

Abstract
The BCR-ABL1 kinase expressed in acute lymphoblastic leukemia (ALL) drives malignant transformation of human pre–B cells. Comparing genome-wide gene expression profiles of BCR-ABL1 + pre–B ALL and normal bone marrow pre–B cells by serial analysis of gene expression, many genes involved in pre–B cell receptor signaling are silenced in the leukemia cells. Although normal pre–B cells are selected for the expression of a functional pre–B cell receptor, BCR-ABL1 + ALL cells mostly do not harbor a productively rearranged IGH allele. In these cases, we identified traces of secondary VH gene rearrangements, which may have rendered an initially productive VH region gene nonfunctional. Even BCR-ABL1 + ALL cells harboring a functional VH region gene are unresponsive to pre–B cell receptor engagement and exhibit autonomous oscillatory Ca2+ signaling activity. Conversely, leukemia subclones surviving inhibition of BCR-ABL1 by STI571 restore responsiveness to antigen receptor engagement and differentiate into immature B cells expressing immunoglobulin light chains. BCR-ABL1 kinase activity is linked to defective pre–B cell receptor signaling and the expression of a truncated isoform of the pre–B cell receptor–associated linker molecule SLP65. Also in primary leukemia cells, truncated SLP65 is expressed before but not after treatment of the patients with STI571. We conclude that inhibition of BCR-ABL1 reconstitutes selection for leukemia cells expressing a functional (pre–) B cell receptor.
Keywords: immunoglobulin, SLP65, differentiation, STI571, V(D)J recombination
Introduction
Several transcription factors, including PAX5, E2A, and EBF, guide commitment of hematopoietic stem cells to the B cell lineage (1). Committed B cell precursors undergo a sequence of Ig gene rearrangements defining distinct stages of early B cell development (2). During their early development within the bone marrow, B cell precursors have to pass checkpoints, at which only cells carrying functional Ig gene rearrangements are selected for further development along the B cell lineage (3). For instance, the presence of a productive IGH gene rearrangement is a prerequisite for the expression of the Ig μ heavy chain as a component of the pre–B cell receptor on the surface of a pre–B cell (4). If these cells initially fail to express a functional pre–B cell receptor on their surface because of a nonproductive IGH gene rearrangement on one allele, they can continue to rearrange Ig V region genes on the second IGH allele or undergo secondary VH gene recombination on the nonproductively rearranged allele (5). Pre–B cells are destined to die by apoptosis unless they are rescued by survival signals through their pre–B cell receptor. The pre–B cell receptor complex including the Ig μ heavy chain, the surrogate light chain composed of λ5 and VpreB, CD19, and the Igα and Igβ signal chains does not only convey survival signals but also terminates the rearrangement process within the IGH locus. A critical component in the pre–B cell receptor signaling cascade is the adaptor molecule SLP65, which links SYK to downstream effector pathways, including PLCγ2, BTK, and VAV (6). Although somatic SLP65 deficiency is a frequent aberration in pre–B acute lymphoblastic leukemia (ALL) in humans (7), SLP65 −/− mutant mice exhibit a differentiation block at the pre–B cell stage (8), and also show autonomous proliferation, ongoing rearrangement of IGH V region genes, and development of leukemia (9).
In most cases of ALL, pre–B cells represent the normal counterpart of the malignant clone, which in many cases carries specific oncogenic gene rearrangements defining both biological and clinical subentities (10). Among these translocation events, the t(9;22) (q34;q11) results in a fusion of the BCR and ABL1 genes (11), which codes for a potent tyrosine kinase and represents the most frequent recurrent genetic aberration leading to ALL in adults (12). Comparing pre–B ALL carrying a BCR-ABL1 gene rearrangement to pre–B ALL harboring other translocations, recent studies using the cDNA microarray technique identified differentially expressed genes predicting a BCR-ABL1 fusion. Aiming at molecular classification of leukemia, these works searched for differentially expressed genes discriminating between various subentities of ALL (13, 14). In an alternative approach using the serial analysis of gene expression (SAGE) technique, we compared genome-wide gene expression profiles of normal hematopoietic bone marrow populations, including pre–B cells, hematopoietic progenitor cells, myeloid progenitor cells, T lymphoid precursors (TLPs), and BCR-ABL1+ pre–B ALL, which are thought to represent the malignant outgrowth of pre–B cells (12). The leukemia cells were also compared with mature B cell populations, including naive B cells (NBCs), germinal center B cells (GCBs), memory B cells (MBCs), and plasma cells (PCs). These comparisons were meant to (a) identify novel target genes of BCR-ABL1–mediated transformation, and (b) to elucidate how the oncogenic BCR-ABL1 kinase may interfere with normal pre–B cell receptor signaling.
Materials and Methods
Patient Samples and Cell Purification.
In total, 19 cases of BCR-ABL1 + pre–B ALL were studied. For cases I–IX (Table I), BCR-ABL1 + pre–B ALL cells obtained from bone marrow samples of untreated patients had a purity of >95% as assessed by bone marrow morphology. Cases X–XII (Table I) correspond to BCR-ABL1 + pre–B ALL cell lines, termed SUP-B15, BV173, and NALM1 (DSMZ), respectively. Purification of ALL cells from cases XIII–XIX (Table I) was described previously (15). In these cases, SLP65 isoform expression before and after treatment with the BCR-ABL1 inhibitor STI571 and expression of CD10, CD19, and CD34 was studied (Fig. 4; Table I, cases XIII–XIX). For nine primary cases (including two cases analyzed by SAGE: II and IX) and three cell lines, the configuration of IGH loci was examined (Table II, cases I–XII). For all cases, breakpoints within the major or minor breakpoint cluster region of the BCR gene, leading to p210 and p190 fusion molecules, respectively, were detected by PCR using primers and PCR conditions as described previously (16). Clinical data for all 19 cases are given in Table I. Normal hematopoietic cell populations, including CD34+ CD38low CD133+ Lin− hematopoietic progenitor cells (HSCs), CD15+ common myeloid progenitor cells (CMPs), CD7+ CD10+ T lymphoid precursors (TLPs), CD19+ VpreB− Igμ chain− pro–B cells (no SAGE profile available), and CD10+ CD19+ Igμ chain+ pre–B cells were enriched from human bone marrow by depletion of unwanted cells and enrichment of the populations of interest using immunomagnetic beads and cell sorting. Mature B cell populations, including CD19+ CD27− NBCs, CD19+ CD27+ MBCs, and CD19+ CD138+ PCs were from pooled peripheral blood samples of 12 healthy donors. CD20+ CD77+ GCBs were isolated from human tonsillectomy resectates. HSCs, CMPs, TLPs, pre–B cells, NBCs, MBCs, and PCs were subjected to SAGE analysis as described previously (17–19). SAGE data on GBCs were provided by I. Schwering and R. Küppers (University of Cologne, Cologne, Germany and University of Essen, Essen, Germany, respectively).
Table I.
Clinical Data on BCR-ABL1+ Pre–B ALL Cases Studied
| Case | Age | Sex | BCR-ABL1 fusion | Clinical course |
|---|---|---|---|---|
| I | 34 | m | p190 | complete remission after BMT |
| II | 32 | m | p190 | died after BMT |
| III | 76 | m | p190 | chemotherapy, died 3 wk after onset of therapy |
| IV | 30 | m | p190 | complete remission after BMT, currently GVHD |
| V | 66 | f | p190 | complete remission after chemotherapy |
| VI | 10 | f | p190 | died after BMT |
| VII | 56 | m | p190 | chemotherapy, currently in remission |
| VIII | 10 | f | p190 | died after BMT, GVHD |
| IX | 16 | m | p210 | complete remission after BMT |
| X | 9 | m | p190 | died after relapse (SUP-B15 cell line) |
| XI | 45 | m | p210 | died after relapse (BV173 cell line) |
| XII | 3 | f | p210 | died after relapse (NALM1 cell line) |
| XIII | 39 | m | p210 | relapse after STI571 treatment |
| XIV | 54 | f | p190 | relapse after STI571 treatment |
| XV | 61 | f | p190 | relapse after STI571 treatment |
| XVI | 51 | m | p190 | relapse after STI571 treatment |
| XVII | N.K. | N.K. | p190 | relapse after STI571 treatment |
| XVIII | 65 | m | p190 | relapse after STI571 treatment |
| XIX | 47 | f | p190 | relapse after STI571 treatment |
Sequencing analysis for IGH alleles, cases I–XII. SAGE analysis, cases II and IX. Comparison of SLP65 isoform expression before and after STI571 treatment, cases XIII–XIX.
m, male. f, female. BMT, bone marrow transplantation. N.K., not known. GVHD, graft versus host disease.
Figure 4.

Inhibition of BCR-ABL1 in pre–B ALL cells results in selection for subclones differentiating to immature B cells with preferential expression of Ig λ light chains. BCR-ABL1 + SUP-B15, BV173, and NALM1 pre–B ALL cells were cultured in the presence or absence of 10 μmol/l STI571 for 2 d and surviving cells were analyzed for surface expression of Ig μ heavy chains, Vpre B surrogate light chains (top), Ig κ light chains (middle), and Ig λ light chains (bottom). Inhibition of BCR-ABL1 by STI571 results in preferential outgrowth of Igμhigh cells, which down-regulate VpreB as compared with Igμlow cells and express conventional, predominantly λ light chains.
Table II.
Sequence Analysis of IGH Alleles in BCR-ABL1+ Pre–B ALL Cases
| Case | Allele | Configuration of IGH loci | Coding capacity | Indication for secondary VH gene rearrangement |
|
|---|---|---|---|---|---|
| I | 1 | VH4.34-DH6.13-JH5 | − | out of frame | + (previously rearranged: VH1.8) |
| 2 | DH5-JH | ||||
| II | 1 | VH4.61-DH2.2-JH4 | − | out of frame; two stop codons in junction | + (previously rearranged: VH1.58) |
| 2 | DH2.4-JH4 | ||||
| III | 1 | VH3.33-DH3.22-JH4 | − | out of frame | − |
| 2 | DH7.27-JH; germline | ||||
| IV | 1 | VH5.51-DH3.22-JH4 | − | in frame; stop codon in junction | − |
| 2 | VH5.51-DH6.19-JH3 | − | out of frame | − | |
| V | 1 | VH5.51-DH1.26-JH1 | − | in frame; stop codon in junction | + (previously rearranged: VH2.5) |
| 2 | DH7.27-JH; germline | ||||
| VI | 1 | VH3.30-DH3.22-JH3 | − | out of frame | + (previously rearranged: VH1.2) |
| 2 | DH5-JH | ||||
| VII | 1 | VH1.46-DH6.13-JH4 | − | out of frame | + (previously rearranged: VH2.5) |
| 2 | DH5-JH | ||||
| VIII | 1 | VH2.5-DH3.22-JH4 | − | in frame; two stop codons in junction | + (previously rearranged: VH2.70) |
| 2 | DH7.27-JH; germline | ||||
| IX | 1 | VH1.46-DH3.10-JH5 | + | in frame | + (previously rearranged: VH1.24) |
| 2 | DH3.9-JH6 | ||||
| Xa | 1 | VH3.53-DH2.8-JH6 | + | in frame | + (previously rearranged: VH3.38) |
| 2 | ND | ||||
| XIa | 1 | VH3.48-DH2.15-JH3 | − | out of frame, stop codon in junction | + (previously rearranged: VH3.38) |
| 2 | DH2-JH | ||||
| XIIa | 1 | VH3.9-DH2.21-JH3 | + | in frame | − |
| 2 | DH4-JH | ||||
Cases X–XII correspond to BCR-ABL1 + pre–B ALL cell lines SUP-B15, BV173, and NALM1, respectively.
SAGE Analysis.
cDNA synthesis, SAGE analysis, and cloning and sequencing of SAGE concatemers was performed as described previously (17–19). The UniGene reference database (March 2001) was obtained at http://www.sagenet.org/SAGEDatabases/unigene.htm. A total of 592,000 SAGE tags were collected for 10 SAGE profiles. 106,000 tags were analyzed for the HSC library, 99,000 for CMPs, 110,000 for pre–B cells, 96,500 for T lymphoid precursors, and each of ∼30,000 tags for two cases of bone marrow–derived pre–B ALL carrying a BCR-ABL1 gene rearrangement, NBCs, GBCs, MBCs, and PCs. All SAGE libraries were normalized to 100,000 tags. SAGE data were graphically visualized using the Cluster and Treeview software (http://rana.lbl.gov) and sorted according to the ratio between SAGE-tag counts in pre–B cells and in BCR-ABL1 + ALL cases. In a comprehensive search for pre–B cell receptor components and pre–B cell receptor–related signaling molecules in PubMed, UniGene (http://www.ncbi.nlm.nih.gov/UniGene), and OMIM (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db= OMIM), we identified 54 genes in at least one of the SAGE-libraries analyzed, for which a role in the assembly or regulation of the pre–B cell receptor was shown.
Semi-quantitative RT-PCR Analysis and Verification of Quantitative Accuracy of SAGE Data.
To corroborate quantitative differences in gene expression as determined by SAGE, semi-quantitative RT-PCR analysis was performed for a set of 45 selected genes differentially expressed between HSCs and pre–B cells and 41 genes differentially expressed between NBCs and MBCs as described previously (17, 18).
To verify differential gene expression observed by SAGE-analysis of pre–B cells and the BCR-ABL1 + pre–B ALL cell line SUP-B15, we performed semi-quantitative RT-PCR using the following primers: GAPDH (for standardization), 5′-TTAGCACCCCTGGCCAAG-3′ and 5′-CTTACTCCTTGGAGGCCATG-3′; AML1, 5′-AATGATGAAAACTACTCGGCT-3′ and 5′-TTGGTCTGATCATCTAGTTTC-3′; GATA1, 5′-AGTCTTTCAGGTGTACCCAT-3′ and 5′-AAAGAAGGTACTGGAAAAGTC-3′; EBF, 5′-AAGGAAAAAGAAGCCAACAG-3′ and 5′-GGACAAACATGTTATCAGGA-3′; E2A, 5′-CTGAAAGCAAGCAACAAAAC-3′ and 5′-CACGGGGTCTTTTTAATACA-3′; PAX5, 5′-CCATGTAAATACCTTCTTGC-3′ and 5′-ATTCTGCTTCGGAAAAGTAG-3′; OCT2, 5′-CAAAATAAGACCTCCCCATT-3′ and 5′-TGAGGTAGCTGGAATAGATT-3′; OBF1, 5′-GGCTTCAAAGAGAAAAGGCA-3′ and 5′-TCTGTCGTGACATTGGTGAT-3′; and IRF4, 5′-CAAGAGCAATGACTTTGAGG-3′ and 5′-TGGGACATTGGTACGGGAT-3′. The same primer sets were also used for RT-PCR analysis of STI571-treated SUP-B15 ALL cells after MACS enrichment of μ+ and μ− subclones.
In addition, quantitative differences between the SAGE libraries for pre–B cells and BCR-ABL1 + pre–B ALL cases were verified at the protein level for 10 surface molecules by flow cytometry using antibodies against CD19, IL7Rα, CD10, CD38, CD40, CD150/SLAM, CD72, VpreB, Igα, and CD34 (BD Biosciences; unpublished data).
Sequence Analysis of IGH Loci.
To characterize the configuration of the IGH loci in nine primary BCR-ABL1 + pre–B ALL cases (Table II, cases I–IX) and three cell lines (SUP-B15, BV173, and NALM1; Table II, cases X–XII), three primer sets were used to amplify germline configuration, D-J and V-DJ gene rearrangements from both alleles of the IGH locus. Clonal VH gene rearrangements were amplified in a single round of PCR amplification using 35 PCR cycles at a detection limit of ∼104 clonal cells in 5 × 106 total cells. PCR primers and conditions were used as described previously (20) and amplification products were directly sequenced and aligned according to Matsuda et al. (21) using the IMGT software at http://www.dnaplot.de/input/human_v.html. To identify footprints from formerly rearranged VH gene segments within potential secondary VDJ rearrangements, a list of VH and DH germline gene segments was obtained at http://imgt.cines.fr and the cryptic recombination signal sequence (cRSS) was identified as described previously (22).
Measurement of pre–B Cell Receptor Responsiveness.
Primary human CD19+ bone marrow mononuclear cells were enriched from bone marrow samples of four healthy donors using immunomagnetic MACS beads as described previously (17). SUP-B15, BV173, and NALM1 pre–B ALL cells (DMSZ) carrying a BCR-ABL1 gene rearrangement (Table II, cases X–XII) were cultured with 20% fetal calf serum in RPMI 1640 medium in the presence or absence of 10 μM STI571 (Novartis) for the times indicated. Among STI571-treated leukemia cells, Igμ+ and Igμ− cells were separated using a nonstimulating, biotin-conjugated anti-μ antibody (BD Biosciences) together with streptavidin microbeads (Miltenyi Biotech). After MACS enrichment or preincubation, cells were washed and stained with Fluo-3 dye (Calbiochem) for 30 min. Changes of cytosolic Ca2+ were measured by laser scans using confocal microscopy (18, 19). After 10–30 s of measurement, antibodies against human μ chains (Jackson ImmunoResearch Laboratories), VpreB (BD Biosciences), or Igκ- together with Igλ-light chains (BD Biosciences) were added to CD19+ bone marrow mononuclear cells or preincubated pre–B ALL cells (in the presence or absence of STI571; cases X–XII). Cytosolic Ca2+ concentrations were determined as described previously (18). As a negative control, cells were also treated with an antibody against CD3 (BD Biosciences), which induces Ca2+ mobilization in T but not B lineage cells.
Flow Cytometry.
Surface expression of VpreB surrogate or Igκ- or Igλ-light chains, Ig μ-chains and CD19 on BV173, NALM1, and SUP-B15 pre–B ALL cells in the presence or absence of 10 μM STI571 was monitored using antibodies against VpreB, Igκ- or Igλ-light chains, the Ig μ heavy chain and CD19 (BD Biosciences) after the incubation times indicated. Apoptotic or dead cells were identified by staining with anti–Annexin V antibodies and propidium iodide (BD Biosciences).
RT-PCR Analysis of SLP65 Isoform Expression.
cDNA amounts were normalized by OD measurements and amplification of a specific fragment of the GAPDH gene using 5′-TTAGCACCCCTGGCCAAGG-3′ as forward and 5′-CTTACTCCTTGGAGGCCATG-3′ as reverse primers. For amplification of SLP65 isoforms, forward primers specific for exon I, 5′-TGGACAGTTATTCGTGTCTCTT-3′, and additional exon IIIb, 5′-AGAGTGTGTTGACCTTGGTG-3′, were used together with a reverse primer specific for additional exon IIIb, 5′-TTGGCTTAGAGGGTTTTGG-3′, and exon VII, 5′-GTGAACTGCTTTCTGTGGGA-3′, as described previously (8). For amplification of another additional exon IIIa, the forward primer, 5′-TTTAATCTCTCCTGGAATGCAG-3′ was used. However, a splice variant including exon IIIa (8) could not be amplified with this primer (unpublished data). cDNAs derived from human pre–B cells, BCR-ABL1 + pre–B ALL cells in the presence or absence of STI571 and MACS-sorted μ-chain+ and μ-chain− BCR-ABL1 + pre–B ALL cells after treatment with 10 μM STI571, were used as RT-PCR templates.
Results and Discussion
Pre–B Cell Receptor-associated Signaling Molecules Are Silenced in BCR-ABL1+ pre–B ALL Cells.
To identify target genes of the oncogenic BCR-ABL1 kinase, we compared genome-wide gene expression profiles of two cases of BCR-ABL1 + pre–B ALL and normal hematopoietic populations using the SAGE technique. For the SAGE analysis of normal hematopoietic populations, a total of 592,000 SAGE tags have been collected, including CD10+ CD19+ human bone marrow pre–B cells as the normal precursors of pre–B ALL, CD7+ CD10+ TLPs, CD15+ CMPs, CD34+ CD38low HSCs, CD19+ CD27− NBCs, CD20+ CD77+ GCBs, and CD19+ CD27+ MBCs, and CD19+ CD138+ PCs (Fig. 1). Comparing the gene expression pattern in BCR-ABL1 + pre–B ALL and normal human pre–B cells, many genes conferring B cell lineage commitment and signal transduction through the pre–B cell receptor were transcriptionally silenced in the leukemic cells (Figs. 1 and 2). Loss of pre–B cell receptor–related molecules in the pre–B ALL cells involves nuclear transcription factors (Fig. 2, OBF1, PAX5, E2A, OCT2, EBF, and IRF4), cytoplasmic kinases and linker molecules (Fig. 1, LYN, BLK, BTK, BRAG, SLP65, SYK, BAP37, IgαBP1, BRDG1, PLCγ2, VAV1-3, HPK1, LCK, FYN, BAM32, AKT, SHC1, SAP, p62DOK, CIN85, NIK, and IKK), and membrane-associated receptor molecules (Fig. 1, CD19, IGHμ, VpreB, Igβ, and Igα). Conversely, transcription factors related to primitive hematopoiesis, including AML1 and GATA1, are up-regulated in the leukemia cells as compared with their normal counterpart (Fig. 2). Also, signaling molecules related to NF-κB, JAK-STAT, GAB2, and GRB2 pathways are expressed in the leukemia cells at similar or higher levels than in pre–B cells (Fig. 1, bottom). However, NF-κB activation (23) and GAB2/GRB2 phosphorylation (24, 25), together with expression of JAK and STAT proteins (26), reflect oncogenic BCR-ABL1 kinase activity or requirements for transformation by BCR-ABL1 rather than pre–B cell receptor signaling in the leukemia cells.
Figure 1.

Expression of pre–B cell receptor–related molecules in BCR-ABL1+ pre–B ALL cells. Using the SAGE method, 10 genome-wide gene expression profiles have been generated for CD34+ CD38low hematopoietic progenitor cells (HSCs), CD15+ common myeloid progenitor cells (CMPs), CD7+ CD10+ T cell lineage precursors (TLPs), CD10+ CD19+ Igμ+ pre–B cells (pre-Bs), CD19+ CD27− naive B cells (NBCs), CD20+ CD77+ germinal center B cells (GCBs), CD19+ CD27+ memory B cells (MBCs), and CD19+ CD138+ plasma cells (PCs), and compared with two cases of pre–B ALL-carrying a BCR-ABL1 gene rearrangement (ALL, cases II and IX). SAGE-tag counts are given as numbers and depicted in colors, with red indicating high and green indicating low levels or no expression. SAGE data were sorted according to the ratio of SAGE-tag counts for pre–B cells and SAGE-tag counts in BCR-ABL1 + ALL cases. In addition to gene names, annotations of proposed gene functions (including a reference) are given.
Figure 2.

Inhibition of BCR-ABL1 in pre–B ALL cells can restore expression of B cell–specific transcription factors. SAGE-tag counts of hematopoietic stem cell–related transcription factors AML1 and GATA1 and B cell–specific transcription factors E2A, EBF, OBF1, OCT2, IRF4, and PAX5 are compared between normal pre–B cells and two cases of BCR-ABL1 + pre–B ALL (cases II and IX). Differential expression of these transcription factors in pre–B cells as compared with leukemia cells was verified by semi-quantitative RT-PCR using cDNAs from normal pre–B cells and BCR-ABL1 + SUP-B15 cells. Inhibition of BCR-ABL1 by STI571 could restore a pre–B cell–like expression pattern of these transcription factors in Igμ+ but not in Igμ− leukemia cells. Igμ+ and Igμ− leukemia cells were separated by MACS.
BCR-ABL1+ pre–B ALL Cells Carry Nonproductive VH Gene Rearrangements in Most Cases.
These findings suggest that BCR-ABL1 + pre–B ALL cells as opposed to normal bone marrow pre–B cells do not express an active pre–B cell receptor. Therefore, we investigated whether the genomic configuration of the IGH loci in BCR-ABL1 + pre–B ALL cells is compatible with the expression of a functional pre–B cell receptor. Only 3 out of 12 BCR-ABL1 + pre–B ALL cases harbored a potentially functional IGH gene rearrangement (Table II). In the remaining nine cases, the coding capacity of IGH alleles was compromised by loss of the reading frame or generation of stop codons within the junction between V, D, and J segments during the rearrangement process. To formally exclude that in these cases a productive V H-DHJH rearrangement on the second allele might have been missed during the PCR amplification, we also amplified the second IGH allele using primers specific for IGH germline configuration or D H-JH rearrangements (Table II). In one case (Table II, case IV), two IGH VDJ rearrangements were amplified, both of which were nonfunctional.
Hence, the majority of pre–B ALL clones carrying the oncogenic BCR-ABL1 gene rearrangement can survive and even clonally expand in the absence of pre–B cell receptor survival signals. We conclude that BCR-ABL1 + leukemia cells can bypass selection for the expression of a pre–B cell receptor.
However, because clonal VH gene rearrangements were amplified in a single round of PCR amplification at a relatively low sensitivity (detection limit of ∼104 clonal cells in 5 × 106 total cells), we cannot exclude that small subclones carrying a productive IGH allele were among the leukemia cells. Of note, the two leukemia cases analyzed by SAGE also differed with respect to the coding capacity of their IGH alleles. However, irrespective of whether the leukemia cells harbored a potentially productive IGH allele or not, mRNA expression of pre–B cell receptor–related signaling molecules was invariably down-regulated or missing in both cases (Fig. 1).
Inhibition of BCR-ABL1 Kinase Activity Reconstitutes Selection of pre–B ALL Cells Expressing Ig μ-chains at High Levels.
To determine whether the BCR-ABL1 kinase has an effect on the expression of the pre–B cell receptor, we measured expression levels of surface Ig μ heavy chain on three BCR-ABL1 + pre–B ALL cell lines (SUP-B15, BV173, and NALM1) in the presence and absence of STI571, a specific inhibitor of BCR-ABL1 kinase activity. CD19+ bone marrow B lineage cells exhibit surface Ig μ heavy chain expression at variable levels (Fig. 3 A). In contrast, Ig μ heavy chain protein was detectable only at very low levels on the surface of SUP-B15 cells (Fig. 3 B) and NALM1 cells (not depicted). On BV173 cells, Ig μ heavy chains were detected neither on the cell membrane nor in the cytoplasm (not depicted), which is consistent with the absence of a productive IGH allele in these cells (Table II, case XI). However, after 19 h of STI571 treatment, a small subset of ∼3% of the leukemia cells expressed surface Ig μ heavy chain at high levels (Fig. 3 C). Only few ALL cells survived continued STI571 treatment, which further increased the fraction of Ig μhigh cells to 23% after 4 d (Fig. 3 F) and 48% after 6 d (Fig. 3 D). In STI571-treated leukemia cells, expression of Ig μ heavy chain surface correlated with viability of the cells (i.e., lack of both annexin V membrane expression and uptake of propidium iodide; Fig. 3 F). Conversely, almost no Ig μ heavy chain protein could be detected on the surface of cells undergoing apoptosis induced by STI571 (Fig. 3 F). During 6 d of STI571 treatment, >95% of all leukemia cells died by apoptosis. Thus, the relative gain of Ig μhigh cells can be fully attributed to selective deletion of Ig μ− leukemia cells. We conclude that ablation of BCR-ABL1 kinase activity by STI571 induces selection of leukemia subclones expressing high levels of μ-chains on their surface.
Figure 3.

Inhibition of BCR-ABL1 induces selection for Ig μhigh leukemia cells. Normal bone marrow mononuclear cells (MNCs) include B lineage cells expressing Ig μ heavy chains at variable levels, depending on their differentiation stage as pro–B cells (Igμ−), pre–B cells (Igμlow), or immature B cells (A, Igμhigh). BCR-ABL1 + SUP-B15 (B–F), BV173 (not depicted), and NALM1 (not depicted) pre–B ALL cells were incubated in the absence (B and E) or presence of 10 μmol/l STI571 (C, D, and F) for 19 h (B and C), 2 (E and F) or 6 d (D) and measured for surface expression of CD19 (A–D), Annexin V (E and F), and Ig μ chains (A–F). Comparing BCR-ABL1 + SUP-B15 pre–B ALL cells to BV173 and NALM1 cells, similar results were obtained (not depicted).
In STI571-treated patients, expression of μ-chains on few leukemia subclones may confer a survival advantage and, hence, represent an escape mechanism through which some leukemic cells can temporarily evade STI571-induced apoptosis. In the absence of BCR-ABL1 kinase activity, the malignant cells would undergo apoptosis unless they successfully reactivate other survival signals by default (e.g., through the pre–B cell receptor).
After extended STI571 treatment, Ig μhigh cells were not only seen in SUP-B15 cells and NALM1 cells, which both carry a functional IGH allele (Table II, cases X and XII). Unexpectedly, they were also seen in BV173 cells, from which we amplified two nonproductive IGH alleles that gave rise to Ig μhigh cells after prolonged exposure to STI571. This could be explained by selective outgrowth of few Ig μhigh subclones of the BV173 cell line that already existed before STI571 treatment (e.g., as a result of spontaneous secondary IGH gene rearrangement). Alternatively, secondary IGH gene rearrangements might be induced by STI571-mediated inhibition of BCR-ABL1. Therefore, remedy of compromised coding capacity for a functional pre–B cell receptor by BCR-ABL1-inhibition in the case of BV173 cells exemplifies that survival signals through the pre–B cell receptor would indeed allow leukemia cells to temporarily evade STI571-mediated apoptosis.
BCR-ABL1 Kinase Activity Overrides pre–B Cell Receptor Signaling in Leukemia Cells.
To elucidate whether the BCR-ABL1 kinase activity in leukemic pre–B cells cannot only bypass selection for but also interfere with pre–B cell receptor signaling, we compared antigen receptor responsiveness in human bone marrow pre–B cells (VpreB+) and immature B cells (Igκ+ or Igλ+) and three BCR-ABL1 + leukemia cell lines. Two out of the three cell lines (SUP-B15 and NALM1; Table II, cases X and XII) indeed carry a productive IGH allele and express surface Ig μ heavy chain at low levels together with VpreB (Fig. 4). To this end, we measured Ca2+ release in response to antigen receptor engagement by anti-VpreB, anti-κ, anti-λ, and anti-μ antibodies in normal pre–B and immature B cells and BCR-ABL1 + leukemia cells at the single cell level. Pre–B cells and immature B cells showed the typical increase of cytoplasmic Ca2+ reaching a plateau ∼1 min after pre–B cell receptor cross-linking (Fig. 5 A). Comparing engagement of pre–B cell receptors by anti-VpreB and cross-linking of B cell receptors by anti-κ or anti-λ, the Ca2+ signal initiated by the pre–B cell receptor had a lower amplitude and was slightly delayed (Fig. 5 A). In contrast, BCR-ABL1 + SUP-B15 leukemia cells did not respond to anti–Ig μ trigger at all, even if fivefold-higher antibody concentrations were used. Instead, the leukemia cells displayed a peculiar autonomous oscillatory Ca2+ signal activity (Fig. 5 B). Autonomous Ca2+ oscillations were not only seen in SUP-B15 cells (Fig. 5 B) but also in BCR-ABL1 + BV173 and NALM1 leukemia cells (not depicted). To test whether autonomous undulatory Ca2+ signals are causally linked to BCR-ABL1 kinase activity, we treated BCR-ABL1 + SUP-B15, BV173, and NALM1 cells with STI571. In the presence of STI571, the leukemia cells did not exhibit any autonomous Ca2+ signal activity. Instead, antigen receptor responsiveness was partially restored in STI571-treated leukemia cells. Upon pre–B or B cell receptor engagement by anti-μ chain antibodies, some of the STI571-treated leukemia cells responded by release of Ca2+ from cytoplasmic stores, whereas many other leukemia cells did not respond to antigen receptor cross-linking (unpublished data).
Figure 5.
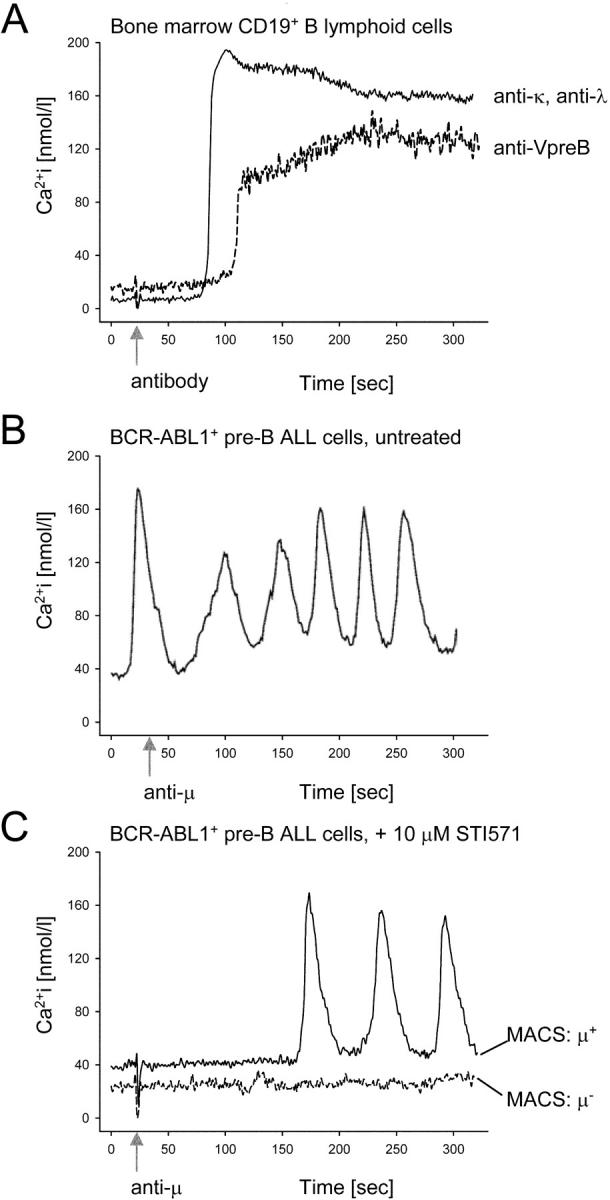
Inhibition of BCR-ABL1 restores (pre) B cell receptor responsiveness in pre–B ALL cells. CD19+ B lineage cells were purified from bone marrow mononuclear cells using immunomagnetic beads. Release of Ca2+ from cytoplasmic stores in these cells in response to pre–B or B cell receptor engagement by anti-VpreB or a mixture of anti-κ and anti-λ chain antibodies (arrow) was measured using a laser scanning microscope (A). Untreated BCR-ABL1 + SUP-B15 (B), BV173, and NALM1 (not depicted) pre–B ALL cells and BCR-ABL1 + pre–B ALL cells incubated with 10 μmol/l STI571 for 24 h (C) were stimulated using an anti-μ chain antibody, which can cross-link both pre–B and B cell receptors. Among STI571-treated leukemia cells, Igμ+ cells were separated from Igμ− cells and analyzed independently (C). In B, fivefold higher concentrations of the anti-μ antibody than in A and C were used. The data shown here refer to SUP-B15 cells. Analyzing BV173 and NALM1 cells, similar results were obtained (not depicted).
To determine whether regained responsiveness of pre–B cell receptors was restricted to differentiating Ig μhigh cells among the STI571-treated leukemia cells, Ig μhigh and Ig μ− cells were separated by MACS after prolonged STI571 treatment. Although 48 out of 56 Ig μhigh STI571-treated cells responded by an oscillatory Ca2+ signal, all 45 Ig μ− STI571-treated cells remained silent upon pre–B cell receptor engagement (Fig. 5 C).
We conclude that the BCR-ABL1 kinase establishes an unusual autonomous signaling activity that overrides signal transduction initiated by the pre–B cell receptor. Although the kinetics of the Ca2+ signal in STI571-treated leukemia cells still clearly differs from the signaling pattern in normal pre–B and immature B cells, inhibition of BCR-ABL1 kinase activity restores responsiveness of the pre–B cell receptor in these leukemia cells.
BCR-ABL1+ Leukemia Cells Surviving STI571 Treatment Differentiate into Immature B Cells.
Assuming a pre–B cell origin of the three BCR-ABL1 + leukemia cell lines analyzed, Ig μ heavy chains on the surface of leukemia cells surviving extended STI571 treatment were expressed at surprisingly high levels (Fig. 3, D and E), which are characteristic for μ chain expression in the context of a B cell receptor rather than a pre–B cell receptor (12). Therefore, we tested whether cells surviving prolonged STI571 treatment were in fact differentiating into immature B cells, which express high levels of surface μ-chain as part of a B cell receptor. As shown in Fig. 4, Ig μhigh leukemia cells down-regulated VpreB as compared with Ig μlow cells (Fig. 4, top). Indeed, surrogate light chains on Ig μhigh leukemia cells were replaced with conventional light chains (Fig. 4, middle and bottom). We conclude that the three BCR-ABL1 + leukemia cell lines treated with STI571 exhibit preferential outgrowth of subclones differentiating into immature B cells. These findings are in agreement with a recent paper on the effect of STI571 on v-abl–mediated differentiation arrest in murine pre–B cells (27). However, in the three BCR-ABL1 + leukemia cell lines tested, the pattern of light chain expression was heavily biased in favor of λ-light chains (Fig. 4, bottom) and only few κ-expressing cells could be detected (Fig. 4, middle), whereas Igκ+ cells in control stainings can easily be identified by the antibody we used (not depicted).
Extensive κ-deleting Element (KDE) Rearrangement Results in Preferential λ-light Chain Expression on STI571-surviving Leukemia Cells.
Searching for a potential explanation for predominant λ-light chain expression on differentiating leukemia cells surviving STI571 treatment, we investigated whether rearrangement of the so-called KDE may contribute to reduced κ light chain usage in STI571-treated leukemia cells. KDE rearrangement may lead to inactivation of productively recombined Vκ-Jκ joints and, hence, represents another level at which Ig light chain expression can be regulated (28).
We amplified specific DNA fragments for KDE-germline configuration, KDE rearrangement to an RSS within the Jκ intron, and KDE rearrangement to RSS sites flanking Vκ gene segments from three BCR-ABL1 + leukemia cells in the absence or presence of STI571. Although KDE was found in germline configuration in untreated leukemia cells, STI571-mediated inhibition of BCR-ABL1 was associated with rearrangement of KDE to Jκ intron RSS sites and, to a lesser extent, to Vκ RSS sites (not depicted). Thus, predominant expression of λ light chains is consistent with extensive KDE rearrangement observed in our studies. In the absence of BCR-ABL1 kinase activity, differentiating leukemia cells seem to undergo multiple rounds of rearrangement targeting both κ and λ light chain loci, which ultimately leads to preferential λ light chain expression due to KDE-mediated deletion of potentially productive Vκ-Jκ joints.
Ig μhigh Leukemia Cells Surviving STI571 Treatment Restore Expression of B Cell–specific Transcription Factors.
Propensity of STI571-treated leukemia cells to differentiate into light chain–expressing immature B cells suggests that these cells exhibit a more mature phenotype as compared with untreated leukemia cells. Therefore, we tested whether STI571-treated differentiating leukemia cells can restore expression of B cell–specific transcription factors, which were down-regulated in the untreated leukemia cells as compared with normal pre–B cells (Fig. 2, SAGE data). BCR-ABL1 + SUP-B15 pre–B ALL cells were cultured either in the presence or absence of STI571. Among STI571-treated leukemia cells, Ig μhigh and Ig μ− cells were separated by MACS using immunomagnetic beads. From normal pre–B cells, untreated ALL cells, STI571-treated Ig μhigh cells, and STI571-treated Ig μ− cells, cDNAs were synthesized and subjected to semi-quantitative RT-PCR analysis for mRNA levels of B cell–specific transcription factors (E2A, EBF, OBF1, OCT2, IRF4, and PAX5) and the stem cell–related transcription factors AML1 and GATA1 (Fig. 2). The expression patterns of these transcription factors in STI571-treated differentiating Ig μhigh and normal pre–B cells were similar, indicating that STI571-treated Ig μhigh leukemia cells indeed reversed the dedifferentiated stem cell–like phenotype seen in untreated leukemia cells and STI571-treated Ig μ− leukemia cells (Fig. 2).
Ablation of BCR-ABL1 Kinase Activity Corrects Expression of Truncated SLP65 in pre–B ALL Cells from STI571-treated Patients.
An earlier work demonstrated that in the human, functional SLP65 is required for the expression of a pre–B cell receptor; B cell precursors in a patient carrying a deleterious mutation of the SLP65 gene are arrested at the pro–B cell stage of development (29). In mice, SLP65 deficiency does not prevent expression of a pre–B cell receptor, but results in a partial differentiation block at the pre–B cell stage (7). Of note, in ∼50% of childhood pre–B ALL cases, SLP65 expression is defective, which results in compromised pre–B cell receptor signaling (8). In these cases, either no SLP65 was expressed or an aberrant splice variant including one or two additional exons was inserted between SLP65 exons III and IV (8). Inclusion of additional exons, termed IIIa and IIIb introduced a premature translation stop resulting in loss of the COOH terminus of SLP65 (SLP65 ΔC). The COOH terminus of SLP65 encompasses tyrosine 96, whose phosphorylation is critical for SLP65 activity in mice (8). Therefore, we tested whether SLP65 expression is deranged in BCR-ABL1 + pre–B ALL cells and whether SLP65 deficiency is linked to the BCR-ABL1 kinase activity. To this end, we analyzed SLP65 isoform expression in primary pre–B cells from the bone marrow of four healthy donors and STI571-treated and untreated BCR-ABL1 + pre–B ALL cells (Fig. 6 A). Although normal pre–B cells expressed high levels of the canonical SLP65 isoform and only very little alternatively spliced SLP65 ΔC, this pattern was almost inverted in BCR-ABL1 + pre–B ALL cells. Also, when BCR-ABL1 + pre–B ALL cells were incubated in the presence of STI571 for 48 h, the leukemia cells expressed the SLP65 ΔC transcript, although at lower levels. Given that short-term treatment with STI571 partially restored (pre–B cell receptor responsiveness; Fig. 5 C) and induced selection for differentiating leukemia cells expressing high levels of Ig μ heavy chains (Fig. 3), we sorted Ig μ+ leukemia cells. STI571-treated leukemia cells, which have recovered pre–B cell receptor expression, also regained normal SLP65 expression (Fig. 6 A) in parallel with restored pre–B cell receptor responsiveness (Fig. 5 C). In humans, deleterious mutation of the SLP65 gene prevents expression and function of the pre–B cell receptor on mutant B cell precursors (29). However, inactivating mutations within pre–B cell receptor–related genes (including IGHCμ, Igα, Igβ, VpreB, and λ5) do not interfere with SLP65 expression (29). Therefore, deranged SLP65 expression is likely the cause rather than the consequence of compromised pre–B cell receptor signaling in BCR-ABL1 + pre–B ALL cells.
Figure 6.

Expression of SLP65 isoforms in BCR-ABL1+ pre–B ALL cells. SLP65 cDNA fragments were amplified from normal human CD19+ CD34+ Igμ− pro-B, CD10+ CD19+ Igμlow pre–B, CD10− CD19+ CD27+ Igμhigh memory B cells (C), and BCR-ABL1 + ALL cells, including a BCR-ABL1 + cell line in the presence or absence of STI571 (A) and enriched ALL cells from seven leukemia patients (Table I, cases XIII–XIX) before and after treatment with STI571 (B). cDNA amounts were normalized using GAPDH as a standard (A–C). The primers used amplified full-length SLP65 and an isoform encoding truncated SLP65 (SLP65 ΔC). For leukemia cases XIII, XIV, XVI, XVII, and XIX described in B, FACS® data on coexpression of CD10 and/or CD34 on leukemic CD19+ cells was available before and after STI571 treatment. a, Percentage of CD19+ leukemia cells coexpressing CD10 or CD34.
To determine whether deranged SLP65 expression is linked to BCR-ABL1 kinase activity also in primary BCR-ABL1 + pre–B ALL leukemia cells, we analyzed the pattern of SLP65 isoform expression in enriched leukemia cells from seven patients before and after treatment with STI571. Comparing matched pairs of leukemia samples, we identified three patients in which the leukemia cells expressed SLP65 only at low levels, if at all (cases XVI, XVII, and XIX; Fig. 6 B). In these cases, treatment by STI571 had no effect on SLP65 isoform expression. Given that the leukemia samples had a purity of ∼80 percent, weak SLP65 expression in these cases can be attributed to few contaminating normal bystander B cells. In four cases, expression of the canonical SLP65 isoform encoding the full-length protein was accompanied by the expression of SLP65 ΔC. Treatment with STI571 not only increased mRNA levels of full-length SLP65 but also abolished expression of the truncated splice variant (Fig. 6 B).
Given that STI571 treatment of the BCR-ABL1 + ALL cell lines BV173, NALM1, and SUP-B15 resulted in the preferential outgrowth of more differentiated subclones (Figs. 2 and 4), which further differentiate into Ig μhigh light chain–expressing immature B cells (Fig. 4), we wondered whether also in patients the BCR-ABL1 + leukemia cells surviving STI571 treatment tend to exhibit a more mature phenotype. Therefore, we analyzed primary B lymphoid bone marrow cells from seven leukemia patients before and after STI571 treatment by flow cytometry and calculated the percentage of CD19+ cells, which coexpress CD34 and/or CD10. In the human, CD19+ CD34+ cells are considered pro–B cells and CD19+ CD10+ CD34− cells include pre–B cells and immature B cells, whereas mature B cell subsets in human bone marrow and the peripheral blood typically express CD19 but neither CD10 nor CD34 (12). Based on this rough classification, from 46 to 98% of untreated BCR-ABL1 + leukemia cells coexpress CD34 and, thus, exhibit a pro–B cell–like phenotype (Fig. 6 B). After STI571 treatment, the percentage of cells displaying a pro–B cell–like phenotype remains high in three cases (XVI, XVII, and XIX). However, in two other cases, the fraction of pro–B cell–like leukemia cells drops to 2 and 8% (cases XIII and XIV; Fig. 6 B, bottom). The remaining cells show a more mature phenotype resembling pre–B or immature B cells. Of note, STI571-induced conversion of primary leukemia cases from a pro–B cell–like to a pre–B cell–/immature B cell–like phenotype correlates with therapy-related correction of SLP65 isoform expression in these cases (Fig. 6 B). Three cases in which STI571 treatment had no effect on SLP65 mRNA expression also did not convert to a more differentiated phenotype in response to STI571 therapy (cases XVI, XVII, and XIX; Fig. 6 B). From two cases, no flow cytometry data was available.
SLP65 ΔC is nonfunctional, and by competing with full-length SLP65 for binding to upstream kinases (e.g., SYK), it may have an inhibitory effect on signal transduction initiated from the pre–B cell receptor in pre–B ALL cells. Although the negative regulatory effect of truncated SLP65 is not fully elucidated, our results indicate that its expression is linked to BCR-ABL1 kinase activity.
A Potential Link between SLP65 Deficiency, Secondary VH Gene Rearrangement, and Loss of Coding Capacity for a μ Heavy Chain in BCR-ABL1+ pre–B ALL Cells.
SLP65 also functions as a sensor for the expression of a productive VH region gene within a pre–B cell receptor complex and halts the recombination machinery to prevent further, potentially inactivating, rearrangement of IGH gene segments (1). In the absence of functional SLP65, the recombination machinery remains active and generates secondary VH region gene rearrangements even if V, D, and J segments have been productively rearranged (9). Assuming that SLP65 deficiency is a common feature of BCR-ABL1 + ALL, one would predict that in most if not all cases, the leukemia clone has undergone one or more rounds of secondary VH gene rearrangement. Such secondary rearrangements can occur by replacement of an already rearranged VH gene segment by another (in most cases upstream) VH gene segment (22). VH gene replacement typically uses a cRSS, which is present in the 3′ part of 40 out of 44 functional VH gene segments (22). A hallmark for VH gene replacement are short sequences (5–7 bp) within the VH-DH junction that are foreign to the actually rearranged VH gene segment but are matching exactly the 3′ end adjacent to the cRSS of another previously rearranged VH gene. It is very unlikely that 5–7 bp stretches matching the extreme 3′ part of another VH gene within the VH-DH joint can be randomly generated as a by-product of junctional diversification (P = 0.009; reference 22). Therefore, these short sequences are considered vestiges of a previously rearranged VH gene segment. Analyzing the IGH VDJ rearrangements for footprints of secondary rearrangements, we found indication of VH gene replacement in 9 out of 12 leukemia cases (Table III). In eight out of nine cases, the potential secondary rearrangement uses a donor VH segment located upstream from the initially rearranged (recipient) VH gene segment (Table III). One potential VH replacement might reflect an inversion or transrecombination event as described previously (22). A comprehensive analysis of IGH VDJ rearrangements amplified from normal human immature B cells identified traces of VH gene replacement in 16 out of 343 sequences (5%; reference 22). Hence, the frequency of VH gene replacement in BCR-ABL1 + ALL cases is, at ∼75%, surprisingly high.
Table III.
Indication of Potential VH Gene Replacement in VH Region Genes of BCR-ABL1+ Pre–B ALL Clones
| Case | 3′ part of recipient VH | 3′ part of donor VH | VH-DH | DH | |||
|---|---|---|---|---|---|---|---|
| I | VH1.8 | CGTGTATTACTGTGCGAGAGG | VH4.34 | GTGTATTACTGTGCG | AGAGGGGGGTA | GGGAATAGCA | DH6.13-JH5 |
| II | VH1.58 | CGTGTATTACTGTG CGGCAGA | VH4.61 | GTGTATTACTGTGCGAGAGA | CACGGCAGATG | TATTGTAGTAGTACCCCC | DH2.2-JH4 |
| V | VH2.5 | CACATATTACTGTG CACACAGACC | VH5.51 | ATGTATTACTGTGCG | AGACACACAGT | GAGAGAAACCAGCC | DH1.26-JH1 |
| VI | VH1.2a | CGTGTATTACTGTG CGAGAGA | VH3.30 | GTGTATTACTGTGCG | CGAGAGATCCCCCTTC | TATTACTATGA | DH3.22-JH3 |
| VII | VH2.5 | CACATATTACTGTGCACACAGACC | VH1.46 | GTGTATTACTGTGCGAG | AGACCGCT | GGGTATAGCAGCAGCTG | DH6.13-JH4 |
| VIII | VH2.70b | CACATATTACTGTG CACACAGAC | VH2.5 | ACATATTACTGTG CA | CACAGCCCCGGGATCCC | CGGGATAGTAGTG | DH3.22-JH4 |
| IX | VH1.24 | CGTGTATTACTGTG CAACAGA | VH1.46 | GTGTATTACTGTGCGAGA | CCAACAG | ACCGTGGTTCGGGGA | DH3.10-JH5 |
| X | VH3.38 | CGTGTATTACTG TGCCAGATATAc | VH3.53 | GTGTATTACTGTGCGAGA | GTTGCCAGGGGG | TGGTGTATGCTATACC | DH2.8-JH6 |
| XI | VH3.38 | CGTGTATTACTGTGCCAGATATA | VH3.48 | GTGTATTACTGTGGCGA | GCCAGATATTGT | AGTGGTGGTAGCT | DH2.15-JH3 |
cRSS motifs (bold). Footprints of recipient VH gene segments (underline).
The footprint of this potential VH replacement may be also derived from VH1.3, VH1.18, VH3.7, VH3.11, VH3.20, VH3.21, and VH4.4 gene segments.
Likely generated by inversion or transrecombination events.
The footprint of VH replacement in this case also involves the last two nucleotides of the cRSS motif.
Therefore, defective SLP65 expression (Fig. 6) is associated with and potentially the reason for secondary, mostly nonproductive, VH gene rearrangements in 9 out of the 12 leukemia cases we analyzed (Table II). In agreement with a pre–B cell origin of BCR-ABL1 + ALL, one might envision that the precursor of the leukemia clone underwent malignant transformation after having passed the pre–B cell receptor checkpoint but subsequently lost coding capacity of an initially productive Ig VH region gene by a deleterious secondary VH gene rearrangement.
Assuming that BCR-ABL1 can relieve the selection pressure for survival signals through the pre–B cell receptor, next we tested whether truncated SLP65 is also expressed in normal pro–B cells, in which selection for the expression of a functional pre–B cell receptor is not operative. Although CD19+ Ig μ chain− VpreB− pro–B cells express full-length SLP65 at similar levels as in CD19+ Ig μ chain+ Vpre-B+ pre–B cells and CD19+ CD27+ MBC cells, SLP65 ΔC is only expressed in pro–B cells (Fig. 6 C). As SLP65 isoform expression is similar in BCR-ABL1 + pre–B ALL and normal human CD19+ Ig μ− VpreB− pro–B cells, we conclude that SLP65 ΔC is not a marker for B lineage leukemia cells, but rather for B lymphoid cells that are not selected for the expression of an active pre–B cell receptor.
Perspective.
A broad range of pre–B cell receptor–related signaling molecules and transcription factors is transcriptionally silenced in BCR-ABL1 + pre–B ALL as compared with human pre–B cells. Loss of pre–B cell receptor–related molecules as observed by SAGE (Figs. 1 and 2) may reflect that the BCR-ABL1 kinase may have relieved the selection pressure for the expression of these molecules. In the presence of autonomous BCR-ABL1 kinase activity, pre–B ALL cells become nonresponsive to pre–B cell receptor engagement (Fig. 5). As a consequence, molecules needed for transduction of survival signals through the pre–B cell receptor, disappear from the gene expression program of the leukemic cells (Fig. 1, SAGE). Conversely, inhibition of BCR-ABL1 reconstitutes dependence on survival signals initiated from surface receptors, including the pre–B cell receptor (Fig. 3). It is even conceivable that default signaling through the pre–B cell receptor represents a mechanism to acquire drug resistance that is available to BCR-ABL1 + pre–B ALL cells but not to other malignant cells carrying a BCR-ABL1 gene rearrangement (e.g., chronic myeloid leukemia [CML] cells). In this regard, it is interesting to note that treatment failure of STI571 is frequent in pre–B ALL but not in CMLs harboring a BCR-ABL1 fusion gene (30). Moreover, STI571 resistant as opposed to STI571-sensitive BCR-ABL1 + pre–B ALL cells exhibit high expression levels of Bruton's tyrosine kinase, which represents a critical component of pre–B cell receptor signal transduction cascade (15). Therefore, resumption of pre–B cell receptor signaling may represent a mechanism to escape immediate apoptosis induced by STI571 and open a time frame during which secondary transforming events that confer permanent STI571 resistance (e.g., mutations within the ATP-binding site of the ABL1 kinase domain) can occur.
Acknowledgments
We would like to thank K. Rajewsky, J.D. Rowley, M. Fischer, H. Jumaa, and M. Reth for critical discussions; I. Schwering and R. Küppers for sharing SAGE data; J.D. Rowley and M. Krönke for continuous support and discussions; J.L. Mooster for critically reading the manuscript; and S. Jauch, J.L. Mooster, and P. Wurst for excellent technical assistance.
F. Klein is supported by scholarships of the Studienstiftung des Deutschen Volkes and the Köln Fortune program of the Faculty of Medicine at the University of Cologne. H. Wang is supported by the German Academic Exchange Service (DAAD) Bioscience program. M. Müschen is supported by the Deutsche Forschungsgemeinschaft through the Emmy-Noether Programm. This work was supported by the Deutsche Forschungsgemeinschaft through grants MU1616/2-1 and MU1616/3-1 (to M. Müschen), the German José Carreras Leukemia Foundation (grant to M. Müschen), the Deutsche Krebshilfe 10-1643-Si1 (grant to R. Siebert), and the Ministry of Science and Research for North Rhine-Westphalia through the Stem Cell Network North Rhine-Westphalia (to M. Müschen).
Abbreviations used in this paper: ALL, acute lymphoblastic leukemia; CMP, common myeloid progenitor cell; cRSS, cryptic recombination signal sequence; GCB, germinal center B cell; HSC, hematopoietic progenitor cell; KDE, κ deleting element; MBC, memory B cell; NBC, naive B cell; SAGE, serial analysis of gene expression; TLP, T lymphoid precursor.
References
- 1.Schebesta, M., P.L. Pfeffer, and M. Busslinger. 2002. Control of pre-BCR signaling by Pax5-dependent activation of the BLNK gene. Immunity. 17:473–485. [DOI] [PubMed] [Google Scholar]
- 2.Hardy, R.R., C.E. Carmack, S.A. Shinton, J.D. Kemp, and K. Hayakawa. 1991. Resolution and characterization of pro-B and pre-pro–B cell stages in normal mouse bone marrow. J. Exp. Med. 173:1213–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rajewsky, K. 1996. Clonal selection and learning in the antibody system. Nature. 381:751–758. [DOI] [PubMed] [Google Scholar]
- 4.Thevenin, C., S.L. Nutt, and M. Busslinger. 1998. Early function of PAX5 (BSAP) before the pre–B cell receptor stage of B lymphopoiesis. J. Exp. Med. 188:735–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nussenzweig, M.C. 1998. Immune receptor editing: revise and select. Cell. 95:875–878. [DOI] [PubMed] [Google Scholar]
- 6.Saijo, K., C. Schmedt, I.H. Su, H. Karasuyama, C.A. Lowell, M. Reth, T. Adachi, A. Patke, A. Santana, and A. Tarakhovsky. 2003. Essential role of Src-family protein tyrosine kinases in NF-κB activation during B cell development. Nat. Immunol. 4:274–279. [DOI] [PubMed] [Google Scholar]
- 7.Jumaa, H., M. Mitterer, M. Reth, and P.J. Nielsen. 2001. The absence of SLP65 and BTK blocks B cell development at the preB cell receptor-positive stage. Eur. J. Immunol. 31:2164–2169. [DOI] [PubMed] [Google Scholar]
- 8.Jumaa, H., L. Bossaller, K. Portugal, B. Storch, M. Lotz, A. Flemming, V. Postila, P. Riikonen, J. Pelkonen, C.M. Niemeyer, and M. Reth. 2003. Deficiency of the adaptor SLP65 in pre-B-cell acute lymphoblastic leukaemia. Nature. 423:452–456. [DOI] [PubMed] [Google Scholar]
- 9.Flemming, A., T. Brummer, M. Reth, and H. Jumaa. 2003. The adaptor protein SLP65 acts as a tumor suppressor that limits pre-B cell expansion. Nat. Immunol. 4:38–43. [DOI] [PubMed] [Google Scholar]
- 10.Look, A.T. 1997. Oncogenic transcription factors in the human acute leukemias. Science. 278:1059–1064. [DOI] [PubMed] [Google Scholar]
- 11.Rowley, J.D. 1973. A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 243:290–293. [DOI] [PubMed] [Google Scholar]
- 12.LeBien, T.W. 2000. Fates of human B-cell precursors. Blood. 96:9–23. [PubMed] [Google Scholar]
- 13.Yeoh, E.J., M.E. Ross, S.A. Shurtleff, W.K. Williams, D. Patel, R. Mahfouz, F.G. Behm, S.C. Raimondi, M.V. Relling, A. Patel, et al. 2002. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. 1:133–143. [DOI] [PubMed] [Google Scholar]
- 14.Armstrong, S.A., J.E. Staunton, L.B. Silverman, R. Pieters, M.L. den Boer, M.D. Minden, S.E. Sallan, E.S. Lander, T.R. Golub, and S.J. Korsmeyer. 2002. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat. Genet. 30:41–47. [DOI] [PubMed] [Google Scholar]
- 15.Hofmann, W.K., S. de Vos, D. Elashoff, H. Gschaidmeier, D. Hoelzer, H.P. Koeffler, and O.G. Ottmann. 2002. Relation between resistance of Philadelphia-chromosome-positive acute lymphoblastic leukaemia to the tyrosine kinase inhibitor STI571 and gene-expression profiles: a gene-expression study. Lancet. 359:481–486. [DOI] [PubMed] [Google Scholar]
- 16.Radich, J., G. Gehly, A. Lee, R. Avery, E. Bryant, S. Edmands, T. Gooley, P. Kessler, J. Kirk, P. Ladne, et al. 1997. Detection of BCR-ABL transcripts in Philadelphia chromosome-positive acute lymphoblastic leukemia after marrow transplantation. Blood. 89:2602–2609. [PubMed] [Google Scholar]
- 17.Müschen, M., S. Lee, G. Zhou, N. Feldhahn, V.S. Barath, J. Chen, C. Moers, M. Krönke, J.D. Rowley, and S.M. Wang. 2002. Molecular portraits of B cell lineage commitment. Proc. Natl. Acad. Sci. USA. 99:10014–10019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feldhahn, N., I. Schwering, S. Lee, M. Wartenberg, F. Klein, H. Wang, G. Zhou, S.M. Wang, J.D. Rowley, J. Hescheler, et al. 2002. Silencing of B cell receptor signals in human naive B cells. J. Exp. Med. 196:1291–1305. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19.Klein, F., N. Feldhahn, S. Lee, H. Wang, F. Ciuffi, M. von Elstermann, M.L. Toribio, H. Sauer, M. Wartenberg, V.S. Barath, et al. 2003. T lymphoid differentiation in human bone marrow. Proc. Natl. Acad. Sci. USA. 100:6747–6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Müschen, M., K. Rajewsky, A. Bräuninger, A.S. Baur, J.J. Oudejans, A. Roers, M.L. Hansmann, and R. Küppers. 2000. Rare occurrence of classical Hodgkin's disease as a T cell lymphoma. J. Exp. Med. 191:387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsuda, F., K. Ishii, P. Bourvagnet, K. Kuma, H. Hayashida, T. Miyata, and T. Honjo. 1998. The complete nucleotide sequence of the human immunoglobulin heavy chain variable region locus. J. Exp. Med. 188:2151–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang, Z., M. Zemlin, Y.H. Wang, D. Munfus, L.E. Huye, H.W. Findley, S.L. Bridges, D.B. Roth, P.D. Burrows, and M.D. Cooper. 2003. Contribution of VH gene replacement to the primary B cell repertoire. Immunity. 19:21–31. [DOI] [PubMed] [Google Scholar]
- 23.Reuther, J.Y., G.W. Reuther, D. Cortez, A.M. Pendergast, and A.S. Baldwin, Jr. 1998. A requirement for NF-κB activation in Bcr-Abl-mediated transformation. Genes Dev. 12:968–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sattler, M., M.G. Mohi, Y.B. Pride, L.R. Quinnan, N.A. Malouf, K. Podar, F. Gesbert, H. Iwasaki, S. Li, R.A. Van Etten, et al. 2002. Critical role for GAB2 in transformation by BCR/ABL. Cancer Cell. 1:479–492. [DOI] [PubMed] [Google Scholar]
- 25.Li, S., A.D. Couvillon, B.B. Brasher, and R.A. Van Etten. 2001. Tyrosine phosphorylation of GRB2 by Bcr/Abl and epidermal growth factor receptor: a novel regulatory mechanism for tyrosine kinase signaling. EMBO J. 20:6793–6804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Danial, N.N., and P. Rothman. 2000. JAK-STAT signaling activated by Abl oncogenes. Oncogene. 19:2523–2531. [DOI] [PubMed] [Google Scholar]
- 27.Muljo, S.A., and M.S. Schlissel. 2003. A small molecule Abl kinase inhibitor induces differentiation of Abelson virus-transformed pre-B cell lines. Nat. Immunol. 4:31–37. [DOI] [PubMed] [Google Scholar]
- 28.Siminovitch, K.A., A. Bakhshi, P. Goldman, and S.J. Korsmeyer. 1985. A uniform deleting element mediates the loss of kappa genes in human B cells. Nature. 316:260–262. [DOI] [PubMed] [Google Scholar]
- 29.Minegishi, Y., J. Rohrer, E. Coustan-Smith, H.M. Lederman, R. Pappu, D. Campana, A.C. Chan, and M.E. Conley. 1999. An essential role for BLNK in human B cell development. Science. 286:1954–1957. [DOI] [PubMed] [Google Scholar]
- 30.Druker, B.J., C.L. Sawyers, H. Kantarjian, D.J. Resta, S.F. Reese, J.M. Ford, R. Capdeville, and M. Talpaz. 2001. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N. Engl. J. Med. 344:1038–1042. [DOI] [PubMed] [Google Scholar]