

Abstract
Experiments were performed to characterize and identify the cellular sources of the secondary interleukin (IL)-4 response to a T cell–dependent antigen. Mice were primed by immunization with goat anti–mouse immunoglobulin (Ig)D antibody (GaMD), which stimulates naive CD4+ T cells to secrete IL-4 in 3–4 d. When challenged with goat serum 14 d after immunization, GaMD-primed mice generated an IL-4 response that exceeded the primary response by ∼100-fold, started in <2 h, and lasted for 4 d. Studies with 4get mice, in which cells with an accessible Il4 gene express a green fluorescent protein (GFP), revealed CD4+ memory T cells, natural killer T cells, basophils, mast cells, and eosinophils as possible rapid producers of IL-4. GFP+CD4+ T cells and basophils expanded more in the spleen than the other cell types during the primary response to GaMD. Quantitation of in vivo IL-4 production by the in vivo cytokine capture assay after individual cell types were selectively stimulated or deleted demonstrated that basophils and memory CD4+ T cells account for most of the secondary IL-4 response, with basophils initiating that response through IgE/FcɛRI-mediated signaling but secreting IL-4 for <4 h and memory T cells secreting IL-4 within 4 h and continuing to secrete this cytokine for 4 d.
Keywords: cytokine, allergy, NKT cell, eosinophil, mast cell
Introduction
IL-4 both protects hosts against intestinal worm infections (1) and contributes to the pathogenesis of allergy (2). Several cell types have been reported to produce IL-4, including conventional CD4+ and CD8+ T cells (3, 4), NKT cells (5), basophils (6), mast cells (7) and eosinophils (8). CD4+ T cells appear to be critical for the generation of a primary IL-4 response: the large IL-4 responses produced in mice treated with the potent TD antigen goat anti–mouse IgD antibody (GaMD) or in mice infected with the intestinal nematode parasite Nippostrongylus brasiliensis are almost totally blocked if these mice are depleted of CD4+ T cells by treating them with anti-CD4 mAb (9, 10). In contrast with the CD4+ T cell dependence of a primary IL-4 response, little is known about the relative contributions of different cells types to the production of IL-4 during a secondary immune response or chronic immune stimulation. Identification of the cellular sources of IL-4 in the secondary response is important because the chronic nature of most allergic disorders suggests that patterns of IL-4 production in patients with these disorders will resemble those generated during a secondary, rather than a primary, response. Indeed, studies of nasal and bronchial cells from patients with allergic rhinitis and atopic asthma have identified IL-4–producing basophils, mast cells, and eosinophils, as well as T cells (8, 11), and some of these studies suggest that most IL-4 is produced by the non–T cells.
The importance of non–T cells as sources of IL-4 production is also suggested by studies performed with two strains of transgenic mice: G4 mice, in which the first exon and a portion of the first intron of the Il4 gene have been replaced by the Gfp gene that encodes enhanced GFP (12) and C.129-IL4tmILK4 mice (4get mice) in which the Il4 gene was modified by the 3′ addition of an internal ribosomal entry sequence (4get mice; reference 13). Studies with G4 mice now demonstrate that IL-4 is produced by T cells and basophils after intestinal worm infection (14), whereas studies with 4get mice have additionally suggested that eosinophils may be important IL-4–producing cells (15). Complicating the interpretation of these studies has been a concern that both stability and regulation of translation may differ for GFP mRNA versus IL-4 mRNA in both mouse strains and that internal ribosomal entry sequence–regulated mRNA and protein expression in 4get mice may correlate more with Il4 gene accessibility than with actual Il4 gene transcription and translation (16, 17). As a result, the relative roles of T cells, basophils, mast cells, and eosinophils as sources of IL-4 during a chronic or secondary Th2 response remain controversial.
To better understand this issue, we have studied a system in which initial immunization of mice with GaMD induces a strong, CD4+ T cell–dependent IgG1 and IgE antibody response that is accompanied by an ∼100-fold increase in CD4+ T cell Il4 gene expression and protein secretion (18, 19). Although antibody and IL-4 production generally return to near baseline levels by 2 wk after the initial GaMD immunization, we find that challenge of previously immunized mice with goat serum induces a dramatic, rapid IL-4 response that can last for several days. We now characterize this response further by studying the effect of primary GaMD immunization on GFP expression in 4get mice; by evaluating the importance of mast cells, eosinophils, basophils, conventional CD4+ T cells, NKT cells, IgE, and FcɛRI in the secondary IL-4 response; and by comparing the IL-4 response generated by challenging goat IgG-immune mice with normal goat serum to that induced by challenging these mice with an anti-IgE mAb.
Materials and Methods
Mice.
BALB/c mice were purchased from the National Cancer Institute. Mast cell–deficient WBB6F1-Kit W/KitW-v (W/Wv) mice, (WBB6F1-Kit W/KitW-v × WBB6F1-+/+)F1 (W/+) mice (which have a normal phenotype), and 4get mice were purchased from The Jackson Laboratory. CD1/CD2-deficient and CD1/CD2+/− mice were a gift from A. Bendelac (University of Chicago, Chicago, IL), who produced them by backcrossing the original C57BL/6 × 129 CD1/CD2-deficient mice (20) to BALB/c mice for 12 generations (21). FcɛRIα-deficient mice (22) were a gift from J.-P. Kinet and IgE-deficient mice (23) were a gift from P. Leder (both of Harvard University, Boston, MA).
Reagents.
The following antisera, antibodies, and mAbs were prepared as described: goat antisera to mouse IgD (GaMD) and keyhole limpet hemocyanin (GaKLH; reference 24); Hδa/1 (mouse IgG2b anti–mouse IgDa; reference 25); FF1-4D5 (mouse IgG2a anti-IgDa; reference 26); GK1.5 (rat IgG2b anti–mouse CD4; reference 27); 2.43 (rat IgG2b anti-CD8; reference 28); J1.2, GL113, and GL117 (rat IgG2b anti-NP, rat IgG1, and IgG2a anti–Escherichia coli β-galactosidase, used as controls; a gift from J. Abrams, DNAX, Palo Alto, CA); EM-95 (rat IgG2a anti–mouse IgE; reference 29); 24G2 (rat IgG2b anti–mouse FcγRII/RIII; reference 30); IgEL2a (mouse IgE anti-TNP; reference 31); RB6-8C5 (rat IgG2b anti-Ly6G/C; reference 32); and 145-2C11 (Armenian hamster IgG1 anti-CD3; reference 33). The following mAbs to mouse cytokines were obtained from BD Biosciences (the original references for each of these mAbs are available in the BD Biosciences catalogue): biotin-JES6-5H4 (rat IgG2b anti–IL-2), JES6-1A12 (rat IgG2a anti–IL-2), biotin-MP2-8F8 (rat IgG1 anti–IL-3), MP2-43D11 (rat IgG2a anti–IL-3), biotin-BVD4-1D11 (rat IgG2b anti–mouse IL-4), BVD6-24G2.3 (rat IgG1 anti–IL-4), biotin-TRFK-4 (rat IgG2a anti–IL-5), JES1-39D10 (rat IgG2a anti–human/mouse IL-5), biotin-R46A2 (rat IgG1 anti–IFN-γ), AN-18 (rat IgG1 anti–IFN-γ), biotin-TN3 (Armenian hamster IgG1 anti-TNF), G281-2626 (rat IgG1 anti-TNF), DX5 (rat IgM anti–mouse Ly49b), GL3 (Armenian hamster IgG2 anti-TCRγ), and H57-597 (Armenian hamster IgG2 anti-TCRβ). The mAb 83101 (rat IgG2a anti-CCR3) and affinity purified goat anti–mouse IL-13 were purchased from R&D Systems. 11B11 (rat IgG1 anti–mouse IL-4; reference 34) was purchased from Verax. C531 (rat IgG anti–mouse IL-13) was a gift from S. Visvanathan (Centocor, Malvern, PA). CD1/α-galactosylceramide (α-gal-cer) tetramers were a gift from A. Bendelac and were prepared and used to identify NKT cells as described previously (35). Recombinant murine IL-3, IL-4, and IL-9 were purchased from PeproTech.
Administration of Cytokines.
IL-3 and IL-4 were administered as complexes with the neutralizing mAbs MP2-8F8 and 11B11, respectively. These complexes (IL-3C and IL-4C), which are prepared by mixing the cytokine and anticytokine mAb at a 2:1 molar ratio, slowly dissociating in vivo, and releasing the free cytokine. A single injection of IL-4C or IL-3C maintains activity of the relevant cytokine for ∼3 d. These complexes do not fix C, bind more avidly than free IgG to FcγRs, or interact simultaneously with FcγRs and cytokine receptors (36).
Measurement of Mouse Mast Cell Protease 1 (MMCP1).
Serum levels of MMCP1, an indicator of mucosal mast cell degranulation (37), were measured with a kit purchased from Moredun according to the manufacturer's directions.
Immunofluorescence Staining and Flow Cytometry.
Four-color flow cytometry was performed with a BD FACScalibur equipped with argon and red diode lasers (BD Biosciences). Fluorescein, GFP, or Alexafluor 488; phycoerythrin; PerCP; and allophycocyanin, Cy5, or Alexafluor 647 were used as the four fluorochromes. Cells stained with anti-IgE mAb were pretreated with IgE mAb in vitro in the presence of anti-FcɛRII mAb to load FcɛRI. Data were analyzed with CELLQuest software (BD Biosciences).
In Vivo Cytokine Capture Assay (IVCCA).
In vivo cytokine production was analyzed with the IVCCA, which increases the sensitivity of cytokine detection ∼100-fold (19, 38). Mice are injected with 10 μg of biotin-labeled, neutralizing anticytokine mAb, which binds secreted cytokine. Cytokine-biotin–anti-cytokine mAb complexes accumulate to much higher levels than free cytokines in serum and are measured by ELISA, using microtiter plate wells coated with mAb to a second, noncompeting epitope on the cytokine molecule to capture the complex and a horseradish peroxidase–streptavidin conjugate (Pierce Chemical Co.) and a luminogenic substrate for horseradish peroxidase (SuperSignal ELISA femto-substrate; Pierce Chemical Co.) to detect the captured complex. Luminescence is measured with a Fluoroskan Ascent FL microtiter plate luminometer/fluorometer (Labsystems).
Immunization and Challenge.
Mice were primed in most experiments by injecting them i.p. with 0.2 ml of GaMD. Goat IgG in GaMD binds to B cell membrane IgD and activates these cells. It is also internalized, processed, and presented to goat IgG-specific T cells. The huge number of goat IgG-presenting activated B cells acts as a potent stimulus for the activation of goat IgG-specific CD4+ T cells and induces a substantial effector T cell response that is characterized by predominantly Th2 cytokine secretion and later, a substantial memory T cell response. GaKLH does not directly activate B cells or indirectly induce a large primary T cell response; however, it contains the same antigenic determinants as GaMD. This allows GaKLH to be used as the challenge antigen for GaMD-primed mice.
Online Supplemental Material.
Fig. S1 illustrates the gating strategy used to define: GFP+CD4+ T cells (gates R1–R3), eosinophils (gates R4 and R5), and basophils (gates R4 and R6). Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20040598/DC1.
Results
Comparison of Primary and Secondary IL-4 Responses to GaMD.
Activation of CD4+ T cells during the primary response to GaMD induced IL-4 production (Fig. 1 A) that peaked 5–7 d after immunization (27–29). To determine whether GaMD immunization primes for a memory IL-4 response, GaMD-primed mice were challenged 14 d after priming with GaKLH and IL-4 production was followed by IVCCA. An IL-4 response developed <2 h after challenge and peaked, at a level 50–100-fold greater than the primary response, at 4–6 h. IL-4 production subsequently decreased but remained elevated for 4–5 d (Fig. 1 B and not depicted). Thus, mice immunized with a TD antigen can rapidly produce IL-4 during a secondary response.
Figure 1.
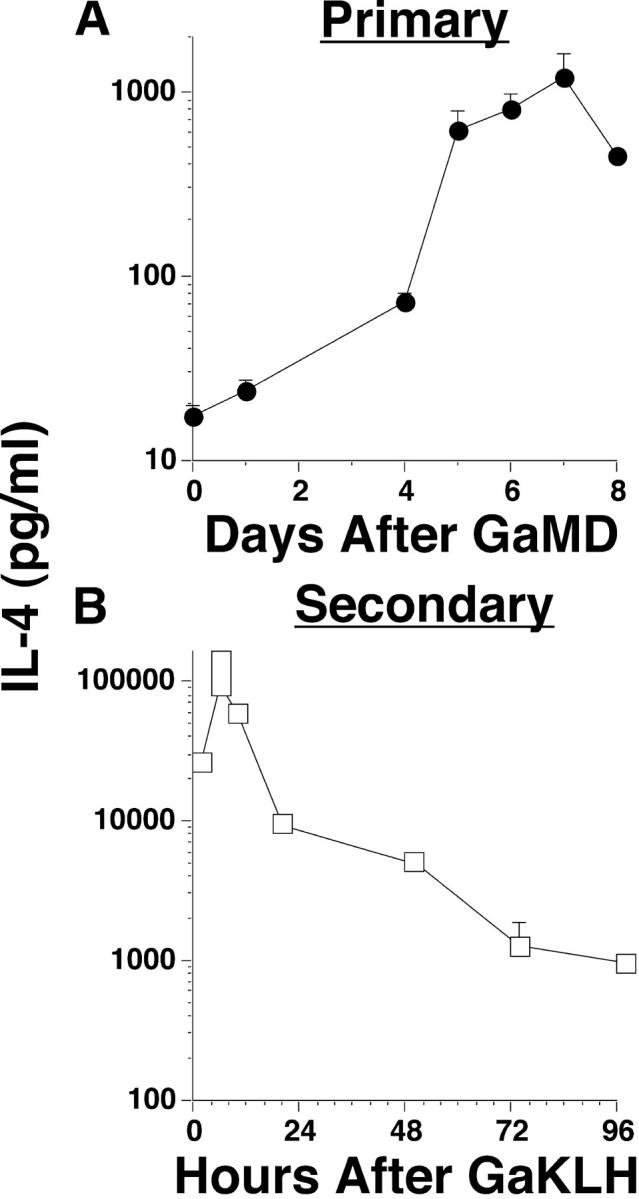
Comparison of primary and secondary responses to GaMD. (A) BALB/c mice (5/group in this and other experiments unless otherwise indicated) were left untreated or were immunized i.p. with GaMD. All mice were injected i.v. with 10 μg biotin-BVD4-1D11 anti–IL-4 mAb (BαIL-4) and were bled 1 d later at the time points shown for GaMD-immunized mice. Serum IL-4 levels were determined by IVCCA. Geometric means and SE are shown in this and in subsequent figures. (B) BALB/c mice were immunized with GaMD and challenged with 0.2 ml GaKLH 14 d later. Challenged mice were also injected with BaIL-4 0, 4, 8, 20, 48, 72, or 96 h later and were bled 2 h after that, at the time points shown. Thus, the first point shows IL-4 produced between 0 and 2 h after challenge. IL-4 levels were not determined in this experiment before challenge on day 14, but have been <200 pg/ml in several other experiments.
Identification of Cells That Have an Accessible Il4 Gene in GaMD-immunized Mice.
Studies were performed with GaMD-immunized 4get mice to identify cell types that might be able to rapidly produce IL-4 in response to antigen challenge. GFP in 4get mice is not expressed by naive, conventional CD4+ T cells, but is constitutively expressed by most basophils, eosinophils, and NKT cells (14, 15, 39), suggesting that the Il4 gene is constitutively accessible in these latter cell types. Most GFP+ cells in unstimulated 4get mice must secrete little or no IL-4 because IL-4 levels are low in these mice, as measured by the IVCCA (unpublished data); however, the open configuration of the Il4 gene in these cells implies that they can rapidly secrete IL-4 when appropriately activated.
With this in mind, we evaluated the effects of primary GaMD immunization of 4get mice on splenic GFP+ cell populations. These studies (Figs. 2 and 3 and Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20040598/DC1) demonstrated a large increase in the number of GFP+CD4+TCRβ+TCRγ− T cells, which peaked 5–8 d after GaMD immunization (Figs. 2 A and 3, A and B). Although 80–90% of GFP+ T cells in unimmunized 4get mice can be identified as NKT cells by virtue of their binding of CD1/α-gal-cer tetramers and most NKT cells are GFP+ even in unimmunized mice (Figs. 2 A and 3 A and reference 39), and although both conventional (CD1/α-gal-cer tetramer nonbinding) and NKT cells become activated during the response to GaMD as reflected by increased forward light scatter (Fig. 3 A), only the conventional CD4+GFP+ T cell population increases in number during this response (Fig. 3 A). Conventional GFP+CD4+ T cell number is still increased several-fold over baseline 14 d after GaMD immunization, but forward light scatter by these cells has returned to baseline (Fig. 3, A and C), suggesting that they have memory, rather than effector, function at this time. This supposition is consistent with evidence, shown below, that IL-4 production has returned to baseline by 14 d after GaMD immunization in most experiments.
Figure 2.
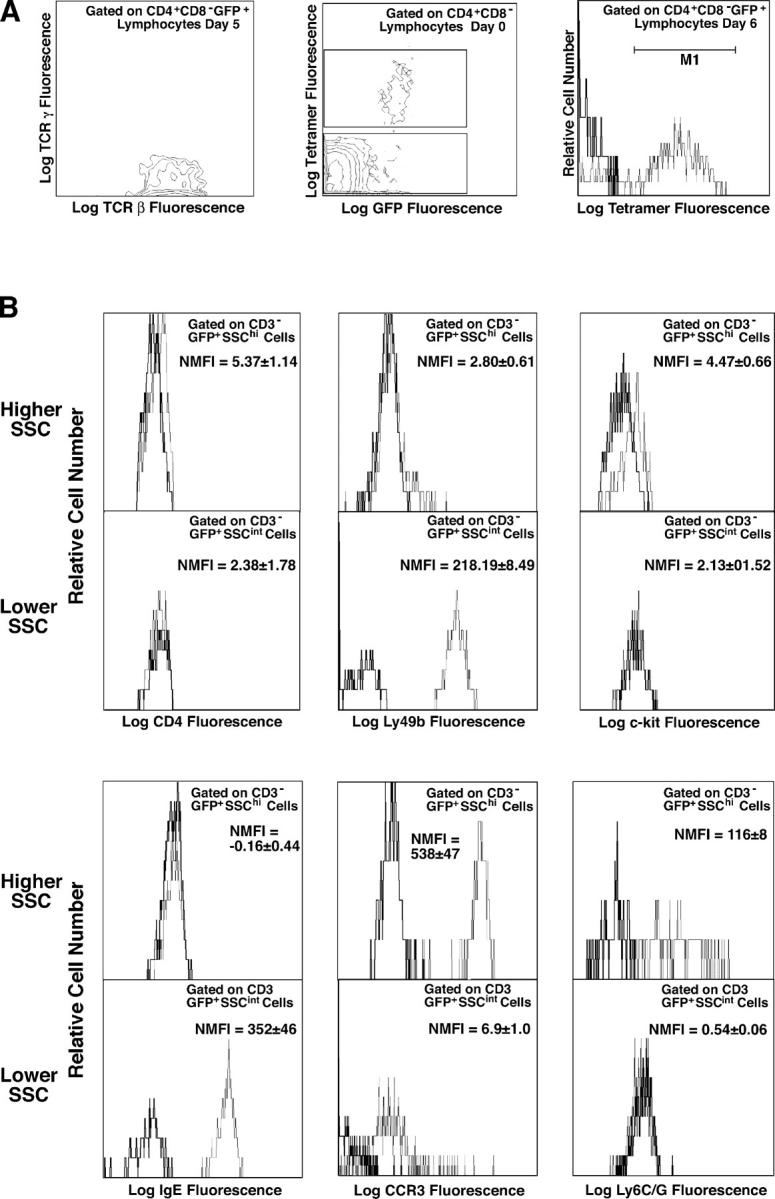
Changes in GFP-expressing spleen cells during the primary response to GaMD in 4get mice. 4get and wild-type mice (4/group) were injected with saline or GaMD and killed 1–14 d later. Spleen cells were stained with combinations of fluorochrome-labeled mAbs to CD3, CD4, CD8, IgE, Ly49b, TCRβ, TCRγ, c-kit, CCR3, and Ly6G/C and with CD1/ α-gal-cer tetramers. Stained cells were analyzed by four-color, dual laser flow cytometry. Histograms show staining with a specific mAb as a fine line; staining with a control mAb or no mAb as a bold line. Gating strategies are shown in Fig. S1. (A) CD4+CD8− spleen cells were analyzed for GFP expression, and GFP+CD4+ cells were analyzed for expression of TCRβ and TCRγ and staining with CD1/α-gal-cer tetramers. The text within the panels indicates gating and time after GaMD immunization. (B) GFP+CD3− spleen cells were analyzed for FSC and side scatter (SSC) of incident light. GFP+CD3− spleen cells with moderate or high SSC were analyzed for expression of surface marker expression. The text within the panels shows normalized median fluorescence intensity of staining (NMFI), defined as (median fluorescence intensity of specifically stained cells) − (median fluorescence intensity of cells not stained with the relevant mAb).
Figure 3.
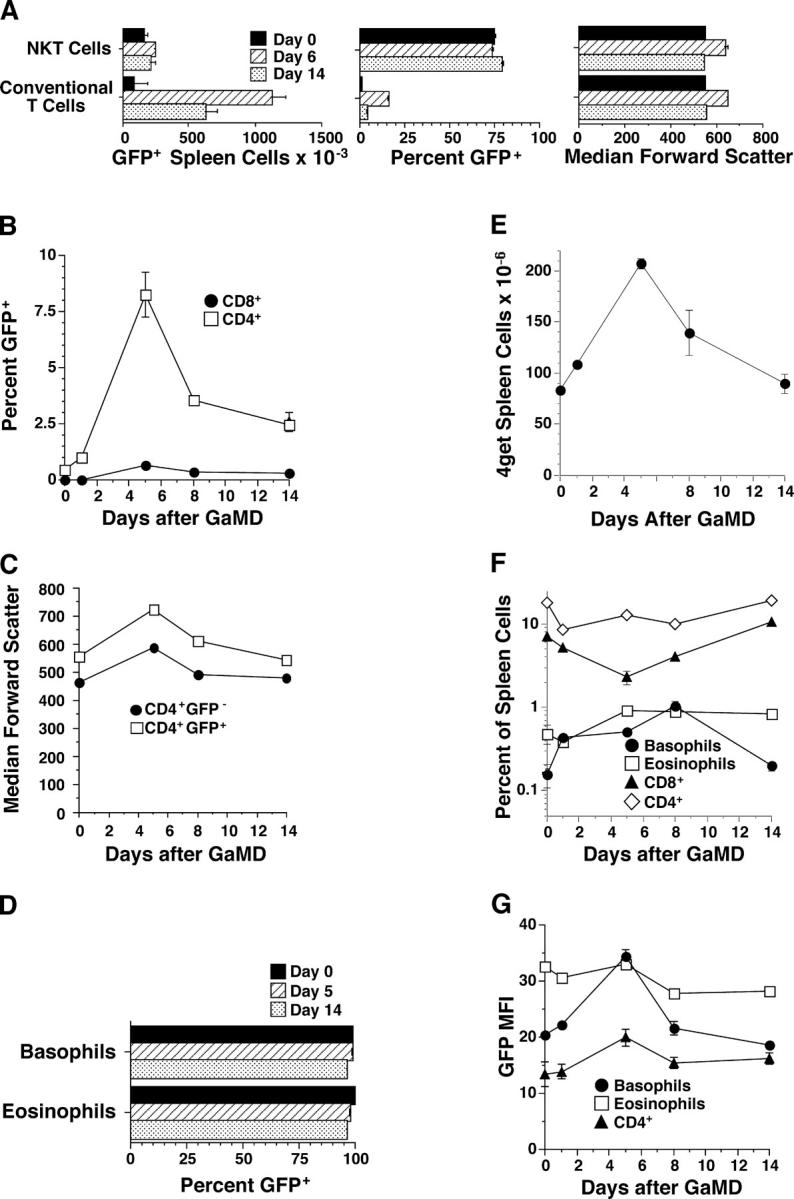
Characterization of GFP+ spleen cells during the primary response to GaMD. Data obtained from the experiments described in Fig. 2 were used to calculate the following. (A) Numbers of GFP+ splenic conventional CD4+ T cells and NKT cells (left), percentages of splenic conventional CD4+ and NKT cells that expressed GFP (middle), and median forward scatter of splenic conventional CD4+ T cells and NKT cells (right). (B) Percentages of CD4+ and CD8+ T cells that expressed GFP. (C) Median FSC of CD4+GFP+ and CD4+GFP− spleen cells. (D) Percentages of splenic basophils and eosinophils that expressed GFP. (E) Total spleen cell number. (F) Percentages of splenic basophils, eosinophils, CD8+ T cells, and CD4+ T cells. (G) Median GFP fluorescence intensity of basophils, eosinophils, and GFP+CD4+ T cells.
Two splenic cell types, in addition to CD4+ T cells, express GFP in GaMD-immunized 4get mice. One cell type can be identified as eosinophils (SSChighCCR3highLy6G/C+c-kitlowLy49b−IgE−) (13, 40); the other can be identified as basophils (SSCintermediateLy49bhighCCR3lowIgE+c-kit− CD4−Ly6G/C−) (Fig. 2 B; reference 14; unpublished data). In contrast, GaMD immunization does not elicit demonstrable numbers of GFP+ cells with a CD4−c-kit+IgE+ mast cell phenotype. Nearly all eosinophils and basophils in 4get mice are GFP+ before immunization (Fig. 3 D). Both basophils and eosinophils increase in percentage and absolute number in the spleen after GaMD immunization (Fig. 3, E and F), but basophils increase considerably more than eosinophils and basophils, but not eosinophils, exhibit an increase in the amount of GFP expressed per cell (Fig. 3, F and G).
Effects of IL-3, IL-4, and IL-9 on GFP-expressing Cells in 4get Mice.
Because GaMD immunization stimulates the production of IL-3, IL-4, and IL-9 (but not IL-5; reference 18) and each of these cytokines can activate basophils, mast cells, T cells, and/or eosinophils (41), we evaluated the effects of in vivo treatment of 4get mice with each cytokine on GFP+ spleen cells and additionally compared GFP+ spleen cell populations in 4get versus 4get mice that also expressed an IL-5 transgene (Fig. 4). IL-3, IL-4, and IL-9 each induced a two- to threefold increase in the number of splenic basophils, and IL-3 and IL-9, but not IL-4, stimulated a substantial increase in splenic eosinophil number (Fig. 4 B). Transgenic overproduction of IL-5 induced massive splenic eosinophilia, but had no significant effect on basophils (Fig. 4 B).
Figure 4.
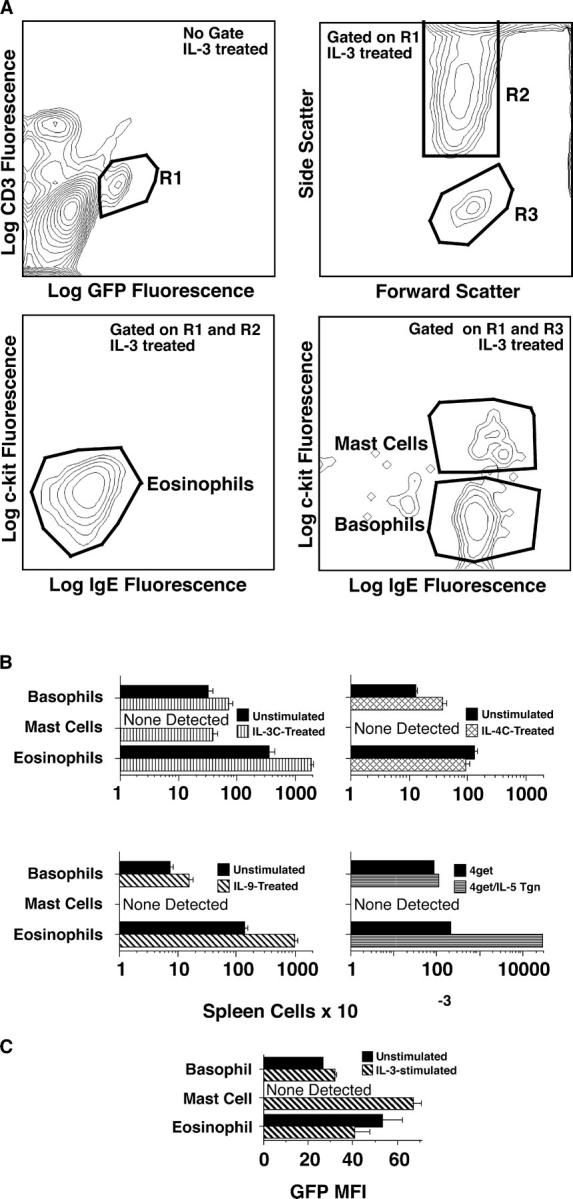
Cytokine effects on spleen cell number and GFP expression in 4get mice. In three separate experiments, 4get mice (4/group) were injected i.v. with saline, IL-3C (10 μg of IL-3 + 50 μg of anti–IL-3 mAb on days 0, 3, and 6), or IL-4C (2 μg IL-4 + 10 μg anti–IL-4 mAb on days 0, 3, and 6), or i.p. with IL-9 (10 μg/d for 7 d). Mice were killed on day 8, and spleen cells from individual mice were counted with a Coulter Counter and stained with fluorochrome-labeled mAbs to CD3, IgE, and c-kit and analyzed by flow cytometry. In a fourth experiment, spleen cells from untreated 4get+/− mice and 4get+/−IL-5 Tgn+/− mice were compared. GFP+c-kitlowIgE−CD3−SSChigh spleen cells were classified as eosinophils; GFP+c-kit−IgE+CD3−SSCintermediate spleen cells were classified as basophils, and GFP+c-kit+IgE+CD3−SSCintermediate spleen cells were classified as mast cells. (A) Gating strategies. (B) Numbers of GFP+ cells of each non–T cell type. Separate groups of control (saline-treated) mice were used for each experiment. Mice differed in age and sex between experiments, explaining modest differences among control groups, but were age and sex matched within each experiment. (C) Median fluorescence intensity of basophil, mast cell, and eosinophil GFP staining in untreated and IL-3–treated mice.
Only IL-3 elicited substantial numbers of GFP+IgE+ c-kit+ spleen cells, which are most likely mast cells. These cells expressed more GFP/cell than the IgE+c-kit− basophils (Fig. 4 C) and scattered light somewhat more than the IgE+c-kit− basophils (not depicted). No cytokine tested increased CD4+ T cell GFP expression (unpublished data). Thus, although increased cytokine production may account for some of the increase in splenic basophils and eosinophils in GaMD-immunized mice, other effects of GaMD immunization must account for the increase in GFP+CD4+ T cells and most of the increase in splenic basophils.
Basophils and Conventional CD4+ T Cells Are the Main Sources of IL-4 in the Secondary Response to Goat Serum.
The studies depicted in Figs. 2–4 implicate CD4+ T cells, eosinophils, basophils, and, possibly, nonsplenic mast cells as possible sources of the rapid IL-4 response to goat serum challenge in GaMD-primed mice. In vivo studies (Figs. 5–7) were performed to evaluate the importance of each possible cell type, using GaMD, GaKLH, or anti-IgE mAb to elicit an IL-4 response, the IVCCA to measure in vivo IL-4 production, anti-CD4 mAb to block the response of conventional CD4+ T cells (42), CD1 deficiency to block NKT development (43), anti-IgE mAb to desensitize the basophil IL-4 response to antigen challenge (44), c-kit deficiency to block mast cell development and survival (45), and anti-Ly6G/C mAb to deplete eosinophils (46). Although treatment with anti-CD4 mAb 1 d after GaMD immunization inhibited the IL-4 response to challenge with goat serum by >99%, treatment of GaMD-primed mice with anti-CD4 mAb 1 d before goat serum challenge had little effect on the initial (4 h) IL-4 response (Fig. 5 A). Thus, conventional CD4+ T cells appear to be absolutely required for priming for the secondary IL-4 response, but not for the effector phase of that response. Treatment with anti-IgE mAb 1 d before challenge had a considerably greater inhibitory effect on the IL-4 response made during the first 4 h after antigen challenge than did pretreatment with anti-CD4 mAb (Fig. 5 B), suggesting that basophils and/or mast cells are a more important source of the initial secondary IL-4 response than conventional CD4+ T cells. Consistent with this, the initial IL-4 response to goat serum challenge in GaMD-primed mice was reduced by ∼90% in mice that lacked FcɛRI (Fig. 5 C). Pretreatment with both anti-IgE and anti-CD4 mAbs completely abolished the initial secondary IL-4 response to antigen challenge (Fig. 5 B), demonstrating that CD4+ T cells and IgE-expressing cells (basophils/mast cells) together account for the entire response.
Figure 5.

IL-4 secretion during the secondary response to goat serum in GaMD-primed mice. (A) BALB/c mice were immunized with GaMD and injected i.v. 1 or 13 d after GaMD immunization with 1 mg anti-CD4 mAb or control mAb. Mice were challenged with GaKLH on day 14 and injected at the time of challenge with BαIL-4 and bled 4 h later. (B) BALB/c mice were primed with GaMD. Some were injected 13 d later with anti-CD4 mAb and/or i.p. with 200 μg anti-IgE mAb or control mAbs. Mice were left unchallenged or were injected i.p. on day 14 with GaMD or GaKLH and were injected at the same time with BαIL-4 and bled 4 h later. (C) Wild-type or FcɛRI-deficient mice on a BALB/c background were primed with GaMD. Some mice were injected 13 d later with anti-CD4 mAb or control mAb. Mice were challenged on day 14 with saline or GaKLH and injected with BαIL-4 at the time of challenge and bled 2 h later. (D) BALB/c mice were primed with GaMD. Some were injected 13 d later with 200 μg anti-IgE mAb or control mAb. One group was challenged with saline (“time 0” group); all others were challenged with GaKLH on day 14. Saline-challenged mice were injected with BαIL-4 at the time of challenge; GaKLH-challenged mice were injected with BαIL-4 at the time of challenge or 2, 4, 48, or 96 h later. All mice were bled 2 h after BαIL-4 injection. (E) An experiment identical to that described in D was performed, with the exception that some mice were injected on day 13 with 1 mg anti-CD4 mAb or isotype-matched mAb instead of anti-IgE mAb and that an additional group of mice was assayed for IL-4 production 24 h after challenge. (F) Unprimed BALB/c mice were injected on day 0 with anti-CD4 mAb or control mAb. Mice were injected with BαIL-4 ± 10 μg anti-CD3 mAb and bled 2 h later. (G) Wild-type or CD1-deficient mice were immunized with GaMD. All mice were injected with 100 μg anti-IgE mAb 13 d later, challenged with GaKLH, injected with BαIL-4 on day 14, and bled 6 h later. (H) BALB/c mice (5/group) were immunized i.p. with 1 mg OVA/d on days 0–5, treated with saline, 1 mg GK1.5, 200 μg EM-95, or 1 mg GK1.5 + 200 μg EM-95 i.p. on day 13, and challenged i.p. with saline or 2 mg OVA on day 14. All mice were injected with BαIL-4 mAb at the time of challenge and bled 4 h later. Similar results were observed when mice were immunized twice i.p. with OVA/alum and challenged i.p. with soluble OVA (not depicted).
Figure 7.

Eosinophil depletion does not inhibit the secondary IL-4 response. BALB/c mice were primed with GaMD and injected 13 d later with 1 mg anti-Ly6G/C or control mAb. Some mice were killed on day 14 and their spleen cells were counted and stained for CD3 and CCR3. (A) Gating strategy. (B) Number of SSChighCCR3+CD3− cells/spleen. (C) Additional mice were left unchallenged or were challenged with GaKLH on day 14, injected with 10 μg BαIL-4 at the time of antigen challenge or 4 or 24 h later and bled 4 h after BαIL-4 injection. (D) Additional mice were treated with saline, anti-CD4 and anti-IgE mAbs, or anti-CD4, anti-IgE, and anti-Ly6C/G mAbs on day 13. Mice were challenged with GaKLH on day 14 or were left unchallenged. IL-4 produced 0–4 h, 4–8 h, and 24–28 h after challenge was determined. ND, not determined.
Although both CD4+ T cells and FcɛRI+ cells contribute to the secondary IL-4 response, kinetics of IL-4 production by these cell types differ. Anti-IgE mAb, but not anti-CD4 mAb, inhibited the initial (2 h) IL-4 response, as was shown in our earlier experiment, whereas anti-CD4 mAb, but not anti-IgE mAb, blocked responses made ≥6 h after antigen challenge (Fig. 5, D and E). Thus, although FcɛRI+ cells are responsible for the initial secondary IL-4 response, IL-4 production by these cells is short lived and rapidly replaced by IL-4 production by memory CD4+ T cells.
Because NKT cells, as well as conventional T cells, can express CD4 and can rapidly produce IL-4 in response to appropriate stimulation (5), we evaluated whether NKT cells contribute to the secondary IL-4 response in GaMD-primed mice. Because the in vivo IL-4 response to anti-CD3 mAb is made by NKT cells and anti-CD4 mAb blocks the conventional T cell response to antigen challenge, we studied whether anti-CD4 mAb would inhibit the IL-4 response to anti-CD3 mAb. No inhibition was detected (Fig. 5 F). Because CD1-deficient mice have few NKT cells (43), we compared IL-4 responses of GaMD-primed wild-type versus CD1-deficient mice to goat serum challenge when the basophil/mast cell response was blocked by pretreatment with anti-IgE mAb. Similar secondary IgE-independent IL-4 responses were made by both mouse strains (Fig. 5 G). Thus, conventional CD4+ T cells appear to be more important that NKT cells as a source of the secondary IL-4 response to a TD antigen.
Because GaMD is an unusually potent T cell–dependent antigen, it was possible that our observations with GaMD-immunized mice might not extend to mice immunized with more conventional antigens. To evaluate this possibility, we immunized mice with six consecutive daily i.p. injections of OVA and studied their IL-4 responses to OVA challenge 14 d after the initial immunization (Fig. 5 H). Although the secondary IL-4 response was much smaller in OVA-primed and challenged mice than in mice primed and challenged with GaMD, the secondary responses in the GaMD and OVA systems were qualitatively similar; basophils and CD4+ T cells contributed substantially to IL-4 production and accounted for nearly all of the IL-4 produced in both systems.
Characterization of the IL-4 Response to FcɛRI Cross-linking in GaMD-primed Mice.
To further define the roles of FcɛRI+ cells in the secondary IL-4 response, studies were performed in which anti-IgE mAb was used to elicit IL-4 secretion. Challenge of GaMD-primed mice with anti-IgE mAb induced production of IL-2, IL-3, IL-4, IL-5, IL-13, and TNF, but not IFN-γ (Fig. 6 A). IL-4 and IL-13 responses appeared particularly large, with levels increasing ∼1,000-fold over background. IL-4 responses made by anti-IgE mAb-challenged unprimed or GaKLH-primed mice were less, by a factor of 10–50, than responses made after priming with anti-IgD antibody (Fig. 6 B). No IL-4 responses were made by anti-IgE, mAb-challenged, FcɛRIα-deficient mice (Fig. 6 C), or IgE-deficient mice, unless the latter mice were first injected with IgE (Fig. 6 D). Although basophils produce IL-4 in vitro in response to immobilized IgG (44), in vivo treatment with 24G2, a mAb to FcγRII and FcγRIII, neither induced IL-4 production nor inhibited anti-IgE mAb-induced IL-4 production (Fig. 6 E).
Figure 6.

In vivo cytokine responses to anti-IgE mAb. (A) BALB/c mice were primed with GaMD and challenged 14 d later with anti-IgE mAb. IL-2, IL-3, IL-4, IL-5, IL-13, TNF, and IFN-γ production during the subsequent 24 h were measured by IVCCA. Quantities of different cytokines detected are not comparable; the assays for IL-3 and IL-5 are less efficient than the other assays. (B) BALB/c mice were left unprimed or were primed with GaMD, GaKLH, allo-anti-IgDa mAbs (Hδa/1 and FF1-4D5), or GaKLH + Hδa/1 and FF1-4D5. All mice were challenged 14 d later with anti-IgE mAb and injected at the same time with BαIL-4 and bled 4 h after challenge. (C) Wild-type and FcɛRI-deficient mice on a BALB/c background were primed with GaMD. Mice were challenged 14 d later with 200 μg anti-IgE mAb and injected with BαIL-4 at the same time. Mice were bled 4 h later. (D) Unprimed wild-type and IgE-deficient mice were challenged with anti-IgE mAb or were first injected with IgE and challenged with anti-IgE mAb 1 d later. All mice were injected with BαIL-4 at the time of challenge and bled 24 h later. (E) BALB/c mice were primed with αδ mAbs and challenged 14 d later with anti-IgE mAb, anti-FcγRII/RIII mAb, or anti-IgE + anti-FcγRII/RIII mAb. All mice were injected with BαIL-4 at the time of challenge and bled 3 h later. (F) c-kit–deficient W/Wv mice or age-matched W/+ mice were primed with GaMD and were left unchallenged or were challenged with anti-IgE mAb 14 d later. All mice were injected with BαIL-4 at the time of challenge and were bled 4 h later. (G) BALB/c mice were primed with GaMD and challenged 14 d later with saline or 1–100 μg anti-IgE mAb. All mice were injected with BαIL-4 at the time of challenge and bled 4 h later. Serum MMCP1 levels were determined by ELISA. (H) BALB/c mice were primed with GaMD. (top) Mice were challenged 14 d later with anti-IgE mAb; injected with BαIL-4 2 h, 1 h and 45 min, 1 h and 30 min, or 1 h before challenge or at the time of challenge; and bled 2 h after BαIL-4 injection. (bottom) Mice were challenged with saline or anti-IgE mAb 14 d after priming. Challenged mice were injected with BαIL-4 at the time of challenge or 4 or 8 h later. Mice were bled 4 h after BαIL-4 injection. (I) BALB/c mice were primed with GaMD and challenged 14 d later. Some were challenged with 2 μg anti-IgE mAb and assayed for IL-4 production during the subsequent 4 h. Some mice were initially challenged with 2 μg or 100 μg anti-IgE mAb 12 or 24 h before a second challenge with an additional 100 μg anti-IgE mAb. All mice were injected with 10 μg BαIL-4 at the time of the final challenge and bled 4 h later. (J) BALB/c mice were primed with GaMD. Some were injected with anti-CD4 mAb or a control mAb 12 d later. Mice were left unchallenged or were challenged with anti-IgE mAb on day 14 and were simultaneously injected with BαIL-4 and bled 4 h later. Treatment with anti-CD4 mAb at the time of GaMD priming almost totally blocked the anti-IgE mAb-induced IL-4 response (not depicted).
Although the Il4 gene is accessible in mast cells (Fig. 4 C) and intestinal mastocytosis develops in GaMD-treated mice (unpublished data), GaMD-primed mast cell–deficient W/Wv mice and mast cell–sufficient mice made similar IL-4 responses to anti-IgE mAb challenge (Fig. 6 F). Dose–response studies with anti-IgE mAb also dissociated mast cells from the IL-4 response to anti-IgE mAb (Fig. 6 G). The IL-4 response to anti-IgE mAb was barely evident in mice treated with 2 μg of anti-IgE mAb, but nearly full-blown in mice injected with 4 μg of this mAb. In contrast, mast cell degranulation, as demonstrated by an increased level of MMCP1, did not develop until mice received 8 μg of anti-IgE mAb and this response increased further as the dose of anti-IgE mAb was raised. Together, these observations suggest that basophils contribute more than mast cells to the anti-IgE mAb-induced mast cell response and indicate that less FcɛRI cross-linking is required to induce basophil IL-4 secretion than to induce mast cell degranulation.
Kinetic studies indicated that the IL-4 response to anti-IgE mAb begins rapidly and is short-lived (Fig. 6 H). Increased IL-4 production was observed 1 h after anti-IgE mAb injection and reached a high level by 2 h, but was complete by 4 h. Treatment of mice with as little as 2 μg anti-IgE mAb blocked the IL-4 response to 100 μg of anti-IgE mAb administered 12–24 h later (Fig. 6 I). In contrast, treatment with anti-CD4 mAb 2 d before challenge with anti-IgE mAb left most of the IL-4 response intact (Fig. 6 J).
Together with the data shown in Fig. 5, these observations demonstrate that cross-linking of basophil FcɛRI is responsible for the initial, large, IL-4 response produced by GaMD-primed mice upon challenge with the relevant antigen, but that this response terminates quickly and is replaced by a more persistent response made by memory CD4+ T cells.
Eosinophils Are Not Required for the Secondary IL-4 Response in GaMD-primed Mice.
Eosinophils are the largest GFP+ cell population in the spleens of 4get mice (Figs. 2 and 3) and increase in number in GaMD-immunized mice (Fig. 3 B). Although mouse eosinophils lack FcɛRI (47), their expression of FcγRIII (48) provides a potential mechanism for activation by IgG-containing immune complexes. Although the nearly complete inhibition of the secondary IL-4 response to antigen challenge by anti-CD4 and anti-IgE mAbs (Fig. 5 B) suggested that eosinophils are not involved in this response, our observation that eosinophils may express a small amount of CD4 (Fig. 2 C) raised the possibility that anti-CD4 mAb might inhibit the secondary IL-4 response in part through an affect on eosinophils. To directly determine whether eosinophils make a large contribution to the secondary IL-4 response, GaMD-primed mice were treated with a cytotoxic anti-Ly6G/C mAb that eliminates most neutrophils and eosinophils (Fig. 7, A and B). This treatment had no effect on the secondary IL-4 response to antigen challenge, whether measured at 0–4, 4–8, or 24–28 h (Fig. 7 C). Anti-Ly6G/C mAb treatment also had no significant effect on the secondary IL-4 response when the CD4+ T cell and basophil components of this response were blocked (Fig. 7 D). Thus, eosinophils do not appear to contribute substantially to this response.
Discussion
Once our initial studies established that the secondary IL-4 response to a TD antigen is much larger and faster than the primary IL-4 response to the same antigen, experiments were performed to identify the cell types that participate in the secondary response and to characterize their secretion of IL-4. Studies with 4get mice, in which cells with an accessible Il4 gene express GFP, identified memory CD4+ T cells, NKT cells, basophils, mast cells, and eosinophils in spleen as cells that might rapidly produce IL-4 if appropriately stimulated. The IVCCA was used to identify which of these cell types contribute substantially to IL-4 production during the secondary response. GaMD was used to prime for the secondary IL-4 response in these experiments because it rapidly induces a large Th2 response that stimulates goat IgG-specific IgE production, mast cells, basophils, and goat IgG-specific memory B and T cells (18, 49, 50).
Studies that used goat serum to elicit the secondary IL-4 response in GaMD-primed mice and the IVCCA to measure this response revealed that CD4+ T cells are required to prime for the response, whereas both conventional CD4+ T cells and FcɛRI+ basophils are important sources of IL-4. The importance of basophils in an in vivo IL-4 response and the requirement for CD4+ T cells for generation of the basophil response have also been established in studies in which Th2 cytokine production was induced by infecting mice with the intestinal nematode parasite N. brasiliensis (reference 14; unpublished data).
Although both CD4+ T cells and basophils were important sources of the secondary IL-4 response in our studies, IL-4 secretion by these cell types differed kinetically. FcɛRI+ cells secreted maximal amounts of IL-4 within 2–4 h of antigen challenge, after which time they secreted little IL-4. In contrast, induction of maximal IL-4 secretion by conventional CD4+ T cells was slightly slower than induction of maximal IL-4 secretion by FcɛRI+ cells, albeit much faster than induction of IL-4 secretion by naive CD4+ T cells during a primary immune response. Although NKT cells can rapidly produce large quantities of IL-4, they did not appear to participate to a great extent in the secondary IL-4 response to goat serum, most likely because they respond to glycolipid antigens presented by CD1 (35), whereas processing of goat IgG produces peptides that are presented by MHC class II antigens.
Studies in which anti-IgE mAb was used instead of goat serum to induce IL-4 secretion in GaMD-primed mice confirmed the rapid induction and short duration of IL-4 secretion by FcɛRI+ cells and revealed that most of these cells are basophils rather than mast cells (the IL-4 response was not decreased in W/Wv mice). However, we cannot rule out the possibility that mast cells produce a small percentage of the secondary IL-4 response.
These experiments also revealed quantitative differences in the extent of FcɛRI cross-linking required to induce basophil IL-4 secretion, as opposed to induction of mast cell degranulation. Stimulation of basophil IL-4 secretion was considerably more sensitive than induction of mast cell degranulation and may have been an all-or-nothing phenomenon because cells triggered with a small quantity of anti-IgE mAb failed to secrete more IL-4 if restimulated within 24 h with a much larger amount of anti-IgE mAb. In contrast, the amount of mast cell degranulation, as measured by serum levels of MMCP1, increased more gradually as larger quantities of anti-IgE mAb were injected.
Together, our results suggest that three cell types, naive CD4+ T cells, basophils, and memory CD4+ T cells contribute substantially to IL-4 secretion during in vivo responses to TD antigens but contribute to this response in different ways. Naive T cells require considerable time to initiate IL-4 production during a primary immune response to a TD antigen, most likely because their Il4 genes are initially inaccessible. The relatively small quantities of IL-4 that these cells secrete for several days once they become activated should allow autocrine and paracrine delivery of the IL-4 that drives Th2 cell differentiation and B cell isotype switching to IgE. In contrast, the rapid, easily triggered, short-lived production of much larger quantities of IL-4 (and additional cytokines) by basophils appears well adapted to the activation of nonimmune cells, including vascular endothelium, smooth muscle, and mucosal epithelial cells, which modify their function to promote the expulsion of enteric nematode parasites when stimulated with large amounts of IL-4 (1, 51).
Because in vivo secretion of massive amounts of IL-4 by basophils appears to be predominantly or entirely dependent on IgE-mediated FcɛRI cross-linking and antigen-specific IgE would not be available for several days after immunization or infection, massive release of IL-4 by basophils might serve as a specialized mechanism that promotes resistance to reinfection with nematode parasites. In this regard, the greater sensitivity of basophils to FcɛRI cross-linking, as compared with mast cells, might cause basophil IL-4 secretion to precede mast cell degranulation during infection with enteric worms. This difference in sensitivity may promote host immunity because preexposure to IL-4 greatly sensitizes nonbone marrow–derived cells, such as intestinal cells, to mediators released by mast cells that promote rapid expulsion of parasites such as Trichinella spiralis (52, 53). Indeed, T. spiralis expulsion requires mast cells, IL-4, or IL-13, and nonbone marrow–derived cells that are IL-4/IL-13 responsive (53). However, it should be noted that others have shown that basophils can be stimulated by CD4+ T cells to secrete relatively small amounts of IL-4 even in the absence of Ig and B cells (14). Thus, basophils may be able to contribute to IL-4–mediated immunity and inflammation through two distinct mechanisms: an IgE-independent mechanism that induces persistent production of small amounts of IL-4 and an IgE-dependent mechanism that rapidly induces the secretion of massive amounts of this cytokine.
Conventional CD4+ T cells that have differentiated into IL-4–producing memory cells share characteristics with both naive CD4+ T cells and basophils. Like basophils, they rapidly produce large amounts of IL-4 when appropriately stimulated; indeed, the short delay in IL-4 production by these cells probably reflects the time required for antigen to be bound and processed by APCs. Like naive conventional CD4+ T cells, memory T cells, once activated, produce IL-4 for a long time. Thus, IL-4–secreting memory CD4+ T cells may take up where FcɛRI-activated basophils leave off in a secondary immune response to sustain IL-4 effects on nonbone marrow–derived cells and provide a relatively stable source of IL-4 that should promote additional isotype switching and Th2 differentiation.
Our observations by no means indicate that the other GFP+ cells in 4get mice, NKT cells, mast cells and eosinophils, are not sources of IL-4 in vivo. NKT cells produce large amounts of IL-4 in mice injected with anti-CD3 mAb or α-gal-cer and, like basophils, secrete IL-4 for only a few hours (54). The adaptive significance of this response, which is accompanied by massive IFN-γ production, is uncertain.
Although eosinophils are the largest population of constitutively GFP+ spleen cells in 4get mice, depletion of this population had no detectable effect on IL-4 responses in our model. Furthermore, in vivo IL-4 production, as detected by IVCCA, is only modestly increased in IL-5 transgenic mice, which have greatly increased numbers of eosinophils (55), and basal IL-4 levels are normal in IL-5–deficient mice, which have relatively few eosinophils (reference 56; unpublished data). These observations indicate that eosinophils, like other cell types that have an accessible Il4 gene, must be appropriately activated to secrete IL-4. To date, only contact with activated CD4+ T cells has been shown to provide a physiological stimulus that induces eosinophils to secrete IL-4 (15). Thus, eosinophils may contribute little to IL-4 production by themselves, but may amplify IL-4 responses made by CD4+ T cells.
Even less can be said about the possible contributions of mast cells to secondary in vivo IL-4 responses. Although treatment with exogenous IL-3 stimulates the appearance of GFP+ spleen cells that are probably mast cells, it is unknown whether the Il4 gene is in an accessible state in the vascular mast cells that are present in unstimulated mice or in mucosal mast cells that increase in response to GaMD immunization and worm infection. It is also not known whether secretion of IL-4 by mouse mast cells, if it occurs, is regulated similarly to mast cell degranulation. These issues are important to address because mast cells are located at sites where IL-4 secretion could have important effects on vascular permeability and parasite expulsion.
Acknowledgments
We are very grateful to A. Bendelac for his help with NKT cell studies and to B. Min, W. Paul, and S. Galli for sharing their unpublished data and for their helpful comments.
This work was supported, in part, by the Veterans administration and by National Institutes of Health grant nos. RO1 AI45766, RO1 AI55848, RO1 AI44971, and PPG1 HL076383.
The authors have no conflicting financial interests.
Abbreviations used in this paper: GaMD, goat anti–mouse IgD antibody; IVCCA, in vivo cytokine capture assay; MMCP1, mouse mast cell protease 1.
References
- 1.Finkelman, F.D., T. Shea-Donohue, J. Goldhill, C.A. Sullivan, S.C. Morris, K.B. Madden, W.C. Gause, and J.F. Urban Jr. 1997. Cytokine regulation of host defense against parasitic gastrointestinal nematodes: lessons from studies with rodent models. Annu. Rev. Immunol. 15:505–533. [DOI] [PubMed] [Google Scholar]
- 2.Wills-Karp, M., S.H. Gavett, B. Schofield, and F. Finkelman. 1996. Role of interleukin-4 in the development of allergic airway inflammation and airway hyperresponsiveness. Adv. Exp. Med. Biol. 409:343–347. [DOI] [PubMed] [Google Scholar]
- 3.Paliard, X., R. de Waal Malefijt, H. Yssel, D. Blanchard, I. Chretien, J. Abrams, J. de Vries, and H. Spits. 1988. Simultaneous production of IL-2, IL-4, and IFN-γ by activated human CD4+ and CD8+ T cell clones. J. Immunol. 141:849–855. [PubMed] [Google Scholar]
- 4.Seder, R.A., J.L. Boulay, F. Finkelman, S. Barbier, S.Z. Ben-Sasson, G. Le Gros, and W.E. Paul. 1992. CD8+ T cells can be primed in vitro to produce IL-4. J. Immunol. 148:1652–1656. [PubMed] [Google Scholar]
- 5.Yoshimoto, T., A. Bendelac, C. Watson, J. Hu-Li, and W.E. Paul. 1995. Role of NK1.1+ T cells in a TH2 response and in immunoglobulin E production. Science. 270:1845–1847. [DOI] [PubMed] [Google Scholar]
- 6.Seder, R.A., W.E. Paul, A.M. Dvorak, S.J. Sharkis, A. Kagey-Sobotka, Y. Niv, F.D. Finkelman, S.A. Barbieri, S.J. Galli, and M. Plaut. 1991. Mouse splenic and bone marrow cell populations that express high-affinity Fcɛ receptors and produce interleukin 4 are highly enriched in basophils. Proc. Natl. Acad. Sci. USA. 88:2835–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bradding, P., I.H. Feather, P.H. Howarth, R. Mueller, J.A. Roberts, K. Britten, J.P. Bews, T.C. Hunt, Y. Okayama, C.H. Heusser, et al. 1992. Interleukin 4 is localized to and released by human mast cells. J. Exp. Med. 176:1381–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moqbel, R., S. Ying, J. Barkans, T.M. Newman, P. Kimmitt, M. Wakelin, L. Taborda-Barata, Q. Meng, C.J. Corrigan, S.R. Durham, et al. 1995. Identification of messenger RNA for IL-4 in human eosinophils with granule localization and release of the translated product. J. Immunol. 155:4939–4947. [PubMed] [Google Scholar]
- 9.Finkelman, F.D., J. Holmes, J.F. Urban Jr., W.E. Paul, and I.M. Katona. 1989. T help requirements for the generation of an in vivo IgE response: a late acting form of T cell help other than IL-4 is required for IgE but not for IgG1 production. J. Immunol. 142:403–408. [PubMed] [Google Scholar]
- 10.Urban, J.F., Jr., I.M. Katona, and F.D. Finkelman. 1991. Heligmosomoides polygyrus: CD4+ but not CD8+ T cells regulate the IgE response and protective immunity in mice. Exp. Parasitol. 73:500–511. [DOI] [PubMed] [Google Scholar]
- 11.Kay, A.B., S. Ying, and S.R. Durham. 1995. Phenotype of cells positive for interleukin-4 and interleukin-5 mRNA in allergic tissue reactions. Int. Arch. Allergy Immunol. 107:208–210. [DOI] [PubMed] [Google Scholar]
- 12.Hu-Li, J., C. Pannetier, L. Guo, M. Lohning, H. Gu, C. Watson, M. Assenmacher, A. Radbruch, and W.E. Paul. 2001. Regulation of expression of IL-4 alleles: analysis using a chimeric GFP/IL-4 gene. Immunity. 14:1–11. [DOI] [PubMed] [Google Scholar]
- 13.Mohrs, M., K. Shinkai, K. Mohrs, and R.M. Locksley. 2001. Analysis of type 2 immunity in vivo with a bicistronic IL-4 reporter. Immunity. 15:303–311. [DOI] [PubMed] [Google Scholar]
- 14.Min, B., M. Prout, J. Hu-Li, J. Zhu, D. Jankovic, E.S. Morgan, J.F. Urban Jr., A.M. Dvorak, F.D. Finkelman, G. LeGros, and W.E. Paul. Basophils produce IL-4 and accumulate in tissues after infection with a Th2-inducing parasite. J. Exp. Med. 200:507–517. [DOI] [PMC free article] [PubMed]
- 15.Shinkai, K., M. Mohrs, and R.M. Locksley. 2002. Helper T cells regulate type-2 innate immunity in vivo. Nature. 420:825–829. [DOI] [PubMed] [Google Scholar]
- 16.Han, B., and J.T. Zhang. 2002. Regulation of gene expression by internal ribosome entry sites or cryptic promoters: the eIF4G story. Mol. Cell. Biol. 22:7372–7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pozner, A., D. Goldenberg, V. Negreanu, S.Y. Le, O. Elroy-Stein, D. Levanon, and Y. Groner. 2000. Transcription-coupled translation control of AML1/RUNX1 is mediated by cap- and internal ribosome entry site-dependent mechanisms. Mol. Cell. Biol. 20:2297–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Svetic, A., F.D. Finkelman, Y.C. Jian, C.W. Dieffenbach, D.E. Scott, K.F. McCarthy, A.D. Steinberg, and W.C. Gause. 1991. Cytokine gene expression after in vivo primary immunization with goat antibody to mouse IgD antibody. J. Immunol. 147:2391–2397. [PubMed] [Google Scholar]
- 19.Finkelman, F.D., and S.C. Morris. 1999. Development of an assay to measure in vivo cytokine production in the mouse. Int. Immunol. 11:1811–1818. [DOI] [PubMed] [Google Scholar]
- 20.Chen, Y.H., N.M. Chiu, M. Mandal, N. Wang, and C.R. Wang. 1997. Impaired NK1+ T cell development and early IL-4 production in CD1-deficient mice. Immunity. 6:459–467. [DOI] [PubMed] [Google Scholar]
- 21.Park, S.H., D. Guy-Grand, F.A. Lemonnier, C.R. Wang, A. Bendelac, and B. Jabri. 1999. Selection and expansion of CD8α/α1 T cell receptor α/β1 intestinal intraepithelial lymphocytes in the absence of both classical major histocompatibility complex class I and nonclassical CD1 molecules. J. Exp. Med. 190:885–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dombrowicz, D., V. Flamand, K.K. Brigman, B.H. Koller, and J.P. Kinet. 1993. Abolition of anaphylaxis by targeted disruption of the high affinity immunoglobulin E receptor α chain gene. Cell. 75:969–976. [DOI] [PubMed] [Google Scholar]
- 23.Oettgen, H.C., T.R. Martin, A. Wynshaw-Boris, C. Deng, J.M. Drazen, and P. Leder. 1994. Active anaphylaxis in IgE-deficient mice. Nature. 370:367–370. [DOI] [PubMed] [Google Scholar]
- 24.Finkelman, F.D., S.W. Kessler, J.F. Mushinski, and M. Potter. 1981. IgD-secreting murine plasmacytomas: identification and partial characterization of two IgD myeloma proteins. J. Immunol. 126:680–687. [PubMed] [Google Scholar]
- 25.Zitron, I.M., and B.L. Clevinger. 1980. Regulation of murine B cells through surface immunoglobulin. I. Monoclonal anti-δ antibody that induces allotype-specific proliferation. J. Exp. Med. 152:1135–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goroff, D.K., A. Stall, J.J. Mond, and F.D. Finkelman. 1986. In vitro and in vivo B lymphocyte-activating properties of monoclonal anti-δ antibodies. I. Determinants of B lymphocyte-activating properties. J. Immunol. 136:2382–2392. [PubMed] [Google Scholar]
- 27.Wilde, D.B., P. Marrack, J. Kappler, D.P. Dialynas, and F.W. Fitch. 1983. Evidence implicating L3T4 in class II MHC antigen reactivity: monoclonal antibody GK1.5 (anti-L3T4a) blocks class II MHC antigen-specific proliferation, release of lymphokines, and binding by cloned murine helper T lymphocyte lines. J. Immunol. 131:2178–2183. [PubMed] [Google Scholar]
- 28.Sarmiento, M.A., A.L. Glasebrook, and F.W. Fitch. 1980. IgG or IgM monoclonal antibodies reactive with different determinants on the molecular complex bearing Ly2 antigen block T cell-mediated cytolysis in the absence of complement. J. Immunol. 125:2665–2672. [PubMed] [Google Scholar]
- 29.Baniyash, M., and Z. Eshhar. 1984. Inhibition of IgE binding to mast cells and basophils by monoclonal antibodies to murine IgE. Eur. J. Immunol. 14:799–807. [DOI] [PubMed] [Google Scholar]
- 30.Unkeless, J.C. 1979. Characterization of a monoclonal antibody directed against mouse macrophage and lymphocyte Fc receptors. J. Exp. Med. 150:580–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kofler, H., I. Schnegg, S. Geley, A. Helmberg, J.M. Varga, and R. Kofler. 1992. Mechanism of allergic cross-reactions–III. cDNA cloning and variable-region sequence analysis of two IgE antibodies specific for trinitrophenyl. Mol. Immunol. 29:161–166. [DOI] [PubMed] [Google Scholar]
- 32.Fleming, T.J., M.L. Fleming, and T.R. Malek. 1993. Selective expression of Ly-6G on myeloid lineage cells in mouse bone marrow. RB6-8C5 mAb to granulocyte-differentiation antigen (Gr-1) detects members of the Ly-6 family. J. Immunol. 151:2399–2408. [PubMed] [Google Scholar]
- 33.Leo, O., M. Foo, D.H. Sachs, L.E. Samelson, and J.A. Bluestone. 1987. Identification of a monoclonal antibody specific for a murine T3 polypeptide. Proc. Natl. Acad. Sci. USA. 84:1374–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ohara, J., and W.E. Paul. 1985. Production of a monoclonal antibody to and molecular characterization of B-cell stimulatory factor-1. Nature. 315:333–336. [DOI] [PubMed] [Google Scholar]
- 35.Benlagha, K., A. Weiss, A. Beavis, L. Teyton, and A. Bendelac. 2000. In vivo identification of glycolipid antigen-specific T cells using fluorescent CD1d tetramers. J. Exp. Med. 191:1895–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Finkelman, F.D., K.B. Madden, S.C. Morris, J.M. Holmes, N. Boiani, I.M. Katona, and C.R. Maliszewski. 1993. Anti-cytokine antibodies as carrier proteins. Prolongation of in vivo effects of exogenous cytokines by injection of cytokine-anti-cytokine antibody complexes. J. Immunol. 151:1235–1244. [PubMed] [Google Scholar]
- 37.Knight, P.A., S.H. Wright, C.E. Lawrence, Y.Y. Paterson, and H.R. Miller. 2000. Delayed expulsion of the nematode Trichinella spiralis in mice lacking the mucosal mast cell–specific granule chymase, mouse mast cell protease-1. J. Exp. Med. 192:1849–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Finkelman, F.D., S.C. Morris, T. Orekhova, and D. Sehy. 2003. The in vivo cytokine capture assay (IVCCA) for measurement of in vivo cytokine production in the mouse. Current Protocols in Immunology. J.E. Coligan, A.M. Kruisbeek, D.H. Margulies, E.M. Shevach, and W. Strober, editors. John Wiley & Sons Inc., New York. 6.28.1–6.28.10.
- 39.Stetson, D.B., M. Mohrs, R.L. Reinhardt, J.L. Baron, Z.E. Wang, L. Gapin, M. Kronenberg, and R.M. Locksley. 2003. Constitutive cytokine mRNAs mark natural killer (NK) and NK T cells poised for rapid effector function. J. Exp. Med. 198:1069–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oliveira, S.H., D.D. Taub, J. Nagel, R. Smith, C.M. Hogaboam, A. Berlin, and N.W. Lukacs. 2002. Stem cell factor induces eosinophil activation and degranulation: mediator release and gene array analysis. Blood. 100:4291–4297. [DOI] [PubMed] [Google Scholar]
- 41.Finkelman, F. 2001. Cytokine regulation of type 2 immunity. Samter's Immunologic Diseases. K.F. Austen, M.M. Frank, J.P. Atkinson, and H. Cantor, editors. Lippincott Williams & Wilkins, Philadelphia. 111–126.
- 42.Tite, J.P., A. Sloan, and C.A. Janeway Jr. 1986. The role of L3T4 in T cell activation: L3T4 may be both an Ia-binding protein and a receptor that transduces a negative signal. J. Mol. Cell. Immunol. 2:179–190. [PubMed] [Google Scholar]
- 43.Smiley, S.T., M.H. Kaplan, and M.J. Grusby. 1997. Immunoglobulin E production in the absence of interleukin-4-secreting CD1-dependent cells. Science. 275:977–979. [DOI] [PubMed] [Google Scholar]
- 44.Seder, R.A., W.E. Paul, S.Z. Ben-Sasson, G.S. LeGros, A. Kagey-Sobotka, F.D. Finkelman, J.H. Pierce, and M. Plaut. 1991. Production of interleukin-4 and other cytokines following stimulation of mast cell lines and in vivo mast cells/basophils. Int. Arch. Allergy Appl. Immunol. 94:137–140. [DOI] [PubMed] [Google Scholar]
- 45.Dvorak, A.M., R.A. Seder, W.E. Paul, E.S. Morgan, and S.J. Galli. 1994. Effects of interleukin-3 with or without the c-kit ligand, stem cell factor, on the survival and cytoplasmic granule formation of mouse basophils and mast cells in vitro. Am. J. Pathol. 144:160–170. [PMC free article] [PubMed] [Google Scholar]
- 46.Singer, M., J. Lefort, and B.B. Vargaftig. 2002. Granulocyte depletion and dexamethasone differentially modulate airways hyperreactivity, inflammation, mucus accumulation, and secretion induced by rmIL-13 or antigen. Am. J. Respir. Cell Mol. Biol. 26:74–84. [DOI] [PubMed] [Google Scholar]
- 47.Jones, R.E., F.D. Finkelman, R.B. Hester, and S.G. Kayes. 1994. Toxocara canis: failure to find IgE receptors (FcɛR) on eosinophils from infected mice suggests that murine eosinophils do not kill helminth larvae by an IgE-dependent mechanism. Exp. Parasitol. 78:64–75. [DOI] [PubMed] [Google Scholar]
- 48.de Andres, B., B. Cardaba, V. del Pozo, E. Martin-Orozco, S. Gallardo, P. Tramon, P. Palomino, and C. Lahoz. 1994. Modulation of the FcγRII and FcγRIII induced by GM-CSF, IFN-γ and IL-4 on murine eosinophils. Immunology. 83:155–160. [PMC free article] [PubMed] [Google Scholar]
- 49.Finkelman, F.D., N. Villacreses, and J.M. Holmes. 1992. Role of antigen-specific T cell help in the generation of in vivo antibody responses. II. Sustained antigen-specific T cell help is required to induce a specific antibody response. J. Immunol. 149:3845–3850. [PubMed] [Google Scholar]
- 50.Finkelman, F.D., S.C. Morris, T. Orekhova, M. Mori, D. Donaldson, S.L. Reiner, N.L. Reilly, L. Schopf, and J.F. Urban Jr. 2000. Stat6 regulation of in vivo IL-4 responses. J. Immunol. 164:2303–2310. [DOI] [PubMed] [Google Scholar]
- 51.Madden, K.B., L. Whitman, C. Sullivan, W.C. Gause, J.F. Urban Jr., I.M. Katona, F.D. Finkelman, and T. Shea-Donohue. 2002. Role of STAT6 and mast cells in IL-4- and IL-13-induced alterations in murine intestinal epithelial cell function. J. Immunol. 169:4417–4422. [DOI] [PubMed] [Google Scholar]
- 52.Strait, R.T., S.C. Morris, K. Smiley, J.F. Urban Jr., and F.D. Finkelman. 2003. IL-4 exacerbates anaphylaxis. J. Immunol. 170:3835–3842. [DOI] [PubMed] [Google Scholar]
- 53.Urban, J.F., Jr., N. Noben-Trauth, L. Schopf, K.B. Madden, and F.D. Finkelman. 2001. Cutting edge: IL-4 receptor expression by non-bone marrow-derived cells is required to expel gastrointestinal nematode parasites. J. Immunol. 167:6078–6081. [DOI] [PubMed] [Google Scholar]
- 54.Yoshimoto, T., and W.E. Paul. 1994. CD4+, NK1.1+ T cells promptly produce interleukin 4 in response to in vivo challenge with anti-CD3. J. Exp. Med. 179:1285–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dent, L.A., M. Strath, A.L. Mellor, and C.J. Sanderson. 1990. Eosinophilia in transgenic mice expressing interleukin 5. J. Exp. Med. 172:1425–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kopf, M., F. Brombacher, P.D. Hodgkin, A.J. Ramsay, E.A. Milbourne, W.J. Dai, K.S. Ovington, C.A. Behm, G. Kohler, I.G. Young, and K.I. Matthaei. 1996. IL-5-deficient mice have a developmental defect in CD5+ B-1 cells and lack eosinophilia but have normal antibody and cytotoxic T cell responses. Immunity. 4:15–24. [DOI] [PubMed] [Google Scholar]