

Abstract
Ig class switch recombination (CSR) requires expression of activation-induced deaminase (AID) and production of germline transcripts to target S regions for recombination. However, the mechanism of CSR remains unclear. Here we show that an extrachromosomal S plasmid assay is AID dependent and that a single consensus repeat is both necessary and sufficient for isotype-specific CSR. Transfected switch substrates specific for μ→γ3 and μ→γ1 are stimulated to switch with lipopolysaccharide (LPS) alone or LPS and interleukin-4, respectively. An Sγ3/Sγ1 substrate containing only three Sγ3-associated nucleotides reconstituted LPS responsiveness and permitted mapping of a functional recombination motif specific for μ→γ3 CSR. This functional recombination motif colocalized with a binding site for NF-κB p50, and p50 binding to this site was previously established. We show a p50 requirement for plasmid-based μ→γ3 CSR using p50-deficient B cells. Switch junctions from p50-deficient B cells showed decreased lengths of microhomology between Sμ and Sγ3 relative to wild-type cells, indicating a function for p50 in the mechanics of CSR. We note a striking parallel between the affects of p50 and Msh2 deficiency on Sμ/Sγ3 junctions. The data suggest that p50 may be the isotype-specific factor in μ→γ3 CSR and epistatic with Msh2.
Keywords: AID, B lymphocyte, immunoglobulin, NF-κB p50, class switch
Introduction
Ig class switch recombination (CSR) promotes the expression of antibody molecules with different constant (CH) regions permitting diversification of effector function while maintaining the original antigen-binding specificity arising from V(D)J joining. CSR is mediated by an intrachromosomal DNA rearrangement that focuses on stretches of repetitive DNA sequences termed switch (S) regions, which are located upstream of all the CH genes except Cδ (for reviews see references 1, 2). It is clear that mechanism of CSR requires the expression of activation-induced deaminase (AID) (3–5, and for review see reference 6) and germline transcription (gt) through participating S regions (for reviews see references 1, 2). Evidence suggests that AID functions by deaminating cytosine residues and converting them to dU (7–9). Removal of the uracil by the base excision repair pathway enzyme uracil-DNA glycosylase is required for CSR and somatic hypermutation (10). Staggered double strand breaks (DSBs) could emerge after deamination of closely spaced dC residues located on opposing strands in the S region. Blunt and staggered DSBs have been observed in S DNA, and their formation is both AID and uracil-DNA glycosylase dependent, demonstrating that they are intermediates in CSR (11–14). S DNA may become accessible to AID-generated lesions through gt expression (15, 16). Indeed, S regions have the unusual propensity to form R loops in vitro and in vivo when transcribed along the C-rich strand, which would provide ssDNA substrate to AID (17–20). Resection of the S/S junctions is likely to be dependent on the nonhomologous end joining proteins, Ku70/Ku80, DNA-PKcs (with the exception of μ→γ1 CSR) (21–24), several of the mismatch repair (MMR) proteins (25–28), and the histone H2AX (29–31).
Although it is clear that gt expression contributes to isotype specificity in CSR, evidence suggests that other factors are also involved in directing isotype choice. B cells deficient for specific transcription factors express AID and appropriate gts but do not switch to particular isotypes (32–34), suggesting that the isotype specificity arises through other factors not yet enumerated. Isotype-specific switch substrates used in transient transfection studies provide evidence for four independent switching activities (35, 36). Switching activity at endogenous loci is strictly correlated with AID expression, the coordinate expression of the appropriate gt, and isotype-specific switch plasmid activity, indicating that transacting switching activities are integral to the process of CSR (36).
We report here that the extrachromosomal switch substrates do not function in AID-deficient LPS-activated B cells, confirming the physiological relevance of these switch substrates. We found that single Sγ3 or Sγ1 tandem repeats are sufficient to support μ→γ3 or μ→γ1 CSR, respectively, demonstrating that isotype specificity is encoded in a single repeat unit. Deletion mapping of the Sγ3 consensus repeat and analysis of chimeric Sγ3/Sγ1 repeats indicates that the specificity for Sγ3 in LPS B cells is dependent on the integrity of a binding site for NF-κB p50. Previous studies showed that endogenous μ→γ3 CSR is abolished in p50-deficient B cells (33, 34, 37). We demonstrate that the switch substrate specific for μ→γ3 CSR is not functional in mitogen-activated p50-deficient B cells. Finally, a reduction of microhomology in Sμ/Sγ3 junctions was observed in p50-deficient but not in WT B cells, demonstrating that NF-κB p50 plays a role in the mechanics of CSR. The data raise the intriguing possibility that p50 is the isotype-specific factor mediating μ→γ3 CSR.
Materials and Methods
Cell Culture, Mice, Transfection, and Cloning of Sμ/Sγ3 Junctions.
The 1.B4.B6 cell line was grown in culture as described previously (36). Splenic B cells derived from Balb/c nu/nu mice were prepared and activated with LPS + anti–δ-dextran (αδdex) + IL-4 + IL-5 + TGFβ or LPS alone as previously described (34, 35). αδdex was a gift from Dr. C. Snapper (Uniformed Services University of the Health Sciences, Bethesda, MD). The nfkb1+/− (p50+/−), nfkb1−/− (p50−/−) mice were littermates and were backcrossed to C57Bl6/J (37). Enrichment of B cells from WT (129 × B6), nfkb1+/− (p50+/−), nfkb1−/− (p50−/−), and AID−/− mouse spleens was accomplished using Cellect Immunocolumns (Cedarlane Laboratories) according to the manufacturer's instructions. AID−/− mice were a gift from T. Honjo (Kyoto University, Kyoto, Japan), and nfkb1 and WT mice were purchased from Jackson Labs. The purity of the cell population was confirmed by FACS® analysis. In transfections experiments, the cells were electroporated in the presence of 8 μg of plasmid at 300 V/950 μF on day 3 of culture. After an additional 3 d in culture, nuclei were isolated using the Blood and Cell Culture DNA Preparation kit (QIAGEN), and DNA was prepared with the Puregene Genomic DNA Purification kit (Gentra Systems). PCR amplification and cloning of Sμ/Sγ3 hybrid molecules was performed as described previously (38) except that primer μ-1.2 (5′-GCTGGGGTGAGCTCAGCTATGCTACGC-3′) was used, which anneals to positions 5307–5333 at the 5′ end of the germline Sμ (MUSIGCD07).
RT-PCR, Quantitative RT-PCR, Digestion Circularization–PCR, and Bacterial Transformation Assays.
RT-PCR for γ3 germline transcripts and GAPDH was performed as described (33, 34). Digestion circularization (DC)–PCR for detection of endogenous and plasmid-based μ→γ3 and μ→γ1 CSR was performed as described previously, respectively (34, 35). The primers used for plasmid-specific DC-PCR were the same for all the switch plasmids studied. Radioactivity was assessed using a PhosphorImager and ImageQuant software for quantitation. Bacterial transformation assays were performed as described (35, 36). Briefly, DNA recovered from nuclei of transfected cells was either left untreated or digested with EcoRI and then transformed into bacteria. The EcoRI-resistant (EcoRIr) colonies denotes plasmid resistant to EcoRI digestion. EcoRIr colonies were prepared as minipreps and analyzed by restriction mapping to identify S/S recombinant plasmids. The percentage of EcoRIr is obtained by dividing the total number of EcoRIr transformants by the number of Ampr transformants and then multiplying by 100.
Construction of Plasmids.
Construction of pG3.1 and pG3.01s were described previously (35). To obtain pG3.02s, pG3.025s, and pG3.045s a cassette was constructed that contained TK, Iα, and the degenerate Sγ3 repeats found in pG3.01s (35). The cloned cassette is referred to as L34 and contains a BamHI site located between the Iα and the degenerate Sγ3 repeats. Two partially complementary oligos containing the consensus Sγ3 repeat were synthesized: G3N1, 5′-TTGTGGGGACCAGGCTGAGCAGCTCTCAGGGAGCTGGGGAGGTGGAGTTGTG-3′ and G3N2, 5′-CACAATGAACCTACCCCAGCTCCCCCAGAGCTGCCCAGC-TTGGTCCCCACAACTCCACC-3′. The oligos were annealed, filled-in with Klenow DNA polymerase (Promega), and cloned into the filled-in BamHI site of L34. Subclones containing 1, 2, or 6 Sγ3 consensus repeats were isolated, and DNA sequence was verified. Complementary consensus Sγ1 or chimeric Sγ1.m1 oligos were synthesized, annealed, and cloned into the Klenow filled-in BamHI site of L34. The G-rich strand for the Sγ1 consensus repeat was 5′-GGTGACCCAGGCAGAGCAGCTCCAGGGGAGCCAGGACAGGTGGAAGTGT-3′. The G-rich strand for the Sγ1.m1 repeat was 5′-GGGGACCAGGCTGAGCAGCTCCAGGGGAGCCAGGACAGGTGGAAGTGT-3′. The cassettes from the appropriate L34 subclones were isolated as EcoRI-NotI restriction fragments and then directionally cloned into a switch plasmid vector containing neo, μ intronic enhancer and VH promoter, and Sμ with EcoRI-NotI ends.
Online Supplemental Material.
Fig. S1 shows a comparison of Sμ/Sγ3 junctions derived from recombinant pG3.1, pG3.02s, and pG1.m1.02s. Switch plasmids were transfected into LPS-activated B cells, and recombinant plasmids were recovered using the bacterial transformation assay. Switch junctions were identified by automated DNA sequence analysis. Fig. S2 shows that reduced microhomology is found in Sμ/Sγ3 junctions from p50-deficient mice. Sμ/Sγ3 junctions from p50+/+ (A), p50+/− (B), and p50−/− (C) mice were PCR amplified from B cells stimulated with LPS + αδdex + IL-4 + IL-5 + TGFβ and cloned. At least two independent DNA samples were used as amplification substrates to generate junctions for each group analyzed. Switch junctions were identified by automated DNA sequence analysis. Fig. S3 shows that the expression of MMR transcripts is not altered by NF-κB p50 deficiency in activated B cells. Quantitative SYBER Green PCR assays were developed for Mlh1, Pms2, Msh2, Msh3, Msh6, and GAPDH. At least two independent RNA samples from each activation state and cell type were reverse transcribed to cDNA and used in the real-time PCR assay. Figs. S1–S3 are available at http://www.jem.org/cgi/content/full/jem.20031935/DC1.
Results
Switch Plasmid Recombination Is Dependent on AID Expression.
Our previous studies using extrachromosomal switch substrates demonstrated distinct isotype switching activities in B cells capable of endogenous CSR (35, 36). AID is a critical mediator of CSR, and its deficiency is characterized by a profound block in CSR to all isotypes, whereas gt expression remains unaffected (3). To further explore the physiological relevance of the switch plasmid assay, the switch substrates were tested for AID dependence. AID+/+ and AID−/− B cells were T cell depleted, LPS activated, and the induction of γ3 gts and endogenous μ→γ3 CSR were examined by RT-PCR and DC-PCR (Fig. 1 A), respectively. In the RT-PCR assay, GAPDH was used as an internal control for cDNA input (Fig. 1 B). After 72 h of activation with LPS, AID+/+ and AID−/− B cells expressed the γ3 gt equally well, indicating that all the cells were successfully stimulated. In the DC-PCR assay, the nonrearranging acetylcholine receptor (nAChR) gene was used as a control for the digestion and ligation reactions, and the nAchR DC-PCR product was found for all samples (Fig. 1 C). AID+/+ B cells activated with LPS for 5 d were positive for the Sμ/Sγ3 DC-PCR product, whereas this product was undetectable in DNA isolated from AID-deficient B cells. The normal expression of gts and the absence of μ→γ3 switching is fully consistent with the AID-deficient phenotype described previously (3, 4).
Figure 1.

Switch plasmid-based μ→γ3 CSR in LPS B cells is AID dependent. (A) A diagram of the DC-PCR strategy for endogenous loci is shown. A portion of the IgH locus is diagramed before and after μ→γ3 recombination. EcoRI sites (RI) flank the 5′ and 3′ ends of the Sμ and Sγ3 regions, respectively, and are preserved after CSR. After digestion with EcoRI, the DNA is ligated. Nested primer sets specific for sites at the 5′ end of Sμ (dc-μ.1, dc-μ.2) and the 3′ end of Sγ3 (dc-γ3.1, dc-γ3.2) amplify the region spanning the circle joint and yield a specific S/S DC-PCR product. The positions and orientations of the primer sets are shown before and after ligation. The nAChR gene serves as an internal control for digestion and ligation. (B–D) B cells from AID+/+ and AID−/− spleens were activated in culture with LPS for 3 (B) or 5 d (C) or were activated with LPS for 3 d and then transfected with pG3.1 and grown in LPS culture for an additional 3 d (D). (B) RT-PCR for detection of GAPDH and the γ3 germline transcript (γ3 GT) was performed on RNA of LPS-activated B cells from AID+/+ (lanes 1 and 2) or AID−/− (lanes 3–5) mice. (C) DC-PCR was used to detect endogenous μ→γ3 switching in DNA from LPS-activated AID+/+ or AID−/− B cells (top). The nAchR gene was used as a positive control (bottom). (D) LPS activated B cells from AID+/+ or AID−/− mice were transfected with the pG3.1 plasmid and used in a plasmid-based DC-PCR assay. The RRL for pG3.1 is the ratio of the radioactivity associated with the 180-bp (S/S) fragment to that of the 510-bp (vector) fragment. Intact pG3.1 (0.5 pg) demonstrated to be in the linear range of detection in the DC-PCR assay (not depicted) was used as a negative control (lane 6), and its RRL was set to 1 (right). The RRL values for the AID+/+ and AID−/− transfections were normalized to the negative control. In the right panel AID+/+ and AID−/− samples are designated +/+ and −/−, respectively. The intact negative control is indicated as −. The average RRLs from two independent experiments for pG3.1 is plotted (right), and SDs are shown.
To evaluate the influence of AID on switch plasmid recombination, the relative frequency of CSR events was compared in AID+/+ and AID−/− B cells using a previously devised plasmid-specific and semiquantitative DC-PCR assay (35). AID+/+ and AID−/− B cells were activated with LPS for 3 d, transfected with pG3.1, which detects μ→γ3 CSR, and cultured in the presence of LPS for an additional 3 d. Amplification of the plasmid-specific vector and S/S recombinant fragments after SacI digestion and ligation yields a 510- and a 180-bp PCR product, respectively. The linear range of detection was established using twofold serial dilutions of pG3.1 into 1 μg of genomic DNA followed by DC-PCR in the presence of radiolabeled nucleotides (unpublished data) as described previously (35). S/S recombinant and intact pG3.1 spiked into genomic DNA as well as mock-transfected DNA were used as positive, negative, and specificity controls for the DC-PCR, respectively (Fig. 1 D). In two independent transfection experiments, the 510-bp vector-associated DC-PCR product was found for all the cells analyzed, whereas the 180-bp product resulting from the presence of composite Sμ/Sγ3 DNA was found only in plasmid transfected into AID+/+ B cells but not AID−/− B cells (Fig. 1 D, left). The relative recombination level (RRL) is expressed as the ratio of the S/S signal to the vector signal. Comparison of RRLs for pG3.1 indicates a <80-fold recombinational activity in AID+/+ B cells than in the AID−/− B cells (Fig. 1 D, right). These findings demonstrate the AID dependency of the switch plasmid assay and provide important confirmation that the switch substrates, analyzed in a transient transfection format, reflect physiological CSR.
A Single Tandem Repeat Supports CSR in Switch Substrates.
The detection of isotype-specific switching activities suggests that molecular recognition of S regions may be a feature of CSR. The pG3.1 plasmid contains a 2.0-kb insert of Sγ3 DNA, which represents the complete genomic version of this S region. Our recent studies indicated that Sγ3 can be reduced from 44 tandem repeats to 5 repeat units without adversely affecting plasmid-based CSR frequency (36). In contrast, pG3.01s, which contains two degenerate tandem repeats comprised of 125 bp derived from the 3′ end of the Sγ3 region, does not support CSR, indicating that the presence of consensus S DNA is crucial for CSR (35). To define the minimum S DNA target required for recombination, structural variants of pG3.01s were constructed which include 1, 2, or 6 consensus tandem repeats and are referred to as, pG3.02s, pG3.025s, and pG3.045s, respectively (Fig. 2 A).
Figure 2.
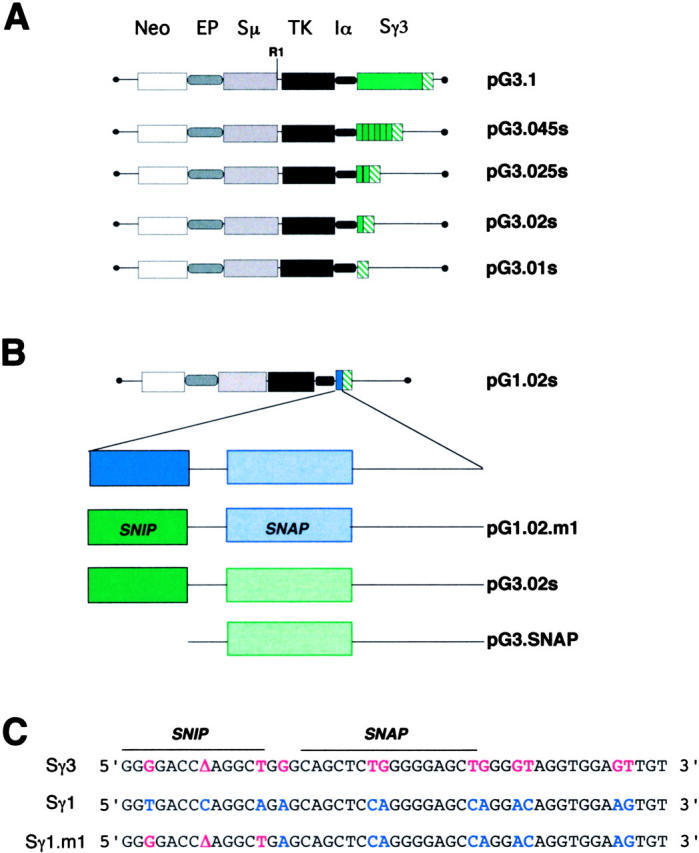
Structure of switch plasmids related to pG3.1. (A) Intact pG3.1 contains a neomycin-resistance gene (neo), the Ig μ intronic enhancer (E), an IgH variable region promoter (P), a thymidine kinase (TK) gene, the promoter for α gts (Iα), and the Sμ and Sγ3 regions. PG3.01s is identical to pG3.1 except for the deletion of the Sγ3 region and retention two nonconsensus tandem repeats from the extreme 3′ end of genomic Sγ3 (35). The plasmids pG3.02s, pG3.025s, and pG3.045s contain 1, 2, and 6 consensus Sγ3 tandem repeats, respectively. The nonconsensus and consensus tandem repeats located in Sγ3 are indicated as green hatched and solid green boxes, respectively. (B) PG1.02s contains an Sγ1 consensus repeat, which is depicted by the solid blue box. The Sγ1 consensus repeat is expanded to show two subsections, SNIP (solid) and SNAP (speckled), separated by a short and long spacer, as indicated. Sγ1 and Sγ3 DNA sequences are shown in blue and green, respectively. PG1.02.m1 contains a chimeric repeat including the Sγ1 repeat backbone and an Sγ3 SNIP site. PG3.SNAP contains a 30-bp truncated Sγ3 consensus repeat centered on the SNAP site. (C) The DNA sequence for Sγ3 and Sγ1 consensus repeats are shown. In the chimeric Sγ1.m1 repeat, the Sγ1 and Sγ3 SNIP sites differ at three nucleotide positions.
DC-PCR analysis was used to determine the level of switching for the minimal plasmids as compared with pG3.1 in 1.B4.B6 cells and in LPS-activated normal splenic B cells (Fig. 3, A and B). 1.B4.B6 cells were shown previously to support endogenous and pG3.1-based μ→γ3 CSR (35, 36). PCR amplification of the S/S composite fragment after SacI digestion and ligation yields 131- and 180-bp fragments for the minimal substrates and for pG3.1, respectively. This analysis demonstrates CSR of pG3.1 in 1.B4.B6 cells and in LPS-activated B cells, whereas essentially no CSR was found for the pG3.01s plasmid, consistent with previous findings (35). The switch substrates, pG3.02s, pG3.025s, and pG3.045s, were active for CSR (Fig. 3, A and B), where pG3.045s showed twofold lower switching activity, and pG3.025s and pG3.02s had about fourfold lower activity than the pG3.1 plasmid as assessed by PhosphorImager analysis (Fig. 3 A, right). As a control, transfected DNA isolated from LPS-activated splenic B cells was digested with the combination SacI and BglI. Under intramolecular ligation conditions, BglI digestion will abolish the 510-bp vector–associated fragment because there are two BglI sites located in the vector backbone. All the switch plasmids also contain a BglI site at the 3′ end of the Sγ3 region, and digestion with SacI and BglI will abolish the S/S DC-PCR product. The absence of the vector and S/S-associated fragments after SacI and BglI digestion indicates that the switch substrates are digested to completion and that ligation is exclusively intramolecular (Fig. 3 B, compare lanes 1–8 and 9–16). These results demonstrate that the efficiency of switching on switch substrates is related to the number of tandem repeats in the Sγ3 region and that a single consensus repeat is sufficient for CSR.
Figure 3.

Switch substrates require a single Sγ consensus repeat to support CSR. (A) Switch plasmids, as indicated, were transfected into 1.B4.B6 cells and analyzed for CSR using the plasmid-based DC-PCR assay (left). The RRLs are the results of five to six transfections from at least two independent experiments, and SDs are shown (right). The RRL is calculated for each plasmid as the ratio of radioactivity associated with the 180-bp S/S fragment to that of the 510-bp vector-associated fragment. The RRLs are not normalized. (B) Switch plasmids were transfected into LPS-activated B cells and analyzed by plasmid-based DC-PCR using either SacI or SacI and BglI digestion. (C) The switching activity of pG3.1, pG3.02s, pG3.SNAP, and pG3.01s were compared in LPS-activated B cells using the bacterial transformation assay. DNA recovered from nuclei of the transfected cells was untreated or digested with EcoRI and then transformed into bacteria. S/S recombinant frequency was as follows: pG3.1 (17/41,540); pG3.02s (10/110,060); pG3.01s (0/231,800); and pG3.SNAP (6/570,400). Switch frequency was obtained by dividing the number of S/S recombinant transformants by the total number of transformants and multiplying by 105. Results are summarized from at least three to six transfections from two to three independent experiments. p-values, derived by χ2 analysis, are positioned above the histograms and indicate the confidence level that the plasmid switch frequency in the pG3.02s was significantly different from that obtained from pG3.01s and pG3.SNAP.
To further evaluate CSR on pG3.02s, the previously described bacterial transformation assay was used to compare plasmid-based CSR frequencies for pG3.1, pG3.02s, and pG3.01s (35). The frequency of CSR for pG3.02s was approximately sixfold lower than pG3.1, whereas no switching was detected for the pG3.01s plasmid (Fig. 3 C). This finding confirms the DC-PCR analysis indicating that a single tandem repeat is sufficient to enable CSR. To determine the minimum length requirements for S DNA in the switch reaction, a new plasmid termed pG3.SNAP was constructed in which the 49-bp consensus repeat found in pG3.02s was truncated to 30 bps and analyzed in LPS-activated B cells (Fig. 2 B). The switching frequency of pG3.SNAP was 41-fold lower than pG3.1 and sixfold lower than pG3.02s, thus demonstrating that reduction of S DNA to less than a single tandem repeat results in a severe diminution of CSR efficiency (Fig. 3 C). The reduced frequency of CSR for pG3.SNAP suggests that sequence important to CSR is located in either or both the SNIP site at the 5′ end and several nucleotides at the 3′ end of the repeat unit.
S/S junctions in recombinant pG3.1 and pG3.02s recovered in the bacterial transformation assay were analyzed by DNA sequence analysis (Fig. S1, A and B, available at http://www.jem.org/cgi/content/full/jem.20031935/DC1). These composite S/S regions were found to have characteristics associated with switch junctions derived from the endogenous locus (39). Similar to physiological switch junctions, the switch plasmid-derived recombination breakpoints were scattered across the breadth of the Sμ and Sγ3 DNA sequences. The structure, degree microhomology, and presence of mutations in the switch junctions derived from pG3.02s conform to the usual parameters associated with CSR and confirms that this minimal switch plasmid is capable of supporting bona fide switching (40–42).
Minimal Switch Substrates Display Isotype Specificity.
Previous studies indicate that a switch substrate specific for μ→γ1 CSR is induced to recombine in splenic B cells activated with LPS + IL-4 but not with LPS alone, demonstrating distinct switching activities for μ→γ3 and μ→γ1 CSR (36). To determine whether isotype specificity is retained in minimal switch plasmids, pG1.02s was constructed and is identical to pG3.02s except for the presence of an Sγ1 consensus repeat (Fig. 2 B). Previous studies indicated that the Sγ tandem repeats contain SNIP and SNAP recognition motifs, which are shown here to provide a point of reference in mapping functional recombination motifs (FRMs) on the switch plasmids (43, 44). To ascertain whether pG1.02s is functional and displays IL-4 dependency for CSR, pG3.02s and pG1.02s were transfected into normal B cells activated with LPS in the presence or absence of IL-4. The DC-PCR assay indicates that pG3.02s undergoes CSR in LPS-activated B cells, whereas switching was at background levels for pG1.02s and pG3.01s under these conditions (Fig. 4 A). Furthermore, the absence of CSR in the pG1.02s and pG3.01s and its presence in pG3.02s after transfection into LPS B cells was confirmed using the bacterial transformation assay (see Materials and Methods). In contrast, both pG3.02s and pG1.02s undergo CSR in B cells stimulated with LPS + IL-4. These results demonstrate that Sγ1-specific switching activity is IL-4 inducible, distinct from Sγ3 switching activity, and that switch substrates containing a single consensus repeat recapitulate full-length switch plasmid function.
Figure 4.

Substitution of three nucleotides in the Sγ1 SNIP site leads to reconstitution of switching in LPS B cells for the pG1.02s.m1 plasmid. (A) The plasmids, pG3.01s, pG3.02s, and pG1.02s, were transfected into B cells activated with LPS in the presence or absence of IL-4 and then harvested and analyzed by DC-PCR. The 510-bp vector (V) and the 180-bp S/S PCR fragments are shown. (B) The plasmids, pG3.01s, pG3.02s, pG1.02s, and pG1.02s.m1, were transfected into B cells activated with LPS then harvested and analyzed using the bacterial transformation assay. S/S recombinant frequency was as follows: pG3.02s (10/110,060); pG3.01s (0/231,800); pG1.02s (0/138,800); and pG1.02.m1 (9/212,080). Switch frequency was obtained by dividing the number of S/S transformants by the total number of transformants and multiplying by 105. Results are summarized from at least three to six transfections from two to three independent experiments. p-values are positioned above the histograms. Values of p were derived by χ2 analysis.
Mapping Functional Recombination Motifs Using Sγ3/Sγ1 Chimeric Switch Substrates.
Retention of isotype specificity by minimal switch plasmids demonstrates that all of the information required for molecular recognition of the Sγ3 and Sγ1 regions is encoded in a single consensus tandem repeat. Comparison of the Sγ3 and Sγ1 tandem repeats reveals differences at only 12 out of 49 nucleotides (Fig. 2 C). Systematic substitution of Sγ3-associated nucleotides into the Sγ1 sequence might lead to reconstitution of the motifs necessary for μ→γ3 plasmid-based CSR in LPS B cells. To test this hypothesis, a new minimal plasmid, referred to as pG1.02.m1, was constructed and contains a Sγ1 repeat altered at three nucleotide positions (T→G, T→A, and C→Δ, where Δ represents a deletion) located at the 5′ end of the repeat (Fig. 2 C). Several other constructs containing additional combinations of S/S chimeric repeats were attempted and proved to be unclonable.
The pG3.02s, pG3.01s, pG1.02s, and pG1.02.m1 plasmids were transfected into LPS B cells and analyzed using the bacterial transformation assay for CSR activity (Fig. 4 B). In this study, pG3.02s and pG3.01s functioned as positive and negative controls, respectively. The pG1.02s plasmid did not undergo CSR in LPS-activated B cells, confirming the DC-PCR analysis of this switch plasmid, whereas pG1.02.m1 was able to recombine under these conditions (Fig. 4 B). The S/S junctions derived from pG1.02.m1 were isolated in the bacterial transformation assay, submitted to DNA sequence analysis, and found to have features similar to those found for pG3.02s, demonstrating that this plasmid is capable of bona fide CSR (Fig. S1 C). Together these studies show that limited nucleotide changes of the Sγ1 sequence are sufficient to reconstitute μ→γ3 CSR competence on switch plasmids in LPS B cells and suggest that the SNIP binding site or DNA sequence surrounding this site operates as an isotype-specific FRM.
Sγ3 Recombination Breakpoints Are Nonrandom in pG3.02s and pG1.02.m1.
The truncated Sγ regions found in pG3.02s and pG1.02.m1 provide a limited target for CSR. Sγ recombination breakpoints derived from pG3.02s and pG1.02.m1 associated switch junctions were found to be nonrandomly distributed (Fig. S1 D). The breakpoints are located in several subregions found in both the Sγ3 consensus and degenerate repeats and are closely flanked by RGYW hotspots. Most of the breakpoints are located in a section of the degenerate repeat replete with RGYW motifs. The absence, truncation, or replacement of the Sγ3-associated SNIP site in pG3.01s, pG3.SNAP, and pG1.02s, respectively, led to loss of CSR in response to LPS in activated B cells, demonstrating that the SNIP motif, located in the consensus repeat, is involved in a critical step of the recombination reaction. The location of recombination breakpoints in the downstream degenerate repeats implies that if CSR initiates in the consensus repeat then processing of staggered DSBs must occur. Genetic evidence suggests that MMR proteins are involved in processing of broken DNA ends in CSR (26, 27, 45) and modulate the efficiency of switching (25, 46).
NF-κB p50 Expression Is Required for Plasmid-based μ→γ3 CSR.
The localization of the FRM to a site containing a NF-κB p50 binding site suggests that p50 may be a mediator of μ→γ3 CSR. To further explore the functional involvement of p50 in μ→γ3 CSR, WT and p50-deficient B cells were activated with LPS or with LPS + αδdex + IL-4 + IL-5 + TGFβ (34) for 3 d and then transfected with the pG3.1 or the pG.1 plasmids, respectively. The LPS + αδdex + IL-4 + IL-5 + TGFβ conditions were used, since these provided robust switching μ→γ1 (47), whereas LPS + IL-4 stimuli did not work well for the p50−/− B cells. Sha et al. reported that resting B cells respond poorly to LPS (37). However, we have found that unfractionated splenic B cells from WT and p50-deficient mice proliferate equally well in response to LPS for at least the first 3 d of culture. Thereafter, p50-deficient B cells do not survive as well as WT so that at the end of 6 d survival of the p50−/− B cells is about half that of the p50+/+ B cells as assessed by trypan blue exclusion (unpublished data). Nonetheless, plasmid-based CSR should be easily detectable even in cell cultures with a 50% reduction of cell survival if switching frequency is unperturbed. Standard curves indicating that the concentration of switch plasmid in the DC-PCR (0.5 pg indicated by the arrow) produces a vector signal in the linear range of detection are shown (Fig. 5, A and B, rightmost panels). These standards are included in all the DC-PCR assays reported here. DC-PCR and RRL analysis of transfected pG3.1 and pG.1 indicates that whereas p50 deficiency has a profound negative impact on μ→γ3 plasmid-based switching, μ→γ1 CSR is intact (Fig. 5, A and B). As an additional control, endogenous μ→γ3 and μ→γ1 CSR was evaluated by DC-PCR in the transfected samples. We found that the WT samples support switching to both loci, whereas the p50-deficient samples are capable of μ→γ1 but not μ→γ3, as expected (Fig. 5, C and D). Endogenous μ→γ1 DC-PCR samples were compared after fourfold serial dilution and show that CSR is reduced three- to fourfold in p50−/− B cells as shown in representative examples (Fig. 5 D, right). Nonetheless, the p50−/− B cells still clearly undergo CSR. These findings demonstrate a striking parallel in the isotype specificity and p50 dependency of both plasmid-based and endogenous CSR and confirm that μ→γ3 plasmid-based CSR is dependent on the presence of p50 as predicted.
Figure 5.

Plasmid-based μ→γ3 CSR is abolished in NF-kB p50-deficient B cells. (A and B) B cells from NF-κB p50 WT and knockout (KO) spleens were T cell depleted, activated with LPS or LPS + αδdex + IL-4 + IL-5 + TGFβ for 3 d, then transfected with pG3.1 (A, left) or pG1 (B, left), respectively, grown in culture for an additional 3 d, and then used in a plasmid-based DC-PCR assay. The PCR products representing the switched plasmid (S/S) and the vector backbone are shown from independent transfected samples. The average RRLs from two independent experiments for pG3.1 (A) and pG1 (B) is plotted (middle), and SDs are shown. Standard curves (fivefold dilutions) for vector DC-PCR products are shown (right). The arrow indicates the concentration of plasmid (0.5 pg) used in the DC-PCR assays shown here. (C) Endogenous DC-PCR assays for μ→γ3 CSR in LPS-activated B cells from p50 WT (lanes 1 and 2) and KO (lanes 3–7) are shown. The nAChR locus is used as a control for sample loading and ligation. (D) Endogenous DC-PCR assays for μ→γ3 CSR in LPS + αδdex + IL-4 + IL-5 + TGFβ activated B cells are shown for p50 WT (lanes 1 and 2) and KO (lanes 3–6) in the left panel. Fourfold serial dilution of representative DC-PCR samples are shown in the right panel for WT (lanes 1–4) and KO (lanes 5–8). The nAChR locus is used as a control for sample loading and ligation.
Switch Junctions Exhibit Reduced Microhomology in p50-deficient B Cells.
Switch junctions display characteristic features including short stretches of identity or microhomology between the Sμ donor and Sγ acceptor in ∼50% of junctions. To determine whether p50 plays a role in the mechanics of endogenous μ→γ3 CSR, B cells were cultured in the presence of LPS + αδdex + IL-4 + IL-5 + TGFβ for 5 d, and Sμ/Sγ3 junctions isolated from p50 WT (+/+), heterozygous (+/−), and deficient (−/−) B cells were examined by DNA sequence analysis (36, 38). Under these activation conditions, the absence of μ→γ3 switching in the p50−/− B cells was not due to a proliferative insufficiency or reduced expression of γ3 gt (34). Despite the significant reduction of μ→γ3 CSR in p50-deficient B cells, rare Sμ/Sγ3 junctions were successfully amplified and analyzed by automated DNA sequence analysis (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20031935/DC1). Comparison of the lengths of identity between Sμ and Sγ3 indicates that junctions from p50+/+ and p50+/− mice have significantly longer overlaps than those from the p50−/− mice (P = 0.017) (Table I). Mismatch repair proteins (MMR) were shown recently to reduce the frequency of CSR (two- to fourfold) and alter the length of microhomology at switch junctions (27). It is striking that Sμ/Sγ3 junctions derived from the Msh2−/− B cells display a similar reduction of microhomology as found in the p50−/− junctions (Table I).
Table I.
Microhomology Is Reduced in Sμ/Sγ3 Junctions Derived from p50−/− Mice
|
|
Percentage of Sμ/Sγ3 junctions with indicated microhomology
|
p-valuea
|
Number of junctions
|
Reference
|
|||
|---|---|---|---|---|---|---|---|
| Mouse | 0 bp | 1–3 bp | 4–8 bp | 9–11 bp | |||
| p50+/+ b | 28 | 52 | 16 | 4 | 25 | This paper | |
| p50+/− b | 33 | 28 | 39 | 0 | NS | 18 | This paper |
| p50−/− b | 50 | 50 | 0 | 0 | 0.017 | 22 | This paper |
| Msh2+/+ c | 28 | 56 | 16 | 0 | 32 | Reference 27 | |
| Msh2−/− c | 50 | 50 | 0 | 0 | 0.004 | 32 | Reference 27 |
NS, not significantly different.
The significance of difference between +/+ and +/− to −/− was calculated using a one-tail Student's t test.
Cells were activated with αδdex + LPS + IL-4 + IL-5 + TGFβ for 5 d, and DNA was isolated. The percentage of junctions with designated overlaps is shown.
Cells were activated with LPS for 5 d, and DNA was isolated.
NF-κB p50 is a transcription factor that is important in B cell activation responses, thus raising the possibility that p50 deficiency leads to reduction of MMR gene expression. This is important since the MMR proteins form heterodimers with distinct functions (for reviews see references 48, 49) and individual protein levels can influence heterodimer formation (48–51). Studies in human cell lines indicate a linear relationship between MMR mRNA and protein, suggesting that the level of each protein is determined by transcription (52). To determine whether p50 deficiency influences MMR levels, real-time RT-PCR assays were devised for Msh2, Msh3, Msh6, Pms2, and Mlh1 transcripts (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20031935/DC1). There was no discernible difference in the level of these transcripts for p50+/+ and p50−/− B cells, indicating that alteration of MMR gene expression is unlikely to have caused the reduction of microhomology at Sμ/Sγ3 junctions derived from p50-deficient B cells.
Discussion
In studies reported here, recombination of a switch plasmid specific for μ→γ3 switching was demonstrated to be AID dependent in LPS-activated splenic B cells. AID is a crucial mediator of CSR and has been shown to be the sole B cell–specific gene required to support CSR in non–B cells (53). Thus, the requirement for AID expression to enable successful substrate-based CSR confirms the physiological relevance of the switch plasmid assay. Four distinct switching activities for μ→γ3, μ→γ1, μ→ɛ, and μ→α CSR have been detected based on the differential capacity of isotype-specific switch substrates to undergo recombination in switching B cell lines and in mitogen-activated splenic B cells (35, 36). In switching B cell lines, endogenous CSR was strictly correlated with coordinate expression of the appropriate gt and isotype-specific switching activity, indicating that transacting switch factors are integral to the recombination reaction.
In an effort to define parameters governing isotype-specific recognition of S regions, we noted that Sγ3 can be reduced from 44 to 5 repeat units without adversely affecting plasmid-based CSR frequency, whereas substrates containing two degenerate tandem repeats were incapable of CSR (35, 36). Our current studies demonstrate that a single 49-bp repeat unit is necessary and sufficient for plasmid-based CSR, whereas reduction of the Sγ3 consensus repeat from 49 to 30 bps severely diminishes plasmid-based switching. This leads to the question: to what extent is endogenous CSR dependent on S region sequence? Analysis of endogenous switching in the Sμ2/− mouse, which retains 15 GAGCT motifs located in the Sμ flanking regions, demonstrates reduced but detectable CSR, indicating that a very limited number of Sμ motifs are sufficient to support CSR (54). The region upstream of the Sμ tandem repeats (TRs) is also a target for AID catalyzed dC deamination, as assessed by the accumulation of mutations, and may account for the residual recombination in the absence of the SμTRs (29, 31, 55, 56), whereas these mutations are essentially undetectable in germline Sγ3 and germline Sγ1 (57). Deletion of the entire endogenous Iγ1-Cγ1 intron containing the Sγ1 region led to undetectable μ→γ1 CSR (58). Replacement of Sγ1 with a 1-kb random G-rich sequence by targeted homologous recombination produced a 93% reduction of μ→γ1 CSR, indicating that non–S region sequence could only marginally support CSR. It is possible that the reintroduced G-rich sequence contains rare motifs capable of supporting CSR, thus explaining the barely detectable switching for that locus. The strict requirement of μ→γ1 switching on the presence of the Sγ1TRs strongly argues that there is sequence dependency for the downstream S regions.
Isotype specificity is a distinctive feature of our switch substrates, which contain extensive tracts of S region DNA (36). It is noteworthy that switch substrates containing a single Sγ1 consensus repeat retain isotype specificity since pG1.02s undergoes μ→γ1 CSR in B cells activated with LPS and IL-4 but not with LPS alone. This observation demonstrates that the minimal switch plasmids recapitulate functions associated with full-length switch substrates (36). The pG3.SNAP plasmid, containing a truncated consensus repeat, which excludes the SNIP site and several nucleotides from the 3′ end, shows a severe diminution of CSR frequency. This phenotype is consistent with the localization of an FRM within either or both ends of the consensus repeat. A chimeric repeat was constructed in which three nucleotide substitutions were introduced into the Sγ1 SNIP site transforming it into a canonical Sγ3 SNIP site. Restoration of switching function to the chimeric switch plasmid in LPS B cells identifies the Sγ3 SNIP motif as containing an isotype-specific FRM. These studies also provide the first clear evidence that S DNA sequence is a major contributing factor in isotype-specific CSR.
Targeted disruption of the NF-κB p105 gene is associated with loss of μ→γ3 CSR (33, 34, 37). We report here that plasmid-based μ→γ3 switching is essentially abolished in p50-deficient B cells. The finding that Sμ/Sγ3 junctions from p50-deficient B cells contained reduced microhomology compared with WT provides genetic evidence that p50 has a role in determining the mechanics of the recombination reaction. Furthermore, in vivo footprinting studies demonstrate p50-dependent protein interactions at Sγ3 in B cells (34, 43). These findings support the hypothesis that p50 is a mediator of μ→γ3 CSR but do not indicate the level of p50 involvement. Deletion mapping and mutational analysis of Sγ3 DNA in switch substrates demonstrate that an isotype-specific FRM colocalizes with the SNIP site, a p50 recognition motif. NF-κB p50 homodimer binding to Sγ3 SNIP sites was demonstrated previously using nuclear extracts from LPS-activated splenic B cells in gel shift, supershift, and chemical footprinting studies (43). However, it is still possible that DNA binding proteins other than p50 could interact with the FRM. Nonetheless, the weight of evidence supports the view that p50 functions directly in the μ→γ3 reaction.
Single-stranded S DNA is substrate for AID (8, 15, 16, 59, 60). Recombination on our switch plasmids is not correlated with the level of transcription of S regions (35, 61). It is possible that the use of superhelically coiled plasmids in our transient transfection experiments provides sufficient ssDNA target for AID to promote switching in the absence of high levels of transcription. Thus, it is unlikely that the absence of p50 leading to transcription insufficiency is the reason for the loss of plasmid switching in p50−/− B cells.
MMR proteins have been implicated in regulating both the frequency and the process of CSR (25–28, 46). Msh2 deficiency is associated with reduced microhomology at Sμ/Sγ3 junctions (27), and this protein can participate in the removal of nonhomologus DNA ends during DSB repair in yeast (62, 63). Our findings highlight a striking reduction in the extent of microhomology at Sμ/Sγ3 junctions from p50−/− and Msh2−/− B cells and are consistent with the hypothesis that p50 is epistatic with Msh2. However, p50 deficiency has a much greater impact on CSR frequency than does Mhs2. These results support a model in which p50 recruits Msh2 and other components of the recombination machinery to Sγ3 DNA.
Given that parameters defining the mechanism of CSR have begun to emerge, it is useful to consider the role FRMs and isotype-specific factors might play in the CSR reaction (for review see reference 6). It is likely that AID initiates CSR by deaminating cytosine residues in genomic S DNA and converting them to dU (7–9, 15). The mutagenic potential of uracil is very high since it can be efficiently replicated like normal thymine to yield C to T transition mutations (64). Indeed, ectopic expression of AID in non–B cells leads to a promiscuous mutator phenotype and tumorigenesis (65–67) and indicates that B cells must have highly specific targeting mechanisms to avoid the introduction of mutations by capricious AID. These considerations point to two scenarios in which isotype-specific FRMs may function in the mechanism of CSR. First, it is possible that isotype-specific factors are adaptor proteins that tether AID to S regions via FRMs. Gt expression, which renders an S target accessible and isotype-specific adaptors used to recruit AID to the FRMs, would together initiate CSR and provide two levels of protection against the promiscuous mutator function associated with AID. Second, isotype-specific activities and their FRMs might be involved in the resolution phase of CSR in which DNA lesions located in S regions are targeted to repair foci specifically engineered for S/S recombination. More work is needed to define the function of FRMS and isotype-specific activities in the mechanism of CSR.
Acknowledgments
We thank Drs. T. Honjo for the AID−/− mice, J. Stavnezer for the critical reading of this manuscript, and C. Snapper for reagents. This work was supported by the National Institutes of Health grant AI 45045 (to A.L. Kenter).
The online version of this article contains supplemental material.
A. Shanmugam's present address is Box 8134, Dept. of Psychiatry, Washington University, 660 S. Euclid Ave., Saint Louis, MO 63110-1010.
Abbreviations used in this study: αδdex, anti–δ dextran; AID, activation-induced deaminase; CSR, class switch recombination; DC, Digestion circularization; DSB, double strand break; FRM, functional recombination motif; gt, germline transcription; MMR, mismatch repair; nAChR, nonrearranging acetylcholine receptor; RRL, relative recombination level; TR, tandem repeat.
References
- 1.Stavnezer, J. 2000. Molecular processes that regulate class switching. Curr. Top. Microbiol. Immunol. 245:127–168. [DOI] [PubMed] [Google Scholar]
- 2.Manis, J.P., M. Tian, and F.W. Alt. 2002. Mechanism and control of class-switch recombination. Trends Immunol. 23:31–39. [DOI] [PubMed] [Google Scholar]
- 3.Muramatsu, M., K. Kinoshita, S. Fagarasan, S. Yamada, Y. Shinkai, and T. Honjo. 2000. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 102:553–563. [DOI] [PubMed] [Google Scholar]
- 4.Muramatsu, M., V.S. Sankaranand, S. Anant, M. Sugai, K. Kinoshita, N.O. Davidson, and T. Honjo. 1999. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J. Biol. Chem. 274:18470–18476. [DOI] [PubMed] [Google Scholar]
- 5.Revy, P., T. Muto, Y. Levy, F. Geissmann, A. Plebani, O. Sanal, N. Catalan, M. Forveille, R. Dufourcq-Labelouse, A. Gennery, et al. 2000. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell. 102:565–575. [DOI] [PubMed] [Google Scholar]
- 6.Kenter, A.L. 2003. Class-switch recombination: after the dawn of AID. Curr. Opin. Immunol. 15:190–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Noia, J., and M.S. Neuberger. 2002. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature. 419:43–48. [DOI] [PubMed] [Google Scholar]
- 8.Bransteitter, R., P. Pham, M.D. Scharff, and M.F. Goodman. 2003. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc. Natl. Acad. Sci. USA. 100:4102–4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petersen-Mahrt, S.K., R.S. Harris, and M.S. Neuberger. 2002. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 418:99–103. [DOI] [PubMed] [Google Scholar]
- 10.Rada, C., G.T. Williams, H. Nilsen, D.E. Barnes, T. Lindahl, and M.S. Neuberger. 2002. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr. Biol. 12:1748–1755. [DOI] [PubMed] [Google Scholar]
- 11.Wuerffel, R.A., J. Du, R.J. Thompson, and A.L. Kenter. 1997. Ig Sγ3 DNA-specific double strand breaks are induced in mitogen-activated B cells and are implicated in switch recombination. J. Immunol. 159:4139–4144. [PubMed] [Google Scholar]
- 12.Chen, X., K. Kinoshita, and T. Honjo. 2001. Variable deletion and duplication at recombination junction ends: implication for staggered double-strand cleavage in class-switch recombination. Proc. Natl. Acad. Sci. USA. 98:13860–13865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Catalan, N., F. Selz, K. Imai, P. Revy, A. Fischer, and A. Durandy. 2003. The block in immunoglobulin class switch recombination caused by activation-induced cytidine deaminase deficiency occurs prior to the generation of DNA double strand breaks in switch mu region. J. Immunol. 171:2504–2509. [DOI] [PubMed] [Google Scholar]
- 14.Imai, K., G. Slupphaug, W.I. Lee, P. Revy, S. Nonoyama, N. Catalan, L. Yel, M. Forveille, B. Kavli, H.E. Krokan, et al. 2003. Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nat. Immunol. 4:1023–1028. [DOI] [PubMed] [Google Scholar]
- 15.Chaudhuri, J., M. Tian, C. Khuong, K. Chua, E. Pinaud, and F.W. Alt. 2003. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature. 422:726–730. [DOI] [PubMed] [Google Scholar]
- 16.Ramiro, A.R., P. Stavropoulos, M. Jankovic, and M.C. Nussenzweig. 2003. Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat. Immunol. 4:452–456. [DOI] [PubMed] [Google Scholar]
- 17.Reaban, M.E., and J.A. Griffin. 1990. Induction of RNA-stabilized DNA conformers by transcription of an immunoglobulin switch region. Nature. 348:342–344. [DOI] [PubMed] [Google Scholar]
- 18.Reaban, M.E., J. Lebowitz, and J.A. Griffin. 1994. Transcription induces the formation of a stable RNA.DNA hybrid in the immunoglobulin alpha switch region. J. Biol. Chem. 269:21850–21857. [PubMed] [Google Scholar]
- 19.Daniels, G.A., and M.R. Lieber. 1995. RNA:DNA complex formation upon transcription of immunoglobulin switch regions: implications for the mechanism and regulation of class switch recombination. Nucleic Acids Res. 23:5006–5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu, K., F. Chedin, C.L. Hsieh, T.E. Wilson, and M.R. Lieber. 2003. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat. Immunol. 4:442–451. [DOI] [PubMed] [Google Scholar]
- 21.Casellas, R., A. Nussenzweig, R. Wuerffel, R. Pelanda, A. Reichlin, H. Suh, X.-F. Qin, E. Besmer, A. Kenter, K. Rajewsky, and M.C. Nussenzweig. 1998. Ku80 is required for immunoglobulin isotype switching. EMBO J. 17:2404–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manis, J., Y. Gu, R. Lansford, E. Sonoda, R. Ferrini, L. Davidson, K. Rajewsky, and F. Alt. 1998. Ku70 Is required for late B cell development and immunoglobulin heavy chain class switching. J. Exp. Med. 187:2081–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Manis, J.P., D. Dudley, L. Kaylor, and F.W. Alt. 2002. IgH class switch recombination to IgG1 in DNA-PKcs-deficient B cells. Immunity. 16:607–617. [DOI] [PubMed] [Google Scholar]
- 24.Bosma, G.C., J. Kim, T. Urich, D.M. Fath, M.G. Cotticelli, N.R. Ruetsch, M.Z. Radic, and M.J. Bosma. 2002. DNA-dependent protein kinase activity is not required for immunoglobulin class switching. J. Exp. Med. 196:1483–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ehrenstein, M.R., and M.S. Neuberger. 1999. Deficiency in Msh2 affects the efficiency and local sequence specificity of immunoglobulin class switch recombination. EMBO J. 18:3484–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ehrenstein, M.R., C. Rada, A.M. Jones, C. Milstein, and M.S. Neuberger. 2001. Switch junction sequences in PMS2-deficient mice reveal a microhomology-mediated mechanism of Ig class switch recombination. Proc. Natl. Acad. Sci. USA. 98:14553–14558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schrader, C.E., J. Vardo, and J. Stavnezer. 2002. Role for mismatch repair proteins Msh2, Mlh1, and Pms2 in immunoglobulin class switching shown by sequence analysis of recombination junctions. J. Exp. Med. 195:367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schrader, C.E., J. Vardo, and J. Stavnezer. 2003. Mlh1 can function in antibody class switch recombination independently of Msh2. J. Exp. Med. 197:1377–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reina-San-Martin, B., S. Difilippantonio, L. Hanitsch, R.F. Masilamani, A. Nussenzweig, and M.C. Nussenzweig. 2003. H2AX is required for recombination between immunoglobulin switch regions but not for intra-switch region recombination or somatic hypermutation. J. Exp. Med. 197:1767–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Celeste, A., S. Petersen, P.J. Romanienko, O. Fernandez-Capetillo, H.T. Chen, O.A. Sedelnikova, B. Reina-San-Martin, V. Coppola, E. Meffre, M.J. Difilippantonio, et al. 2002. Genomic instability in mice lacking histone H2AX. Science. 296:922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petersen, S., R. Casellas, B. Reina-San-Martin, H.T. Chen, M.J. Difilippantonio, P.C. Wilson, L. Hanitsch, A. Celeste, M. Muramatsu, D.R. Pilch, et al. 2001. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature. 414:660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zelazowski, P., D. Carrasco, F. Rosas, M. Moorman, R. Bravo, and C. Snapper. 1997. B cells genetically deficient in the c-Rel transactivation domain have selective defects in germline CH transcription and Ig class switching. J. Immunol. 159:3133–3139. [PubMed] [Google Scholar]
- 33.Snapper, C., P. Zelazowski, F. Rosas, F. Kehry, M. Tian, D. Baltimore, and W. Sha. 1996. B cells from p50/NFκB knockout mice have selective defects in proliferation, differentiation, germline CH transcription and Ig class switching. J. Immunol. 156:183–191. [PubMed] [Google Scholar]
- 34.Wuerffel, R.A., L. Ma, and A.L. Kenter. 2001. NF-κB p50-dependent in vivo footprints at Ig Sγ3 DNA are correlated with μ→γ3 switch recombination. J. Immunol. 166:4552–4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shanmugam, A., M.-J. Shi, L. Yauch, J. Stavnezer, and A. Kenter. 2000. Evidence for class specific factors in immunoglobulin isotype switching. J. Exp. Med. 191:1365–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma, L., H. Wortis, and A.L. Kenter. 2002. Two new isotype specific switching factors detected for Ig class switching. J. Immunol. 168:2835–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sha, W., H.-C. Liou, E. Tuomanen, and D. Baltimore. 1995. Targeted disruption of the p50 subunit of NFkB leads to multifocal defects in immune resposes. Cell. 80:321–330. [DOI] [PubMed] [Google Scholar]
- 38.Du, J., Y. Zu, A. Shanmugam, and A.L. Kenter. 1997. Analysis of immunoglobulin Sγ3 recombination breakpoints by PCR: implications for the mechanism of isotype switching. Nucleic Acids Res. 25:3066–3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gritzmacher, C.A. 1989. Molecular aspects of heavy-chain class switching. Crit. Rev. Immunol. 9:173–200. [PubMed] [Google Scholar]
- 40.Dunnick, W., M. Wilson, and J. Stavnezer. 1989. Mutations, duplication, and deletion of recombined switch regions suggest a role for DNA replication in the immunoglobulin heavy-chain switch. Mol. Cell. Biol. 9:1850–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dunnick, W., G.Z. Hertz, L. Scappino, and C. Gritzmacher. 1993. DNA sequences at immunoglobulin switch region recombination sites. Nucleic Acids Res. 21:365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dunnick, W., and J. Stavnezer. 1990. Copy choice mechanism of immunoglobulin heavy chain switch recombination. Mol. Cell. Biol. 10:397–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wuerffel, R., C.E. Jamieson, L. Morgan, G.V. Merkulov, R. Sen, and A.L. Kenter. 1992. Switch recombination breakpoints are strictly correlated with DNA recognition motifs for immunoglobulin Sγ3 DNA-binding proteins. J. Exp. Med. 176:339–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kenter, A.L., R. Wuerffel, R. Sen, C.E. Jamieson, and G.V. Merkulov. 1993. Switch recombination breakpoints occur at nonrandom positions in the Sγ tandem repeat. J. Immunol. 151:4718–4731. [PubMed] [Google Scholar]
- 45.Min, I.M., C.E. Schrader, J. Vardo, T.M. Luby, N. D'Avirro, J. Stavnezer, and E. Selsing. 2003. The Smu tandem repeat region is critical for Ig isotype switching in the absence of Msh2. Immunity. 19:515–524. [DOI] [PubMed] [Google Scholar]
- 46.Schrader, C.E., W. Edelmann, R. Kucherlapati, and J. Stavnezer. 1999. Reduced isotype switching in splenic B cells from mice deficient in mismatch repair enzymes. J. Exp. Med. 190:323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McIntyre, T.M., M.R. Kehry, and C.M. Snapper. 1995. Novel in vitro model for high-rate IgA class switching. J. Immunol. 154:3156–3161. [PubMed] [Google Scholar]
- 48.Evans, E., and E. Alani. 2000. Roles for mismatch repair factors in regulating genetic recombination. Mol. Cell. Biol. 20:7839–7844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakagawa, T., A. Datta, and R.D. Kolodner. 1999. Multiple functions of MutS- and MutL-related heterocomplexes. Proc. Natl. Acad. Sci. USA. 96:14186–14188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marra, G., I. Iaccarino, T. Lettieri, G. Roscilli, P. Delmastro, and J. Jiricny. 1998. Mismatch repair deficiency associated with overexpression of the MSH3 gene. Proc. Natl. Acad. Sci. USA. 95:8568–8573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Drummond, J.T., J. Genschel, E. Wolf, and P. Modrich. 1997. DHFR/MSH3 amplification in methotrexate-resistant cells alters the hMutSalpha/hMutSbeta ratio and reduces the efficiency of base-base mismatch repair. Proc. Natl. Acad. Sci. USA. 94:10144–10149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chang, D.K., L. Ricciardiello, A. Goel, C.L. Chang, and C.R. Boland. 2000. Steady-state regulation of the human DNA mismatch repair system. J. Biol. Chem. 275:29178. [PubMed] [Google Scholar]
- 53.Okazaki, I.M., K. Kinoshita, M. Muramatsu, K. Yoshikawa, and T. Honjo. 2002. The AID enzyme induces class switch recombination in fibroblasts. Nature. 416:340–345. [DOI] [PubMed] [Google Scholar]
- 54.Luby, T.M., C.E. Schrader, J. Stavnezer, and E. Selsing. 2001. The Sμ switch region tandem repeats are important, but not required, for antibody class switch recombination. J. Exp. Med. 193:159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dudley, D.D., J.P. Manis, A.A. Zarrin, L. Kaylor, M. Tian, and F.W. Alt. 2002. Internal IgH class switch region deletions are position-independent and enhanced by AID expression. Proc. Natl. Acad. Sci. USA. 99:9984–9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nagaoka, H., M. Muramatsu, N. Yamamura, K. Kinoshita, and T. Honjo. 2002. Activation-induced deaminase (AID)-directed hypermutation in the immunoglobulin Smu region: implication of AID involvement in a common step of class switch recombination and somatic hypermutation. J. Exp. Med. 195:529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schrader, C.E., S.P. Bradley, J. Vardo, S.N. Mochegova, E. Flanagan, and J. Stavnezer. 2003. Mutations occur in the Ig S micro region but rarely in Sgamma regions prior to class switch recombination. EMBO J. 22:5893–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shinkura, R., M. Tian, M. Smith, K. Chua, Y. Fujiwara, and F.W. Alt. 2003. The influence of transcriptional orientation on endogenous switch region function. Nat. Immunol. 4:435–441. [DOI] [PubMed] [Google Scholar]
- 59.Pham, P., R. Bransteitter, J. Petruska, and M.F. Goodman. 2003. Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature. 424:103–107. [DOI] [PubMed] [Google Scholar]
- 60.Dickerson, S.K., E. Market, E. Besmer, and F.N. Papavasiliou. 2003. AID mediates hypermutation by deaminating single stranded DNA. J. Exp. Med. 197:1291–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stavnezer, J., S. Bradley, N. Rousseau, T. Pearson, A. Shanmugam, D. Waite, P. Rogers, and A. Kenter. 1999. Switch recombination in a transfected plasmid occurs specifically in a B cell line that undergoes switch recombination of its chromosomal Ig heavy chain genes. J. Immunol. 163:2028–2040. [PubMed] [Google Scholar]
- 62.Paques, F., and J.E. Haber. 1997. Two pathways for removal of nonhomologous DNA ends during double strand break repair in Saccharomyces cerevisiae. Mol. Cell. Biol. 17:6765–6771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sugawara, N., F. Paques, M. Colaiacovo, and J. Haber. 1997. Role of Msh2 and Msh3 repair proteins in double strand break induced recombination. Proc. Natl. Acad. Sci. USA. 94:9214–9219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lindahl, T. 2000. Suppression of spontaneous mutagenesis in human cells by DNA base excision-repair. Mutat. Res. 462:129–135. [DOI] [PubMed] [Google Scholar]
- 65.Yoshikawa, K., I.M. Okazaki, T. Eto, K. Kinoshita, M. Muramatsu, H. Nagaoka, and T. Honjo. 2002. AID enzyme-induced hypermutation in an actively transcribed gene in fibroblasts. Science. 296:2033–2036. [DOI] [PubMed] [Google Scholar]
- 66.Martin, A., P.D. Bardwell, C.J. Woo, M. Fan, M.J. Shulman, and M.D. Scharff. 2002. Activation-induced cytidine deaminase turns on somatic hypermutation in hybridomas. Nature. 415:802–806. [DOI] [PubMed] [Google Scholar]
- 67.Okazaki, I.M., H. Hiai, N. Kakazu, S. Yamada, M. Muramatsu, K. Kinoshita, and T. Honjo. 2003. Constitutive expression of AID leads to tumorigenesis. J. Exp. Med. 197:1173–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]