

Abstract
CD40, a member of the tumor necrosis factor receptor family, and the Epstein-Barr virus–encoded oncoprotein latent membrane protein 1 (LMP1) share several tumor necrosis factor receptor–associated factor (TRAF) adaptor proteins for signaling. Among these, TRAF3 was the first identified to directly bind both receptors, yet its role remains a mystery. To address this, we generated B cell lines deficient in TRAF3 by homologous recombination. We found that CD40 signals were normal in the absence of TRAF3, with the exception of moderately enhanced c-Jun NH2-terminal kinase (JNK) activation and antibody secretion. In sharp contrast, LMP1 signaling was markedly defective in TRAF3−/− B cells. LMP1-induced activation of JNK and nuclear factor κB, up-regulation of CD23 and CD80, and antibody secretion were substantially affected by TRAF3 deficiency. Reconstitution of TRAF3 expression decreased CD40-induced JNK activation and antibody secretion, and fully restored LMP1 signaling. Although TRAF2 is widely believed to be important for LMP1 function, LMP1 signaling was intact in TRAF2−/− B cells. Our data reveal that CD40 and LMP1 unexpectedly use TRAF3 in different ways, and that TRAF3 is required for LMP1-mediated activation of B cells.
Keywords: TNF-R family, Epstein-Barr virus, antibody secretion, signal transduction, B cell
Introduction
TNF-R–associated factor (TRAF) 3 was first identified as a protein associated with CD40 (1, 2). Surprisingly, another work identified the same protein as a factor that binds to latent membrane protein 1 (LMP1), the oncoprotein encoded by the human pathogen EBV (3). This finding provided the first connection between the signal transduction pathways of CD40 and LMP1, two membrane proteins without apparent structural homology.
CD40, a member of the TNF-R superfamily, is a transmembrane receptor on B lymphocytes and other cell types, including dendritic cells, monocytes/macrophages, mast cells, fibroblasts, and endothelial cells, as well as certain activated T lymphocytes (4–7). Its ligand, CD154, is expressed primarily on activated CD4+ T cells (5, 6). The CD40/CD154 interaction is crucial for T cell–dependent humoral immunity, regulating B cell proliferation, Ig isotype switching, formation of germinal centers, and development of memory B cells (4–6). CD40 depends on the TRAF family of cytoplasmic adaptor proteins for signaling (6, 8). TRAF2, -3, and -6 could directly bind to CD40, whereas TRAF1 and -5 may associate with CD40 through interactions with TRAF2 and -3, respectively (4–6). Upon ligand binding, the trimerization of CD40 is triggered by CD154 and allows the association of TRAFs, which function as adaptor proteins to transduce signals to downstream molecules (4–6).
LMP1 is the only EBV-encoded protein that alone can transform rodent fibroblasts in vitro (9, 10). Transgenic mice expressing LMP1 under the control of the mouse IgH enhancer and a VH promoter develop B cell lymphomas at an accelerated rate as they age (11). These findings together with a variety of other analyses indicate that LMP1 is a major contributing factor to the development of EBV-associated lymphoproliferative disease and lymphomas (12, 13). LMP1 is an integral membrane protein with a short cytoplasmic NH2-terminal domain, six transmembrane domains, and a long cytoplasmic COOH-terminal tail. The six transmembrane domains of LMP1 spontaneously aggregate and oligomerize within the plasma membrane, and are responsible for the ligand-independent constitutive activation of the protein. However, the cytoplasmic carboxyl terminus (CCT) appears to be the major region required for signaling itself (13–15). Adaptor proteins able to interact with the CCT of LMP1 were found to be members of the TRAF family, including TRAF1, -2, -3, and -5 (3, 16–18). Subsequently, it was documented that LMP1 closely mimics most of the signaling events and effector functions of CD40 in B lymphocytes, including activation of kinases and transcription factors, up-regulation of adhesion molecules and costimulatory molecules, and secretion of antibodies and cytokines. However, unlike CD40, LMP1 signals to B cells in a dysregulated manner, leading to amplified and sustained B cell activation (13, 19–21).
To understand how CD40 and LMP1 activate B cells, it is important to elucidate how each TRAF molecule participates in signaling. Despite its likely importance, little is yet known about the role of TRAF3. CD40 or LMP1 mutants specifically defective only in TRAF3 binding have not been created because the binding sites for TRAF1, -2, -3, and -5 on CD40 or LMP1 overlap (5, 8, 13, 22). Papers in which overexpressed dominant-negative (DN) or WT TRAF3 was used suggest that TRAF3 plays a negative role in both CD40- and LMP1-mediated signaling (8, 12, 13). However, again, the overlap in TRAF binding sites prevents clear interpretation of these results. Alternatively, TRAF3-deficient mice have been generated in an attempt to better understand its role in signaling. Unfortunately, these mice die within 10 d of birth (23), limiting their use in delineating the specific role of TRAF3 in signaling by CD40, and particularly, by LMP1. Thus, we have used a novel somatic cell gene targeting approach to generate TRAF3−/− mouse B cell lines and have compared CD40 and LMP1 signaling in WT and TRAF3−/− B cells. We elected to focus on B cells because they are the major cell type that express CD40 and are also the principle targets of EBV infection in vivo. Surprisingly, we found that CD40 and LMP1 use TRAF3 in different ways. CD40 signals are either independent of, or negatively regulated by TRAF3, whereas LMP1 requires TRAF3 for c-Jun NH2-terminal kinase (JNK) and nuclear factor κB (NF-κB) activation, antibody secretion, and CD80 up-regulation. We have also generated B cell lines deficient in TRAF2 (24) or TRAF1 (unpublished data), and find, in contrast to previous papers (12, 13), that these TRAFs are not critical for many LMP1-mediated B cell activation events.
Materials and Methods
Cell Lines.
The mouse B cell lines CH12.LX and A20.2J have been described previously (25, 26). Generation and characterization of TRAF2−/− CH12.LX and A20.2J cells has been described in detail elsewhere (24), and production of TRAF1−/− CH12.LX will be reported elsewhere (unpublished data). Cells were cultured in RPMI 1640 supplemented with 10% FCS, 10 μM β-mercaptoethanol, and antibiotics (BCM-10) as described previously (20). Spodoptera frugiperda (Sf9) cells infected with WT baculovirus or recombinant baculovirus expressing mouse or human CD154 were prepared as described previously (27, 28).
Generation of TRAF3−/− Mouse B Cell Lines.
The mTRAF3 gene targeting construct was transfected by electroporation into subclones of the mouse B cell lines CH12.LX and A20.2J, already stably expressing LacR as described previously (29). Transfected cells were selected with geneticin and further screened by genomic PCR as described previously (24) using primer set 1 (neo-C and mT3-BT6) and set 2 (neotailA and mT3-S4) (Fig. 1 B). Correctly targeted clones (TRAF3+/−) were transiently transfected with a Cre expression vector (pBS185, a gift from C. Sigmund, University of Iowa, Iowa City, IA) and subcloned to obtain geneticin-sensitive clones. TRAF3+/− cells that regained sensitivity to geneticin were selected, and the targeting was repeated to disrupt the remaining WT TRAF3 gene. Genomic PCR was performed to screen the clones with both alleles targeted (TRAF3−/−) using primer set 1 (neo-C and mT3-BT6) and set 3 (mT3-U5 and mT3-Y). TRAF3−/− cells were transiently transfected with the Cre expression vector again to remove the neor to allow the use of geneticin selection in subsequent transfections.
Figure 1.

Targeted disruption of the mouse TRAF3 gene. (A) Schematic diagrams of the mTRAF3 gene and spliced mRNA are shown. Exons are depicted (open boxes); lengths of introns are shown. UTR, untranslated region. (B) Schematic diagrams of the targeting vector, the initial targeted locus, and the final targeted locus after transfection with the DNA recombinase Cre are shown. The translation start codon of mTRAF3 (ATG), the neomycin resistance cassette (Neor), the diphtheria toxin subunit A cassette (DT-A), loxP sites (recognition sites for Cre, lox), and an SV40 polyadenylation signal sequence (pA) are indicated. PCR primers used to screen B cell clones for homologous recombination are indicated with arrows. (C) Total cell lysates were prepared from WT (+/+), TRAF3+/− (+/−), and TRAF3−/− (−/−) A20.2J and CH12.LX cells. Protein blots were immunoblotted for TRAF3, stripped, and reimmunoblotted for TRAF2 and TRAF6. (D) WT (TRAF3+/+) and TRAF3−/− A20.2J stably transfected with hCD40LMP1 were analyzed by immunofluorescence flow cytometry using FITC-labeled anti-hCD40 (shaded) or isotype control Ab (unshaded). All transfectants used in this analysis, including those of CH12.LX cells, were expression matched in this manner. Results in all figures were repeated in three independent experiments or as specified.
In Vitro c-Jun Kinase Assay.
A20.2J (4 × 106) and CH12.LX cells (2 × 106) expressing human CD40 (hCD40)LMP1 were stimulated with 10 μg/ml of anti–mouse CD40 (mCD40), anti-hCD40, or isotype control Abs at 37°C for 10 or 20 min as indicated in the figures. Cell lysates were prepared, and JNK activity was measured as described previously (30). Reactions were separated by SDS-PAGE, and phosphorylated GST-c-Jun was visualized by autoradiography of dried gels.
NF-κB Luciferase Reporter Assay.
A20.2J and CH12.LX cell subclones (2 × 107 cells) expressing hCD40LMP1 were electroporated at 225 V and 50 mS with 38 μg 4X NF-κB firefly luciferase (a gift from E. Clark, University of Washington, Seattle, WA) and 2 μg renilla (null) luciferase reporter plasmids (Promega). After transfection, cells were rested in medium containing 15% FCS overnight at 37°C. Cells were washed and resuspended in BCM-10, aliquoted into 24-well plates (2 ml/well), and stimulated with 10 μg of anti-mCD40, anti-hCD40, or isotype control mAbs for 5 h at 37°C. Cell lysates were analyzed for the firefly and renilla luciferase activities with the Dual Luciferase Reporter Assay kit (Promega) on a TD-20/20 Luminometer (Turner Designs) following the manufacturer's protocol.
Surface Molecule Up-regulation.
5 × 105 A20.2J cells were stimulated in six-well plates in a total volume of 5 ml with 2 μg/ml of anti-mCD40, anti-hCD40, or isotype control mAbs for 48 h at 37°C. For detection of CD23 and CD80, cells were stained with FITC-labeled mAb against these surface antigens or FITC-labeled isotype control mAb. For detection of CD95, cells were stained with biotin-labeled anti-CD95 (Jo-2), followed by FITC-labeled streptavidin. For detection of MHC class II, cells were stained with anti-Ek (14–4-4S), followed by FITC-labeled goat anti–mouse IgG2a Ab. Stained cells were subsequently analyzed by immunofluorescence flow cytometry using a FACScan™ benchtop flow cytometer (Becton Dickinson). Mean channel fluorescence was determined using WinMDI 2.8 software (http://facs.scripps.edu).
Ab Secretion Assay.
CH12.LX and its transfected subclones express surface IgM specific for phosphatidylcholine, an Ag found on the surface of sheep red blood cells (31). Enumeration of SRBC-specific IgM-secreting cells was by direct plaque assay as described previously (32). In brief, 1.5 × 103 cells were stimulated in 96-well plates in a total volume of 200 μl for 72 h. For reconstituted TRAF3−/− cells, TRAF3 expression was induced by preincubation with BCM-10 containing 100 μM isopropyl-β-d-thiogalactopyranoside (IPTG) at 37°C for 24 h, and stimuli were added for a subsequent 48 h. Ab-secreting cells were measured as cells capable of forming lytic plaques on a lawn of SRBCs in the presence of complement and quantitated as plaque-forming cells (Pfcs) per 106 viable cells recovered from replicate cultures.
Cytokine ELISAs.
For TNFα secretion, 5 × 105 CH12.LX cells were stimulated with Sf9 cells infected with WT baculovirus, or a recombinant baculovirus expressing mouse or human CD154 at a ratio of B cells/Sf9 cells of 10:1, or 10 μg/ml of anti-mCD40, anti-hCD40, or isotype control Ab in an anti-TNF–coated ELISA plate for 3 h at 37°C. The amount of TNFα in the supernatant was measured by ELISA as described previously (33). For IL-6 secretion, 105 CH12.LX cells were stimulated with Sf9 cells infected with WT baculovirus, or a recombinant baculovirus expressing mouse or human CD154 at an 8:1 ratio in a 96-well plate for 48 h. The amount of IL-6 in the supernatant was determined by ELISA as described previously (34).
TRAF Recruitment to Receptors in Detergent-insoluble Microdomains (Rafts) and Immunoprecipitation.
A20.2J and CH12.LX subclones (2 × 107 cells) expressing hCD40LMP1 were stimulated in a total volume of 1 ml with 10 μg of anti-mCD40 (1C10), anti-hCD40 (G28–5), or isotype control mAbs for 10 min at 37°C to induce recruitment of TRAFs to membrane rafts and allow formation of CD40 or LMP1 signaling complexes. To induce raft recruitment of TRAFs by CD40 signaling in the presence of LMP1, CH12.LX subclones expressing an IPTG-inducible LMP1 were preincubated in BCM-10 containing 100 μM IPTG for 24 h and cells were stimulated with anti-mCD40 Ab as aforementioned. Cells were chilled on ice, lysed in 400 μl of ice-cold 1% Brij 58 lysis buffer (35), and incubated for 30 min on ice. Detergent-soluble and insoluble fractions were separated by centrifugation at 14,000 g for 30 min. The 1% Brij 58–insoluble pellets were resolubilized in 400 μl of octylglucopyranoside lysis buffer (36) and sonicated, followed by a 30-min incubation on ice. The octylglucopyranoside lysates (raft lysates) were clarified by centrifugation at 14,000 g for 10 min to remove the remaining insoluble materials. Samples (40 μl) of 1% Brij 58–soluble lysates as well as the raft lysates were reserved and the remainder of the lysates (360 μl) were immunoprecipitated with protein G–Sepharose beads (Amersham Biosciences) prearmed with anti-mCD40 (an equal mixture of 1C10 and 4F11), or anti-hCD40 (G28–5) for 3 h at 4°C. The immunoprecipitation complexes were washed four times with lysis buffer and left in a final volume of 50 μl. Aliquots of lysates and the immunoprecipitates were separated by SDS-PAGE and analyzed by immunoblot.
Online Supplemental Material.
We provide here a detailed description of the mTRAF3 gene targeting construct, methods of stable transfection of mouse B cell lines and immunoblot analysis, PCR primers, and antibodies used in this paper. In addition, we include further results that support those described here, including PCR screening, titration experiments, cytokine ELISA, quantitation data, detailed results of the second cell line, and FACS® profiles of surface molecule up-regulation. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20031255/DC1.
Results
Generation of TRAF3−/− B Cell Lines.
To generate TRAF3−/− B cells, we first cloned ∼23 kb of the mTRAF3 gene using a PCR-based chromosomal walking technique (Fig. 1 A). We generated TRAF3−/− A20.2J mouse B lymphoblastic leukemia cells and CH12.LX mouse B lymphoma cells by successive homologous recombination using a mTRAF3 gene targeting construct as described in Materials and Methods (Fig. 1 B and Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20031255/DC1). Neither full-length nor truncated TRAF3 were detected in TRAF3−/− B cells by immunoblot analysis (Fig. 1 C). The abundance of other TRAF molecules, such as TRAF2 and TRAF6, was not affected by TRAF3 deficiency (Fig. 1 C).
Effects of TRAF3 Deficiency on Early Signaling Events.
LMP1 self-aggregates through its six transmembrane domains and, thus, is constitutively active when it is expressed on cells (8, 12, 13). It has been shown previously that only the CCT of LMP1 is required for postaggregation delivery of signals and that LMP1 signals similarly in both human and mouse B cells (11, 13, 20). To study LMP1 signaling, we stably transfected A20.2J and CH12.LX cells with an expression vector encoding a chimeric molecule (hCD40LMP1) composed of the extracellular and transmembrane domains of hCD40 and the CCT of LMP1. This chimeric molecule signals indistinguishably from LMP1, but with controllable initiation, and like LMP1, its aggregation localizes the hybrid receptor to plasma membrane rafts (21, 36). The extracellular domains of hCD40 and mCD40 are not cross-reactive with species–specific antibodies, so hCD40LMP1 and the endogenous mCD40 can be engaged differentially (21, 36). WT and TRAF3− / − B cells stably expressing matched levels of hCD40LMP1 were selected by immunofluorescence flow cytometry (Fig. 1 D) and used in the following study.
First, we examined activation of the MAPK family kinases and the transcription factor NF-κB, reproducible proximal signaling events induced by both CD40 and LMP1 in B cells (5, 12, 37). Interestingly, TRAF3−/− B cells displayed moderately enhanced CD40-mediated JNK activation as compared with TRAF3+/+ B cells. This was seen using either agonistic Abs (Fig. 2 A) or CD154 (Figs. S2 A and S3, available at http://www.jem.org/cgi/content/full/jem.20031255/DC1) as a stimulus. CD40-mediated activation of p38, NF-κB, Akt, and ERK was unaltered in both TRAF3−/− B cell lines (Fig. S2 A and not depicted). Previous papers in which DN or WT TRAF3 were overexpressed suggest that TRAF3 does not affect LMP1-mediated JNK activation and inhibits LMP1-mediated NF-κB activation (17, 38). Surprisingly, we found that activation of JNK by LMP1 signaling was markedly defective in TRAF3−/− B cells as measured by immunoblot analysis as well as by an in vitro c-Jun kinase assay (Fig. 2, B and C, and Figs. S4 A and S5 A, available at http://www.jem.org/cgi/content/full/jem.20031255/DC1). Furthermore, activation of p38 and degradation of IκBα (the inhibitory subunit of NF-κB) induced by LMP1 signaling were severely impaired in TRAF3−/− B cells (Fig. 2 B and Fig. S4 A). In contrast, activation of Akt and ERK by LMP1 signaling was normal in TRAF3−/− B cells (Fig. S5 B). We confirmed the inhibitory effect of TRAF3 deficiency on LMP1-mediated NF-κB activation (a consistent twofold inhibition compared with WT cells) by a luciferase reporter assay (Fig. 2 D).
Figure 2.
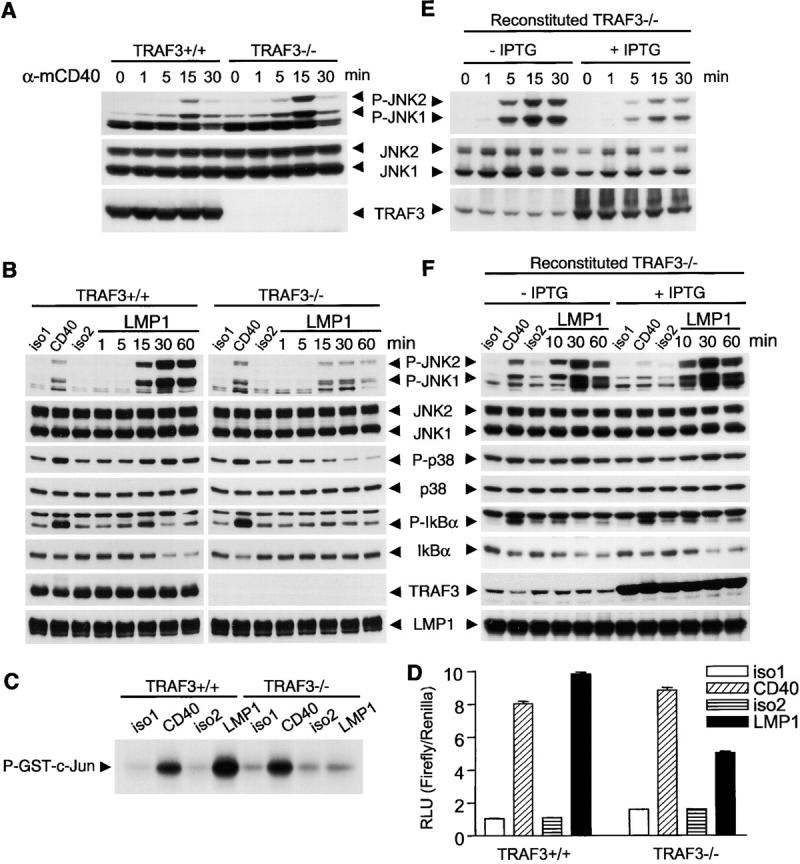
Effects of TRAF3 deficiency on activation of JNK, p38, and NF-κB by CD40 and LMP1 signaling. (A) A20.2J cells were stimulated with anti-mCD40 Ab (α-mCD40) for the indicated times. Lysates were immunoblotted for phosphorylated JNK (P-JNK), total JNK (JNK), and TRAF3. (B) A20.2J cells stably transfected with hCD40LMP1 were stimulated with isotype control Abs (iso1 and iso2) or anti-mCD40 Ab (CD40) for 5 min, or anti-hCD40 Ab to trigger signaling through hCD40LMP1 (LMP1) for the indicated times. Lysates were immunoblotted for phosphorylated (P-) or total JNK, p38, or IκBα, followed by TRAF3 and LMP1. (C) Cells as in B were stimulated with anti-mCD40 Ab (CD40) for 10 min, anti-hCD40 Ab (LMP1) for 20 min, or isotype control Abs (iso1 and iso2). JNK activity was determined by an in vitro c-Jun kinase assay and indicated by the level of phosphorylated GST-c-Jun. (D) Cells as in B were transiently transfected with a NF-κB firefly reporter plasmid and a renilla control plasmid, stimulated with antibodies as in B for 5 h. Relative light units (RLUs) were calculated by normalizing the firefly activity to the renilla activity in each sample. (E) Reconstituted TRAF3−/− A20.2J cells were preincubated in the absence (-IPTG) or presence of IPTG (+IPTG) for 24 h, stimulated with anti-mCD40 Ab (α-mCD40) for the indicated times. (F) Reconstituted TRAF3−/− A20.2J cells were preincubated in the absence (-IPTG) or presence of IPTG (+IPTG) for 24 h, stimulated with isotype control Abs (iso1 and iso2) or anti-mCD40 Ab (CD40) for 10 min, or anti-hCD40 Ab to trigger signaling through hCD40LMP1 (LMP1) for the indicated times. Activation of JNK, p38, and NF-κB were analyzed as in B.
To verify that the reduced ability of TRAF3−/− B cells to activate JNK, p38, and NF-κB in response to LMP1 signaling was caused by the loss of TRAF3, we stably transfected an IPTG-inducible TRAF3 expression construct into TRAF3−/− B cells. Even in the absence of IPTG, a low level of TRAF3 expression restored the activation of JNK, p38, and NF-κB in response to LMP1 signaling (Fig. 2 F and Fig. S4 B). Restoration of TRAF3 expression to WT levels with IPTG induction further enhanced JNK activation induced by LMP1 signaling (Fig. 2 F and Fig. S4 B), but modestly inhibited that induced by CD40 engagement in both B cell lines (Fig. 2 E and Figs. S2 B and S3). Titration of the stimuli used in these experiments confirmed the conclusion that TRAF3 is required for LMP1- but not CD40-induced JNK activation (Fig. S6, available at http://www.jem.org/cgi/content/full/jem.20031255/DC1). Together, our data define a negative role for normal levels of TRAF3 in CD40-mediated JNK activation, but in contrast, an unexpected requirement for TRAF3 in LMP1-induced activation of JNK, p38, and NF-κB. These findings suggest that TRAF3 expressed at endogenous levels plays roles in signaling that are not revealed by studies using overexpression of TRAF3.
Effects of TRAF3 Deficiency on Effector Functions.
Next, we determined if the effects of TRAF3 deficiency on early signaling pathways of CD40 and LMP1 are predictive of downstream B cell effector functions, including up-regulation of surface molecules and secretion of cytokines and antibodies. After CD40 engagement, TRAF3-deficient A20.2J cells exhibited up-regulation of CD23 and CD95 similar to that of WT cells (summarized in Fig. 3 A; representative histograms shown in Fig. S7, available at http://www.jem.org/cgi/content/full/jem.20031255/DC1). Although we consistently observed a partial decrease in CD40-mediated up-regulation of CD80 in TRAF3−/− B cells (Fig. 3 A), it may be due to clonal variation because different A20.2J subclones vary in this response (not depicted). Using the mCD40-elicited response as an internal control for each subclone, we found that LMP1-induced up-regulation of CD23 and CD80 was reduced approximately two- and threefold, respectively, whereas up-regulation of CD95 and MHC class II was not affected by TRAF3 deficiency in A20.2J cells (Fig. 3 A and Fig. S7). For detection of cytokine secretion, the membrane ligands for hCD40 and mCD40 (hCD154, mCD154, respectively) were also used, as they are more potent activators of this specific function of CD40. Although mCD154 is capable of activating hCD40 to a certain extent (39), cross-reaction between hCD154 and mCD40 is negligible (21). Neither CD40- nor LMP1-mediated secretion of TNFα and IL-6 was affected by TRAF3 deficiency in CH12.LX cells (Fig. S8, available at http://www.jem.org/cgi/content/full/jem.20031255/DC1). Interestingly, TRAF3−/− CH12.LX cells exhibited enhanced IgM secretion after CD40 engagement, but profoundly suppressed IgM secretion in response to LMP1 signaling as compared with TRAF3+/+ cells (Fig. 3 B). We further verified this observation by examining TRAF3−/− CH12.LX cells reconstituted with an IPTG-inducible TRAF3. Induction of TRAF3 expression with IPTG completely restored IgM secretion in response to LMP1 signaling, while decreasing IgM secretion mediated by CD40 signaling (Fig. 3, C and D). These results extend our previous finding that overexpression of TRAF3 decreases CD40-mediated IgM secretion (29), and in contrast, highlight the importance and indispensability of TRAF3 in LMP1-induced B cell activation. Furthermore, we also stably transfected an IPTG-inducible full length LMP1 into WT and TRAF3−/− B cells. A similar requirement for TRAF3 was observed when the signal was delivered by inducible full-length LMP1 (unpublished data).
Figure 3.

Effects of TRAF3 deficiency on effector functions of CD40 and LMP1. (A) A20.2J cells stably transfected with hCD40LMP1 were stimulated with 2 μg/ml anti-mCD40 Ab (CD40), anti-hCD40 Ab to trigger signaling through hCD40LMP1 (LMP1), or isotype control Abs for each (iso1 and iso2) for 48 h. Expression of CD23, CD80, and CD95 was determined by immunofluorescence staining and analyzed by flow cytometry. MCF, mean channel fluorescence. Values presented are the mean ± SEM of three independent experiments. (B) CH12.LX cells stably transfected with hCD40LMP1 were stimulated with Abs as in A for 72 h and assayed for IgM secretion. Plaque-forming cells (Pfcs, IgM-secreting cells) per 106 viable recovered cells are shown. Values presented are the mean ± SE of replicate samples. Data are representative of three independent experiments. (C) TRAF3−/− CH12.LX cells stably transfected with hCD40LMP1 were stably supertransfected with an IPTG-inducible TRAF3 (Reconstituted TRAF3−/−). Reconstituted cells were preincubated in the absence (−IPTG) or presence of IPTG (+IPTG) for 24 h and stimulated with Abs as in B in the absence or presence of IPTG, whereas TRAF3+/+ cells were stimulated with Abs as in B in the absence of IPTG. IgM-secreting cells were enumerated as in B. Values presented are the mean ± SE of replicate samples. Data are representative of three independent experiments. (D) Expression of TRAF3 in the reconstituted TRAF3−/− CH12.LX cells (Recon.) in the absence (−) or presence of IPTG (+IPTG), compared with that of WT (+/+) and TRAF3−/− (−/−) cells, was analyzed by immunoblotting. An immunoblot for actin served as loading control.
TRAF2 Recruitment and Degradation in TRAF3−/− B Cells.
We found previously that both TRAF2 and TRAF3 are recruited to membrane rafts upon CD40 or LMP1 signaling (21, 35). To begin to understand how TRAF3 differentially regulates CD40 and LMP1 signaling, we examined whether the amount of TRAF2 recruitment by CD40 or LMP1 signaling is changed in the absence of TRAF3. In TRAF3−/− B cells, quantification of Western blot band density showed that ∼60% more TRAF2 (as compared with WT cells) was recruited to detergent-insoluble raft fractions and coimmunoprecipitated with CD40 upon CD40 engagement (Fig. 4 A). Considering that TRAF2 is important for CD40-induced JNK activation and IgM secretion (24), our finding suggests that TRAF3 may exert its inhibitory effects on CD40 signaling by competing with TRAF2 for association with CD40.
Figure 4.

Recruitment and degradation of TRAFs in TRAF3−/− B cells. (A) A20.2J cells were stimulated with anti-mCD40 Ab to trigger signaling through endogenous CD40 (CD40) or an isotype control Ab (iso) for 10 min. Detergent soluble (S) and insoluble raft (I) lysates were prepared. 90% of the lysates were incubated with anti-mCD40 Ab (an equal mixture of 1C10 and 4F11) to immunoprecipitate mCD40. The lysates and immunoprecipitates were analyzed by immunoblotting for TRAF2 and TRAF3. (B) A20.2J cells stably transfected with hCD40LMP1 were stimulated with anti-hCD40 Ab to trigger signaling through hCD40LMP1 (LMP1) or an isotype control Ab (iso) for 10 min. Detergent soluble (S) and insoluble raft (I) lysates were prepared. 90% of the lysates were incubated with anti-hCD40 Ab (G28-5) to immunoprecipitate hCD40LMP1. The lysates and immunoprecipitates were analyzed by immunoblotting for TRAF2, TRAF3, and LMP1. (C) Cells as in B were stimulated with isotype control Abs (iso1 and iso2) or anti-mCD40 Ab (CD40) for 4 h, or anti-hCD40 Ab to trigger signaling through hCD40LMP1 (LMP1) for the indicated times. Lysates were immunoblotted for TRAF2, followed by TRAF3, LMP1, and actin. Similar results were obtained with CH12.LX cells.
In light of numerous findings that TRAF2 is important for LMP1-mediated NF-κB and JNK activation (for reviews see references 12, 13), we investigated the possibility that defective LMP1 signaling in TRAF3−/− B cells was an indirect result of alterations in the interaction between TRAF2 and LMP1. However, we found no defects in the association of TRAF2 with LMP1 in TRAF3−/− B cells as measured by recruitment of TRAF2 to detergent-insoluble membrane rafts or by coimmunoprecipitation of TRAF2 with LMP1 (Fig. 4 B). We have reported previously that both TRAFs are degraded after CD40- but not LMP1-induced signaling, suggesting that failure to degrade TRAFs may contribute to the more pronounced and sustained effect of LMP1 on B cell activation (21, 40). Thus, we also investigated the possibility that TRAF2 might undergo degradation in response to LMP1 signaling in the absence of TRAF3. However, this was not evident (Fig. 4 C), indicating that the functional defects in LMP1 signaling were directly related to the loss of TRAF3.
TRAF2-independence of LMP1 Signaling to B Cells.
Previous studies in which DN or WT TRAF2 were overexpressed have led to the conclusion that TRAF2 is essential for LMP1-induced activation signals (12, 13). However, it is notable that in WT B cells, only ∼5% of cellular TRAF2, but the majority (∼80%) of cellular TRAF3, was recruited to detergent-insoluble membrane rafts upon LMP1 signaling (Fig. 4 B). This is consistent with the observation that in EBV-transformed B lymphocytes, most of TRAF3 and very little TRAF2 are associated with LMP1 (17, 41). This prompted us to test the requirement for TRAF2 in LMP1 signaling using TRAF2−/− B cell lines that we have generated (24). Surprisingly, LMP1-mediated activation of JNK and NF-κB in both TRAF2−/− B cell lines was as robust as that observed in WT cells (Fig. 5, A and B, and Fig. S9, available at http://www.jem.org/cgi/content/full/jem.20031255/DC1), although CD40-mediated JNK activation was compromised by TRAF2 deficiency (Fig. 5 A and Fig. S9; reference 24). Furthermore, LMP1-mediated IgM secretion was also intact in TRAF2−/− CH12.LX cells (Fig. 5 C), although CD40-mediated IgM secretion was decreased in these cells (Fig. 5 C; reference 24). Our data indicate that TRAF2 appears dispensable for many LMP1-induced B cell activation events.
Figure 5.

TRAF2-independent LMP1 signaling in B cells. (A) A20.2J cells stably transfected with hCD40LMP1 were stimulated with isotype control Abs (iso1 and iso2) or anti-mCD40 Ab (CD40) for 10 min, or anti-hCD40 Ab for the indicated times to trigger signaling through hCD40LMP1 (LMP1). Cell lysates were analyzed by immunoblotting for phosphorylated (P-) or total JNK or IκBα, followed by TRAF2 and LMP1. (B) Cells as in A were transiently transfected with a NF-κB firefly luciferase reporter plasmid and a renilla control plasmid, stimulated with isotype control Abs (iso1 and iso2), anti-mCD40 Ab (CD40), or anti-hCD40 Ab to trigger signaling through hCD40LMP1 (LMP1) for 5 h. RLU, relative light unit. Values presented are the mean ± SE of triplicate samples. Data are representative of three independent experiments. (C) CH12.LX cells stably transfected with hCD40LMP1 were stimulated with Abs as in B for 72 h, and assayed for IgM secretion. Plaque-forming cells (Pfcs) per 106 viable recovered cells are shown. Values presented are the mean ± SE of replicate samples. Data are representative of three independent experiments.
TRAF1 Is Not Required for LMP1-induced JNK Activation.
A recent work has implied a critical role for TRAF1 in LMP1-induced JNK activation (38). We noticed a striking difference in the expression level of TRAF1 between the two B cell lines we studied. Expression of TRAF1 is barely detectable but inducible by CD40 and LMP1 signaling in A20.2J cells (Fig. 6 A), whereas CH12.LX cells constitutively express higher levels of TRAF1 (Fig. 6 B). Nevertheless, both cell lines activate JNK similarly in response to LMP1 signaling, which is inconsistent with a critical role for TRAF1 in this function. However, it may be that only small amounts of TRAF1 are sufficient for signaling. Thus, we further tested the role of TRAF1 in LMP1-induced JNK activation by examining TRAF1−/− CH12.LX cells (unpublished data), generated using the same approach as that used to produce TRAF2−/− and TRAF3−/− cells. In TRAF1−/− CH12.LX cells, activation of JNK by either CD40 or LMP1 signaling was intact (Fig. 6 B). Our observation is consistent with the previous finding that B cells isolated from TRAF1−/− mice activate JNK normally in response to engagement of CD40 (42), and indicates that, at least in B cells, TRAF1 is not essential for LMP1-mediated JNK activation.
Figure 6.
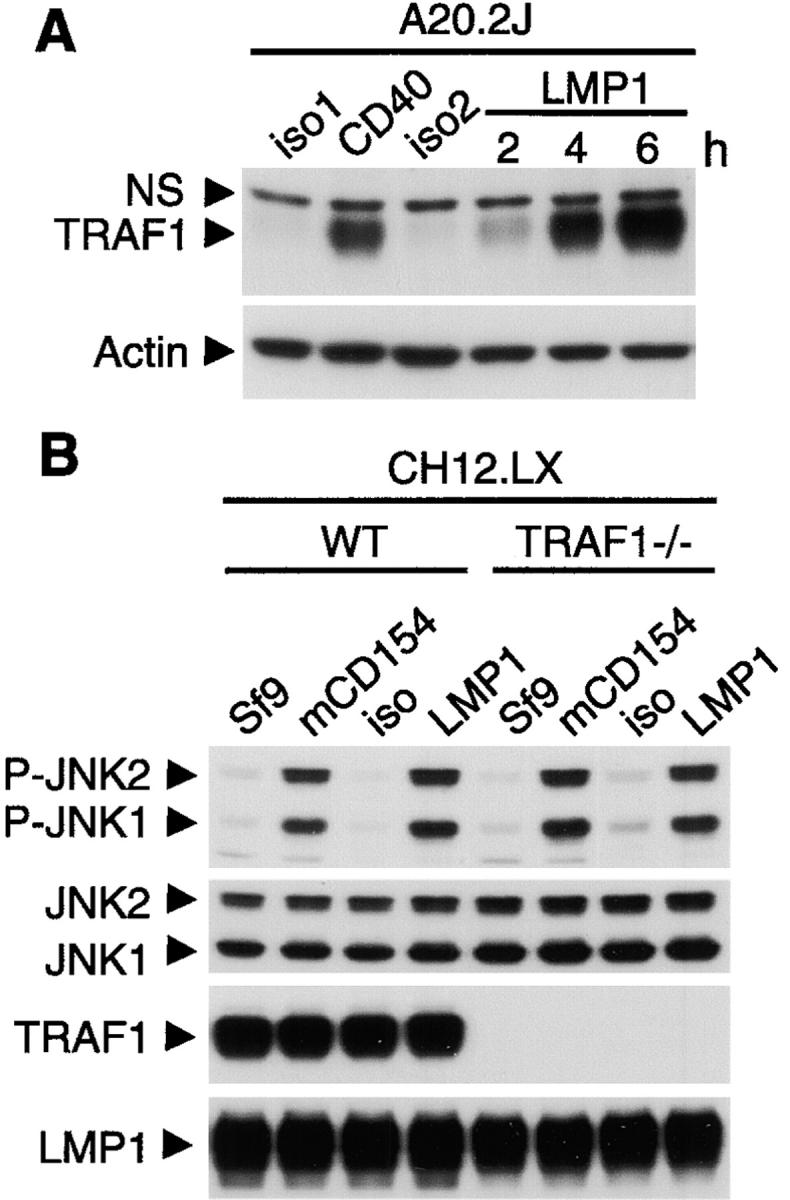
TRAF1 is inducible but not required for JNK activation by LMP1 signaling in B cells. (A) A20.2J cells stably transfected with hCD40LMP1 were stimulated with isotype control Abs (iso1 and iso2) or anti-mCD40 Ab (CD40) for 4 h, or anti-hCD40 Ab to trigger signaling through hCD40LMP1 (LMP1) for the indicated times. Lysates were immunoblotted for TRAF1, followed by actin. A nonspecific band (NS) in the TRAF1 blot is indicated. (B) WT and TRAF1−/− CH12.LX cells stably transfected with hCD40LMP1 were stimulated with Sf9 cells infected with WT baculovirus (Sf9) or a recombinant baculovirus-expressing mouse CD154 (mCD154), an isotype control Ab (iso), or anti-hCD40 Ab to trigger signaling through hCD40LMP1 (LMP1) for 30 min. Cell lysates were analyzed by immunoblotting for phosphorylated (P-) or total JNK, followed by TRAF1 and LMP1.
Effects of TRAF3 Deficiency on Simultaneous CD40 and LMP1 Signaling in B Cells.
Because simultaneous CD40 and LMP1 signaling may occur in EBV-infected B cells in vivo in situations where CD154 is available, it would be interesting to know how TRAF3 deficiency affects simultaneous LMP1 and CD40 signaling. We found that CD40 and LMP1 signaling synergistically induced JNK activation in WT B cells at early time points (10 and 30 min) after engagement. In contrast, such synergistic effects observed in WT B cells were inhibited in TRAF3− / − cells by approximately threefold (Fig. 7 A and Fig. S10, available at http://www.jem.org/cgi/content/full/jem.20031255/DC1). CD40 and LMP1 additively induced NF-κB activation in both WT and TRAF3−/− B cells. However, the level of NF-κB activation induced by simultaneous CD40 and LMP1 signaling observed in TRAF3−/− B cells is ∼60% of that of WT B cells due to the inhibitory effect of TRAF3 deficiency on LMP1-mediated NF-κB activation (Fig. 7 B). We have reported previously that CD40 and LMP1 cooperated to induce antibody secretion in WT B cells (20). Interestingly, such enhancing effects of CD40 and LMP1 on antibody secretion were completely abolished in TRAF3−/− B cells (Fig. 7 C). Together, our data indicate that deletion of TRAF3 can dramatically affect the outcome of simultaneous CD40 and LMP1 signaling.
Figure 7.

Effects of TRAF3 deficiency on simultaneous CD40 and LMP1 signaling in B cells. (A) A20.2J cells stably transfected with hCD40LMP1 were stimulated with 1 μg/ml of isotype control Abs (iso), anti-mCD40 Ab (CD40), anti-hCD40 Ab to trigger signaling through hCD40LMP1 (LMP1), alone or in combination for the indicated times. Cell lysates were analyzed by immunoblotting for phosphorylated (P-) or total JNK, followed by TRAF3 and LMP1. (B) Cells as in A were transiently transfected with a NF-κB firefly luciferase reporter plasmid and a renilla control plasmid and stimulated with Abs as in A for 5 h. RLU, relative light unit. Values presented are the mean ± SE of replicate samples. Data are representative of three independent experiments. (C) CH12.LX cells stably transfected with hCD40LMP1 were stimulated with Abs (250 ng/ml of each Ab) as in A for 72 h and assayed for IgM secretion. Plaque-forming cells (Pfcs) per 106 viable recovered cells are shown. Values presented are the mean ± SE of replicate samples. Data are representative of two independent experiments.
TRAF Recruitment by CD40 Signaling in the Presence of LMP1 in B Cells.
In light of the evidence that LMP1 signaling may sequester most cellular TRAF3 (Fig. 4 B; references 17, 41), we predict that ongoing LMP1 signaling may cause changes in CD40-mediated TRAF recruitment and that such changes in TRAF recruitment may contribute to the synergistic effects of simultaneous CD40 and LMP1 signaling observed in TRAF3+/+ B cells. To address this possibility, we measured TRAF recruitment by CD40 signaling in the presence of LMP1 signaling by using B cell lines stably transfected with an IPTG-inducible full-length LMP1. As shown in Fig. 8, in LMP1-expressing B cells, ∼50% more TRAF2 (as compared with untransfected cells, measuring band intensity by a low-light imaging system) was recruited to detergent-insoluble raft fraction and coimmunoprecipitated with CD40, whereas ∼70% less TRAF3 was coimmunoprecipitated with CD40 upon CD40 engagement (Fig. 8). Considering that TRAF2 plays positive roles, whereas TRAF3 plays negative roles in CD40-induced JNK activation and IgM secretion (Figs. 2 and 3 and Figs. S2 and S3; reference 24), the changes in TRAF2 and TRAF3 recruitment upon CD40 engagement in the presence of LMP1 signaling suggest amplified CD40 signals and, thus, may partially account for the synergistic effects of simultaneous CD40 and LMP1 signaling observed in WT B cells.
Figure 8.

Recruitment of TRAFs by CD40 signaling in the presence of LMP1. CH12.LX cells stably transfected with an IPTG-inducible wild-type full length LMP1 (WT LMP1) or untransfected control cells (CH12.LX) were preincubated with BCM-10 containing 100 μM IPTG for 24 h and stimulated with anti-mCD40 Ab to trigger signaling through endogenous CD40 (CD40) or an isotype control Ab (iso) for 10 min. Detergent soluble (S) and insoluble raft (I) lysates were prepared. 90% of the lysates were incubated with anti-mCD40 Ab (an equal mixture of 1C10 and 4F11) to immunoprecipitate mCD40. The lysates and immunoprecipitates were analyzed by immunoblotting for TRAF2, TRAF3, and LMP1.
Discussion
The functional contribution of TRAF3, an adaptor protein used by both CD40 and its viral oncogenic mimic LMP1, has long remained unclear (13, 37). To better understand the biologic roles played by TRAF3 while circumventing the early lethality of TRAF3−/− mice (23), we have generated two TRAF3−/− mouse B cell lines by applying a novel somatic cell gene targeting strategy. Detailed comparison of CD40 and LMP1 signaling between WT and TRAF3−/− B cells has allowed us to delineate the specific roles of TRAF3.
The role of TRAF3 in CD40-mediated signaling has been controversial. When fetal liver cells from TRAF3−/− embryos were used to reconstitute lethally irradiated mice, TRAF3−/− B cells purified from spleens of these chimeric mice respond to CD154 normally in proliferation or up-regulation of CD23 and CD80 (23). Therefore, TRAF3 appears dispensable for these CD40 function in B cells, but more detailed analysis was not performed. The Ramos B cell line stably transfected with DN TRAF3 shows decreased CD40-mediated activation of JNK and p38, cytokine secretion, and Ig production compared with untransfected parent cells (43). We have shown previously that inducible overexpression of either full-length or DN TRAF3 inhibits CD40-mediated IgM secretion, suggesting that TRAF3 exerts this effect by competition with TRAF2 (29). In the present paper, we demonstrated that CD40-mediated JNK activation and IgM production were enhanced, whereas CD40-induced activation of p38 and secretion of TNFα and IL-6 were unaltered by TRAF3 deficiency in B cells. Our results support a negative or neutral role for TRAF3 in CD40 signaling. The inhibitory effect of DN TRAF3 on CD40-mediated signaling in Ramos B cells (43) may result from an inhibition of the association of other TRAF molecules (such as TRAF1, -2, and -5) with CD40.
Little was known previously about the role of TRAF3 in LMP1 signaling, although it was known that among all TRAFs, TRAF3 binds to LMP1 most avidly (8, 13, 17, 44). Previous analyses in which WT or DN TRAF3 were overexpressed suggest a negative role for TRAF3 in LMP1-medated activation of NF-κB and induction of gene expression (17, 45). However, here we provided several lines of evidence demonstrating that TRAF3 plays positive roles and is indispensable for LMP1 signaling. Our results indicate that TRAF3 is critical for LMP1-induced activation of JNK, p38, and NF-κB, up-regulation of surface molecules CD23 and CD80, as well as antibody secretion. Notably, we also observed that activation of ERK and Akt, up-regulation of CD95, as well as secretion of TNFα and IL-6 in response to LMP1 signaling were intact in TRAF3−/− B cells, suggesting that this subset of LMP1 signaling events is TRAF3-independent or that redundancy exists. Further study is needed to identify the factors that are responsible for the residual LMP1 signaling observed in TRAF3−/− B cells. Candidate molecules include other adaptor proteins shown previously to be able to interact with LMP1, such as TRAF5, receptor-interacting protein, and TNF-R–associated DD protein (18, 46), and perhaps TRAF1 and -2 may share overlapping functions with these factors.
The general belief that TRAF2 but not TRAF3 is important for LMP1-mediated signaling arose from studies in which overexpression of WT or DN TRAF molecules was used, mostly with the human embryonic kidney cell line 293 (for reviews see references 12, 13). Because TRAF2 is well known as a powerful activator of positive signals by the receptors to which it binds (47, 48), the conclusion that TRAF2 is the major mediator of LMP1-induced activation seemed reasonable and expected. These overexpression approaches provided valuable initial direction, but verification with endogenous molecules in the specific cell type of interest is also necessary (8, 13). In the present work, evaluation of LMP1 signaling in two TRAF2−/− B cell lines has revealed that LMP1-mediated activation of JNK and NF-κB as well as antibody secretion were intact in the absence of TRAF2. In contrast, CD40-mediated JNK activation and IgM secretion were dramatically inhibited by TRAF2 deficiency (24). Our findings indicate that TRAF2 plays important roles in CD40 signaling, but is dispensable for many LMP1-mediated signaling events in B cells. Therefore, by characterizing B cell lines individually deficient in TRAF2 and TRAF3, we have demonstrated that CD40 and LMP1 differentially use TRAF2 and TRAF3, and that TRAF3 is required and critical for LMP1 signaling.
How TRAF3 differentially regulates and participates in CD40 and LMP1 signaling awaits further investigation, although our data suggest that TRAF3 may exert its inhibitory effects on CD40 signaling by competing with TRAF2 for association with CD40. Although CD40 appears to have much greater avidity for TRAF2 compared with LMP1, LMP1 binds TRAF3 with high avidity (8, 13, 17, 44). These differences may play an important role in TRAF3's differential regulation of the two receptors. One previous paper has shown that forced localization of TRAF3 to the cell membrane by NH2-terminal myristoylation is sufficient to convert it into an activator of JNK in 293 cells (49). However, both CD40 and LMP1 recruit TRAF3 to membrane rafts (21, 35), yet expression of normal levels of TRAF3 promoted LMP1-induced JNK activation in B cells, whereas it inhibited JNK activation induced by CD40. Therefore, it appears that translocation to membrane rafts is necessary to enable TRAF3 to regulate JNK activation, but is not sufficient to determine the outcome of TRAF3 binding. Distinct structural features of CD40 and LMP1 cytoplasmic tails and/or additional factors present in the signaling rafts of these two receptors may be essential in determining the nature of TRAF3 function. Identification of such structural determinants of CD40 and LMP1, and delineation of the profiles of factors assembled in the signaling rafts will be key to further elucidate the precise role of TRAF3 in CD40 and LMP1 signaling.
In addition to CD40 and LMP1, members of the large and diverse TNF-R superfamily have been shown to interact with one or more of the TRAF family members, and members of the Toll/interleukin-1 receptor family also bind to TRAF6 (47, 48, 50), emphasizing the broad importance of this family of adaptor proteins in immune regulation. For example, TRAF3 is also used by CD27, CD30, LTβ-R, RANK, ATAR, OX40, 4–1BB, and BAFF-R (47, 48, 50, 51). Cell lines deficient in specific TRAFs should be valuable in dissecting the specific roles of each TRAF molecule in the signaling pathways of these receptors. Additionally, this approach will allow in the future the production of cell lines deficient in multiple TRAFs, thus enabling the delineation of pathways in which TRAFs play overlapping or partially redundant roles.
In summary, the present work illustrates the power of our new approach in revealing an unexpected, critical role for TRAF3 in mediating signaling to B cells by the viral oncogenic mimic of CD40, LMP1. Our findings could have important biological implications for how LMP1 transforms B cells. This viral oncoprotein can use, and potentially sequester a TRAF that normally inhibits signaling. Furthermore, LMP1 can be independent of the levels of TRAF2 available, so the tighter binding of TRAF2 by other receptors will not limit LMP1 signals. Because LMP1 does not induce degradation of TRAF3 after its binding, as does CD40 (21), the TRAF3-mediated signals can be further sustained. These sharp contrasts in the way a receptor and its viral mimic use the same TRAFs may make critical contributions to EBV-mediated pathogenesis, and raise the intriguing possibility that TRAF3 may serve as a potential therapeutic target in EBV-associated malignancies.
Acknowledgments
We thank Drs. L. Stunz and J. Harty for critical review of this paper, and L. Ramirez for his excellent technical assistance.
P. Xie is a Special Fellow of the Leukemia and Lymphoma Society. This paper was supported by National Institutes of Health grants AI28847, AI49993, CA099997; Veterans Administration Merit Review 383 (to G.A. Bishop); and a grant from the American Heart Association (to B.S. Hostager).
The online version of this article includes supplemental material.
Abbreviations used in this paper: CCT, cytoplasmic carboxyl terminus; DN, dominant-negative; hCD40, human CD40; IPTG, isopropyl-β-d-thiogalactopyranoside; JNK, c-Jun NH2-terminal kinase; LMP1, latent membrane protein 1; mCD40, mouse CD40; NF-κB, nuclear factor κB; Pfc, plaque-forming cell; Sf9, Spodoptera frugiperda; TRAF, TNF-R–associated factor.
References
- 1.Cheng, G., A.M. Cleary, Z.S. Ye, D.I. Hong, S. Lederman, and D. Baltimore. 1995. Involvement of CRAF1, a relative of TRAF, in CD40 signaling. Science. 267:1494–1498. [DOI] [PubMed] [Google Scholar]
- 2.Hu, H.M., K. O'Rourke, M.S. Boguski, and V.M. Dixit. 1994. A novel RING finger protein interacts with the cytoplasmic domain of CD40. J. Biol. Chem. 269:30069–30072. [PubMed] [Google Scholar]
- 3.Mosialos, G., M. Birkenbach, R. Yalamanchili, T. VanArsdale, C. Ware, and E. Kieff. 1995. The EBV transforming protein LMP1 engages signaling proteins for the TNF-R family. Cell. 80:389–399. [DOI] [PubMed] [Google Scholar]
- 4.Bishop, G.A., and B.S. Hostager. 2001. Molecular mechanisms of CD40 signaling. Arch. Immunol. Ther. Exp. 49:129–137. [PubMed] [Google Scholar]
- 5.Grammer, A.C., and P.E. Lipsky. 2000. CD40-mediated regulation of immune responses by TRAF-dependent and TRAF-independent signaling mechanisms. Adv. Immunol. 76:61–178. [DOI] [PubMed] [Google Scholar]
- 6.Schonbeck, U., and P. Libby. 2001. The CD40/CD154 receptor/ligand dyad. Cell. Mol. Life Sci. 58:4–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bourgeois, C., B. Rocha, and C. Tanchot. 2002. A role for CD40 expression on CD8+ T cells in the generation of CD8+ T cell memory. Science. 297:2060–2063. [DOI] [PubMed] [Google Scholar]
- 8.Bishop, G.A., B.S. Hostager, and K.D. Brown. 2002. Mechanisms of TNF receptor-associated factor (TRAF) regulation in B lymphocytes. J. Leukoc. Biol. 72:19–23. [PubMed] [Google Scholar]
- 9.Cahir McFarland, E.D., K.M. Izumi, and G. Mosialos. 1999. EBV transformation: involvement of LMP1-mediated activation of NF-kB. Oncogene. 18:6959–6964. [DOI] [PubMed] [Google Scholar]
- 10.Knecht, H., P. Brousset, E. Bachmann, K. Sandvej, and B.F. Odermatt. 1993. LMP1: a key oncogene in EBV-related carcinogenesis? Acta. Haematol. 90:167–171. [DOI] [PubMed] [Google Scholar]
- 11.Kulwichit, W., R.H. Edwards, E.M. Davenport, J.F. Baskar, V. Godfrey, and N. Raab-Traub. 1998. Expression of the EBV LMP1 induces B cell lymphoma in transgenic mice. Proc. Natl. Acad. Sci. USA. 95:11963–11968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eliopoulos, A.G., and L.S. Young. 2001. LMP1 structure and signal transduction. Semin. Cancer Biol. 11:435–444. [DOI] [PubMed] [Google Scholar]
- 13.Hatzivassiliou, E., and G. Mosialos. 2002. Cellular signaling pathways engaged by the EBV transforming protein LMP1. Front. Biosci. 7:d319–d329. [DOI] [PubMed] [Google Scholar]
- 14.Eliopoulos, A.G., and A.B. Rickinson. 1998. EBV: LMP1 masquerades as an active receptor. Curr. Biol. 8:R196–R198. [DOI] [PubMed] [Google Scholar]
- 15.Farrell, P.J. 1998. Signal transduction from the EBV LMP-1 transforming protein. Trends Microbiol. 6:175–178. [DOI] [PubMed] [Google Scholar]
- 16.Kaye, K.M., O. Devergne, J.N. Harada, K.M. Izumi, R. Yalamanchili, E. Kieff, and G. Mosialos. 1996. TRAF2 is a mediator of NF-kB activation by LMP1, the EBV transforming protein. Proc. Natl. Acad. Sci. USA. 93:11085–11090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Devergne, O., E. Hatzivassiliou, K.M. Izumi, K.M. Kaye, M.F. Kleijnen, E. Kieff, and G. Mosialos. 1996. Association of TRAF1, TRAF2, and TRAF3 with an EBV LMP1 domain important for B-lymphocyte transformation: role in NF-κB activation. Mol. Cell. Biol. 16:7098–7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brodeur, S.R., G. Cheng, D. Baltimore, and D.A. Thorley-Lawson. 1997. Localization of the major NF-kB-activating site and the sole TRAF3 binding site of LMP-1 defines two distinct signaling motifs. J. Biol. Chem. 272:19777–19784. [DOI] [PubMed] [Google Scholar]
- 19.Uchida, J., T. Yasui, Y. Takaoka-Shichijo, M. Muraoka, W. Kulwichit, N. Raab-Traub, and H. Kikutani. 1999. Mimicry of CD40 signals by EBV LMP1 in B lymphocyte responses. Science. 286:300–303. [DOI] [PubMed] [Google Scholar]
- 20.Busch, L.K., and G.A. Bishop. 1999. The EBV transforming protein, LMP1, mimics and cooperates with CD40 signaling in B lymphocytes. J. Immunol. 162:2555–2561. [PubMed] [Google Scholar]
- 21.Brown, K.D., B.S. Hostager, and G.A. Bishop. 2001. Differential signaling and TRAF degradation mediated by CD40 and the EBV oncoprotein LMP1. J. Exp. Med. 193:943–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Kooten, C., and J. Banchereau. 2000. CD40-CD40 ligand. J. Leukoc. Biol. 67:2–17. [DOI] [PubMed] [Google Scholar]
- 23.Xu, Y., G. Cheng, and D. Baltimore. 1996. Targeted disruption of TRAF3 leads to postnatal lethality and defective T-dependent immune responses. Immunity. 5:407–415. [DOI] [PubMed] [Google Scholar]
- 24.Hostager, B.S., S.A. Haxhinasto, S.L. Rowland, and G.A. Bishop. 2003. TRAF2-deficient B lymphocytes reveal novel roles for TRAF2 in CD40 signaling. J. Biol. Chem. 278:45382–45390. [DOI] [PubMed] [Google Scholar]
- 25.Bishop, G.A., and G. Haughton. 1986. Induced differentiation of a transformed clone of Ly-1+ B cells by clonal T cells and antigen. Proc. Natl. Acad. Sci. USA. 83:7410–7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Catlett, I.M., and G.A. Bishop. 1999. Cutting edge: a novel mechanism for rescue of B cells from CD95/Fas-mediated apoptosis. J. Immunol. 163:2378–2381. [PubMed] [Google Scholar]
- 27.Hostager, B.S., Y. Hsing, D.E. Harms, and G.A. Bishop. 1996. Different CD40-mediated signaling events require distinct CD40 structural features. J. Immunol. 157:1047–1053. [PubMed] [Google Scholar]
- 28.Baccam, M., S.Y. Woo, C. Vinson, and G.A. Bishop. 2003. CD40-mediated transcriptional regulation of the IL-6 gene in B lymphocytes: Involvement of NF-kB, AP-1, and C/EBP. J. Immunol. 170:3099–3108. [DOI] [PubMed] [Google Scholar]
- 29.Hostager, B.S., and G.A. Bishop. 1999. Cutting edge: contrasting roles of TRAF2 and TRAF3 in CD40-activated B lymphocyte differentiation. J. Immunol. 162:6307–6311. [PubMed] [Google Scholar]
- 30.Hsing, Y., and G.A. Bishop. 1999. Requirement for NF-kB activation by a distinct subset of CD40-mediated effector functions in B lymphocytes. J. Immunol. 162:2804–2811. [PubMed] [Google Scholar]
- 31.Mercolino, T.J., L.W. Arnold, and G. Haughton. 1986. Phosphatidyl choline is recognized by a series of Ly-1+ murine B cell lymphomas specific for erythrocyte membranes. J. Exp. Med. 163:155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bishop, G.A. 1991. Requirements of class II-mediated B cell differentiation for class II cross-linking and cyclic AMP. J. Immunol. 147:1107–1114. [PubMed] [Google Scholar]
- 33.Hostager, B.S., and G.A. Bishop. 2002. Role of TRAF2 in the activation of IgM secretion by CD40 and CD120b. J. Immunol. 168:3318–3322. [DOI] [PubMed] [Google Scholar]
- 34.Baccam, M., and G.A. Bishop. 1999. Membrane-bound CD154, but not CD40-specific antibody, mediates NF-kB-independent IL-6 production in B cells. Eur. J. Immunol. 29:3855–3866. [DOI] [PubMed] [Google Scholar]
- 35.Hostager, B.S., I.M. Catlett, and G.A. Bishop. 2000. Recruitment of CD40 and TRAFs 2 and 3 to membrane microdomains during CD40 signaling. J. Biol. Chem. 275:15392–15398. [DOI] [PubMed] [Google Scholar]
- 36.Busch, L.K., and G.A. Bishop. 2001. Multiple carboxyl-terminal regions of the EBV oncoprotein, LMP1, cooperatively regulate signaling to B lymphocytes via TRAF-dependent and TRAF-independent mechanisms. J. Immunol. 167:5805–5813. [DOI] [PubMed] [Google Scholar]
- 37.Bishop, G.A., and B.S. Hostager. 2001. Signaling by CD40 and its mimics in B cell activation. Immunol. Res. 24:97–109. [DOI] [PubMed] [Google Scholar]
- 38.Eliopoulos, A.G., E.R. Waites, S.M. Blake, C. Davies, P. Murray, and L.S. Young. 2003. TRAF1 is a critical regulator of JNK signaling by the TRAF-binding domain of the EBV-encoded LMP1 but not CD40. J. Virol. 77:1316–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ahonen, C., E. Manning, L.D. Erickson, B. O'Connor, E.F. Lind, S.S. Pullen, M.R. Kehry, and R.J. Noelle. 2002. The CD40-TRAF6 axis controls affinity maturation and the generation of long-lived plasma cells. Nat. Immunol. 3:451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brown, K.D., B.S. Hostager, and G.A. Bishop. 2002. Regulation of TRAF2 signaling by self-induced degradation. J. Biol. Chem. 277:19433–19438. [DOI] [PubMed] [Google Scholar]
- 41.Ardila-Osorio, H., B. Clausse, Z. Mishal, J. Wiels, T. Tursz, and P. Busson. 1999. Evidence of LMP1-TRAF3 interactions in glycosphingolipid-rich complexes of lymphoblastoid and nasopharyngeal carcinoma cells. Int. J. Cancer. 81:645–649. [DOI] [PubMed] [Google Scholar]
- 42.Tsitsikov, E.N., D. Laouini, I.F. Dunn, T.Y. Sannikova, L. Davidson, F.W. Alt, and R.S. Geha. 2001. TRAF1 is a negative regulator of TNF signaling. Enhanced TNF signaling in TRAF1-deficient mice. Immunity. 15:647–657. [DOI] [PubMed] [Google Scholar]
- 43.Grammer, A.C., J.L. Swantek, R.D. McFarland, Y. Miura, T. Geppert, and P.E. Lipsky. 1998. TRAF3 signaling mediates activation of p38 and JNK, cytokine secretion, and Ig production following ligation of CD40 on human B cells. J. Immunol. 161:1183–1193. [PubMed] [Google Scholar]
- 44.Sandberg, M., W. Hammerschmidt, and B. Sugden. 1997. Characterization of LMP-1's association with TRAF1, TRAF2, and TRAF3. J. Virol. 71:4649–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller, W.E., G. Mosialos, E. Kieff, and N. Raab-Traub. 1997. EBV LMP1 induction of the epidermal growth factor receptor is mediated through a TRAF signaling pathway distinct from NF-kB activation. J. Virol. 71:586–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Izumi, K.M., E.D. Cahir McFarland, A.T. Ting, E.A. Riley, B. Seed, and E.D. Kieff. 1999. The EBV oncoprotein LMP1 engages the TNF-R-associated proteins TRADD and receptor-interacting protein (RIP) but does not induce apoptosis or require RIP for NF-kB activation. Mol. Cell. Biol. 19:5759-5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wajant, H., F. Henkler, and P. Scheurich. 2001. The TRAF family. Scaffold molecules for cytokine receptors, kinases and their regulators. Cell. Signal. 13:389–400. [DOI] [PubMed] [Google Scholar]
- 48.Inoue, J., T. Ishida, N. Tsukamoto, N. Kobayashi, A. Naito, S. Azuma, and T. Yamamoto. 2000. TRAF family: adapter proteins that mediate cytokine signaling. Exp. Cell Res. 254:14–24. [DOI] [PubMed] [Google Scholar]
- 49.Dadgostar, H., and G. Cheng. 2000. Membrane localization of TRAF3 enables JNK activation. J. Biol. Chem. 275:2539–2544. [DOI] [PubMed] [Google Scholar]
- 50.Bishop, G.A., and B.S. Hostager. 2001. B lymphocyte activation by contact-mediated interactions with T lymphocytes. Curr. Opin. Immunol. 13:278–285. [DOI] [PubMed] [Google Scholar]
- 51.Xu, L.G., and H.B. Shu. 2002. TRAF3 is associated with BAFF-R and negatively regulates BAFF-R-mediated NF-kB activation and IL-10 production. J. Immunol. 169:6883–6889. [DOI] [PubMed] [Google Scholar]