

Abstract
Leukocyte migration is a key event both in host defense against invading pathogens as well as in inflammation. Bacteria generate chemoattractants primarily by excretion (formylated peptides), complement activation (C5a), and subsequently through activation of leukocytes (e.g., leukotriene B4, platelet-activating factor, and interleukin 8). Here we describe a new protein secreted by Staphylococcus aureus that specifically impairs the response of neutrophils and monocytes to formylated peptides and C5a. This chemotaxis inhibitory protein of S. aureus (CHIPS) is a 14.1-kD protein encoded on a bacteriophage and is found in >60% of clinical isolates. CHIPS reduces the neutrophil recruitment toward C5a in a mouse peritonitis model, even though its activity is much more potent on human than on mouse cells. These findings suggest a new immune escape mechanism of S. aureus and put forward CHIPS as a potential new antiinflammatory therapeutic compound.
Keywords: Staphylococcus aureus, phagocytes, chemotaxis, C5a, formylated peptides
Introduction
Staphylococcus aureus is a Gram-positive pathogen and a major cause of wound and nosocomial infections in humans causing high morbidity and mortality (1). S. aureus produces a wide variety of exoproteins that contribute to its ability to colonize and cause disease in the host (2). The recent increase in septic S. aureus diseases and in strains resistant to virtually all available antibiotics puts urgency to a better understanding of staphylococcal virulence strategies and new means to combat S. aureus infections (3).
An immediate host response toward bacterial infection is the migration of leukocytes from the circulation into the sites of infection. Numerous exogenously and endogenously produced leukocyte chemoattractants that initiate leukocyte migration and activation have been identified. They include both the “classical” chemoattractants, like formylated peptides, C5a, C3a, platelet-activating factor (PAF), and leukotriene B4, as well as the superfamily of chemokines that selectively induce infiltration, trafficking, and homing of leukocytes (4, 5). The effects of all of these chemoattractants on their target cells are mediated through specific cell surface receptors, which belong to the superfamily of seven transmembrane, heterotrimeric G protein–coupled receptors (GPCRs; 6). Neutrophils bear several members of this receptor family on their surface including the receptors for formylated peptides, C5a, C3a, PAF, and leukotriene B4, as well as the chemokine receptors CXCR1 and CXCR2 that recognize IL-8 and NAP/GRO/ENA, respectively. After activation, these receptors are desensitized to repeated stimulation with the same or other agonists, which is called homologous and heterologous desensitization, respectively. Desensitization might be critical for maintaining the capacity of the cells to sense a chemoattractant gradient (7).
Although the ability of S. aureus to escape from human host defenses is a well-known characteristic, the underlying mechanisms have remained unclear. Early reports suggest that some S. aureus strains possess an activity to delay leukocyte migration toward the site of infection. In 1967, Agarwal (8) discovered a strong strain-dependent difference in the ability to produce a purulent lesion after subcutaneous injection of staphylococci in mice. Mice challenged with “virulent” strains showed a 2-h delay in leukocyte influx and accumulation of edema fluid compared with “nonvirulent” strains. It was postulated that these virulent strains inhibit early leukocyte migration toward the site of infection (8). Hill (9) reported in 1968 that in contrast to nonvirulent strains, isolated cell wall fractions of “mouse-virulent” strains of S. aureus inhibited the accumulation of edema fluid at the site of injection. In 1976, Russell et al. (10) found chemotaxis-inhibiting properties for neutrophils and monocytes of several S. aureus strains in a modified Boyden chamber system.
These reports prompted us to look further into the proposed chemotaxis-inhibiting properties of S. aureus. Recently, we described an extracellular component produced by S. aureus that specifically impaired neutrophil chemotaxis toward fMLP, a synthetic formylated peptide, and C5a (11). In this study, we isolated and identified this substance as a new protein, chemotaxis inhibitory protein of S. aureus (CHIPS), which is able to specifically inhibit C5a- and fMLP-induced responses of neutrophils and monocytes. By constructing an S. aureus CHIPS knockout and complemented strain, we show that the activity completely matched with the presence of the CHIPS gene. CHIPS is produced by S. aureus in vivo and is also able to reduce the C5a-induced recruitment of neutrophils into the peritoneal cavity in a mouse model.
Materials and Methods
Reagents.
fMLP and C5a were obtained from Sigma-Aldrich. IL-8 and macrophage inflammatory protein 1α (MIP-1α) were purchased from PeproTech. Boron dipyrromethane (BODIPY)- and FITC-labeled fMLP (BODIPY-N-formyl-Nle-Leu-Phe-Nle-Tyr-Lys), BCECF-AM, and Fluo-3-AM were obtained from Molecular Probes. S5/1 mAb (anti-CD88; C5aR) was provided by O. Götze (University Göttingen, Göttingen, Germany). SE-2 (anti-CD128w; IL-8RA) and anti-PAFR mAb were from Qbiogene. The secondary FITC-labeled goat F(ab′)2 anti–mouse Ig was from DakoCytomation. MMK-1 peptide (LESIFRSLLFRVM) was synthesized by Sigma-Genosys.
Chromatography.
Chromatographic procedures were performed with FPLC™ equipment from Amersham Biosciences at 4°C. S. aureus Newman or clinical isolate 1690 supernatant (11) was passed over a reactive Yellow 86, a deoxyribonucleic acid cellulose, and a Reactive Green 19 column, respectively (all from Sigma-Aldrich), coupled in tandem. The Reactive Green column was eluted with 2 M NaCl containing 1 mM phenylmethylsulfonylfluoride. The eluate was passed over a Superdex 200 HR column (Amersham Biosciences). Fractions were screened in a flow cytometer for inhibition of binding of BODIPY-labeled formylated peptides to neutrophils as previously described (11). Active fractions were applied to a Mono Q HR anion exchange column (Amersham Biosciences) in 10 mM Tris-HCl, pH 8.0, and eluted with 0–1 M NaCl. Active fractions were analyzed on 12.5% SDS-PAGE.
Receptor Expression on Human Neutrophils and Chemotaxis.
Human neutrophil isolation and receptor expression were performed as previously described (11). In brief, human neutrophils were incubated with increasing concentrations of CHIPS for 30 min at 37°C in RPMI containing 0.05% human serum albumin (HSA; RPMI/0.05% HSA). The samples were then incubated with 10 μg/ml anti-C5aR, anti–IL-8RA, anti-PAFR mAb, or 1 μM BODIPY-labeled fMLP on ice for 30 min. After washing twice, the neutrophils, incubated with mAbs, were incubated with FITC-labeled goat F(ab′)2 anti–mouse Ig for 30 min on ice. Samples were analyzed on a FACScan™ flow cytometer (Becton Dickinson). Chemotaxis of human neutrophils toward chemoattractants C5a and fMLP was determined using BCECF-labeled neutrophils as previously described (11). In brief, neutrophils were incubated with increasing concentrations of CHIPS for 30 min at 37°C in RPMI/0.05% HSA. The cells were then washed once and resuspended in HBSS/1% HSA (2.5 × 106 c/ml). The cells (100 μl) were added to the upper compartment of a Transwell system (3 μm; Costar), which was placed into a 24-well plate containing 600 μl HBSS/1% HSA, 10−7 M fMLP or 10−10 M C5a. After 30 min of incubation at 37°C/5% CO2, the inlays were removed and the fluorescence of the well was read in a CytofluorII multiwell fluorometer (PerSeptive Biosystems).
NH2-terminal Sequencing.
For NH2-terminal sequencing, the purified protein was blotted onto a polyvinylidene difluoride membrane and stained with Coomassie brilliant blue according to the manufacturer's instructions (ProBlott; Applied Biosystems). The band of interest was excised and sequenced by the Sequence Center Utrecht.
DNA Sequencing.
Genomic DNA of S. aureus Newman was isolated as previously described (12). A set of degenerative primers was designed on basis of the first 32 sequenced amino acids taking into account the codon usage of staphylococci and used to directly sequence the genomic S. aureus Newman DNA by cycle sequencing as previously described (13). With primer 5′-GAAAAAGAAAAAGCATATAAAGAA-3′ a sequence was obtained, which was extended by primer walking. The program tblastn with the nonredundant DNA database and the microbial genomes database at http://www.ncbi.nlm.nih.gov was used to perform sequence similarity searches. The chp sequence is available from GenBank/EMBL/DDBJ under accession number AF 285146.
chp Knockout.
To inactivate the chp gene of S. aureus Newman, DNA fragments of 840 bp upstream and 510 bp downstream of chp were amplified by PCR and cloned together with the ermB gene from Tn551 into the temperature-sensitive shuttle plasmid pBT2 as depicted in Fig. 2 A. After cloning in Escherichia coli DH5α, the resulting plasmid pBTΔchp was transformed into S. aureus Newman to achieve integration of the ermB gene into the genome by homologous recombination. Mutants were enriched by cultivation at 42°C in the presence of 2.5 μg/ml erythromycin. The proper integration of ermB, which is considerably larger than chp, was confirmed by PCR analysis. A 290-bp fragment encoding the mature portion of CHIPS was deleted. To complement the knockout strain with the chp gene, plasmid pRBchp was constructed by cloning a 1,293-bp PCR fragment bearing the chp gene together with the putative promoter region (738 bp noncoding upstream DNA) into the shuttle vector pRB473. E. coli– and Staphylococcus-specific plasmid vectors and molecular methods used have been described (13–16).
Figure 2.

S. aureus Newman CHIPS knockout strain. Construction of S. aureus Newman CHIPS knockout results in the total loss of CHIPS activity and the CHIPS protein band in the culture supernatant. A CHIPS knockout was constructed by homologous recombination with plasmid pBTΔchp, whereby an erythromycin resistance cassette replaced chp (A). Binding of BODIPY-labeled formylated peptide to neutrophils after preincubation with different concentrations (vol/vol) of supernatant of S. aureus Newman wild-type (•), S. aureus Newman Δchp (○), and S. aureus Newman Δchp complemented with pBRchp (□) (B) or S. aureus COL wild-type (○) and S. aureus COL transformed with pBRchp (▪) (C). Data are mean ± SEM of three independent experiments. Detection of CHIPS in S. aureus culture supernatants by Western blot analysis using CHIPS-specific polyclonal rabbit antiserum. Lanes 1 and 4 contain proteins from S. aureus Newman wild-type, lane 2 from S. aureus Newman Δchp, lane 3 from S. aureus Newman Δchp-pRBchp, lane 5 from S. aureus COL wild-type, and lane 6 from S. aureus COL-pRBchp (D).
Expression of CHIPS in E. coli.
The chp gene, except for the signal sequence, was amplified by PCR on chromosomal DNA of S. aureus Newman using Pwo DNA polymerase (Roche Diagnostics). The PCR product was cloned into the pTrcHISB vector (Invitrogen) directly downstream the enterokinase cleavage site. The vector was transformed into TOP10 E. coli and recombinant CHIPS was expressed and purified according to the manufacturer's instructions (Invitrogen).
Detection of chp in Clinical S. aureus Isolates.
PCR was performed on the chromosomal DNA of 244 clinical S. aureus isolates using the forward primer 5′-GAAAAAGAAATTAGCAACAACAG-3′ and the reverse primer 5′-CATAAGATGATTTAGACTCTCC-3′.
CHIPS Binding to Blood Cells.
To determine the binding of CHIPS to different cell types, FITC-labeled CHIPS (CHIPS-FITC) was used. FITC-labeling of CHIPS was performed as follows. 500 μg/ml CHIPS was incubated with 50 μg/ml FITC in 0.1 M sodium carbonate buffer, pH 9.6, for 1 h at 37°C. Subsequently, CHIPS-FITC was separated from unbound FITC using a G25-desalting column (Amersham Biosciences). Fractions were collected and tested for the presence of CHIPS (OD280) and FITC (OD496) in a spectrophotometer. Fractions containing CHIPS-FITC were pooled. Human neutrophils and mononuclear cells were incubated with increasing concentrations of CHIPS-FITC for 45 min on ice in the presence of PE-conjugated anti-CD14 mAb (Becton Dickinson). Samples were analyzed on a FACScan™ flow cytometer using scatters and anti-CD14 staining to distinguish the different cell populations. In separate experiments, CHIPS-FITC binding was tested on neutrophils of mice, rabbits, rats, guinea pigs, pigs, and dogs. Leukocytes were isolated from heparinized blood by either red blood cell lysis of whole blood or standard Ficoll-Histopaque isolation as for human cells. Specific binding of CHIPS-FITC was determined to neutrophils identified by their characteristic scatter pattern.
Calcium Mobilization.
Calcium mobilization with isolated human neutrophils and monocytes (17) was performed as previously described (11). In brief, Fluo-3–loaded cells were incubated with or without 1 μg/ml CHIPS. Basal calcium levels were then measured in the FACSCalibur™ flow cytometer (Becton Dickinson), after which agonist (10−9 M fMLP, 10−10 M C5a, 10−10 M IL-8, or 10−10 M MIP-1α) was added. 5 min after the first stimulus, the neutrophils were rechallenged with a second agonist. In separate experiments, different concentrations of CHIPS were tested on mouse and human whole blood leukocytes. Specific neutrophil response to 10−10 M C5a was measured by electronic gating on scatters (human) and specific anti–mouse granulocyte mAb staining (R-PE–conjugated RB6-8C5; Caltag).
CHIPS Production In Vivo.
S. aureus Newman or its chp deletion mutant were grown overnight on a blood agar plate and washed in PBS. C57Bl mice were challenged intraperitoneally with 200 μl (109 CFU) S. aureus Newman or its chp deletion mutant. After 4 h, mice were killed, the peritoneal lavage (2.5 ml) was collected, and the amount of CHIPS was determined with a specific ELISA using a mAb as capture and a polyclonal rabbit IgG as detection antibody. Recombinant CHIPS was used as the reference standard protein. The ELISA has a limit of detection of 100 pg/ml. All animal experiments were performed in compliance with University of Utrecht guidelines and were approved by the institutional animal care and ethics committee.
Mouse Neutrophil Influx Model.
8–12-wk-old C57Bl mice were pretreated by intravenous injection of 100 μl PBS containing 0, 3, 10, or 30 μg CHIPS and after 15 min they were challenged intraperitoneally with 10 μg human C5a. After 3 h, mice were killed, the peritoneal lavage was collected, and the number of emigrated neutrophils was quantified and analyzed by flow cytometry by differential staining using CyChrome-conjugated anti–mouse CD45 (BD Biosciences) and R-PE–conjugated anti–mouse granulocyte (RB6-8C5).
Results
Isolation of CHIPS.
The chemotaxis-inhibiting component in the supernatant of the clinical strain S. aureus 1690 (11) was purified by ligand dye chromatography, gel permeation, and anion exchange chromatography. To detect the samples containing the chemotaxis-inhibiting activity, all samples were screened by flow cytometry for the inhibition of binding of fluorescent-labeled formylated peptides to neutrophils. The final preparation revealed a nearly homogeneous protein band of ∼15 kD on SDS-PAGE (Fig. 1 A). The purified protein exhibited the same activity as the supernatant. It inhibited binding of formylated peptides and anti-C5aR antibodies to neutrophils (Fig. 1 B) as well as neutrophil chemotaxis toward fMLP and C5a (Fig. 1 C). Neither expression of the receptors for IL-8 and PAF were affected nor their chemotactic activity. The protein itself showed no direct chemotactic activity (not shown). NH2-terminal sequencing identified the first 32 amino acids, which showed no major homology with any protein in databases. We named this new protein chemotaxis inhibitory protein of S. aureus (CHIPS).
Figure 1.

Purified CHIPS inhibits neutrophil chemotaxis toward C5a and fMLP. (A) CHIPS was purified by ligand dye chromatography and gel filtration from S. aureus culture supernatant. Active fractions were pooled and concentrated. SDS-PAGE revealed a band of ∼15 kD. The right lane represents a marker. (B) CHIPS affects C5aR and FPR expression on neutrophils in a dose-dependent fashion. Binding of BODIPY-formylated peptide (♦) and anti-C5aR mAb (•) to their receptors on neutrophils is inhibited after 30 min of incubation with different concentrations of CHIPS, whereas binding of anti-PAFR (▪) and anti–IL-8R (▴) mAb is not inhibited. Data are expressed as fluorescence values compared with neutrophils without CHIPS and are mean ± SEM of three separate experiments. (C) CHIPS inhibits chemotaxis of neutrophils toward 10−7 M fMLP (♦) and 10−10 M C5a (•) after incubation of neutrophils with different concentrations of CHIPS. Data are expressed as the percentage of migrated neutrophils added to the upper compartment of the transwell chemotaxis chamber and are mean ± SEM of one representative experiment (n = 2–4). Migration of untreated neutrophils was 79% for C5a and 85% for fMLP.
Identification of the CHIPS Gene.
To identify the CHIPS gene, we performed direct sequencing on genomic DNA of S. aureus Newman, which is also a CHIPS-producing strain. This yielded a DNA sequence encoding a protein of 149 amino acids: MKKKLATTVLALSFLTAGISTHHHSAKA FTFEPFPTNEEIESNKKMLEKEKAYKESFKNSGLP-TTLGKLDERLRNYLKKGTKNSAQFEKMVILTEN-KGYYTVYLNTPLAEDRKNVELLGKMYKTYFFKK-GESKSSYVINGPGKTNEYAY. The NH2-terminal 28 amino acids constituted a signal peptide containing a signal peptidase 1 cleavage site (underlined) followed by amino acids that perfectly matched the NH2-terminal CHIPS sequence (bold). The mature protein had a mass of 14.1 kD. The gene, named chp, was preceded by a Shine Dalgarno sequence and followed by three stop codons. In strain Newman chp was found to be located on the 3′ end of a bacteriophage. This bacteriophage is incorporated in the gene for β-toxin (hlb) and its 3′ end carries, beside chp, the genes for staphylokinase (sak) and staphylococcal enterotoxin A (sea).
CHIPS Knockout S. aureus Strain Lacks Chemotaxis-inhibiting Activity.
To confirm that chp was responsible for the CHIPS activity in the S. aureus supernatant, a CHIPS knockout strain was constructed by replacement of chp with an erythromycin resistance cassette (Fig. 2 A). This resulted in total loss of CHIPS activity and absence of the protein in the culture supernatant (Fig. 2, B and D). When the knockout strain was complemented with plasmid pRBchp, containing chp with its putative promoter region, CHIPS activity was restored. When S. aureus COL (chp negative by PCR) was transformed with pRBchp, it showed CHIPS activity and produced the protein (Fig. 2, C and D). These data show that CHIPS activity correlates with the presence of chp. Additionally, recombinant CHIPS produced by E. coli had exactly the same activity as native S. aureus CHIPS (unpublished data).
Prevalence in Clinical S. aureus Isolates.
To investigate the prevalence of chp in clinical S. aureus isolates, we screened 204 methicillin-sensitive isolates (102 from blood stream infections and 102 from healthy nasal carriers) and 40 methicillin-resistant S. aureus strains by PCR. Of all strains, 62% were positive for chp and the presence of chp neither correlated with the clinical source of the strains nor differed between methicillin-sensitive and methicillin-resistant isolates.
CHIPS Binds to Human Neutrophils and Monocytes.
To examine the binding capacity of CHIPS to different leukocytes, FITC-labeled CHIPS was used. CHIPS specifically bound to human neutrophils and monocytes and not to lymphocytes (Fig. 3), which indicates its specificity for those cells that pose the greatest threat for invading staphylococci.
Figure 3.

CHIPS binds to phagocytes. Dose-dependent binding of CHIPS-FITC to neutrophils (♦), monocytes (•), and lymphocytes (▴). Cells were identified based on scatter parameters and anti-CD14 staining. Data are mean ± SEM of five independent experiments.
CHIPS Specifically Inhibits the fMLP- and C5a-induced Calcium Mobilization.
The hallmark for the GPCR is a rapid and transient increase in free intracellular calcium upon ligand binding (6, 18). The inhibiting effect of CHIPS on the fMLP- and C5a-induced calcium mobilization in human neutrophils was not due to desensitization. CHIPS specifically blocked the calcium mobilization of neutrophils induced by fMLP or C5a, but not IL-8 (Fig. 4). CHIPS-treated cells, triggered with fMLP or C5a, responded with a normal calcium response to a rechallenge with IL-8 (Fig. 4, A and C). Vice versa, CHIPS-treated cells were normally activated by IL-8, but failed to mount a calcium mobilization upon rechallenge with fMLP or C5a (Fig. 4, B and D). Buffer-treated cells were unaffected in their primary response to fMLP, C5a, and IL-8, whereas their secondary response to a heterologous stimulus was lower due to desensitization. The full secondary response to IL-8 of CHIPS-treated cells, preexposed to fMLP or C5a, can be explained by the efficient FPR and C5aR receptor blockade by CHIPS. CHIPS itself did not induce a calcium response. CHIPS also inhibited the fMLP- and C5a-induced calcium mobilization in monocytes, whereas the response to MIP-1α was not affected (Fig. 5). In addition, CHIPS did not inhibit human neutrophils triggered with an FPRL1-specific agonist, the synthetic peptide MMK-1 derived from a random peptide library that does not activate FPR (unpublished data and 19). We conclude that CHIPS affects specifically two members of the GPCR family and is not toxic for cells because the cellular response to MMK-1, IL-8, and MIP-1α remained intact.
Figure 4.

CHIPS specifically inhibits fMLP- and C5a-induced calcium mobilization in neutrophils. The effect of CHIPS on intracellular-free calcium release induced by 10−9 M fMLP, 10−10 M C5a, and 10−10 M IL-8 in neutrophils was determined using Fluo-3–labeled cells and flow cytometry. After preincubating neutrophils with 1 μg/ml CHIPS (gray lines) or buffer (black lines), the basal fluorescence level was measured for each sample before fMLP (A), C5a (C), or IL-8 (B and D) was added (indicated by first arrow). To show receptor specificity, cells were rechallenged with a second stimulus of fMLP (B), C5a (D), or IL-8 (A and C) after 5 min (indicated by second arrow).
Figure 5.

CHIPS specifically inhibits fMLP- and C5a-induced calcium mobilization in monocytes. The effect of CHIPS on intracellular-free calcium release induced by 10−9 M fMLP, 10−10 M C5a, and 10−10 M MIP-1α in monocytes was determined using Fluo-3–labeled cells and flow cytometry. After preincubating monocytes with 1 μg/ml CHIPS (gray lines) or buffer (black lines), the basal fluorescence level was measured for each sample before fMLP (A), C5a (B), or MIP-1α (C) was added (indicated by arrow).
CHIPS Is Human Specific.
As chp is located on a bacteriophage together with the two human-specific virulence genes, sak and sea, we tested the species specificity of CHIPS. Binding of FITC-labeled CHIPS to isolated neutrophils of different animal species revealed a low binding to neutrophils of all species tested when compared with human neutrophils (Fig. 6 A). Furthermore, the efficacy of CHIPS to inhibit the C5a-induced calcium mobilization was evaluated with human and mouse neutrophils. CHIPS showed a 30-fold reduced capacity to inhibit the C5a-induced calcium mobilization in mouse neutrophils as compared with human neutrophils (Fig. 6 B). These results suggest that CHIPS is human specific.
Figure 6.
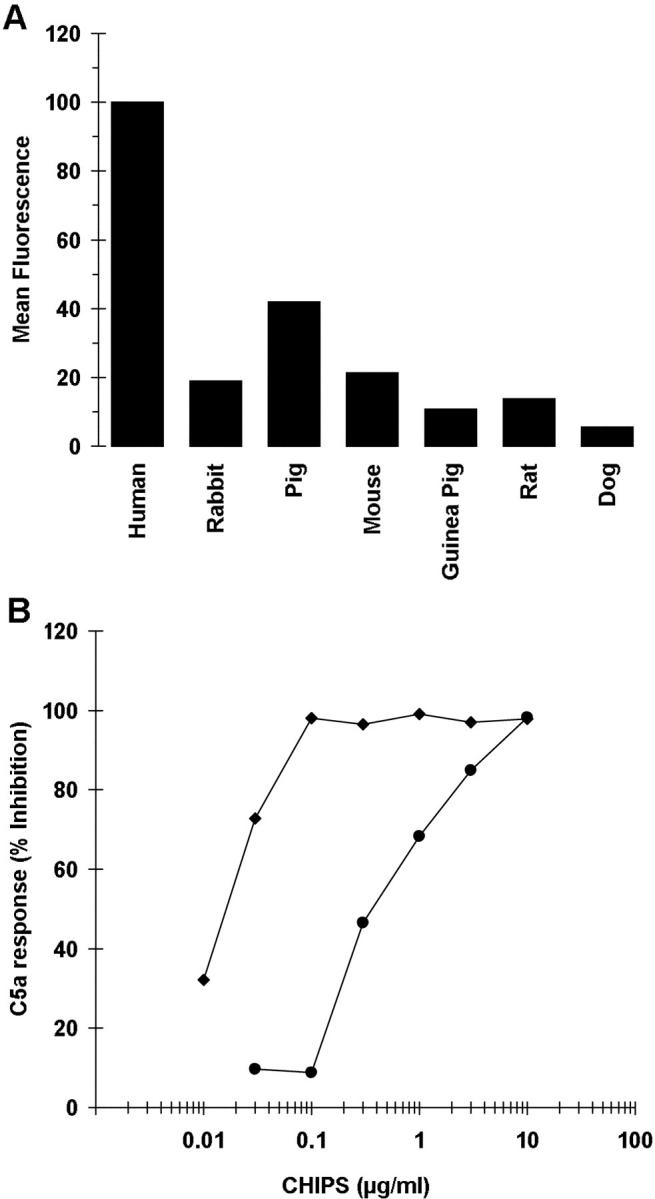
CHIPS binding to neutrophils of different species. (A) Leukocytes of different species were incubated with 1 μg/ml CHIPS-FITC and analyzed by flow cytometry based on scatter parameters to identify neutrophils. Data shown are from one representative experiment. (B) The effect of different concentrations CHIPS on the C5a-induced calcium mobilization in human (♦) and mouse (•) neutrophils. Data are expressed as percentage inhibition of the response of control cells to 10−10 M C5a (n = 2). The response was defined by the difference in mean fluorescence (MFL) of basal level and maximal level after challenge (ΔMFL = 84.3 for human and 22.9 for mouse).
CHIPS Is Produced In Vivo.
CHIPS is secreted by S. aureus in vitro in sufficient amounts to inhibit neutrophil activation. However, to demonstrate a role for CHIPS in the virulence of S. aureus, we examined whether S. aureus produces detectable amounts of CHIPS in vivo. Therefore, mice were injected intraperitoneally with 109 CFU S. aureus Newman or the CHIPS knockout strain. After 4 h, 8.18 ± 2.89 ng CHIPS was recovered from the peritoneal lavage of mice injected with S. aureus Newman (n = 6), whereas no CHIPS was found in knockout strain-injected mice (n = 2). This shows that in a localized infection, S. aureus actively secretes only minimal amounts of CHIPS in the surroundings.
CHIPS Inhibits the Mouse Neutrophil Influx In Vivo.
To investigate the potency of CHIPS to specifically inhibit the neutrophil migration in vivo, mice were intravenously treated with CHIPS and subsequently, after 15 min, intraperitoneally challenged with human C5a. At least 10 μg of CHIPS per mouse was needed to give a 65% inhibition of neutrophil influx (Fig. 7). These results indicate a potential use of CHIPS as an antiinflammatory drug in diseases where influx of neutrophils is more detrimental than beneficial for the host.
Figure 7.

CHIPS inhibits the C5a-induced neutrophil migration in vivo. Mice were pretreated by intravenous injection of different concentrations of CHIPS and after 15 min were challenged intraperitoneally with human C5a. After 3 h, the percentage of neutrophils in the peritoneal lavage was analyzed. Data are expressed as percentage inhibition compared with buffer-treated mice challenged with C5a after correction for control nontreated mice. Data are mean ± SEM of two to three separate experiments (n = 2 mice per treatment).
Discussion
When S. aureus invades the human host, complement is rapidly activated, resulting in opsonization of the bacteria and the generation of large amounts of C5a (20–22). Together with formylated peptides, side products of bacterial translation, generation of C5a constitutes the first trigger of the innate immune system (23). These early signs of bacterial invasion are recognized by the innate immune system through two related receptors on neutrophils, the C5aR and FPR. In vitro, in vivo, and possibly during S. aureus infection, recognition by these two receptors is counteracted by CHIPS as it inhibits specifically the C5a- and fMLP-induced responses in neutrophils and monocytes. Moreover, CHIPS inhibits the neutrophil recruitment toward C5a in a mouse peritonitis model. Because CHIPS did not inhibit receptors for secondary chemoattractants, such as IL-8, we hypothesize that CHIPS plays a major role early in bacterial invasion. Once staphylococci have multiplied abundantly, the overwhelming presence of other chemokines will overcome CHIPS inhibitory effects causing massive influx of neutrophils. This results in the typical hallmark of a staphylococcal infection, a purulent lesion (1). As yet, it is not known how CHIPS expression is regulated, but we would predict from its ligand specificity that CHIPS expression is most abundant at the onset of infection.
As CHIPS is an actively excreted protein of S. aureus, the S. aureus–induced chemotaxis-inhibiting findings of Agarwal (8) might be attributed to the in vivo production of CHIPS by the virulent S. aureus strains. The observation of a 2-h delay in leukocyte migration toward the infected site would correlate with our hypothesis of an early effect of CHIPS inhibiting the leukocyte recruitment toward the two first chemoattractants produced, formylated peptides and C5a. Recently, extracellular adherence protein (Eap) of S. aureus was reported to inhibit leukocyte chemotaxis by its interaction with intercellular adhesion molecule 1 (24). Like CHIPS, Eap was produced by S. aureus Newman in vivo, which suggests that this strain has at least two different mechanisms to avoid leukocyte recruitment (25).
Given its potent action on human phagocytes, CHIPS may represent an important virulence factor. This seems to be in agreement with the abundance of chp (62%) in S. aureus isolates. The presence of chp did not correlate with the development of bactereamia. There are two different explanations for this. We hypothesize that CHIPS is mainly important in the initial interaction with the host and mainly contributes to effective colonization or early stages of infection. In addition, there might be some redundancy in virulence factors, such as Eap, which avoid the innate immune system. Once the bacterium harbors more than a threshold number of these factors, the bacterium becomes pathogenic. This research includes CHIPS as one of these factors.
The human specificity of CHIPS as shown by a 30-fold difference in activity toward human cells as compared with mouse cells, in combination with its low in vivo expression, hampers testing of CHIPS in a mouse infection model or other animal models. Apart from chp, the other adjacent genes sak and sea have also been reported to be highly human specific. The response of mouse T cells to the superantigen staphylococcal enterotoxin A requires 1,000-fold higher concentrations compared with human T cells (26). In addition, the ability of bacterial plasminogen activators to cleave different animal plasminogen is restricted. Staphylokinase could only activate plasminogen of humans, dogs, and baboons (27). The exact role of CHIPS in S. aureus virulence will be a subject for future studies. As an in vivo infection model was not possible due to the human specificity of CHIPS, we included a mouse peritonitis model with purified CHIPS. As we could simply overdose CHIPS in this model, we were able to show a proof of principle for the in vivo anti-chemotactic action of CHIPS.
CHIPS fulfils a unique function, affecting two related receptors with strikingly different ligand specificities. Both the C5aR and FPR belong to the superfamily of seven transmembrane, heterotrimeric GPCRs (28, 29). All members of this family have a comparable architecture and show a 20–30% amino acid homology, mainly in the transmembrane regions, whereas the ligands are highly diverse and receptor specific. Small molecules, such as fMLP, bind to their GPCR in the interhelical region, whereas larger molecules, such as C5a, use a two-site motif for binding their GPCR (30, 31).
How CHIPS interferes with both fMLP and C5a responses is still speculative. It is unlikely that CHIPS affects common downstream signaling events related to the GPCR as was shown for pertussis toxin (32), because the MMK-1, IL-8, and MIP-1α responses were unaffected. Binding of CHIPS-FITC was restricted to phagocytes, in which neutrophils showed a twofold higher binding than monocytes. This coincides with the expression of the C5aR and FPR on the respective cell types (33–36) and could therefore implicate direct binding to these chemokine receptors. The fact that CHIPS binds to phagocytes implies that CHIPS probably is not a protease-like protein cleaving (a part of) the receptor, as was described for other receptors (37, 38). Alternative explanations, in which CHIPS could act via a separate CHIPS receptor or interferes with proper surface exposure of C5aR and FPR by mimicking conserved receptor transmembrane regions (39), do not provide a plausible explanation for its narrow specificity for C5aR and FPR. The exact mechanism of CHIPS inhibiting fMLP- and C5a-induced responses will be further investigated.
We present data that implicate a new virulence strategy of S. aureus and thereby a new potential target in the treatment of S. aureus infection. In addition, the potent capacity of CHIPS to inhibit neutrophil chemotaxis, in vitro and in vivo, makes this new protein a promising candidate antiinflammatory drug for those diseases in which C5a-induced damage by neutrophils plays a pivotal role (40–44).
Acknowledgments
We thank Drs. C.M.J.E. Vandenbroucke-Grauls, W.T.M. Jansen, and T. van der Bruggen for critically reading the manuscript.
C.J.C. de Haas and K.E. Veldkamp contributed equally to this work.
Abbreviations used in this paper: BODIPY, boron dipyrromethane; CHIPS, chemotaxis inhibitory protein of Staphylococcus aureus; Eap, extracellular adherence protein; GPCR, G protein–coupled receptor; HSA, human serum albumin; MIP-1α, macrophage inflammatory protein 1α; PAF, platelet-activating factor.
References
- 1.Lowy, F.D. 1998. Staphylococcus aureus infections. N. Engl. J. Med. 339:520–532. [DOI] [PubMed] [Google Scholar]
- 2.Dinges, M.M., P.M. Orwin, and P.M. Schlievert. 2000. Exotoxins of Staphylococcus aureus. Clin. Microbiol. Rev. 13:16–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sieradzki, K., R.B. Roberts, S.W. Haber, and A. Tomasz. 1999. The development of vancomycin resistance in a patient with methicillin-resistant Staphylococcus aureus infection. N. Engl. J. Med. 340:517–523. [DOI] [PubMed] [Google Scholar]
- 4.Rollins, B.J. 1997. Chemokines. Blood. 90:909–928. [PubMed] [Google Scholar]
- 5.DeVries, M.E., L. Ran, and D.J. Kelvin. 1999. On the edge: the physiological and pathophysiological role of chemokines during inflammatory and immunological responses. Semin. Immunol. 11:95–104. [DOI] [PubMed] [Google Scholar]
- 6.Murdoch, C., and A. Finn. 2000. Chemokine receptors and their role in inflammation and infectious diseases. Blood. 95:3032–3043. [PubMed] [Google Scholar]
- 7.Ferguson, S.S. 2001. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol. Rev. 53:1–24. [PubMed] [Google Scholar]
- 8.Agarwal, D.S. 1967. Subcutaneous staphylococcal infection in mice. II. The inflammatory response to different strains of staphylococci and micrococci. Br. J. Exp. Pathol. 48:468–482. [PMC free article] [PubMed] [Google Scholar]
- 9.Hill, M.J. 1968. A staphylococcal aggressin. J. Med. Microbiol. 1:31–43. [DOI] [PubMed] [Google Scholar]
- 10.Russell, R.J., P.C. Wilkinson, R.J. McInroy, S. McKay, A.C. McCartney, and J.P. Arbuthnott. 1976. Effects of staphylococcal products on locomotion and chemotaxis of human blood neutrophils and monocytes. J. Med. Microbiol. 9:433–439. [DOI] [PubMed] [Google Scholar]
- 11.Veldkamp, K.E., H.C. Heezius, J. Verhoef, J.A. van Strijp, and K.P. van Kessel. 2000. Modulation of neutrophil chemokine receptors by Staphylococcus aureus supernate. Infect. Immun. 68:5908–5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mamur, J. 1961. A procedure for isolation of deoxyribonucleic acid from microorganisms. J. Mol. Biol. 3:208–218. [Google Scholar]
- 13.Peschel, A., and F. Gotz. 1996. Analysis of the Staphylococcus epidermidis genes epiF, -E, and -G involved in epidermin immunity. J. Bacteriol. 178:531–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bruckner, R. 1997. Gene replacement in Staphylococcus carnosus and Staphylococcus xylosus. FEMS Microbiol. Lett. 151:1–8. [DOI] [PubMed] [Google Scholar]
- 15.Bruckner, R. 1992. A series of shuttle vectors for Bacillus subtilis and Escherichia coli. Gene. 122:187–192. [DOI] [PubMed] [Google Scholar]
- 16.Augustin, J., and F. Gotz. 1990. Transformation of Staphylococcus epidermidis and other staphylococcal species with plasmid DNA by electroporation. FEMS Microbiol. Lett. 54:203–207. [DOI] [PubMed] [Google Scholar]
- 17.Boven, L.A., L. Gomes, C. Hery, F. Gray, J. Verhoef, P. Portegies, M. Tardieu, and H.S. Nottet. 1999. Increased peroxynitrite activity in AIDS dementia complex: implications for the neuropathogenesis of HIV-1 infection. J. Immunol. 162:4319–4327. [PubMed] [Google Scholar]
- 18.Bockaert, J., and J.P. Pin. 1999. Molecular tinkering of G protein-coupled receptors: an evolutionary success. EMBO J. 18:1723–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le, Y., P.M. Murphy, and J.M. Wang. 2002. Formyl-peptide receptors revisited. Trends Immunol. 23:541–548. [DOI] [PubMed] [Google Scholar]
- 20.Kawasaki, A., H. Takada, S. Kotani, S. Inai, K. Nagaki, M. Matsumoto, K. Yokogawa, S. Kawata, S. Kusumoto, and T. Shiba. 1987. Activation of the human complement cascade by bacterial cell walls, peptidoglycans, water-soluble peptidoglycan components, and synthetic muramylpeptides–studies on active components and structural requirements. Microbiol. Immunol. 31:551–569. [DOI] [PubMed] [Google Scholar]
- 21.Wilkinson, B.J., Y. Kim, and P.K. Peterson. 1981. Factors affecting complement activation by Staphylococcus aureus cell walls, their components, and mutants altered in teichoic acid. Infect. Immun. 32:216–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neth, O., D.L. Jack, A.W. Dodds, H. Holzel, N.J. Klein, and M.W. Turner. 2000. Mannose-binding lectin binds to a range of clinically relevant microorganisms and promotes complement deposition. Infect. Immun. 68:688–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schiffmann, E., B.A. Corcoran, and S.M. Wahl. 1975. N-formylmethionyl peptides as chemoattractants for leucocytes. Proc. Natl. Acad. Sci. USA. 72:1059–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chavakis, T., M. Hussain, S.M. Kanse, G. Peters, R.G. Bretzel, J.I. Flock, M. Herrmann, and K.T. Preissner. 2002. Staphylococcus aureus extracellular adherence protein serves as anti-inflammatory factor by inhibiting the recruitment of host leukocytes. Nat. Med. 8:687–693. [DOI] [PubMed] [Google Scholar]
- 25.Palma, M., A. Haggar, and J.I. Flock. 1999. Adherence of Staphylococcus aureus is enhanced by an endogenous secreted protein with broad binding activity. J. Bacteriol. 181:2840–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dohlsten, M., M. Bjorklund, A. Sundstedt, G. Hedlund, D. Samson, and T. Kalland. 1993. Immunopharmacology of the superantigen staphylococcal enterotoxin A in T-cell receptor V beta 3 transgenic mice. Immunology. 79:520–527. [PMC free article] [PubMed] [Google Scholar]
- 27.Gladysheva, I.P., R.B. Turner, I.Y. Sazonova, L. Liu, and G.L. Reed. 2003. Coevolutionary patterns in plasminogen activation. Proc. Natl. Acad. Sci. USA. 100:9168–9172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gerard, N.P., and C. Gerard. 1991. The chemotactic receptor for human C5a anaphylatoxin. Nature. 349:614–617. [DOI] [PubMed] [Google Scholar]
- 29.Boulay, F., M. Tardif, L. Brouchon, and P. Vignais. 1990. The human N-formylpeptide receptor. Characterization of two cDNA isolates and evidence for a new subfamily of G-protein-coupled receptors. Biochemistry. 29:11123–11133. [DOI] [PubMed] [Google Scholar]
- 30.Mills, J.S., H.M. Miettinen, D. Barnidge, M.J. Vlases, S. Wimer-Mackin, E.A. Dratz, J. Sunner, and A.J. Jesaitis. 1998. Identification of a ligand binding site in the human neutrophil formyl peptide receptor using a site-specific fluorescent photoaffinity label and mass spectrometry. J. Biol. Chem. 273:10428–10435. [DOI] [PubMed] [Google Scholar]
- 31.Siciliano, S.J., T.E. Rollins, J. DeMartino, Z. Konteatis, L. Malkowitz, G. Van Riper, S. Bondy, H. Rosen, and M.S. Springer. 1994. Two-site binding of C5a by its receptor: an alternative binding paradigm for G protein-coupled receptors. Proc. Natl. Acad. Sci. USA. 91:1214–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katada, T., and M. Ui. 1982. Direct modification of the membrane adenylate cyclase system by islet-activating protein due to ADP-ribosylation of a membrane protein. Proc. Natl. Acad. Sci. USA. 79:3129–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chenoweth, D.E., and T.E. Hugli. 1978. Demonstration of specific C5a receptor on intact human polymorphonuclear leukocytes. Proc. Natl. Acad. Sci. USA. 75:3943–3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Epps, D.E., and D.E. Chenoweth. 1984. Analysis of the binding of fluorescent C5a and C3a to human peripheral blood leukocytes. J. Immunol. 132:2862–2867. [PubMed] [Google Scholar]
- 35.Sklar, L.A., D.A. Finney, Z.G. Oades, A.J. Jesaitis, R.G. Painter, and C.G. Cochrane. 1984. The dynamics of ligand-receptor interactions. Real-time analyses of association, dissociation, and internalization of an N-formyl peptide and its receptors on the human neutrophil. J. Biol. Chem. 259:5661–5669. [PubMed] [Google Scholar]
- 36.Weinberg, J.B., J.J. Muscato, and J.E. Niedel. 1981. Monocyte chemotactic peptide receptor. Functional characteristics and ligand-induced regulation. J. Clin. Invest. 68:621–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kahn, J., B. Walcheck, G.I. Migaki, M.A. Jutila, and T.K. Kishimoto. 1998. Calmodulin regulates L-selectin adhesion molecule expression and function through a protease-dependent mechanism. Cell. 92:809–818. [DOI] [PubMed] [Google Scholar]
- 38.Liu, L.Y., J.B. Sedgwick, M.E. Bates, R.F. Vrtis, J.E. Gern, H. Kita, N.N. Jarjour, W.W. Busse, and E.A. Kelly. 2002. Decreased expression of membrane IL-5 receptor alpha on human eosinophils: II. IL-5 down-modulates its receptor via a proteinase-mediated process. J. Immunol. 169:6459–6466. [DOI] [PubMed] [Google Scholar]
- 39.Tarasova, N.I., W.G. Rice, and C.J. Michejda. 1999. Inhibition of G-protein-coupled receptor function by disruption of transmembrane domain interactions. J. Biol. Chem. 274:34911–34915. [DOI] [PubMed] [Google Scholar]
- 40.Stevens, J.H., P. O'Hanley, J.M. Shapiro, F.G. Mihm, P.S. Satoh, J.A. Collins, and T.A. Raffin. 1986. Effects of anti-C5a antibodies on the adult respiratory distress syndrome in septic primates. J. Clin. Invest. 77:1812–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Czermak, B.J., V. Sarma, C.L. Pierson, R.L. Warner, M. Huber-Lang, N.M. Bless, H. Schmal, H.P. Friedl, and P.A. Ward. 1999. Protective effects of C5a blockade in sepsis. Nat. Med. 5:788–792. [DOI] [PubMed] [Google Scholar]
- 42.Jacob, H.S., and D.E. Hammerschmidt. 1981. Complement-induced granulocyte aggregation. Importance in myocardial infarction and shock lung. JAMA. 245:2013–2017. [DOI] [PubMed] [Google Scholar]
- 43.Pemberton, M., G. Anderson, V. Vetvicka, D.E. Justus, and G.D. Ross. 1993. Microvascular effects of complement blockade with soluble recombinant CR1 on ischemia/reperfusion injury of skeletal muscle. J. Immunol. 150:5104–5113. [PubMed] [Google Scholar]
- 44.Jose, P.J., I.K. Moss, R.N. Maini, and T.J. Williams. 1990. Measurement of the chemotactic complement fragment C5a in rheumatoid synovial fluids by radioimmunoassay: role of C5a in the acute inflammatory phase. Ann. Rheum. Dis. 49:747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]