed in Escherichia coli and purified using standard protocols. GST-CED-4 protein was provided by B. Seiffert (Technical University Munich, Munich, Germany; reference 23). For assessment of proteolytic activity, 400 μg of cytosolic extract was incubated with 0.8 μg GST-BimEL, 0.4 μg GST-CED-4, and 500 μM of proteasome inhibitor MG132 for 1.5 h at 37°C. Western blot analysis was performed as described above.
Assay for Degradation of GFP-BimL Mutants.
BimL was fused to the COOH terminus of EGFP in the vector pEGFP-C1. A number of Bim fragments were generated by PCR and cloned into pEGFP-C2 as COOH-terminal fusions to EGFP (BD Biosciences; details about the cloning procedure are available from the authors upon request). Integrity of PCR products was confirmed by sequencing. Hep2 cells were transfected with the constructs by electroporation (5 × 106 cells with 20 μg DNA at 280 V, 960 μF). Some aliquots were infected with C. trachomatis after 4 h. In some experiments, cells were treated with 500 nM of proteasome inhibitor MG132 (Calbiochem) or 10 μM lactacystin (BIOMOL Research Laboratories, Inc.). 20 h after infection, cells were harvested and GFP fluorescence was detected by flow cytometry with a FACSCalibur (Becton Dickinson). At least 105 cells per sample were recorded.
Online Supplemental Material
Fig. S1 shows micrographs of Hoechst staining of normal or infected cells upon UV irradiation. Fig. S2 demonstrates the inhibition of taxol-induced apoptosis by chlamydial infection. The photos provided in Fig. S3 show the morphology of cells (normal or infected) transfected with an expression vector for Puma. Fig. S4 gives data that show that chlamydial infection also fails to protect cells against short-term (6-h) induction of BimS through tetracyclin. Fig. S5 demonstrates that transfection of BimS causes the mitochondrial release of cytochrome c also in infected cells. Figs. S1–S5 are available at http://www.jem.org/cgi/content/full/jem.20040402/DC1.
Results
Chlamydia Inhibit UV Light–induced Nuclear Apoptosis, Effector Caspase Activation, and Cytochrome C Release.
We chose to study apoptosis induced by UV irradiation in epitheloid cells infected with Chlamydia because this pathway to apoptosis is well investigated and can therefore be studied closely. Infection with Chlamydia has been found to inhibit apoptosis induced by a variety of external stimuli such as staurosporine, etoposide, TNF, or anti-CD95 (9–11, 16). Where investigated, this block was associated with the inhibition of cytochrome c release (9, 11, 16). Although probably most or all stimuli to mitochondrial apoptosis act by activation of BH3-only proteins, it is unclear in most cases which BH3-only protein implements which apoptotic stimulus. It has, however, been shown that UV irradiation induces apoptosis via the activation of Bim, and Bim-dependent events of apoptosis have been described. UV irradiation leads to the release of Bim from the cytoskeleton, which translocates to the mitochondria and causes the activation of Bax. Active Bax in turn causes the release of cytochrome c (24). We first tested whether Chlamydia are able to inhibit apoptosis induced by UV irradiation. Hep2 cells were infected with C. trachomatis or C. pneumoniae, or left uninfected and subjected to UV irradiation. The morphological analysis of nuclear apoptosis 6 h later revealed the expected protection by both chlamydial species. The samples not treated with UV light showed no difference between Chlamydia- and mock-infected cells (Fig. 1 A and Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20040402/DC1). Detection of caspase activity by enzyme assay gave similar results. Upon UV irradiation, caspase 3–like activity and apoptosis were strongly reduced in both C. trachomatis– and C. pneumoniae–infected cells compared with noninfected cells (Fig. 1 A). No difference in inhibition was seen when cycloheximide was included (not depicted). Cytochrome c release upon UV irradiation was further studied by immunostaining of cytochrome c. Samples not subjected to irradiation showed a typical mitochondrial pattern of cytochrome c in both mock- and Chlamydia-infected cells. UV irradiation of uninfected cells caused morphological changes and the release of cytochrome c from mitochondria, resulting in a cytosolic distribution and a reduction in intensity of cytochrome c staining (Fig. 1 B). Infected cells retained the typical mitochondrial pattern of cytochrome c staining. Thus, chlamydial infection blocks UV-induced apoptosis at the level or upstream of cytochrome c release. Apoptosis induced by the cytotoxic drug taxol, which depends on Bim (25), was also blocked in infected cells (Fig. S2). UV irradiation was chosen for the consecutive experiments because it acts much faster than taxol, allowing finer analysis.
Figure 1.
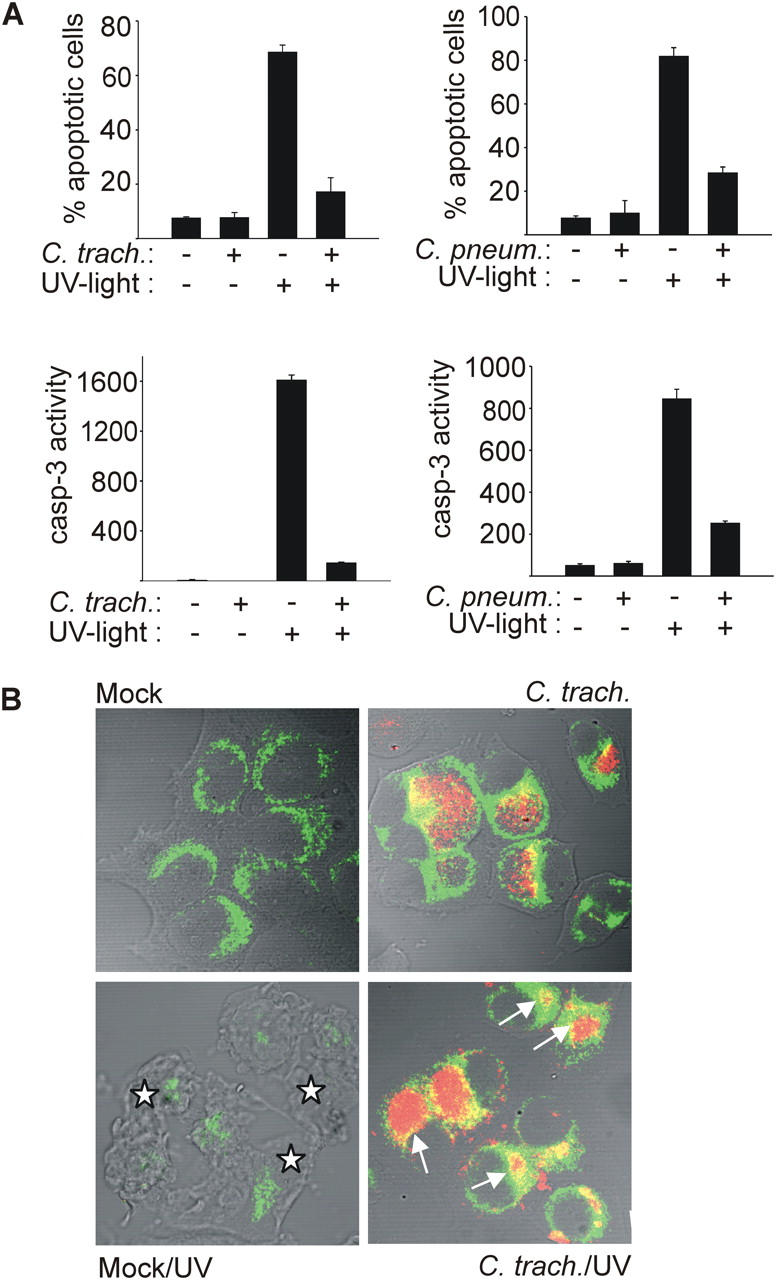
Infection with C. trachomatis or C. pneumoniae inhibits UV light–induced apoptosis. Hep2 cells were infected with Chlamydia or not (mock). After 24 (C. trachomatis) or 48 h (C. pneumoniae), apoptosis was induced by UV irradiation (1,000 J/m2). 6 h later, cells were analyzed. (A) Nuclear apoptosis and caspase 3–like activity. Cells were stained with Hoechst dye and nuclear apoptosis was scored under a fluorescence microscope (top; at least 300 nuclei were counted per sample and values are mean/SD of triplicates) or lysed, and DEVD cleaving activity was measured in cell extracts (bottom; mean/SD of triplicate measurements). (B) Cytochrome c release from mitochondria. Cells were stained for cytochrome c (green) and chlamydial LPS (red), and analyzed by laser-scanning microscopy. Arrows point at chlamydial inclusions in cells that have retained cytochrome c in their mitochondria. Asterisks indicate noninfected cells with typical apoptotic morphology and release of cytochrome c from mitochondria (the cytochrome c signal is diminished upon release). The figure shows results typical of three independent experiments.
Chlamydial Infection Blocks UV Light–induced Activation of Bax/Bak.
Cytochrome c release is the result of the activation of Bax/Bak (these two proteins probably act in the same way and can replace each other; reference 26). Bax/Bak are present in the cytosol and during the induction of apoptosis, translocate to the mitochondrial membrane (27, 28). This activation of Bax/Bak involves a conformational change that can be specifically detected by using antibodies directed against an epitope that is only exposed during activation (19, 29). Cells were infected and UV irradiated, and the activation of Bax/Bak was analyzed by staining with the activation-specific antibodies. Flow cytometry showed that UV irradiation induced positive staining for both Bax (Fig. 2 A, left) and Bak (Fig. 2 A, right) in a high proportion of noninfected HeLa cells, but not in C. trachomatis–infected cells. This inhibition was still seen at late stages of the replicative cycle (Fig. 2 A). Infection with the more virulent species Chlamydia caviae has been found to cause the activation of Bax in infected cells (30). We did not observe such activation with C. trachomatis with either Bak or Bax (Fig. 2), suggesting species-specific differences (the results from later time points should be interpreted with caution, however, as at these points the cells are strongly distorted by the chlamydial inclusion, which might impede staining). Staining for active Bax and the mitochondrial marker MitoTracker Green FM and analysis by confocal laser scanning gave a positive staining and colocalization only in noninfected cells upon treatment with UV light (Fig. 2 C, left). HeLa cells were further infected with a low dose of Chlamydia (MOI: 0.5), UV irradiated, and subjected to anti-active Bax staining. Costaining for chlamydial LPS showed that Bax activation was blocked only in infected cells (Fig. 2 C, right). There was no difference in Bax/Bak protein expression in infected versus noninfected cells, as detected by Western blotting (Fig. 2 B). These results indicate that infection with C. trachomatis blocks apoptosis upstream of Bax activation.
Figure 2.
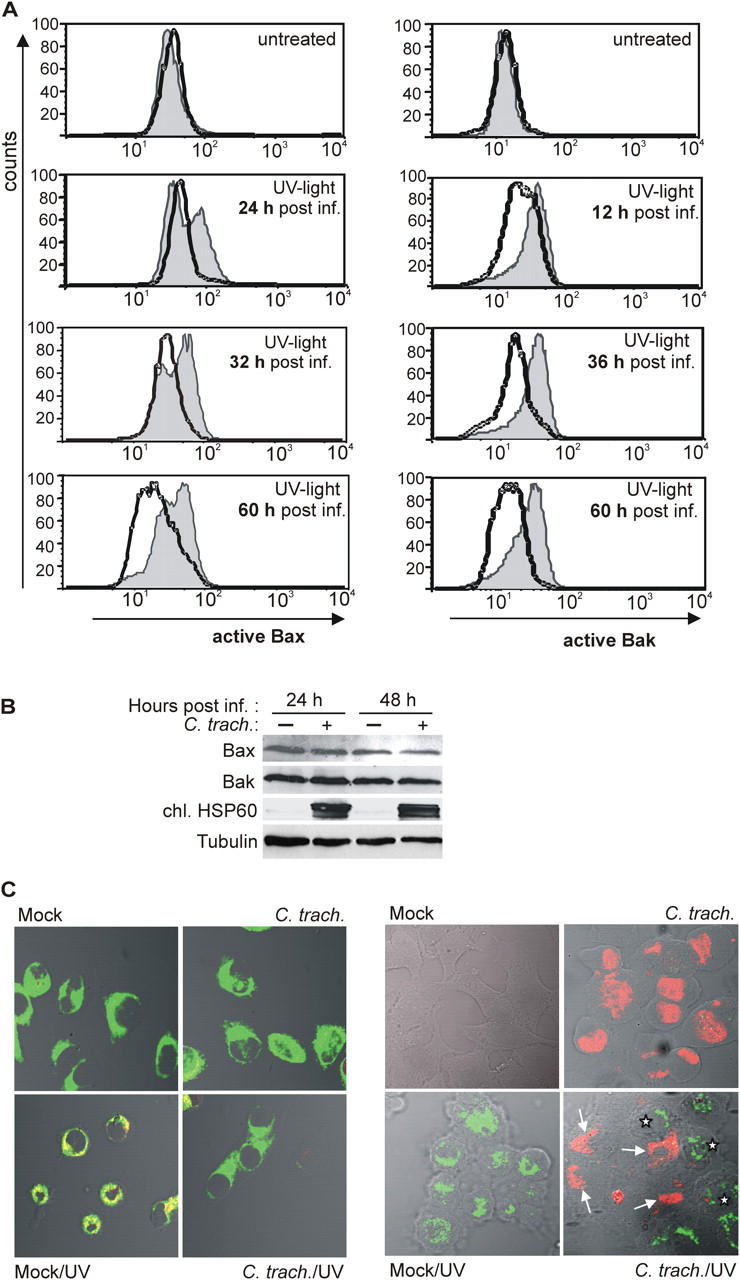
Activation of Bax and Bak by UV light is inhibited in C. trachomatis–infected cells. (A) On the left, detection of active Bax by flow cytometry is shown. HeLa cells were either infected or left uninfected. At the time points indicated, cells were treated with UV light (1,000 J/m2; bottom) or not (top). Cells were collected 6 h later and stained for active Bax. Normal line/shaded area, uninfected cells; bold line, C. trachomatis–infected cells. On the right, detection of active Bak is shown. Data are representative of three experiments. (B) Detection of Bax and Bak by Western blot. HeLa cells were infected or left uninfected. 24 or 48 h later, cells were collected and subjected to Western blotting with antibodies specific for total Bax, total Bak, chlamydial HSP60 (as a control of infection), and tubulin (as a loading control). Similar results were obtained in three independent experiments. (C) Active Bax localizes to mitochondria and is only detectable in noninfected cells. In Hep2 cells with or without (mock) chlamydial infection, apoptosis was induced by UV irradiation (1,000 J/m2). After 6 h, cells were doubly labeled with anti-active Bax (red) and the mitochondrial marker MitoTracker Green FM (left, green; colocalization results in yellow color) or anti-active Bax (green) and anti-chlamydial LPS (right, red). Active Bax was not detected in infected cells (arrows). Asterisks indicate noninfected cells positive for active Bax. For the results shown in the right panel, infectious dose was reduced (MOI: 0.5). Similar results were obtained in three independent experiments.
Chlamydia-infected Cells Retain Sensitivity to BimS- and Puma-induced Cell Death.
The above data show, at least in part, that the Bim-dependent, UV-induced activation of Bax is prevented in cells infected with Chlamydia. Although their activation was prevented, the protein levels of Bax and Bak did not change during infection (Fig. 2 B). This suggested that either active Bim was unable to activate Bax, as would be the case in the presence of high levels of Bcl-2 or a similar protein, or that no active Bim was generated in infected cells. To distinguish between these possibilities, Bim was directly expressed in infected cells. At least three isoforms of Bim exist, all of which promote apoptosis (31). BimEL and BimL are present in normal cells (in HeLa and Hep2 cells BimEL is predominant) and are activated by release from the cytoskeleton. No such posttranslational regulation is known for BimS. This isoform probably induces apoptosis directly upon gene induction (24). We transfected HeLa cells with a construct where BimS is placed under the control of a strong promoter and monitored cell death. Cells were infected or not and cotransfected with a BimS expression vector or empty vector and a LacZ reporter construct to detect transfected cells by β-galactosidase staining (Fig. 3, A and B). Viability of blue (transfected) cells was assessed by morphological criteria (22). Upon expression of BimS, ∼80% of transfected normal HeLa cells were dead. In infected cells, the percentage of dead cells upon BimS expression was only marginally reduced, indicating that chlamydial infection did not protect against the direct Bim activity. Similar results were seen when the BH3-only protein Puma was transfected (Fig. 3 B, top, and Fig. S3). To exclude the possibility that transfection killed the cells before Chlamydia had established the protective mechanism, we further used an inducible expression system. HeLa cells expressing a tet repressor were first transfected with the β-galactosidase reporter and a vector in which either Bim or Puma was placed under a tetracycline-inducible promoter. Cells were then infected with C. trachomatis, 24 h later expression of Bim or Puma was induced, and cells were stained after an additional 20 h. As shown in Fig. 3 B, bottom, the addition of tetracycline caused cell death in a large proportion of reporter-positive cells. Infection failed to protect the cells against cell death caused by the induction of Bim or Puma. Similar results were observed in experiments of short-term induction of Bim (6 h of tetracycline; see Fig. S4). Simultaneous detection of transfection, infection, and nuclear morphology further confirmed that BimS induced apoptosis in cells harboring chlamydial inclusions (Fig. 3, C and D).
Figure 3.
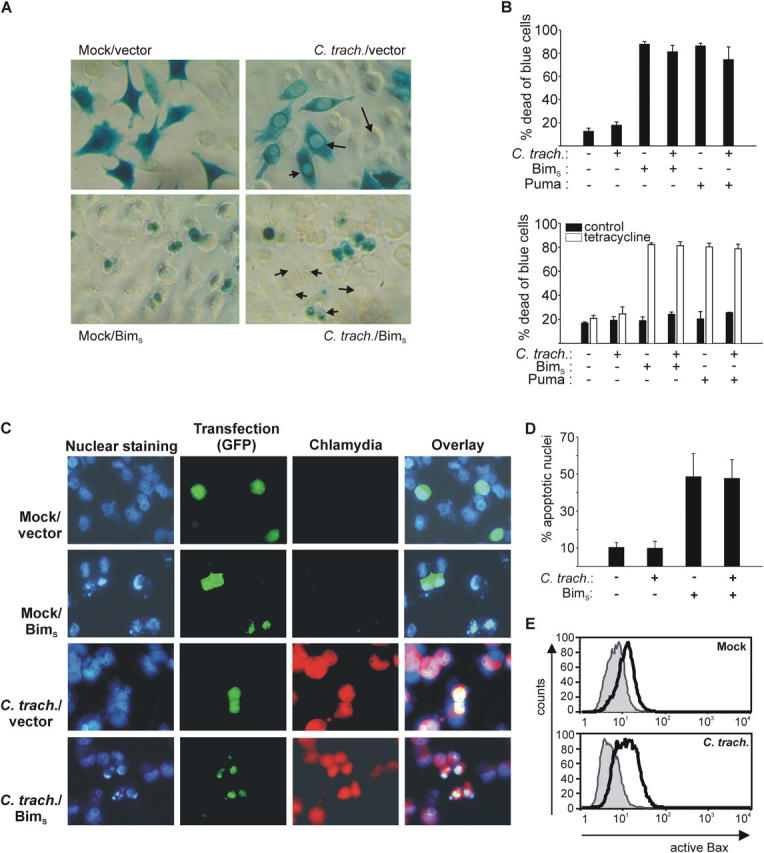
Chlamydia-infected cells retain sensitivity to cell death induced by overexpression of BimS and Puma. (A) HeLa cells were infected with C. trachomatis or not (mock). 4 h after infection, cells were transfected by lipofection with BimS expression vector or control vector together with CMV-LacZ. 3 h later, medium was changed and 12 h later, cells were stained for β-galactosidase activity. Blue cells were viewed under a microscope and scored alive or dead. Examples of chlamydial inclusions are illustrated with arrows. (B) Quantification of dead and blue cells (graphs show percentages of dead cells; mean/SD of triplicate wells). At least 300 cells per sample were counted. On the top, HeLa cells were infected with C. trachomatis and transfected 8 h later. 20 h later, cells were stained for β-galactosidase expression and counted. This experiment was performed three times with similar results. On the bottom, HeLa cells stably expressing the tet repressor were transfected with reporter and a vector where the expression of either BimS or Puma is placed under the control of a tetracycline-inducible promoter. 3 h later, some wells were infected with C. trachomatis. 24 h later, BimS/Puma expression was induced by the addition of tetracycline and cells were fixed and stained as described above after an additional 10 h. This experiment was performed twice with similar results. Although tetracycline may inhibit the growth of Chlamydia, our earlier results indicate that the antiapoptotic activity is long-lived (it was still detectable after 48 h of rifampin treatment; reference 11). (C) BimS induces nuclear condensation in cells infected with C. trachomatis. HeLa cells were infected with C. trachomatis at an MOI of 3 or mock infected as indicated. After 8 h, cultures were cotransfected with EGFP and either empty vector or BimS expression constructs. 16 h later, cells were fixed and stained with antichlamydial LPS antibody to visualize chlamydial inclusions and Hoechst dye to visualize nuclear morphology. The micrographs show EGFP fluorescence (green, as a marker of transfection), Chlamydia (red), and nuclei/DNA (blue). Note the apoptotic morphology in both uninfected and infected cells transfected with BimS. Similar results were obtained in three separate experiments. (D) Quantification of the results illustrated in C. At least 200 GFP+ cells per transfection were counted, and nuclei were scored as normal or apoptotic. For infected cultures, only cells staining positive for chlamydial antigen were counted. Data are mean/SD of three independent experiments. (E) Normal Bax activation by BimS in infected cells. Hep2 cells were infected or not and were cotransfected by electroporation with an EGFP expression plasmid as a marker, and either empty vector or a BimS expression vector 10 h later. 16 h later, cells were stained for active Bax and analyzed by flow cytometry. The histograms show expression of active Bax in EGFP+ cells. Normal line/shaded area, empty vector; bold line, BimS expression vector.
The events caused by Bim expression in infected cells were further investigated. Cells were infected and then cotransfected with an EGFP expression construct and empty vector, or a BimS expression construct. After 16 h, cells were stained for active Bax and analyzed by flow cytometry. As shown in Fig. 3 E, coexpression of BimS caused positive staining for active Bax in EGFP+ (i.e., transfected) cells as compared with transfection with empty vector in both normal and infected cells. In similar experiments, cells were infected, transfected with BimS, and stained for cytochrome c. As shown in Fig. S5, BimS caused the release of cytochrome c also in infected cells.
These data show that active Bim (or Puma) can still cause apoptosis in infected cells. The inhibition of apoptosis must therefore involve a block in the activation of Bim/Puma.
Chlamydial Infection Causes the Disappearance of Bim, Puma, and Bad Proteins.
Next, we tested whether chlamydial infection modified the expression of Bim. mRNA expression profiling of HeLa cells was performed at 2, 4, 12, 24, 36, and 48 h after infection with either C. trachomatis or C. pneumoniae (20). Bim mRNA was also analyzed by quantitative PCR and neither method showed a significant reduction during infection. When measured by PCR, the relative expression was 1.18 at 24 h and 0.9 at 48 h (expression during infection with C. trachomatis/expression during mock infection, mean of two experiments). However, Western blot analysis revealed a near-complete disappearance of Bim protein at 24 h after infection with C. trachomatis. This effect was most pronounced in the epitheloid cell lines HeLa and Hep2 (derivatives of the natural host cells of Chlamydia) and the breast carcinoma line MCF-7, but could also be observed easily in the T cell line Jurkat and the pro-myeloid cell line HL60 (infection blocks apoptosis in all of them; Fig. 4 A; reference 16 and not depicted). To determine whether chlamydial protein synthesis was required for this effect, Hep2 cells were infected in the presence of the antibiotic rifampin. Under this protocol, the Chlamydia-induced disappearance of Bim was strongly reduced (the efficiency of inhibition of the bacterial protein synthesis can be estimated by monitoring the production of bacterial Hsp60; Fig. 4, A and B). Bim levels were also investigated in cells infected with C. pneumoniae. As shown in Fig. 4 B, infection with this species also caused the disappearance of Bim, which was prevented by rifampin. To investigate whether this disappearance was specific for chlamydial infection or also occurred in other bacterial infections, Hep2 cells and Jurkat cells were infected with L. pneumophila. Bim protein expression remained unchanged in Legionella-infected cells. Infection was controlled by induction of apoptosis by Legionella as measured as the activation of effector caspases by enzyme assay (Fig. 4 C; reference 32).
Figure 4.

Disappearance of Bim, Puma, and Bad in Chlamydia-infected cells. (A) Analysis of SDS and Triton X-100 extracts of various cell lines. Cells were infected with C. trachomatis or left uninfected and harvested after 24 h of infection or as indicated. SDS extracts from MCF-7 cells (top) or Triton X-100 extracts (bottom) were subjected to Western blotting with antibodies against Bim (isoform BimEL is detected) and chlamydial HSP60 (as control for chlamydial infection). Detection of tubulin served as a loading control. SDS extracts were used in the initial experiments to exclude the possibility of Bim degradation during extraction. (B) Rifampin treatment prevents degradation of Bim by both chlamydial species. Hep2 cells were infected with C. trachomatis (24 h before analysis) or C. pneumoniae (as indicated) in the presence or absence of 10 μg/ml rifampin. Triton X-100 extracts were analyzed by Western blotting. (C) No disappearance of Bim is seen in Legionella-infected cells. Jurkat and Hep2 cells were infected with L. pneumophila for 8 h. Triton X-100 extracts were analyzed by Western blotting (ns, nonspecific band). Caspase 3–like activity was measured in cell extracts as described above to confirm infection, which is known to cause caspase activation. (D) Kinetics of Bim disappearance. At the indicated time points after infection, Triton X-100 extracts of Hep2 cell samples were taken and analyzed by anti-Bim Western blotting. The figure shows typical results from at least three independent experiments. (E) Disappearance of Puma and Bad in infected cells. Hep2 or HeLa cells were infected with C. trachomatis. 24 h later, the expression of Puma or Bad was assessed by Western blotting.
The disappearance of Bim became noticeable after 14–16 h of infection with C. trachomatis and was complete after ∼24–26 h of infection (Fig. 4 D and not depicted). Bim is one important member of the BH3-only class of proapoptotic proteins, and the loss of Bim could explain some but not all cases where Chlamydia have been found to protect against apoptosis. A second BH3-only protein whose in vivo importance has been demonstrated recently by gene targeting is Puma (33). Puma is known to be regulated transcriptionally, but can also be detected in normal cells in culture (34). We analyzed expression of Puma and found that Puma protein also disappeared during chlamydial infection (Fig. 4 E). The BH3-only protein Bad also disappeared upon infection (Fig. 4 E), suggesting that a common characteristic of BH3-only proteins was targeted.
Chlamydial Infection Targets Bim for Proteasomal Destruction.
These data suggested that Bim was proteolytically destroyed. The finding that no smaller fragments of Bim could be detected by Western blotting (not depicted) further suggested that Bim was degraded by a proteasomal activity. The proteasome is a large multisubunit protein complex that acts to degrade aged and misfolded cellular proteins. To investigate whether Bim was degraded in the proteasome, the proteasome inhibitor MG132 was used. Hep2 cells were infected with C. trachomatis or left uninfected and treated with MG132. As shown in Fig. 5 A, MG132 was able to inhibit Chlamydia-induced degradation of Bim. Although this finding suggested that Bim was degraded by the cellular proteasome, proteasome activity was not enhanced in cytosolic extracts from infected cells (Fig. 5 B). It is further worth noting that although most cellular proteins (for instance tubulin, caspases, Bcl-2, and others) were not degraded in intact cells (11 and not depicted), proteins other than Bim were digested in cytosolic preparations. During incubation in vitro, not only exogenously added, recombinant Bim, but also endogenous tubulin and a control protein (GST-CED-4) were noticeably degraded. This degradation was blocked by MG132, but not by a mix of conventional protease inhibitors, indicating a proteasome-like activity also in these preparations, albeit less specific (Fig. 5 C). Zhong et al. (35) could identify a protein that they named chlamydial proteasome-like activity factor (CPAF), which is secreted from the inclusion into the host cell cytosol and causes the degradation of host transcription factors like RFX5. Under the conditions used here, RFX5 degradation started at 14 h after infection and was complete at ∼16 h of infection. The disappearance of Bim started ∼2 h later (not depicted).
Figure 5.
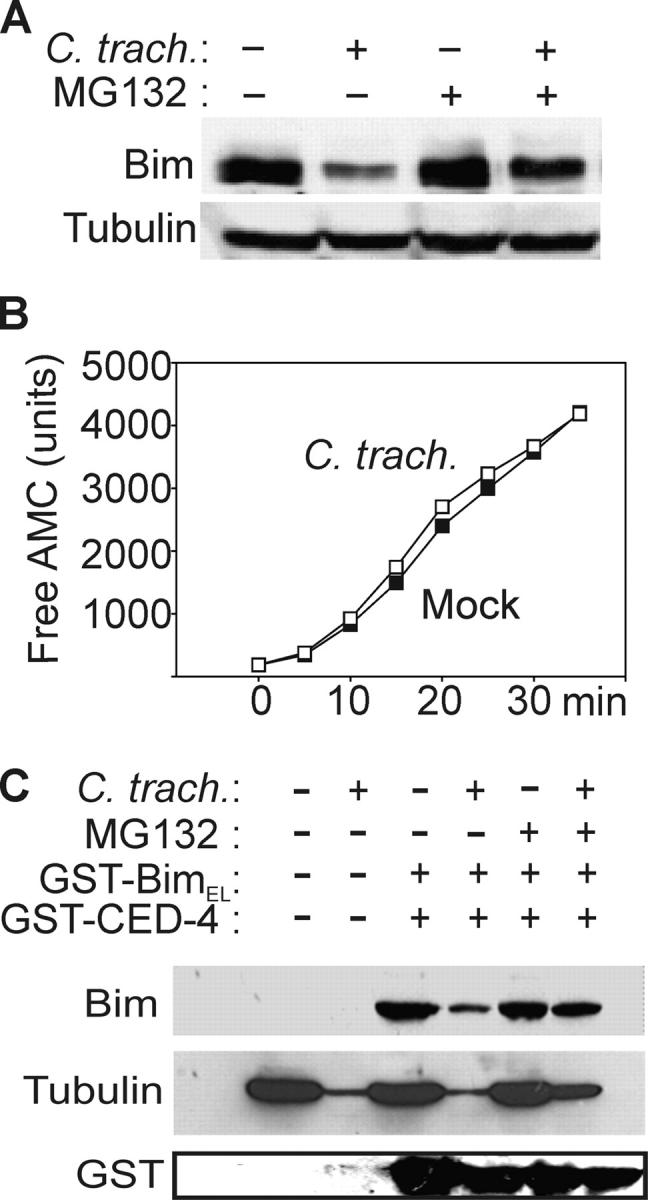
Bim disappearance requires proteasomal activity. (A) Disappearance of Bim is prevented by a proteasome inhibitor. Hep2 cells were infected or not with C. trachomatis and treated with 500 nM MG132 and analyzed by Bim-specific Western blotting. (B) Infection with C. trachomatis does not increase proteasomal activity. As a measure of proteasomal activity, cleavage of Suc-LLVY-AMC was analyzed in cytosolic extracts from C. trachomatis and noninfected cells. ▪, uninfected; □, C. trachomatis–infected. The appearance of free AMC over time is shown. (C) Nonspecific proteasomal degradation of proteins in cytosolic extracts from infected cells. Cytosolic extracts of cells infected with C. trachomatis or not were incubated for 1.5 h at 37°C with GST-BimEL, GST-CED-4, and 500 nM proteasome inhibitor MG132 as indicated. Western blotting was performed using antibodies against Bim, tubulin, or GST.
The BH3 Domain Is Required for Bim Degradation.
Next, an EGFP–BimL fusion protein was expressed by transient transfection in Hep2 cells. Cells were then infected with C. trachomatis and analyzed by flow cytometry for EGFP fluorescence. A population of EGFP–BimL-expressing cells was seen, although the fluorescence intensity was reduced compared with EGFP alone, an effect that may have been due to Bim toxicity that precluded higher expression. Upon infection, the size of the EGFP+ population was strongly reduced in the EGFP–BimL, but not the EGFP-expressing cells (Fig. 6 A). The two inhibitors of proteasome activity, MG132 and lactacystin, prevented this disappearance (Fig. 6 A) These chemicals are highly toxic to cells, which probably explains the lower expression of transfected EGFP–BimL in treated cells. The expression of EGFP alone was also much reduced in the presence of MG132 (not depicted). This suggests that a chlamydial activity is able to target not only Bim, but also EGFP–BimL for proteasomal destruction. Disappearance of EGFP–BimL was also seen by Western blotting (Fig. 6 D).
Figure 6.

The BH3 domain is required for degradation of EGFP–BimL mutants by chlamydial infection. Hep2 cells were transfected with EGFP or EGFP–BimL, or its mutants by electroporation. After 4 h, cells were left uninfected or infected with C. trachomatis. 20 h after infection, cells were collected for the detection of EGFP fluorescence by flow cytometry. (A) Chlamydial infection leads to the proteasome inhibitor–sensitive disappearance of fluorescence of EGFP–BimL. 500 nM MG132 or 10 μM lactacystin (LC) were added at the time of transfection. Normal line/shaded area, uninfected cells; bold line, C. trachomatis–infected cells. (B) EGFP–BimL mutants require the BH3 domain for degradation during chlamydial infection. Normal line/shaded area, uninfected cells; bold line, C. trachomatis–infected cells. The figure shows typical results from five independent experiments. (C) Schematic representation of the constructs used and the susceptibility to Chlamydia-mediated loss of fluorescence. EGFP was fused to the NH2 terminus of all Bim mutants. Numbers represent amino acids in the BimL sequence. Localization of the domains is given at the top. LC8-BD, dynein light chain 8 binding domain; BH3-D, BH3 domain; HD, hydrophobic domain. (D) Disappearance of the EGFP–BimL Western blot band upon infection with C. trachomatis. Hep2 cells were transfected with EGFP–BimL, and one aliquot was infected with C. trachomatis (MOI: 3). 20 h later, cells were lysed and EGFP–BimL was detected by anti-GFP Western blotting. Tubulin served as a loading control. Similar results were obtained in three experiments.
To map the target site of this chlamydial activity, truncation mutants of EGFP–BimL were generated. The following three domains have been described in the Bim structure: the BH3 domain (the site similar in all known BH3-only proteins that is required for their apoptosis-inducing activity; reference 31); the dynein light chain 8 binding domain, which sequesters Bim to the cytoskeleton (24); and a hydrophobic domain at the C terminus (31). We generated constructs for the expression of various truncations of EGFP–BimL, transfected them into Hep2 cells, and monitored their expression during chlamydial infection by flow cytometry. Fig. 6 B shows examples of these results, and Fig. 6 C provides a summary of the results obtained with the constructs tested. Neither the dynein light chain 8 binding domain (see EGFP–BimL 71–141) nor the hydrophobic domain (EGFP–BimL 2–106) were required for the destruction of the fusion protein. However, all constructs that encompassed the BH3 domain appeared to be degraded upon infection (Fig. 6, B and C). The degradation of a mutant that lacked the five COOH-terminal amino acids of the BH3 domain (EGFP–BimL 2–101) was somewhat intermediate. These results strongly suggest that an activity is generated in cells infected with Chlamydia that attacks the BH3-domain of Bim, which thereby leads to the proteasomal destruction of the protein and the protection of the infected cells against apoptosis.
Discussion
The ability of Chlamydia to protect an infected host cell against experimental stimuli to apoptosis is well established. Here, we describe the following mechanism that very likely accounts for this protection: a chlamydial factor targets, upon recognition of a region around its BH3 domain, the proapoptotic protein Bim for proteasomal destruction. This process requires bacterial protein synthesis and operates during infection with both chlamydial species, C. trachomatis and C. pneumoniae. Both species have previously been found to protect against apoptosis in an, as far as investigated, identical manner. The direct expression of Bim under a strong promoter was still able to kill infected cells, indicating that the apoptosis pathway downstream of Bim is intact and that loss of Bim is indeed functionally relevant for the inhibition of apoptosis. Similar results were obtained for Puma. Puma protein disappeared during infection and infection did not protect cells against Puma overexpression. Furthermore, Bad protein also disappeared. Because Puma and Bad also have an active BH3 domain, this suggests that they are targeted by the same mechanism as Bim.
Chlamydial infection can protect a host cell against a large number of stimuli. Chlamydia did not afford protection against the one stimulus tested that does not involve mitochondrial cytochrome c release, which is death receptor– (CD95) dependent apoptosis induction in so-called type I cells (16), supporting the concept of a block confined to mitochondrial apoptosis. Apoptosis induced by all the stimuli against which protection by Chlamydia has been reported is also inhibited by Bcl-2. The molecular function of Bcl-2 is still somewhat under debate, but the most plausible role appears to be connected to its ability to bind and sequester active BH3-only proteins (15).
The allocation of apoptotic stimuli to BH3-only proteins is incomplete. Among the stimuli inhibited by chlamydial infection, UV irradiation is mediated by Bim and possibly Bmf (36), etoposide at least in part by Puma (33), staurosporine by Bim and Puma (25, 33), and TNF and CD95 signal via Bid cleavage (where a mitochondrial contribution is required; references 37 and 38). Our data suggest a model that accounts for the far-reaching antiapoptotic effect of Chlamydia. All BH3-only proteins share the BH3 domain, whose presence is required for the induction of apoptosis. Because the chlamydial activity targets this domain in the Bim protein, all BH3-only proteins might be subjected to proteasomal destruction. This hypothesis was tested on the BH3-only proteins Puma and Bad, which were also found to disappear during chlamydial infection. All of the following proteins of the apoptosis pathway we have investigated do not change in expression: Bax, Bak (above), Bcl-2, Bcl-x, Mcl-1, Bid, caspase 3, caspase 8, caspase 9, Apaf-1, Hsp70, and cytochrome c (11, 16, and not depicted). Up-regulation of the antiapoptotic protein C-IAP2 has been reported during chlamydial infection (39). However, IAP act at the level of caspases and, accordingly, our results do not support a role for IAP in inhibition against apoptosis by Chlamydia.
Genetically modified mice lacking either Bim (25) or Puma (33) show a clear protection against some, but not all, apoptotic stimuli. No double mutant mice have been tested, but the combined deficiency of Bim and Puma might well explain the greater share of apoptosis protection of Chlamydia-infected cells. During CD95 signaling, caspase 8 activation and Bid cleavage were unaffected by chlamydial infection (16). Bid is present in an inactive state and is activated by cleavage through caspase 8. Accordingly, the BH3-domain in Bid is normally inactive and only exposed by this cleavage event (40). Therefore, it might be speculated that the active Bid (tBid), but not the inactive form, is also recognized via its BH3 domain and targeted for destruction, which would explain the protection against CD95-induced apoptosis (the antibody used to detect Bid in the above study did not recognize tBid). Such a general BH3-targeting approach of Chlamydia would explain the almost general protection of infected cells against apoptotic stimuli.
We still do not know what the chlamydial effector is and how degradation is achieved. The inhibition of bacterial protein synthesis with rifampin prevented the disappearance (above) and precluded the development of protection against apoptosis (11). Most likely, this means that rifampin inhibits the production of the chlamydial BH3-only, protein-degrading factor. The sensitivity to MG132 and lactacystin indicates the involvement of a proteasomal activity and immediately suggests targeting to the cellular proteasome. However, a chlamydial factor has been purified and partially characterized, named CPAF. The activity of CPAF has been shown to be both necessary and sufficient to cause degradation of cellular transcription factors such as RFX5, even when purified recombinant proteins were used (35). RFX5 degradation slightly preceded Bim degradation during chlamydial infection. It is conceivable that CPAF also mediates degradation of BH3-only proteins. How CPAF works on a molecular level is still unclear, but it is also inhibited by inhibitors of the proteasome. Recognition by the proteasome requires modification of a protein by ligation with the abundant small cellular protein ubiquitin. Ubiquination involves a series of molecular interactions with ubiquitin-transferring enzymes and factors that convey specificity (41). Bim is targeted at an internal structure and its degradation is probably complete because no fragments could be detected by Western blotting and because the fluorescence of the attached EGFP also disappeared in a lactacystin/MG132-sensitive manner. Therefore, it is possible that the chlamydial activity causes a modification of Bim, which then leads to the destruction by the cellular proteasome. Interference with the cellular ubiquitination/SUMOylation system has been described by an effector protein from Yersinia (42). Although mechanism and effect here appear to be opposite (disruption of proteasomal targeting), it provides an example of where bacteria use the strategy of interfering with this host cell pathway.
Inhibition of apoptosis in infected cells is a mechanism that is often used by viruses. Many viruses carry genes whose products interfere with the host cell's apoptosis machinery (43, 44). Chlamydia share a virus' need for integrity of the host cell. They differ in that unlike Chlamydia, viruses pick up their genetic material from host cells. It is in accordance with this model that Chlamydia use a mechanism to inhibit apoptosis that is not found as a regulatory means in host cells.
It has been suggested that at later stages of chlamydial infection, apoptosis is induced through the activation of Bax (30) and the clearance of Chlamydia during vaginal infection of mice was accelerated in mice lacking Bax (45). Experimentally, Bax could directly (in the absence of BH3-only proteins) be activated through changes in the intracellular milieu such as pH changes or oxidative stress (46, 47). If this occurred at late stages of the infection and led to the uptake of the infected dead cell by phagocytes or surrounding cells, such a mechanism might aid the spreading of the infection.
Why do Chlamydia possess the ability to block cell death? Obviously, there is no need for Chlamydia to protect the cell against stimuli like staurosporine. An antiapoptotic capacity only makes sense if there is apoptosis that occurs during infection and has to be inhibited. It could be argued that Chlamydia need to prolong the lifespan of the host cell; however, the natural host cells of Chlamydia, epithelial cells, probably live longer than a normal replication cycle would take. To us, it appears more likely that Chlamydia also have the potential to induce apoptosis. The cell is probably able to pick up on the presence of Chlamydia and undergo apoptosis. It has been shown that the development of the chlamydial inclusion involves restructuring of the cytoskeleton, including microtubular components (see for instance reference 17). Because Bim is sequestered to the cytoskeleton and initiates apoptosis upon its release from this location, Bim release might be the defense mechanism that a cell uses against chlamydial infection and that has to be overcome by the degradation of Bim.
It is a plausible speculation that the inhibition of apoptosis is important for the growth of the bacteria. Most chlamydial infections clear spontaneously, but there is evidence of chronic infections that may even occur in and contribute to the development of atherosclerotic lesions (48). In vitro studies further suggest that chronic infections might be insensitive to antibiotics (49). Therapies directed at an interference with chlamydial antiapoptotic activities could therefore turn out helpful to prevent sequelae of protracted infections with these bacteria.
Acknowledgments
We thank Dr. Barbara Seiffert for constructs and Dr. Ralph Fritsch for help with the siRNA experiments.
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (to G. Häcker).
The authors have no conflicting financial interests.
Abbreviations used in this paper: CPAF, chlamydial proteasome-like activity factor; EGFP, enhanced GFP; MOI, multiplicity of infection.
References
- 1.Grayston, J.T., and S. Wang. 1975. New knowledge of chlamydiae and the diseases they cause. J. Infect. Dis. 132:87–105. [DOI] [PubMed] [Google Scholar]
- 2.Grayston, J.T. 1992. Chlamydia pneumoniae, strain TWAR pneumonia. Annu. Rev. Med. 43:317–323. [DOI] [PubMed] [Google Scholar]
- 3.Vaux, D.L., G. Haecker, and A. Strasser. 1994. An evolutionary perspective on apoptosis. Cell. 76:777–779. [DOI] [PubMed] [Google Scholar]
- 4.Vaux, D.L., and A. Strasser. 1996. The molecular biology of apoptosis. Proc. Natl. Acad. Sci. USA. 93:2239–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roulston, A., R.C. Marcellus, and P.E. Branton. 1999. Viruses and apoptosis. Annu. Rev. Microbiol. 53:577–628. [DOI] [PubMed] [Google Scholar]
- 6.Everett, H., and G. McFadden. 2002. Poxviruses and apoptosis: a time to die. Curr. Opin. Microbiol. 5:395–402. [DOI] [PubMed] [Google Scholar]
- 7.Muller, A., J. Hacker, and B.C. Brand. 1996. Evidence for apoptosis of human macrophage-like HL-60 cells by Legionella pneumophila infection. Infect. Immun. 64:4900–4906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zychlinsky, A., M.C. Prevost, and P.J. Sansonetti. 1992. Shigella flexneri induces apoptosis in infected macrophages. Nature. 358:167–169. [DOI] [PubMed] [Google Scholar]
- 9.Fan, T., H. Lu, H. Hu, L. Shi, G.A. McClarty, D.M. Nance, A.H. Greenberg, and G. Zhong. 1998. Inhibition of apoptosis in chlamydia-infected cells: blockade of mitochondrial cytochrome c release and caspase activation. J. Exp. Med. 187:487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rajalingam, K., H. Al-Younes, A. Muller, T.F. Meyer, A.J. Szczepek, and T. Rudel. 2001. Epithelial cells infected with Chlamydophila pneumoniae (Chlamydia pneumoniae) are resistant to apoptosis. Infect. Immun. 69:7880–7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fischer, S.F., C. Schwarz, J. Vier, and G. Hacker. 2001. Characterization of antiapoptotic activities of Chlamydia pneumoniae in human cells. Infect. Immun. 69:7121–7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hacker, G., and S.F. Fischer. 2002. Bacterial anti-apoptotic activities. FEMS Microbiol. Lett. 211:1–6. [DOI] [PubMed] [Google Scholar]
- 13.Nicholson, D.W., and N.A. Thornberry. 1997. Caspases: killer proteases. Trends Biochem. Sci. 22:299–306. [DOI] [PubMed] [Google Scholar]
- 14.Green, D.R., and J.C. Reed. 1998. Mitochondria and apoptosis. Science. 281:1309–1312. [DOI] [PubMed] [Google Scholar]
- 15.Bouillet, P., and A. Strasser. 2002. BH3-only proteins-evolutionarily conserved proapoptotic Bcl-2 family members essential for initiating programmed cell death. J. Cell Sci. 115:1567–1574. [DOI] [PubMed] [Google Scholar]
- 16.Fischer, S.F., T. Harlander, J. Vier, and G. Hacker. 2004. Protection against CD95-induced apoptosis by chlamydial infection at a mitochondrial step. Infect. Immun. 72:1107–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clausen, J.D., G. Christiansen, H.U. Holst, and S. Birkelund. 1997. Chlamydia trachomatis utilizes the host cell microtubule network during early events of infection. Mol. Microbiol. 25:441–449. [DOI] [PubMed] [Google Scholar]
- 18.Garduno, R.A., E. Garduno, and P.S. Hoffman. 1998. Surface-associated hsp60 chaperonin of Legionella pneumophila mediates invasion in a HeLa cell model. Infect. Immun. 66:4602–4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsu, Y.T., and R.J. Youle. 1997. Nonionic detergents induce dimerization among members of the Bcl-2 family. J. Biol. Chem. 272:13829–13834. [DOI] [PubMed] [Google Scholar]
- 20.Hess, S., C. Rheinheimer, F. Tidow, G. Bartling, C. Kaps, J. Lauber, J. Buer, and A. Klos. 2001. The reprogrammed host: Chlamydia trachomatis-induced up-regulation of glycoprotein 130 cytokines, transcription factors, and antiapoptotic genes. Arthritis Rheum. 44:2392–2401. [DOI] [PubMed] [Google Scholar]
- 21.Verhagen, A.M., P.G. Ekert, M. Pakusch, J. Silke, L.M. Connolly, G.E. Reid, R.L. Moritz, R.J. Simpson, and D.L. Vaux. 2000. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 102:43–53. [DOI] [PubMed] [Google Scholar]
- 22.Miura, M., H. Zhu, R. Rotello, E.A. Hartwieg, and J. Yuan. 1993. Induction of apoptosis in fibroblasts by IL-1 beta-converting enzyme, a mammalian homolog of the C. elegans cell death gene ced-3. Cell. 75:653–660. [DOI] [PubMed] [Google Scholar]
- 23.Seiffert, B.M., J. Vier, and G. Hacker. 2002. Subcellular localization, oligomerization, and ATP-binding of Caenorhabditis elegans CED-4. Biochem. Biophys. Res. Commun. 290:359–365. [DOI] [PubMed] [Google Scholar]
- 24.Puthalakath, H., D.C. Huang, L.A. O'Reilly, S.M. King, and A. Strasser. 1999. The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol. Cell. 3:287–296. [DOI] [PubMed] [Google Scholar]
- 25.Bouillet, P., D. Metcalf, D.C. Huang, D.M. Tarlinton, T.W. Kay, F. Kontgen, J.M. Adams, and A. Strasser. 1999. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 286:1735–1738. [DOI] [PubMed] [Google Scholar]
- 26.Wei, M.C., W.X. Zong, E.H. Cheng, T. Lindsten, V. Panoutsakopoulou, A.J. Ross, K.A. Roth, G.R. MacGregor, C.B. Thompson, and S.J. Korsmeyer. 2001. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 292:727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Antonsson, B., S. Montessuit, B. Sanchez, and J.C. Martinou. 2001. Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells. J. Biol. Chem. 276:11615–11623. [DOI] [PubMed] [Google Scholar]
- 28.Nechushtan, A., C.L. Smith, I. Lamensdorf, S.H. Yoon, and R.J. Youle. 2001. Bax and Bak coalesce into novel mitochondria-associated clusters during apoptosis. J. Cell Biol. 153:1265–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Griffiths, G.J., L. Dubrez, C.P. Morgan, N.A. Jones, J. Whitehouse, B.M. Corfe, C. Dive, and J.A. Hickman. 1999. Cell damage–induced conformational changes of the pro-apoptotic protein Bak in vivo precede the onset of apoptosis. J. Cell Biol. 144:903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perfettini, J.L., J.C. Reed, N. Israel, J.C. Martinou, A. Dautry-Varsat, and D.M. Ojcius. 2002. Role of Bcl-2 family members in caspase-independent apoptosis during Chlamydia infection. Infect. Immun. 70:55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O'Connor, L., A. Strasser, L.A. O'Reilly, G. Hausmann, J.M. Adams, S. Cory, and D.C. Huang. 1998. Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 17:384–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao, L.Y., and Y. Abu Kwaik. 1999. Activation of caspase 3 during Legionella pneumophila-induced apoptosis. Infect. Immun. 67:4886–4894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Villunger, A., E.M. Michalak, L. Coultas, F. Mullauer, G. Bock, M.J. Ausserlechner, J.M. Adams, and A. Strasser. 2003. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 302:1036–1038. [DOI] [PubMed] [Google Scholar]
- 34.Han, J., C. Flemington, A.B. Houghton, Z. Gu, G.P. Zambetti, R.J. Lutz, L. Zhu, and T. Chittenden. 2001. Expression of bbc3, a pro-apoptotic BH3-only gene, is regulated by diverse cell death and survival signals. Proc. Natl. Acad. Sci. USA. 98:11318–11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhong, G., P. Fan, H. Ji, F. Dong, and Y. Huang. 2001. Identification of a chlamydial protease-like activity factor responsible for the degradation of host transcription factors. J. Exp. Med. 193:935–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Puthalakath, H., A. Villunger, L.A. O'Reilly, J.G. Beaumont, L. Coultas, R.E. Cheney, D.C. Huang, and A. Strasser. 2001. Bmf: a proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science. 293:1829–1832. [DOI] [PubMed] [Google Scholar]
- 37.Li, H., H. Zhu, C.J. Xu, and J. Yuan. 1998. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 94:491–501. [DOI] [PubMed] [Google Scholar]
- 38.Luo, X., I. Budihardjo, H. Zou, C. Slaughter, and X. Wang. 1998. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 94:481–490. [DOI] [PubMed] [Google Scholar]
- 39.Wahl, C., S. Maier, R. Marre, and A. Essig. 2003. Chlamydia pneumoniae induces the expression of inhibitor of apoptosis 2 (c-IAP2) in a human monocytic cell line by an NF-kappaB-dependent pathway. Int. J. Med. Microbiol. 293:377–381. [DOI] [PubMed] [Google Scholar]
- 40.McDonnell, J.M., D. Fushman, C.L. Milliman, S.J. Korsmeyer, and D. Cowburn. 1999. Solution structure of the proapoptotic molecule BID: a structural basis for apoptotic agonists and antagonists. Cell. 96:625–634. [DOI] [PubMed] [Google Scholar]
- 41.Ciechanover, A. 1998. The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J. 17:7151–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Orth, K., Z. Xu, M.B. Mudgett, Z.Q. Bao, L.E. Palmer, J.B. Bliska, W.F. Mangel, B. Staskawicz, and J.E. Dixon. 2000. Disruption of signaling by Yersinia effector YopJ, a ubiquitin-like protein protease. Science. 290:1594–1597. [DOI] [PubMed] [Google Scholar]
- 43.Cuconati, A., and E. White. 2002. Viral homologs of BCL-2: role of apoptosis in the regulation of virus infection. Genes Dev. 16:2465–2478. [DOI] [PubMed] [Google Scholar]
- 44.Bump, N.J., M. Hackett, M. Hugunin, S. Seshagiri, K. Brady, P. Chen, C. Ferenz, S. Franklin, T. Ghayur, P. Li, et al. 1995. Inhibition of ICE family proteases by baculovirus antiapoptotic protein p35. Science. 269:1885–1888. [DOI] [PubMed] [Google Scholar]
- 45.Perfettini, J.L., D.M. Ojcius, C.W. Andrews Jr., S.J. Korsmeyer, R.G. Rank, and T. Darville. 2003. Role of proapoptotic BAX in propagation of Chlamydia muridarum (the mouse pneumonitis strain of Chlamydia trachomatis) and the host inflammatory response. J. Biol. Chem. 278:9496–9502. [DOI] [PubMed] [Google Scholar]
- 46.Khaled, A.R., K. Kim, R. Hofmeister, K. Muegge, and S.K. Durum. 1999. Withdrawal of IL-7 induces Bax translocation from cytosol to mitochondria through a rise in intracellular pH. Proc. Natl. Acad. Sci. USA. 96:14476–14481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jungas, T., I. Motta, F. Duffieux, P. Fanen, V. Stoven, and D.M. Ojcius. 2002. Glutathione levels and BAX activation during apoptosis due to oxidative stress in cells expressing wild-type and mutant cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 277:27912–27918. [DOI] [PubMed] [Google Scholar]
- 48.Leinonen, M., and P. Saikku. 2002. Evidence for infectious agents in cardiovascular disease and atherosclerosis. Lancet Infect. Dis. 2:11–17. [DOI] [PubMed] [Google Scholar]
- 49.Kutlin, A., P.M. Roblin, and M.R. Hammerschlag. 2002. Effect of prolonged treatment with azithromycin, clarithromycin, or levofloxacin on Chlamydia pneumoniae in a continuous-infection model. Antimicrob. Agents Chemother. 46:409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]