

Abstract
Psoriasis is a common T cell–mediated autoimmune disorder where primary onset of skin lesions is followed by chronic relapses. Progress in defining the mechanism for initiation of pathological events has been hampered by the lack of a relevant experimental model in which psoriasis develops spontaneously. We present a new animal model in which skin lesions spontaneously developed when symptomless prepsoriatic human skin was engrafted onto AGR129 mice, deficient in type I and type II interferon receptors and for the recombination activating gene 2. Upon engraftment, resident human T cells in prepsoriatic skin underwent local proliferation. T cell proliferation was crucial for development of a psoriatic phenotype because blocking of T cells led to inhibition of psoriasis development. Tumor necrosis factor-α was a key regulator of local T cell proliferation and subsequent disease development. Our observations highlight the importance of resident T cells in the context of lesional tumor necrosis factor-α production during development of a psoriatic lesion. These findings underline the importance of resident immune cells in psoriasis and will have implications for new therapeutic strategies for psoriasis and other T cell–mediated diseases.
Keywords: autoimmunity, immunotherapy, inflammation, mouse model, skin
Introduction
Psoriasis represents one of the most common T cell–mediated autoimmune diseases (1–5). The initial onset of skin lesions is followed by chronic relapses of disease, not unlike the clinical course for multiple sclerosis or rheumatoid arthritis (6). Based on various animal models (7–9) and indirect clinical observations (1, 6), T cells responsible for psoriatic skin lesions are thought to be confined to the circulating pool of immune cells. However, the role of resident T cells in the development of psoriatic lesions has not been investigated yet, mainly due to the lack of an appropriate animal model. Current psoriasis mouse models using SCID mice necessitate the injection of activated PBMCs with or without superantigen to induce a psoriatic phenotype in the human prepsoriatic skin graft (7, 8). SCID (and recombinase activating gene (RAG)−/−) mice lack T and B cells but show mature NK cells with normal NK cell activity (10–12). Since NK cells are involved in rejection of xenogeneic tissue (13), SCID mice are not ideal acceptors of human skin grafts. To improve graft acceptance, we used mice deficient in type I (A) and type II (G) IFN receptors in addition to being RAG-2−/− (AGR129 mice). AGR129 mice show immature NK cells with severely impaired cytotoxic (cytolytic) activity in vitro and in vivo (unpublished data) due to a deficiency in type I (A) and type II (G) IFN receptors, in addition to lacking T and B cells (RAG-2−/−) (14). Disruption of both IFN receptors has been previously shown to lead to decreased NK cytotoxic (cytolytic) activity in vitro and in vivo (15), whereas type I IFN receptor– or type II IFN receptor–deficient mice demonstrated reduced but fully functional NK cell activity (12, 16). In the current study, we use AGR129 mice as excellent acceptors of human prepsoriatic skin grafts to show spontaneous onset of bona fide psoriatic lesions that do not require any exogenous cells or factor except for those contained within the engrafted skin itself. Our observations establish proliferation of local T cells, dependent on TNF-α production, as essential elements of psoriatic lesion formation.
Materials and Methods
Animals, Patients, and Transplantation Procedure.
AGR129, mice, mice deficient in type I (A) and type II (G) IFN receptors in addition to being RAG-2−/−; AR129 mice, mice deficient in type I (A) IFN receptors in addition to being RAG-2−/−; and GR129 mice, mice deficient in type II (G) IFN receptors in addition to being RAG-2−/− were either on a 129Sv/Ev or C57BL/6 background. Mice were kept pathogen free throughout the study. Animal studies were approved by the Kantonale Veterinaeramt of Zurich and human studies by the Institutional Review Boards of the University Hospital of Zurich. Keratome biopsies (6 × 2 × 0.04 cm) of symptomless prepsoriatic skin (PN skin) were taken from the lower back or buttock of patients with confirmed plaque-type psoriasis after informed consent was obtained. No topical or systemic medication was administered for at least 4 wk before the study. Skin grafts were transplanted to the back of mice using an absorbable tissue seal (Vet-Seal; B. Braun Medical AG).
Immunohistochemistry, Confocal Laser Scanning Microscopy (CLSM) Analysis, and Flow Cytometry Analysis.
Acetone-fixed cryostat sections were stained using standard staining techniques. Unspecific Fc receptor binding of antibodies was measured with isotype-matched controls. Unconjugated antibodies used were monoclonals to CD1c (Immunotech), CD1d (51.1.3, provided by Dr. S.P. Balk, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, MA), CD3 (Dako), CD4 (Dako), CD8 (Dako), CD31 (Dako), CD54 (BD Biosciences), HLA-DR (BD Biosciences), IFN-γ (BD Biosciences), IL-12 (R&D Systems), keratin 16 (Sigma-Aldrich), Ki-67 (Dako), TNF-α (R&D Systems), polyclonals to involucrin (Anawa Trading SA), and conjugated CD3-PE (BD Biosciences), CD69-FITC (BD Biosciences), CD83-PE (Immunotech), and TNF-α–FITC (BD Biosciences). Sensitivity of the flow cytometry analyses was between 1:1,000 and 1:3,000 human to mouse PBMCs for the anti–human CD3-PE mAb (Fig. S1 A, available at http://www.jem.org/cgi/content/full/jem.20031482/DC1).
Histologic Assessment and Quantification Experiments.
Histologic quantification experiments represent the mean of three random fields with a 400-fold magnification. The acanthosis and papillomatosis index was defined as published (17). The indicated values of both indices represent the mean of 10 random areas of each sample.
Quantitative Real-time RT-PCR.
Total RNA was extracted from organs (High Pure RNA Isolation kit; Roche) followed by reverse transcription (AMV First Strand cDNA Synthesis kit; Roche). Real-time PCR was performed in a LightCycler (Roche). Primer sequences were for human GAPDH, 5′-ATTGCCCTCAACGACCACTTTG-3′ and 5′-TTGATGGTACATGACAAGGTGCGG-3′ and mouse GAPDH, 5′-CATCAAGAAGGTGGTGAAGC-3′ and 5′-CCTGTTGCTGT-AGCCGTATT-3′. Sensitivity of the RT-PCR method was between 1:100,000 and 1:1,000,000 human PBMCs to mouse splenocytes for the human GAPDH primer (Fig. S1 B), and its specificity for human cDNA was demonstrated by the lack of amplification of specific cDNA sequences when applied on RNA isolated from spleens of normal C57BL/6 mice.
Neutralization Studies.
Dosage and route of administration of the reagents applied was deduced from therapeutic trials in humans, calculated using the allometric approach, and administered as follows: 17 μg muromonab-CD3 (neutralizing anti–human CD3 mAb, clone OKT3) twice weekly, i.v.; 1,000 μg infliximab (anti–human TNF-α mAb) on days 0, 14, 28, 42, and 56, i.v.; 90 μg etanercept (TNF receptor fusion protein) twice weekly, s.c. Control mice received isotype-matched antibodies or PBS.
Online Supplemental Material.
Fig. S1 shows the detection threshold for flow cytometry and RT-PCR analyses, and Fig. S2 shows TNF-α production by few T cells. Figs. S1 and S2 are available at http://www.jem.org/cgi/content/full/jem.20031482/DC1.
Results and Discussion
Spontaneous Development of a Psoriatic Phenotype upon Transplantation of Symptomless Prepsoriatic Skin onto AGR129 Mice.
PN skin from 12 different patients with confirmed plaque-type psoriasis was transplanted onto AGR129 mice. The skin grafts developed a psoriatic phenotype in 28 out of 31 (90%) grafted mice. Phenotypic conversion started at week 4 and was fully developed at 6–8 wk after engraftment. Appearance of PN skin on the day of transplantation (Fig. 1, A and B) was comparable to normal human skin. In contrast, 8 wk after transplantation onto AGR129 mice, PN skin grafts showed typical features of psoriasis (Fig. 1, C and D). Histology of PN skin after development of a psoriatic phenotype (Fig. 1 C) was comparable to a biopsy of a psoriasis lesion from the same patient donating the PN skin graft (Fig. 1 E). Several different combinations of human skin and mouse strains served to demonstrate that development of a psoriatic phenotype was unique to PN skin transplanted onto AGR129 mice (Fig. 1, F–H). Quantification of psoriatic features in a time-course experiment using papillomatosis and acanthosis indices (17) reflected significant (P = 0.0002) changes of PN skin transplants on AGR129 mice, whereas changes in phenotype were clearly absent from control skin grafts (Fig. 1 H).
Figure 1.
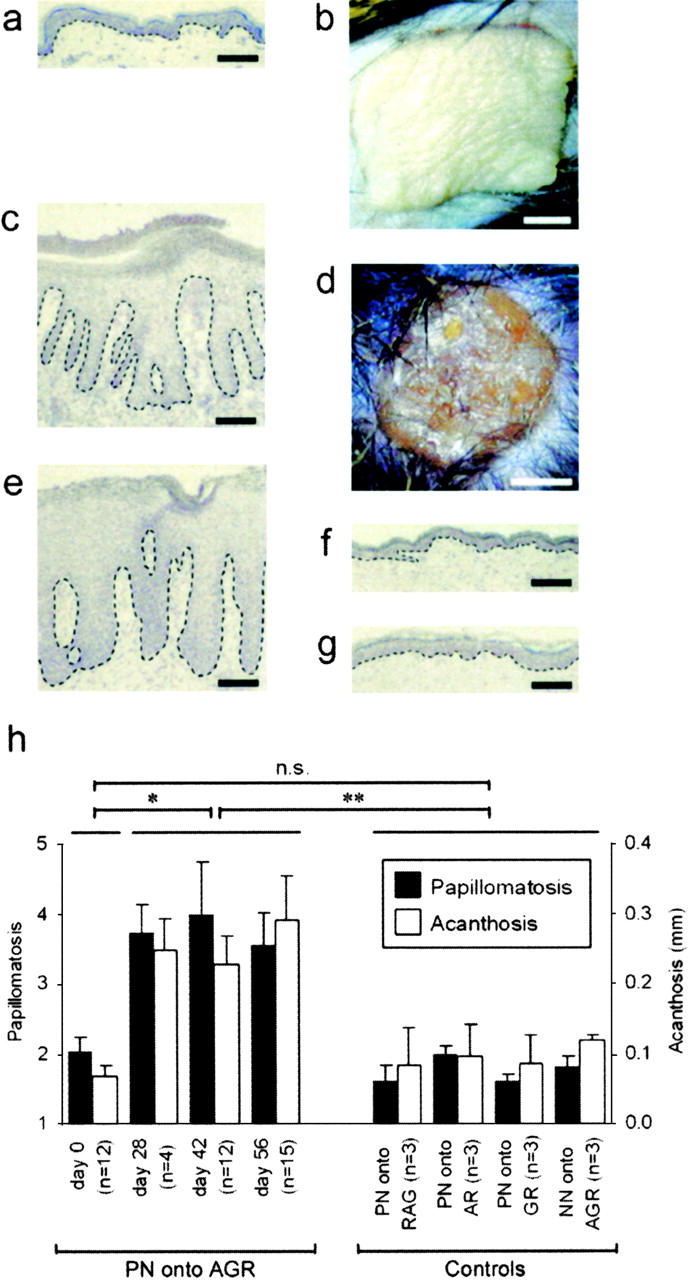
Development of a psoriatic phenotype in symptomless prepsoriatic skin after transplantation onto AGR129 mice. Microscopic (A) and macroscopic (B) appearance of PN skin on the day of transplantation was comparable to normal human skin. PN skin 6–8 wk after transplantation onto AGR129 mice developed typical histological features of psoriasis such as parakeratosis (nucleated keratinocytes in stratum corneum), focal loss of the granular cell layer, acanthosis (hyperplasia of viable epidermis), papillomatosis (elongation of dermal papillae), prominent vessels of the papillary dermis, and numerous mononuclear cells representing immune cells (C). Typical clinical signs of psoriasis such as elevation, erythema, induration, and scaling could be observed (D). Histology of a biopsy of a psoriasis lesion from the same patient donating the PN skin graft (E). Control grafting experiments such as PN skin grafts onto GR129 mice (GR) (F) and skin from healthy individuals (NN skin) onto AGR129 mice (G) 8 wk after transplantation. The microscopic changes were quantified using papillomatosis and acanthosis indices of PN skin transplants onto AGR129 mice before transplantation (day 0) and on day 28, 42, and 56 after transplantation (H). Controls were skin grafts 56 d after transplantation: PN skin transplants onto RAG-2−/− mice (PN onto RAG), AR129 mice (PN onto AR), and GR129 mice (PN onto GR), and NN skin from three healthy individuals onto AGR129 mice (NN onto AGR) (H). Dashed lines indicate border between epidermis above and dermis below. n, number of grafted mice. Error bars represent one SD. Bars: (A, C, and E–G) 100 μm; (B and D) 2 mm. n.s., not significant. *, P = 0.0002; **, P = 0.005, unpaired t test.
To further confirm that the morphologic changes of PN skin transplanted onto AGR129 mice were consistent with psoriasis, immunohistochemical analyses of PN skin before transplantation (Fig. 2, pre) and 6–8 wk after transplanation (Fig. 2, post) was performed. In summary, upon transplantation of PN skin onto AGR129 mice, resident skin cells including epidermal keratinocytes, DCs, endothelial cells, and immune cells became activated to create fully fledged psoriasis.
Figure 2.
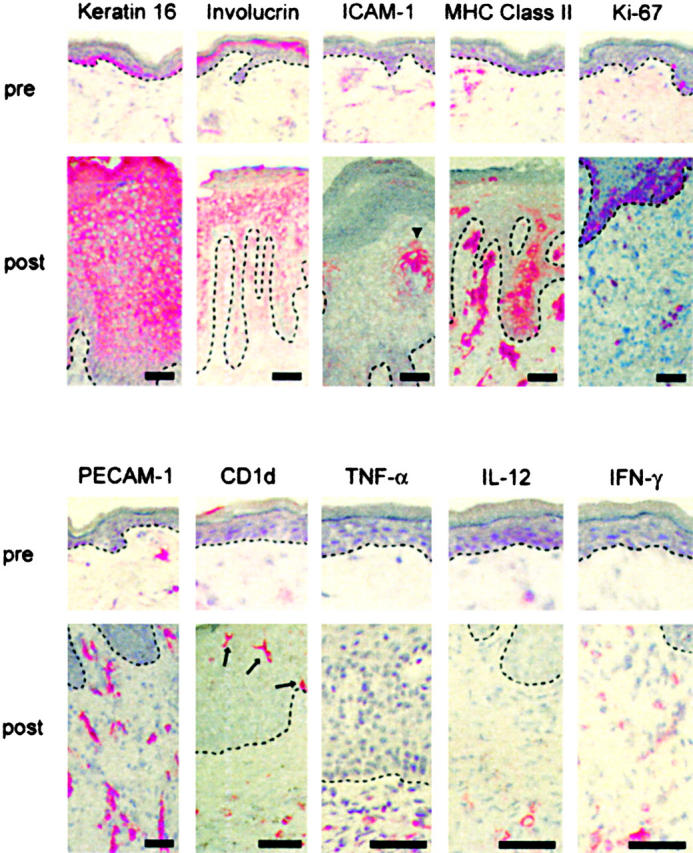
Epidermal hyperproliferation, blood vessel formation, and immune activation. In contrast to the appearance of PN skin before transplantation (pre), 6–8 wk after transplantation onto AGR129 mice (post) keratin 16 was expressed throughout the epidermis and involucrin-positive cell layers were increased, indicating aberrant epidermal differentiation. Whereas before no marked expression of intercellular adhesion molecule-1 (ICAM-1) and MHC class II by keratinocytes could by observed, transplantation onto AGR129 mice and development of a psoriatic phenotype led to up-regulation of these markers, indicating activation of keratinocytes (ICAM-1, arrowhead). Strong suprabasal expression of Ki-67 demonstrated a hyperproliferative epidermal compartment in addition to proliferating dermal cells. Marked new blood vessel formation was shown by platelet endothelial cell adhesion molecule-1 (PECAM-1) staining. An activated immune compartment was observed including expanded CD1d-positive APCs in the epidermis (arrows) and dermis as well as proinflammatory cytokines such as TNF-α, IL-12, and IFN-γ. Dashed lines indicate border between epidermis above and dermis below. Bars, 50 μm.
Resident T Cells Are Essential for Development of Lesions.
Currently existing psoriasis models implicate the importance of circulating T cells being recruited to the site of lesion (7, 8). However, they do not address the role of resident T cells during development of lesions, mainly due to the lack of an appropriate model system. The present psoriasis model allowed us to address this issue. Immunohistochemical analyses of PN skin before transplantation (Fig. 3, A and B, pre), compared with 6–8 wk after transplantation onto AGR129 mice (Fig. 3, A and B, post), revealed a more than twofold increase in total T cell numbers (P < 0.002), which corresponds to an almost fivefold increase in the tissue. We demonstrated a preference of CD4-positive cells for the dermis, whereas CD8-positive cells were located predominantly in the epidermis or the dermo-epidermal junction zone (Fig. 3 A). This finding corresponds to observations in human psoriasis samples (18). Proliferation of lesional CD3-positive cells paralleled disease formation (Fig. 3 B). T cell proliferation predominantly took place in the dermis, peaked at 4 wk after transplantation, and was followed by a decrease in the dermis and a simultaneous increase of T cells in the epidermis (Fig. 3 B). These data suggest a migration of human dermal T cells into the epidermis during psoriasis formation. Increase in the number of lesional T cells could be due to local proliferation or recruitment of T cells recirculating and proliferating extracutaneously in the immunodeficient mice (19). To test the second possibility, we investigated PBMCs, LNs, and spleens of PN skin–transplanted AGR129 mice 8 wk after engraftment for the presence of human T cells. No T cells could be detected in LNs, spleen, or PBMCs neither by immunohistochemistry, flow cytometry, nor RT-PCR analyses (Fig. 3, C–E). The local increase in T cells could reflect a nonrelevant proliferation or indicate a crucial contribution of activated T cells to disease development. T cells expressed the activation marker CD69 during psoriasis development (unpublished data). We blocked T cell function using the monoclonal anti–human CD3 antibody muromonab-CD3 (OKT3) over 8 wk starting after transplantation of PN skin onto AGR129 mice. Injection of muromonab-CD3 inhibited development of a psoriatic phenotype (Fig. 3 F) and compared with mice receiving isotype-matched control antibody induced a significant (P < 0.0001) reduction of the papillomatosis and acanthosis index in 12 out of 12 mice grafted with PN skin from three different patients (Fig. 3 G). We further showed the importance of proliferating T cells during development of lesions by administration of a neutralizing anti–human IL-2 mAb (unpublished data). These experiments clearly show the essential role of locally activated and proliferating resident T cells during psoriasis development in our model system. We cannot rule out an involvement of recruited T cells in psoriatic lesion formation in general. However, due to the high sensitivity of the methods employed (Fig. S1) recirculation and recruitment of human T cells cannot play a major role in our model. In support of our findings, there is evidence from other models of autoimmune-type inflammatory diseases that local proliferation of effector T cells, rather than their recruitment, are important for disease induction (20).
Figure 3.
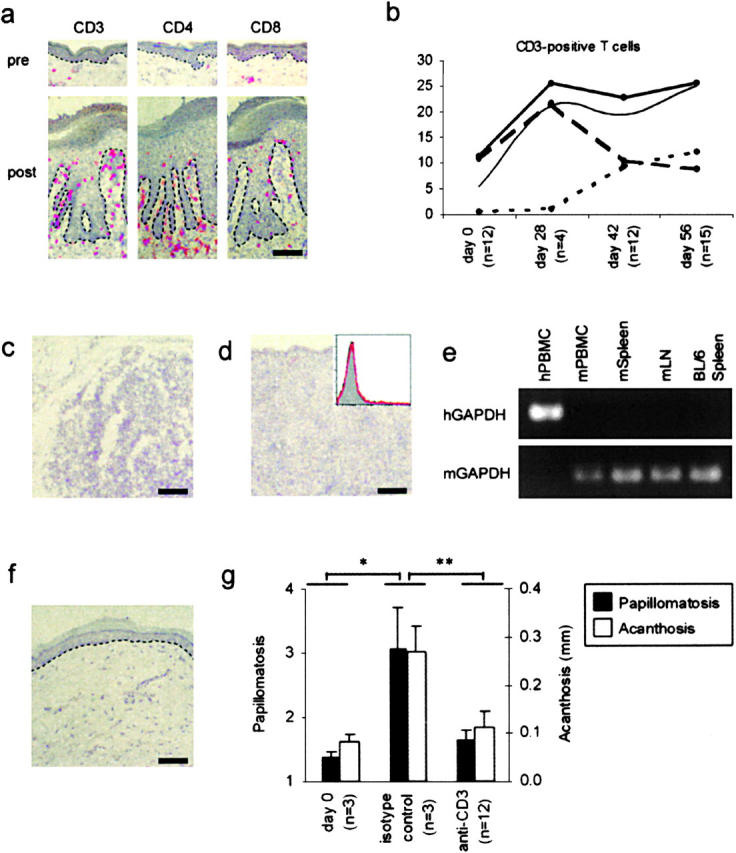
Resident T cells during development of lesions. CD3, CD4, and CD8 immunostaining of PN skin before transplantation (pre) compared with 6–8 wk after transplantation onto AGR129 mice (post) (A). Epidermal (B, dotted line), dermal (B, dashed line), and total (B, thick solid line) CD3 counts in PN skin grafts before transplantation (day 0) and on day 28, 42, and 56 after transplantation onto AGR129 mice. The thin solid line indicates acanthosis index. CD3 immunostaining of LN (C), spleen (D), and flow cytometry of PBMCs (D, insert; isotype control, gray area; CD3 staining, red line) from AGR129 mice 56 d after transplantation of PN skin grafts. RT-PCR analyses of cDNA from human PBMCs (hPBMC), mouse PBMCs (mPBMC), mouse spleen (mSpleen), and mouse LNs (mLN) from AGR129 mice 56 d after transplantation of PN skin using primers for human GAPDH (hGAPDH) and mouse GAPDH (mGAPDH) (E). Spleen of untreated C57BL/6 mice (BL/6 Spleen) served as control to show specificity of the primers used. Hematoxylin staining of PN skin grafted onto AGR129 mouse and blocking of T cells with an anti–human CD3 mAb for 8 wk (F). Papillomatosis and acanthosis indices of PN skin transplants onto AGR129 mice before transplantation (day 0), 56 d after transplantation and administration of isotype-matched antibody (isotype control) or of an anti–human CD3 mAb (anti-CD3) (G). Dashed lines, n, and error bars are defined as in the legend to Fig. 1. Bars, 70 μm. *, P < 0.01; **, P < 0.0001, unpaired t test.
TNF-α Is Crucial for Local T Cell Proliferation and Psoriasis Development.
The concept of an altered cytokine network is central to the pathogenesis of psoriasis and other autoimmune-type inflammatory diseases (21, 22). In recent clinical trials, the usage of a mAb against TNF-α (infliximab) or a soluble TNF receptor fusion protein (etanercept) resulted in disease improvement in psoriasis patients, suggesting a role for TNF-α in the pathogenesis of psoriasis (23, 24). Although supporting the importance of TNF-α in maintaining established psoriatic lesions, these studies did not investigate the role of TNF-α during lesion development. In our model, TNF-α was predominantly localized to APCs (Fig. 4, A and B). TNF-α was also observed in few dermal T cells (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20031482/DC1) but not keratinocytes or fibroblasts. Injection of a neutralizing anti–human TNF-α mAb or a TNF receptor fusion protein over 8 wk inhibited the development of psoriatic phenotype in eight out of nine mice grafted with PN skin from three different patients (Fig. 4, C and D). PN skin grafts in mice receiving anti–TNF-α treatment showed markedly reduced TNF-α levels in contrast to PN skin converted to a psoriatic phenotype (Fig. 4, E and F). Papillomatosis and acanthosis indices were significantly decreased (P < 0.01) in mice treated with an anti–TNF-α treatment (Fig. 4 G). Neutralization of TNF-α was accompanied by a significant decrease (P < 0.004) of T cells in the graft (Fig. 4 H), suggesting a feedback loop between T cell proliferation and TNF-α production as described for monocytes and suggested for T cells (25). These data demonstrate that development of psoriatic lesions is mediated by TNF-α and indicate that proliferation of resident T cells is dependent on local TNF-α production.
Figure 4.
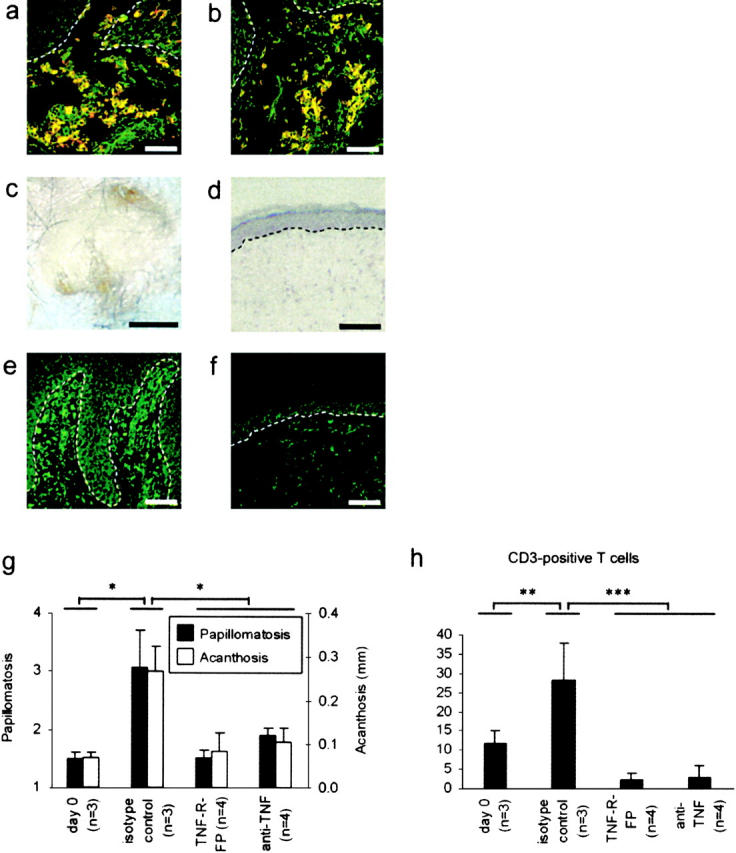
TNF-α is crucial for local T cell proliferation and psoriasis development. APCs appeared yellow in the confocal laser scanning microscope as a consequence of colocalization of specific markers CD1c (A, red) or CD83 (B, red), respectively, and TNF-α (green). Application of neutralizing anti–human TNF-α mAb or TNF receptor fusion protein over 8 wk led to the inhibition of psoriatic phenotype development (C and D). Immunostaining for TNF-α in PN skin grafts 8 wk after transplantation onto AGR129 mice and psoriasis development (E) and in PN skin grafts from mice receiving anti–TNF-α treatment (F). Papillomatosis and acanthosis indices of PN skin transplants onto AGR129 mice before transplantation (day 0) and 56 d after transplantation and administration of isotype-matched antibody (isotype control) of a TNF receptor fusion protein (TNF-R-FP) or an anti–TNF-α mAb (anti-TNF) (G). Total CD3 counts in PN skin grafts before transplantation (day 0) and 56 d after transplantation and administration of isotype-matched antibody (isotype control) of a TNF receptor fusion protein (TNF-R-FP) or an anti–TNF-α mAb (anti-TNF) (H). Dashed lines, n, and error bars are defined as in the legend to Fig. 1. Bars: (A, B, and D–F) 100 μm; (C) 2 mm. *, P = 0.01; **, P < 0.05; ***, P < 0.004, unpaired t test.
In conclusion, our findings, based on a novel autoimmune mouse model, strongly support the view that activation of resident T cells is necessary and sufficient for development of lesions in psoriasis. Local T cells are key players in this process, depending on a cytokine network governed by the primary cytokine TNF-α. These findings could have relevance for other common autoimmune diseases such as multiple sclerosis and rheumatoid arthritis, and new pharmacological agents could be envisioned focusing on resident immune cells.
Acknowledgments
We thank K. Shortman and M. Gilliet for discussions, James P. Di Santo (Institut Pasteur, Paris, France) for sharing unpublished data, and K. Hoek for reading the manuscript. We appreciate the excellent technical assistance by E. Ammann, C. Dudli, and E. Kielhorn. We further would like to thank A. Tassis and B. Odermatt (University Hospital of Zurich) and T. Baechi (Laboratory of Electron Microscopy, Zurich, Switzerland) for their help.
This work was supported by the European Union, the Bundesministerium fuer Bildung und Forschung, the Swiss National Science Foundation, the Bonizzi-Theler-Stiftung, and the National Institutes of Health (AR40065 and AR47307).
The online version of this article includes supplemental material.
H.P. Hefti and C. Conrad contributed equally to this work.
O. Boyman's present address is Dept. of Immunology, IMM4, The Scripps Research Institute, 10550 N. Torrey Pines Rd., La Jolla, CA 92037.
H.P. Hefti's present address is Institute of Virology, University of Zurich, Winterthurerstrasse 266, CH-8057 Zurich, Switzerland.
This work was presented in part at the annual meetings 2002 of the Society of Investigative Dermatology, European Society of Dermatological Research, and the Third International Congress on Psoriasis.
References
- 1.Griffiths, C.E., and J.J. Voorhees. 1996. Psoriasis, T cells and autoimmunity. J. R. Soc. Med. 89:315–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krueger, J.G. 2002. The immunologic basis for the treatment of psoriasis with new biologic agents. J. Am. Acad. Dermatol. 46:1–23. [DOI] [PubMed] [Google Scholar]
- 3.Barker, J.N. 1991. The pathophysiology of psoriasis. Lancet. 338:227–230. [DOI] [PubMed] [Google Scholar]
- 4.Gottlieb, S.L., P. Gilleaudeau, R. Johnson, L. Estes, T.G. Woodworth, A.B. Gottlieb, and J.G. Krueger. 1995. Response of psoriasis to a lymphocyte-selective toxin (DAB389IL-2) suggests a primary immune, but not keratinocyte, pathogenic basis. Nat. Med. 1:442–447. [DOI] [PubMed] [Google Scholar]
- 5.Ellis, C.N., and G.G. Krueger. 2001. Treatment of chronic plaque psoriasis by selective targeting of memory effector T lymphocytes. N. Engl. J. Med. 345:248–255. [DOI] [PubMed] [Google Scholar]
- 6.Christophers, E. 1996. The immunopathology of psoriasis. Int. Arch. Allergy Immunol. 110:199–206. [DOI] [PubMed] [Google Scholar]
- 7.Wrone-Smith, T., and B.J. Nickoloff. 1996. Dermal injection of immunocytes induces psoriasis. J. Clin. Invest. 98:1878–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boehncke, W.H., D. Dressel, T.M. Zollner, and R. Kaufmann. 1996. Pulling the trigger on psoriasis. Nature. 379:777. [DOI] [PubMed] [Google Scholar]
- 9.Schon, M.P., M. Detmar, and C.M. Parker. 1997. Murine psoriasis-like disorder induced by naive CD4+ T cells. Nat. Med. 3:183–188. [DOI] [PubMed] [Google Scholar]
- 10.Biron, C.A., K.B. Nguyen, G.C. Pien, L.P. Cousens, and T.P. Salazar-Mather. 1999. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu. Rev. Immunol. 17:189–220. [DOI] [PubMed] [Google Scholar]
- 11.Fernandez, N.C., A. Lozier, C. Flament, P. Ricciardi-Castagnoli, D. Bellet, M. Suter, M. Perricaudet, T. Tursz, E. Maraskovsky, and L. Zitvogel. 1999. Dendritic cells directly trigger NK cell functions: cross-talk relevant in innate anti-tumor immune responses in vivo. Nat. Med. 5:405–411. [DOI] [PubMed] [Google Scholar]
- 12.Ashkar, A.A., J.P. Di Santo, and B.A. Croy. 2000. Interferon gamma contributes to initiation of uterine vascular modification, decidual integrity, and uterine natural killer cell maturation during normal murine pregnancy. J. Exp. Med. 192:259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gourlay, W.A., W.H. Chambers, A.P. Monaco, and T. Maki. 1998. Importance of natural killer cells in the rejection of hamster skin xenografts. Transplantation. 65:727–734. [DOI] [PubMed] [Google Scholar]
- 14.Grob, P., V.E. Schijns, M.F. van den Broek, S.P. Cox, M. Ackermann, and M. Suter. 1999. Role of the individual interferon systems and specific immunity in mice in controlling systemic dissemination of attenuated pseudorabies virus infection. J. Virol. 73:4748–4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee, C.K., D.T. Rao, R. Gertner, R. Gimeno, A.B. Frey, and D.E. Levy. 2000. Distinct requirements for IFNs and STAT1 in NK cell function. J. Immunol. 165:3571–3577. [DOI] [PubMed] [Google Scholar]
- 16.Muller, U., U. Steinhoff, L.F. Reis, S. Hemmi, J. Pavlovic, R.M. Zinkernagel, and M. Aguet. 1994. Functional role of type I and type II interferons in antiviral defense. Science. 264:1918–1921. [DOI] [PubMed] [Google Scholar]
- 17.Fraki, J.E., R.A. Briggaman, and G.S. Lazarus. 1983. Transplantation of psoriatic skin onto nude mice. J. Invest. Dermatol. 80:31s–35s. [PubMed] [Google Scholar]
- 18.Baker, B.S., A.F. Swain, H. Valdimarsson, and L. Fry. 1984. T-cell subpopulations in the blood and skin of patients with psoriasis. Br. J. Dermatol. 110:37–44. [DOI] [PubMed] [Google Scholar]
- 19.Surh, C.D., and J. Sprent. 2000. Homeostatic T cell proliferation: how far can T cells be activated to self-ligands? J. Exp. Med. 192:F9–F14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Westermann, J., B. Engelhardt, and J.C. Hoffmann. 2001. Migration of T cells in vivo: molecular mechanisms and clinical implications. Ann. Intern. Med. 135:279–295. [DOI] [PubMed] [Google Scholar]
- 21.Nickoloff, B.J., G.D. Karabin, J.N. Barker, C.E. Griffiths, V. Sarma, R.S. Mitra, J.T. Elder, S.L. Kunkel, and V.M. Dixit. 1991. Cellular localization of interleukin-8 and its inducer, tumor necrosis factor-alpha in psoriasis. Am. J. Pathol. 138:129–140. [PMC free article] [PubMed] [Google Scholar]
- 22.Adorini, L., and F. Sinigaglia. 1997. Pathogenesis and immunotherapy of autoimmune diseases. Immunol. Today. 18:209–211. [DOI] [PubMed] [Google Scholar]
- 23.Chaudhari, U., P. Romano, L.D. Mulcahy, L.T. Dooley, D.G. Baker, and A.B. Gottlieb. 2001. Efficacy and safety of infliximab monotherapy for plaque-type psoriasis: a randomised trial. Lancet. 357:1842–1847. [DOI] [PubMed] [Google Scholar]
- 24.Mease, P.J., B.S. Goffe, J. Metz, A. VanderStoep, B. Finck, and D.J. Burge. 2000. Etanercept in the treatment of psoriatic arthritis and psoriasis: a randomised trial. Lancet. 356:385–390. [DOI] [PubMed] [Google Scholar]
- 25.Vassalli, P. 1992. The pathophysiology of tumor necrosis factors. Annu. Rev. Immunol. 10:411–452. [DOI] [PubMed] [Google Scholar]