

Abstract
HLA class I polymorphism creates diversity in epitope specificity and T cell repertoire. We show that HLA polymorphism also controls the choice of Ag presentation pathway. A single amino acid polymorphism that distinguishes HLA-B*4402 (Asp116) from B*4405 (Tyr116) permits B*4405 to constitutively acquire peptides without any detectable incorporation into the transporter associated with Ag presentation (TAP)-associated peptide loading complex even under conditions of extreme peptide starvation. This mode of peptide capture is less susceptible to viral interference than the conventional loading pathway used by HLA-B*4402 that involves assembly of class I molecules within the peptide loading complex. Thus, B*4402 and B*4405 are at opposite extremes of a natural spectrum in HLA class I dependence on the PLC for Ag presentation. These findings unveil a new layer of MHC polymorphism that affects the generic pathway of Ag loading, revealing an unsuspected evolutionary trade-off in selection for optimal HLA class I loading versus effective pathogen evasion.
Keywords: HLA, polymorphism, Ag presentation, tapasin, immune evasion
Introduction
Polymorphism in the HLA genes of the MHC is concentrated in the Ag-binding cleft and is thought to be maintained by natural selection for pathogen immunity (1). Accordingly, these polymorphisms probably provide a heterozygous advantage in presenting diverse ligands to T cells (1) and broadening the immune response to pathogens (2, 3). Some polymorphisms also appear to affect generic aspects of Ag presentation such as class I assembly and rate of transport to the cell surface (4–12). Class I molecules first associate with calnexin in the ER before complexing with β2-microglobulin and then calreticulin (13). Class I–peptide loading is facilitated by tapasin, a molecule that brings the calreticulin–class I–β2m complex into physical association with the heterodimeric transporter associated with Ag presentation (TAP) (14) and the thiol oxidoreductase ERp57, creating the PLC (13, 15). Tapasin also increases steady-state levels of TAP (16), retains empty class I molecules in the ER (11, 17, 18), and partially facilitates peptide loading by stabilizing the empty class I complexes without bridging them to the TAP complex (16). Tapasin is transiently disulfide linked to ERp57 in the PLC, suggesting that disulfide bond isomerization may play a role in peptide loading (19). These functions of tapasin optimize peptide loading of class I molecules, producing a more stable cohort of class I–peptide complexes for presentation to T cells (13, 18, 20).
There is a spectrum in class I dependency on tapasin for loading of peptide in the ER (4, 6, 8–10, 17). Most class I allotypes, such as HLA-B*4402 (6, 17), HLA-B8 (6), and B*1510 (8), associate strongly with the TAP and are highly dependent on tapasin for effective Ag presentation (6, 21). Other class I molecules, such as HLA-B*2705 (6), HLA-B*1501, B*1518 (8), and RT1.Au (7), can load peptides without incorporation into the PLC, even though most of these allotypes are normally incorporated into the PLC in the presence of tapasin (6, 7).
The structural features of class I molecules that confer the capacity for peptide loading without TAP assembly appear to involve the F pocket of the Ag binding cleft, particularly residues 116 (8–10, 18) and 114 (11, 12). However, it is unclear whether class I–peptide loading in the absence of TAP association predominantly reflects (a) a physical inability to associate with TAP under any circumstances, (b) competition for TAP by other endogenous class I molecules (7, 22), or (c) efficient peptide capture mechanisms that preclude further ligand optimization in the PLC. We have suggested previously that phenotypic variation in assembly and transport of class I molecules reflects a level of HLA polymorphism shaped by a host advantage in resisting viral interference with peptide loading (6, 23, 24). Here, we demonstrate a striking difference in Ag presentation between HLA-B*4405 and B*4402. These natural HLA-B alleles are present in diverse human populations with B*4402 being most common (found in ∼11% of caucasians) and B*4405 being relatively rare (<1%) (25). They differ by only a single residue at position 116 (B*4402 116D→ B*4405 116Y) located in the F pocket of the binding cleft (26, 27). This amino acid substitution subtly alters the peptide repertoire of the two allotypes, but more importantly the hydrophobic F pocket of B*4405 is associated with binding of peptide Ag in the absence of detectable association with the PLC. Significantly, this class I–loading phenotype is also less susceptible to viral interference with the supply of peptide Ag.
Materials and Methods
Cell Lines and Flow Cytometry.
721.220 is a human lymphoblastoid cell line that lacks HLA-A and -B and functional tapasin (4, 28). Transfection of 721.220 cells with HLA-B gene constructs, WT, and soluble human tapasin cDNAs was performed as described previously (6, 17, 18). The human B lymphoblastoid cell line Hmy2.C1R is a γ-irradiation mutant that expresses HLA-C and insignificant levels of endogenous HLA-A2 and B35 (29). Hmy2.C1R transfected with HLA-B*4402 and HLA-B*4405 has been described previously (18). Hmy2.C1R transfectants expressing either HLA-B*4402 or HLA-B*4405 and ICP47 were created by transfection of the gene encoding HSV ICP47 (30) into Hmy2.C1R.B*4402 and Hmy2.C1R.B*4405, respectively. Cell culture conditions have been described previously (6, 7).
Antibodies.
HC-10 is a mouse mAb that recognizes free class I heavy chain and poorly conformed HC–β2M complexes (31, 32). Mouse mAb 148.3 recognizes human TAP-1 (33), and RING4C is a rabbit antipeptide antibody to the COOH-terminal region of TAP-1 (34). Giles anti-hTsn (a gift from B. Gao, University of Oxford, Oxford, England, UK) and Rgp48N (a gift from P. Cresswell, Yale University, New Haven, CT) (14) are rabbit antipeptide antibodies raised against the NH2 terminus of human tapasin. For cell surface staining of HLA molecules, the mAbs W6/32 (pan class I) or RM7.9.63 (anti-Bw4) were used as primary antibodies.
Immunoprecipitations and Western Blots to Assess Protein–Protein Interactions.
Metabolic labeling of cells, immunoprecipitation, and endoglycosidase H (Endo H) experiments were performed as described previously (6, 17, 18). After primary immunoprecipitation, immune complexes were washed then boiled in SDS loading buffer (100°C for 10 min), and the cellular proteins (2.25 × 106 cells/lane) were separated on a 10% SDS-PAGE gel before immunoblotting with biotinylated HC-10, RING4C, or Giles anti-hTsn antibodies to detect either class I heavy chain, TAP, or tapasin, respectively. Either HRP-conjugated StreptAvidin (Chemicon) or sheep anti–rabbit Ig (Silenus) was used as a secondary Ab, and proteins were visualized using Renaissance chemiluminescence substrate (NEN).
Purification of Cell Surface-associated HLA-B44 Complexes and Peptide Analysis.
Purification of HLA-B*4402 and B*4405 was performed from 5 × 109 C1R.B*4402 and C1R.B*4405 cells grown in roller bottles, and peptides were recovered as described previously (23). Peptides were separated by RP-HPLC using a SMART system HPLC (Pharmacia Biotech) with a μRPC C2/C18 column (2.1 mm [inside diameter] × 10 cm) and characterized by matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF MS) as described previously (23, 35).
Crystallization, Data Collection, and Structure Determination.
Complexes of B*4405–EEFGRAFSF and B*4403–EEPTVIKKY were prepared from Escherichia coli, refolded, and crystallized with the indicated peptide as described previously (35, 36). The data were processed and scaled using the HKL package (37). The structures were refined using our previously defined B*4402 structure as a starting model (35). The progress of refinement was monitored by the Rfree value (3% of the data) with neither a sigma, nor a low resolution cut off being applied to the data. The structures were refined using the simulated-annealing protocol implemented in CNS (version 1.0) (38). The electrostatic surfaces of the area bounding the F pocket of B*4402 and B*4405 were depicted using the program GRASP (39). A summary of the data collection and refinement statistics is given in Tables I and II, respectively. The pdb accession codes are 1SYS and 1SYV.
Table I.
Crystal Structure Data Collection
| B*4405/EEFGRAFSF | B*4403/EEPTVIKKY | |
|---|---|---|
| Temperature | 100 K | 100 K |
| Space Group | P212121 | P212121 |
| Cell dimensions (Å) (a, b, c) | 50.77, 82.23, 110.00 | 50.8, 82.0, 110.2 |
| Resolution (Å) | 1.7 | 2.4 |
| Total no. observations | 132,999 | 79,815 |
| No. unique observations | 48,317 | 18,236 |
| Multiplicity | 2.75 | 4.4 |
| Data completeness (%) | 93.8 (90.0) | 99.5 (99.8) |
| No. data > 2σI | 77.5 (42.9) | 82.1 |
| I/σI | 22.1 (2.7) | 7.9 (2.3) |
| Rmerge a (%) | 4.9 (41.4) | 6.9 (23.9) |
The values in parentheses are for the highest resolution bin (approximate interval 0.1 Å).
Rmerge = Σ|Ihkl − <Ihkl> | / ΣIhkl.
Table II.
Crystal Structure Refinement Statistics
| B*4405/EEFGRAFSF | B*4403/EEPTVIKKY | |
|---|---|---|
| Nonhydrogen atoms | ||
| Protein | 3,166 | 3,162 |
| Water | 643 | 481 |
| Resolution (Å) | 50 − 1.7 | 45 − 2.4 |
| Rfactor a (%) | 19.9 | 24.4 |
| Rfree b (%) | 22.4 | 27.1 |
| rms deviations from ideality | ||
| Bond lengths (Å) | 0.005 | 0.01 |
| Bond angles (°) | 1.27 | 1.08 |
| Dihedrals (°) | 25.09 | 25.1 |
| Impropers (°) | 0.73 | 0.65 |
| Ramachandran plot | ||
| Most favored and allowed region (%) | 99.4 | 99.1 |
| B factors (Å2) | ||
| Average main chain | 23.1 | 48.5 |
| Average side chain | 25.8 | 50.1 |
| Average water molecule | 39.2 | 48.2 |
| rms deviation bonded Bs | 1.66 | 1.61 |
rms, root mean square.
Rfactor = Σhkl | |Fo| − |Fc| | / Σhkl |Fo| for all data except for 3%, which was used for the
Rfree calculation.
HSV Infection of Human PBMCs.
Human PBMCs (3 × 106 HLA-A2, 29, B*4402, Cw5 and A2, 26, B*4405, 55, Cw2, 3) were inoculated with either HSV-KOS (multiplicity of infection = 4) or with PBS (mock control) in 900 μl serum-free RPMI 1640 for 1 h at room temperature with occasional mixing. PBMCs were subsequently resuspended in medium containing human serum and incubated for 12, 24, 36, 48, 56, and 72 h at 37°C, 5% CO2 before flow cytometric analysis.
Online Supplemental Material.
Supplemental material provides experimental results of TAP peptide translocation assays, expression data for soluble human tapasin transfections, pulse–chase experiments on ICP47 transfectants, metabolic labeling experiments to demonstrate class I–TAP association, a table of contacts between DPα peptide and B*4405, results of peptide elution experiments, and B*4405 expression in the TAP-deficient T2 cell line. Figs. S1–S7 and Tables S1 and S2 are available at http://www.jem.org/cgi/content/full/jem.20031680/DC1.
Results
HLA-B*4405 (116Tyr) but not HLA-B*4402 (116Asp) Is Expressed on the Surface of Tapasin-deficient Cells.
Surface expression of B*4402 is highly dependent on interaction with human tapasin in mouse cells (17) and in tapasin-deficient human 721.220 cells (6). In contrast, HLA-B*4405 containing a single substitution at position 116 (B*4402 116D→B*4405 116Y) is expressed at high levels on the cell surface of mouse L cells and in the 721.220 cell line. Culture of transfected cells at 26°C stabilizes poorly loaded class I molecules permitting their transport to the cell surface. Accordingly, HLA-B*4402 showed almost 10-fold enhancement of cell surface expression at 26°C compared with culture at 37°C (Fig. 1 A). Culture of B*4405+ L cells or 721.220 transfectants at 26°C did not improve cell surface expression of B*4405, indicating that B*4405 molecules were already loaded with peptides in these cells. Moreover, introduction of the human tapasin gene into L cells or 721.220 transfectants rescued surface expression of B*4402 by ∼10-fold, whereas tapasin expression made no significant difference to surface levels of B*4405 (Fig. 1 A). In tapasin-deficient cells, the surface expression of B*4405, but not B*4402, was associated with intact Ag presentation of endogenous virus-specific determinants to CTL (unpublished data), consistent with Ag presentation by B*2705 in tapasin-deficient cells (6). These data demonstrate that unlike B*4402 molecules, HLA-B*4405 retains the capacity for cell surface expression and Ag presentation in cells lacking functional tapasin, a molecule essential for formation of the PLC.
Figure 1.
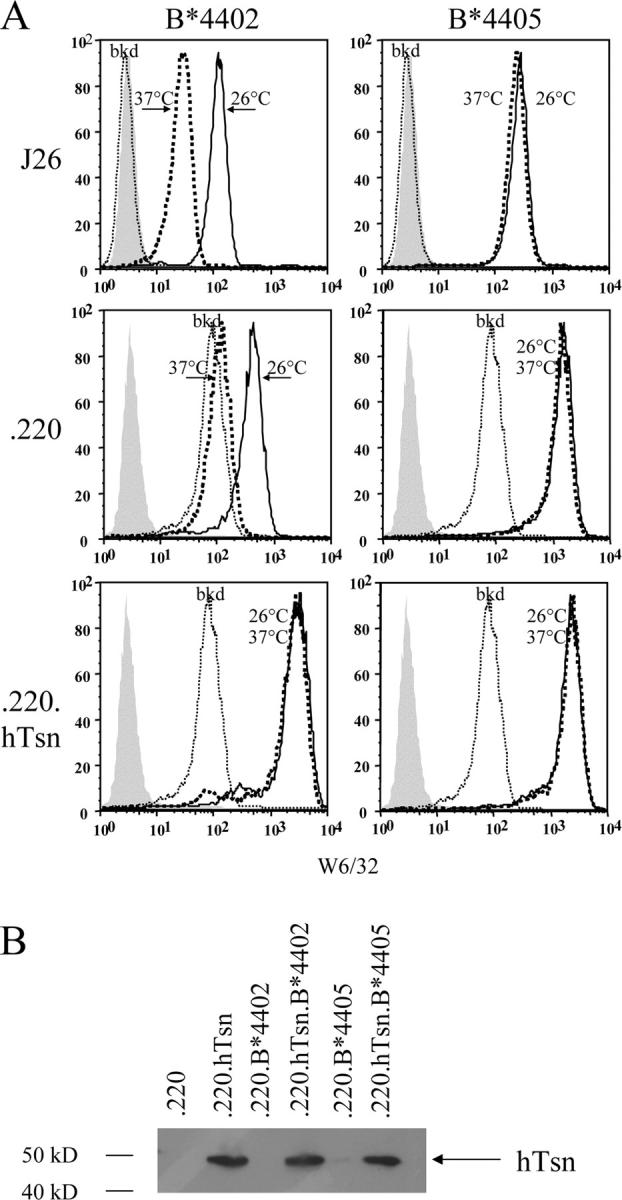
HLA-B*4405 (Tyr 116) but not B*4402 (Asp 116) is expressed on the surface of cell lines deficient in human tapasin. (A) Mouse L cells expressing human β2m (J26) and either the human cell line 721.220 (.220) or transfectants expressing human tapasin (.220.hTsn) were stably transfected with genes encoding either B*4402 or B*4405 and then stained by indirect immunofluorescence with mAb W6/32 (anti-HLA class I) after culture overnight at 26°C (solid lines) or 37°C (bold dotted lines). Flow cytometric histograms also show background staining of untransfected J26 and 721.220 cells (thin dotted lines) and conjugated antibody alone (shaded). (B) Expression of human tapasin (hTsn) was verified in immunoblots of the indicated cell lysates probed with a specific rabbit antiserum.
HLA-B*4405 Is not Detectable in the TAP-associated PLC.
HLA-B*2705 molecules can also load with peptides in the absence of any association with TAP (6, 23); however, in the presence of endogenous tapasin, some B*2705 is incorporated into the PLC and undergoes optimization of its peptide cargo (6, 18). Thus, we evaluated whether B*4405 is also associated with the PLC in cells expressing tapasin. HLA class I heavy chain was not detected in any of the TAP complexes from cells lacking tapasin, verifying the documented role of tapasin in bridging class I–TAP complexes (14, 21) (Fig. 2 A), nor was class I heavy chain identified in the 721.220 cell line expressing human tapasin (.220.hTsn) reflecting the absence of HLA-A and -B. Prolonged exposure of immunoblots revealed a faint band corresponding to normal levels of background HLA-C complexed to TAP in these cells (not depicted). In contrast, in the presence of tapasin class I heavy chains were present in TAP complexes from 721.220 cells expressing B*4402 and B*2705, but not in cells expressing B*4405 despite the presence of tapasin. This observation was confirmed in the class I–deficient lymphoblastoid cell line C1R expressing B*4405, where only very low levels of endogenous class I heavy chains were detected in longer exposures of immunoblotted TAP complexes, reflecting the defect in HLA-A and -B expression by these cells (Fig. 2 B). Although B*4405–TAP complexes were not detectable in C1R cells, there was a significant increase in class I heavy chains detected above background in TAP complexes from C1R cells expressing B*4402 (Fig. 2 B). The mAb HC-10 recognizes free class I heavy chains and heavy chains that are poorly assembled with β2M such as those present in the PLC (32). Therefore, we examined HC-10 immunoprecipitates from C1R transfectants for the presence of any associated TAP (Fig. 2 C) or tapasin (Fig. 2 D). TAP1 molecules were efficiently coprecipitated with B*4402 but were barely detectable in immune precipitates from B*4405-positive C1R cells (Fig. 2 C). Similarly, an association of tapasin with B*4405 could not be detected above the endogenous C1R background in class I immune precipitates from C1R.B*4405 cells but was detected in C1R.B*4402 cell lysates (Fig. 2 D). In metabolic labeling experiments, there were no detectable TAP-associated class I heavy chains above the background of HLA-C molecules in B*4405-transfected 721.220.tapasin or C1R cells (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20031680/DC1). Together, the data are consistent with either very short-lived or negligible incorporation of B*4405 molecules into the PLC.
Figure 2.

No detectable incorporation of HLA-B*4405 into the PLC. (A) Cell lysates (lysate) or TAP immune complexes formed with anti-mAb 148.3 mAb (TAP IP) were immunoblotted for the presence of class I heavy chains using biotinylated HC-10 (anti–class I hc). Untransfected 721.220 cells (untrans) and cells transfected with the indicated genes were compared. (B) Class I–reduced (C1R) B-LCLs expressing either B*4402 or B*4405 were assayed as in A. A prolonged exposure (5 min) of the blot is shown to demonstrate the association of low levels of endogenous HLA-C class I heavy chain with the TAP complex (bottom). (C) Cell lysates (lysate) or class I heavy chain complexes formed with mAb HC-10 (hc IP) were then immunoblotted for the presence of TAP-1 using anti–TAP-1 rabbit antiserum. Immunoblotted lysates of the C1R cells and TAP-negative cell T2 are shown as controls (right). (D) Cell lysates (lysate) or class I heavy chain complexes (hc IP) from C were immunoblotted for the presence of tapasin using anti-hTsn rabbit antiserum. The parental 721.220 (.220) and 721.220.hTsn (.220.hTsn) transfectants are shown as controls.
Rapid Intracellular Transport of B*4405 versus B*4402 Molecules.
Tapasin retains nascent class I molecules in the ER (13, 17, 40) but B*4405 is transported rapidly to the cell surface (41) consistent with its capacity to acquire peptide cargo in the absence of detectable association with the PLC. Hence, we compared the intracellular transport kinetics of B*4402 and B*4405 in the 721.220 and C1R cell lines. Pulse-chased class I molecules were examined for their maturity by digestion with Endo H to distinguish between immature, Endo H–sensitive (Fig. 3, s) proteins in the ER and mature, Endo H resistant (Fig. 3, r) proteins that have transited the Golgi. In tapasin-deficient 721.220 cells, B*4402 molecules fail to load with peptide resulting in their low surface expression and degradation (6). However, B*4405 molecules do load with peptide in the 721.220 cells where their rate of acquisition of Endo H resistance was as rapid in the absence of tapasin as for B*4402 molecules in the presence of tapasin (Fig. 3 A). In tapasin-positive 721.220 cells, the transport of B*4405 molecules was even faster compared with B*4402 molecules (compare the ratio of ‘r’ and ‘s’ at 70 min [Fig. 3 A]). Similar relative maturation kinetics were observed in C1R cells that express endogenous HLA-C, which matures very slowly and is stably associated with TAP (42). Hence, in C1R cells Endo H–resistant class I molecules were detected at 20 min in the B*4405 transfectants versus 45 min in the B*4402 transfectants (Fig. 3 B, compare lanes 1, 4, and 7). Interestingly, tapasin expression accelerates the maturation and transport of B*4405 molecules in the 721.220 transfectants (Fig. 3 A, compare 20 min time points B*4405 with or without tapasin). We reasoned that this enhancement might be due to the increase in peptide translocation after tapasin expression in 721.220 transfectants (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20031680/DC1) or to an influence of free tapasin on B*4405 molecules (16). Therefore, we examined the effect of soluble tapasin on the kinetics of B*4405 maturation (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20031680/DC1). Although soluble tapasin does not bridge TAP and class I proteins or increase TAP expression (16), its expression enhanced the maturation of B*4405 in 721.220 cells to much the same extent as WT tapasin (Fig. 3 C) consistent with a role for tapasin in optimizing peptide cargo independently of the PLC (16, 43). However, peptide optimization of B*4405 even in the presence of tapasin was only partial compared with B*4402 (18). We conclude that although B*4405 is not incorporated into the PLC there is some interaction with free tapasin. The lack of detectable tapasin association with B*4405 in Fig. 2 D suggests a low affinity for this interaction consistent with the detergent-sensitive nature of the PLC (14, 21).
Figure 3.
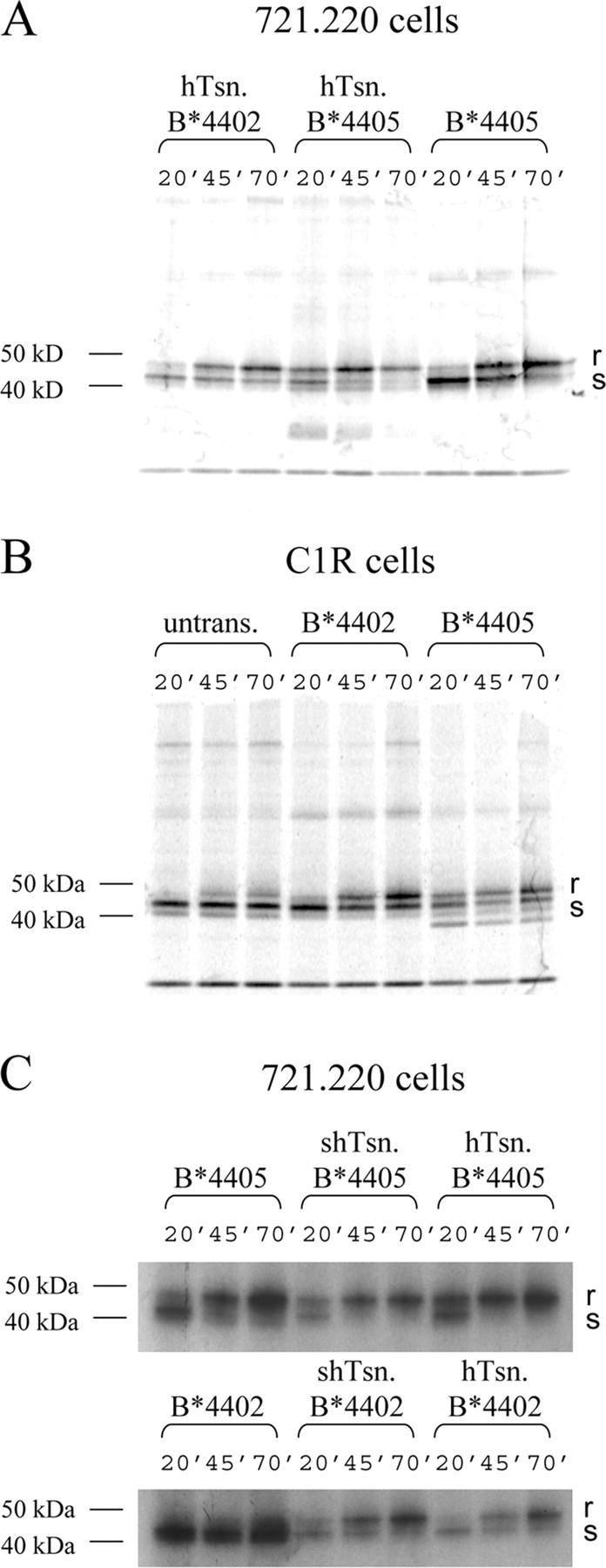
Rapid maturation and transport of HLA-B*4405 compared with HLA-B*4402. Cellular proteins from the indicated 721.220 transfectants expressing or lacking membrane tapasin (A), C1R cells (B), and the indicated 721.220 transfectants expressing or lacking soluble tapasin (C) were labeled with 35S-Met/Cys for 5 min and chased for 20, 45, and 70 min before immunoprecipitating class I/β2M molecules with mAb W6/32. Immune complexes were treated with Endo H to determine the proportion of mature, Endo H–resistant (r), or post-Golgi proteins versus the immature, Endo H–sensitive (s), or ER proteins by SDS-PAGE and fluorography. In the C1R.B*4405 transfectants, there is an additional band of unknown origin migrating faster than the class I proteins. This appears to be a clone-specific background band that is not observed in anti–class I Western blots.
Substitution of 116Asp (B*4402)→116Tyr (B*4405) Biases the P9 Side Chain Preference and Creates a Hydrophobic F Pocket in B*4405.
To better understand the impact of the polymorphism at 116Asp (B*4402)→116Tyr (B*4405), we compared the peptides eluted from B*4402 and B*4405 molecules expressed by C1R cells. The overall recovery of peptides and their ionization intensity was greater for B*4402 than B*4405 (Fig. 4), consistent with the failure of B*4405 to fully optimize its cargo in the PLC (18).
Figure 4.

Overlapping peptide repertoires but altered P9 anchor preference in B*4402 and B*4405. Comparison of B*4402 and B*4405 peptide repertoires by MALDI-TOF mass spectrometry. Total peptide eluates from HLA-B*4402 (positive polarity spectra) and HLA-B*4405 (negative polarity spectra) after a single dimension chromatographic separation. The masses of prominent species are shown. Pool Edman sequence analysis of peptides eluted from HLA-B*4402 and B*4405 revealed dominant anchor residues at P2Glu and P9Tyr/Phe. Phe and Tyr were recovered in equal amounts from P9 of B*4402, but Phe was predominant at this position in B*4405 (Phe:Tyr = 4:1; see Table S1)
Whereas the B pocket preference for P2-Glu was conserved between B*4402 and B*4405, the preference at P9 in the F pocket was not identical in these allotypes. Edman sequencing of pooled peptide eluate from B*4402 contained equal amounts of phenylalanine or tyrosine at P9, but in B*4405 the ratio of Phe:Tyr was 4:1, suggesting a strong preference for Phe in the F pocket of B*4405 (Table S1, available at http://www.jem.org/cgi/content/full/jem.20031680/DC1). Analysis of the peptide repertoire by MALDI-TOF mass spectrometry revealed some peptides were unique to each allotype, but overall there was a substantial overlap in the peptides eluted from B*4405 and B*4402 (Fig. 4). This was confirmed in a fraction by fraction analysis (not depicted) and is reminiscent of the subtle impact of F pocket polymorphism on the peptide repertoire of HLA-B27 subtypes (44, 45).
We next determined the structure of HLA-B*4405 complexed with EEFGRAFSF, an abundant shared ligand derived from HLA-DPα, and compared this with the structure of B*4402/EEFGRAFSF (35) (Fig. 5). A detailed description of the structures including amino acid contacts is given in Table S2 (available at http://www.jem.org/cgi/content/full/jem.20031680/DC1). The polymorphism at 116 (Tyr to Asp) significantly impacts on the characteristics of the respective F pockets (Fig. 5, A–F). The H-bond networks involving residue 116 and the arrangement of aromatic residues in the F pockets of B*4402 and B*4405 are shown in Fig. 5, B and C, respectively. In B*4402, the presence of Asp 116 imparts a markedly electronegative potential on the F pocket (Fig. 5 E), whereas in B*4405 the F pocket is predominantly hydrophobic (Fig. 5 F). Nonetheless, the specificity, mode of P9-Phe binding, and architecture of the F pocket is largely maintained in the two structures (Fig. 5, B and C). The basis for the alternate selection of either Phe or Tyr in the F pocket of B*4402 was addressed by analyzing a new structure of B*4403 complexed to the natural peptide ligand EEPTVIKKY (Fig. 5 D). Both B*4402 and B*4403 have Asp at position116 and are virtually identical in their F pockets (35). In the B*4403–P9 Tyr complex, the carboxylate of Asp 116 formed a H-bond with the P9 TyrOη group (Fig. 5 D). Accordingly, a tyrosine would be much less favorable within the B*4405 F pocket as this polar P9 TyrOη group will not be satiated, thus explaining the bias to Phe at P9 in the B*4405 peptide repertoire.
Figure 5.

The structure of HLA-B*4405 alters peptide specificity and increases hydrophobicity in the F pocket. (A) Overview of the structure of B*4405 highlighting Tyr116 and the electron density of the peptide (Dpα46-54, EEFGRAFSF). The structure of the F pocket in HLA-B*4402/EEFGRAFSF (B) (35) and the new structures B*4405/EEFGRAFSF (C) and HLA-B**4403/EEPTVIKKY (D) are shown as ball and stick representations of F pocket amino acids (gray), residue 116 (yellow), and the peptide ligand (green), including the COOH-terminal peptide residue P9 Phe in B and C and P9 Tyr in D. Dotted lines are H bonds. The α2-helix has been removed for clarity. In the B*4402 structure (B), Asp 116 is part of an intricate polar network involving Asp 114, Asp 156, Arg 97, and several conserved water molecules that additionally bridge contacts to the bound peptide. Asp 116 is orientated in the same direction as the aromatic ring of Tyr 116 in the B*4405 structure. A water molecule fills the “cavity” at position 116 of B*4402 such that it superposes closely to Tyr 116 Oη group in B*4405 and forms a similar role in that it bridges one H bond to Asp 114. In B*4405 (C), the P9 anchor residue Phe projects into a hydrophobic F pocket where it is surrounded by the aromatic rings of Tyr 74, Tyr 116, Tyr123, and Trp 147, as well as making van der Waals contacts with Ile 95 and the aliphatic moiety of Asn77. The main chain of this COOH-terminal residue is tethered by H bonds to Asn77, Tyr84, and Thr143. Tyr 116 forms the base of this pocket, where the aromatic ring points away from the F pocket, such that the Tyr 116 Oη group points toward the P7 pocket where it forms two H bonds with the Asp 114 carboxylate, an H bond to Arg 97 Nɛ, and additionally forms a water-mediated H bond to the backbone of the bound peptide (Phe 7N). The aromatic ring of Tyr 116 also packs against the long aliphatic side chain of Arg 97. The electrostatic surfaces of the area bounding the F pocket of B*4402 (E) and B*4405 (F) are depicted using the program GRASP(39). Electropositive (blue); electronegative (red).
Resistance of B*4405 Molecules to TAP Blockade by the HSV Inhibitor ICP47.
The ability of B*4405 molecules to load with peptides in cells lacking tapasin suggested that nascent B*4405 may acquire peptide very efficiently, thus averting the normal ER retention mechanisms that optimize peptide loading. Given the very short half-life of peptides in the ER (46), this property of B*4405 could have advantages under conditions of limiting peptide supply or disruption of the PLC by pathogen-encoded molecules (47, 48). We tested this idea by examining the impact of the Herpes simplex TAP inhibitor, ICP47 (30), on surface expression of B*4402 and B*4405. Expression of ICP47 markedly reduced peptide delivery to the ER in the transfectants, but notably, it was not completely abolished in either cell line (Fig. S4, available at http://www.jem.org/cgi/content/full/jem.20031680/DC1). Although peptide supply was comparably impaired in the B*4405 and B*4402 transfectants, cell surface expression of B*4402 on C1R cells was almost completely abolished, whereas B*4405 expression was reduced by 10–30-fold in the presence of ICP47 (Fig. 6 A). There was significant improvement in B*4405 but not B*4402 expression after culture of cells at 26°C, reflecting further stabilization of suboptimally loaded molecules (Fig. 6 A). Similar results were observed in multiple independent ICP47 transfectants (not depicted).
Figure 6.
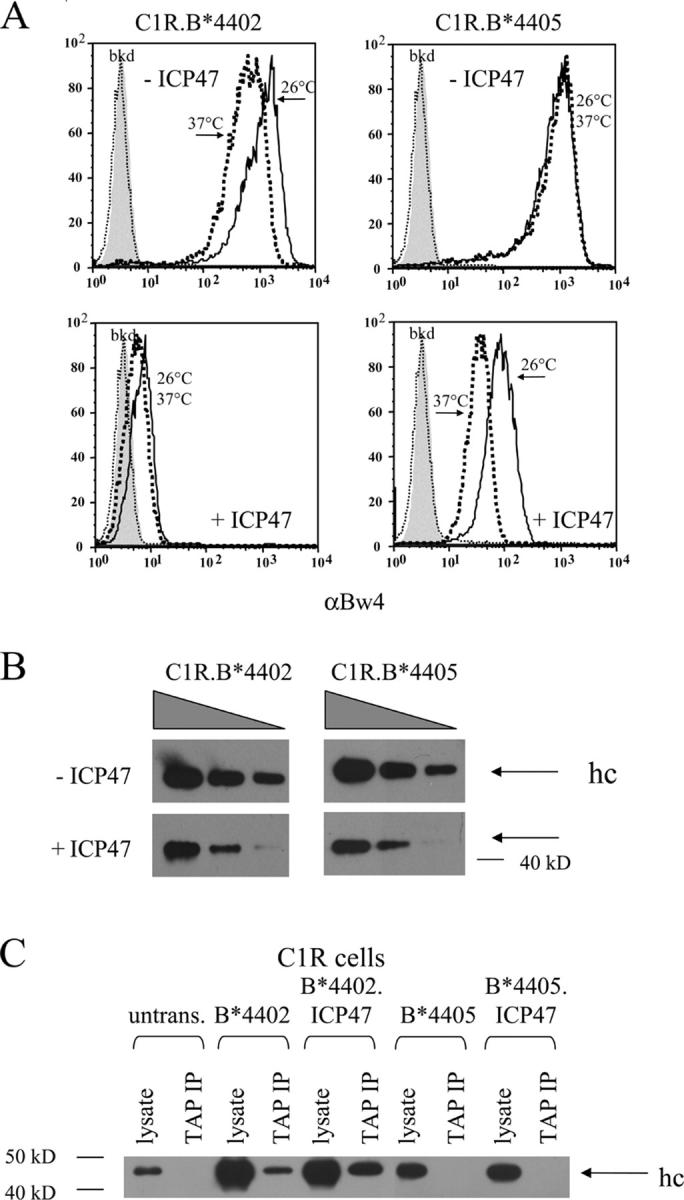
Expression of the HSV TAP inhibitor ICP47 differentially impairs cell surface expression of HLA-B*4402 versus B*4405. (A) Cell surface expression HLA-B*4402 and B*4405 by C1R transfectants in the presence and absence of ICP47. Cells were stained by indirect immunofluorescence for the shared Bw4 determinant after culture at 37°C (bold dotted lines) or 26°C (solid lines). Flow cytometric histograms include staining of untransfected cells (thin dotted lines) and conjugate alone (filled histograms). (B) Steady-state total HLA–class I heavy chain expression in C1R transfectants ± ICP47. Immunoblots using mAb HC-10 were performed on graded cell numbers of transfectants. (C) Lack of detectable B*4405 within the TAP-associated PLC of C1R transfectants expressing ICP47. TAP complexes were immunoprecipitated from C1R transfectants expressing either B*4402 or B*4405 ± ICP47 using mAb 148.3. Immunoprecipitates were subsequently immunoblotted for the presence of class I hc using biotinylated HC-10 and developed as described in Fig. 2.
The peptides loaded by B*4405 in the presence of ICP47 were TAP dependent since surface expression of B*4405 was minimal in the TAP-deficient cell line T2 and peptides eluted from B*4405 transfectants expressing ICP47 largely overlapped with those eluted from C1R cells expressing B*4405 without ICP47 (Figs. S5 and S6, available at http://www.jem.org/cgi/content/full/jem.20031680/DC1). We conclude that B*4405, but not B*4402, can capture ligands under drip feed conditions of peptide delivery due to TAP blockade.
Expression of ICP47 also resulted in reduced steady-state levels of intracellular B*4402 and B*4405 heavy chains (Fig. 6 B) that were mostly retained in the ER or degraded due to impaired peptide supply (references 6, 49; Fig. S7, available at http://www.jem.org/cgi/content/full/jem.20031680/DC1). Retention of peptide-starved B*4405 could involve interaction with calnexin (32), calreticulin (14), or free tapasin (50, 51). Peptide starvation by ICP47 did not drive the association of nascent B*4405 and the PLC but significantly increased the proportion of B*4402 molecules bound to TAP (Fig. 6 C). These findings imply that B*4405 is structurally unsuited to incorporation into the PLC, even under the stress of limiting peptide supply, and suggests rapid capture of peptides through a PLC-independent pathway. This feature of B*4405 is likely to make it less susceptible to pathogen evasion mechanisms that impair peptide supply or that interfere with components of the PLC. We tested this idea further by monitoring the surface expression of B*4402 and B*4405 in PBMCs infected with HSV in vitro (Fig. 7). Infection of PBMCs with HSV resulted in an early up-regulation of surface class I expression, presumably due to induction of cytokines like IFNγ. However, there was also significant down-regulation of surface class I molecules in the infected cell population (10–15% cells; Fig. 7, arrows), initially evident at 24 h and persisting for 36–48 h postinfection. Although there was a 5–10-fold down-regulation of surface B*4402, the effect on surface B*4405 was barely detectable over the same period. The findings confirm that HLA-B*4405 molecules are relatively unaffected by interference with TAP function during HSV infection and suggest that B*4405 will have low susceptibility to diverse viral evasion strategies that disrupt the PLC.
Figure 7.
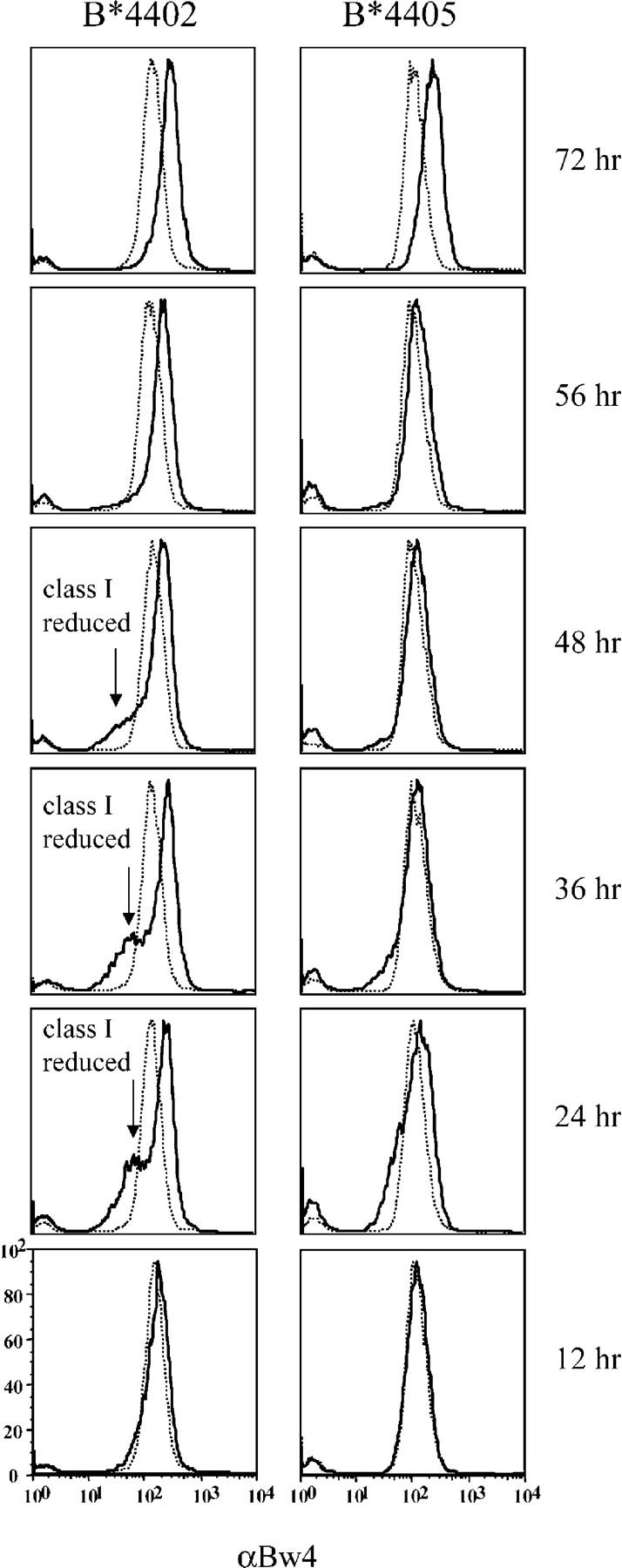
Infection of primary lymphocytes with HSV differentially down-regulates surface expression of B*4402 versus B*4405. Human PBMCs from B*4402- and B*4405-positive donors were inoculated with HSV-KOS at an multiplicity of infection of 4. PBMCs were then stained with biotinylated anti-Bw4 (solid lines) followed by streptavidin APC at the indicated time points after inoculation. Flow cytometric histograms include staining of mock-infected cells (thin dotted lines). One of two independent experiments is shown. The arrows show the population of infected cells with reduced class I expression. The experiment shown used WT HSV for infection to exclude artifacts from the β-gal–tagged virus that was used in other experiments to define the infected cells.
Discussion
In this study, we provide evidence that the functional consequences of some class I polymorphisms extend beyond modulating peptide specificity to substantially affect the generic pathway of Ag presentation. Our findings focus on a natural polymorphism of HLA-B44 but are applicable to other class I polymorphisms showing variable incorporation into the PLC during Ag presentation (4–6, 8–12, 18). Although incorporation of HLA-B*4405 into the PLC is not detectable, free tapasin appears to partially optimize the peptide cargo of B*4405 molecules similar to the rat class I molecule RT1-Au which loads peptides even when excluded from the TAP complex by competition with RT1-Aa (43).
The differences in Ag presentation pathways used by B*4402 and B*4405 are controlled by a single residue at position 116 (Asp→Tyr). Although the peptide repertoire of HLA-B*4405 and B*4402 is overlapping, both allotypes present some unique peptides. The choice of P9 anchor residue (Phe or Tyr) in B*4402 is more constrained in B*4405 (Phe) largely because Asp116 on the floor of the B*4402 F pocket can H bond with P9 Tyr. However, this effect in B*4405 is a bias toward peptides with a P9 Phe rather than exclusion of peptides with a P9 Tyr. The differences in P9 anchor specificity and the natural repertoire of peptides bound by B*4402 and B*4405 are comparable to the reported differences between B*2705 and B*2709 which differ by Asp116→His116 (45). Ag presentation by B*4402 is highly dependent on tapasin, which is essential for the recruitment of class I molecules into the PLC (6, 17). Accordingly, B*4402 molecules bind TAP and undergo substantial optimization of their peptide cargo (18). In contrast, B*4405 molecules do not associate with the PLC and undergo only partial optimization in the presence of tapasin (18). HLA-B*4405 molecules are rapidly transported to the cell surface (41) and the average thermostability of B*4405–peptide complexes expressed by lymphoblastoid cells is lower than those expressed by B*4402 (18). This finding reflects the more efficient PLC-mediated optimization of peptide cargo in B*4402 versus B*4405 (18, 23, 51).
The ability of B*4405 to capture limiting peptides without being incorporated into the PLC is associated with decreased electronegativity and increased hydrophobicity of the F pocket arising from the Asp116→Tyr116 substitution. Nonpolar interactions drive class I–peptide association (52) and are generally unfavorable at solvent-exposed sites (53), so the Tyr at position 116 will exacerbate the need to satiate the hydrophobic F pocket of B*4405. Thus, the hydrophobic F pocket (Tyr 74, Tyr 116, Tyr 123, Trp 147, Ile 95, and the aliphatic moiety of Asn 77) is shielded from solvent when occupied by P9 Phe. The stacking interaction between the side chains of aromatic residues can contribute between 1.3 and 6 kcal/mol to protein stability (54). This property of the F pocket in B*4405 will significantly reduce the entropic penalty of peptide ligation compared with that of B*4402, predicting more favorable assembly kinetics of B*4405 and perhaps explaining the tapasin dependence of B*4402. Of note, recombinant B*4405 assembles more efficiently with the EEFGRAFSF peptide ligand than B*4402 (not depicted). A critical role for F pocket hydrophobicity in controlling peptide capture is also suggested by the tapasin independence of in vitro mutants at F pocket residue 114 of class I molecules (11, 12), natural allotypes of HLA-B15 that differ at residue 116 (8, 10), and site-directed mutants at residue 116 of HLA-B7 (8). However the overall architecture of the F pocket probably determines whether hydrophobically driven peptide ligation is the sole mechanism of acquiring peptides under limiting conditions (9).
The loading of peptide into the class I molecule initiates the folding of “open” class I molecules into their final conformation (32). Initial binding of peptide may involve ligation of the COOH terminus, thus creating folding intermediates suitable for NH2-terminal peptide trimming (55, 56), or peptide exchange. Hence, peptide loading might resemble a zipper, or a venus fly-trap (15), that closes from the peptide COOH terminus to NH2 terminus. In this model, any chemical advantage in the trapping and stabilization of peptide through the F pocket structure might be critical in controlling tapasin dependence. Interestingly, the structural changes in the F pocket of B*4405 also appear to abrogate its incorporation into the PLC, a tapasin-dependent process (13, 21), raising the possibility that in addition to other binding sites tapasin interacts with the F pocket of class I molecules.
Our data suggests that B*4405–peptide complexes assemble very efficiently leading to rapid egress of loaded molecules from the ER rendering them relatively resistant to viral evasion mechanisms that target the conventional loading pathway. Viral interference with Ag presentation is widespread, and several viral proteins specifically affect the incorporation of class I molecules with the PLC (24, 57–59). Adenovirus E19 protein may inhibit tapasin function and retain some molecules in the ER (58), and cytomegalovirus US3 protein impairs class I expression by interfering directly with tapasin (24, 48, 60). The mouse γ Herpes virus also appears to subvert class I molecules assembled in the PLC (47) while simultaneously orchestrating the degradation of tapasin and TAP (59). Viral proteins such as HSV ICP47 can also impair peptide supply to the PLC (30); therefore, we used HSV ICP47 to probe the loading capabilities of B*4405 and B*4402 under the stress of limiting peptide supply. HLA-B*4405 was far more efficient at preserving Ag presentation under ICP47 blockade. These findings were observed both in transfected cell lines and during HSV infection of primary lymphocytes. The findings highlight the advantage of the B*4405 loading pathway that ligates peptides without incorporation into the PLC, allowing some degree of continued Ag presentation despite peptide starvation. By contrast, B*4402 molecules sustain much lower surface expression under the same “drip-feed” conditions of peptide supply. This feature of B*4405, and other allotypes that can load without incorporation into the PLC, might be particularly important during competition for the TAP complex by multiple class I allotypes (7). The capacity to capture peptides without incorporation into the PLC would permit Ag presentation by those class I molecules, whereas slow class I allotypes complexed with the PLC selfishly optimized their peptide Ag (43). HLA-B*4402 and B*4405 appear to represent polarized extremes within a much broader spectrum of tapasin dependence in HLA class I allotypes (5). HLA-B27 expresses an increased number of “empty” molecules on the cell surface (61–63), suggesting that some B27 molecules load suboptimally without incorporation into the loading complex (6), whereas others interact with the TAP complex and create very stable complexes (18). In contrast, HLA-B8 is more dependent on the loading complex for proper peptide presentation but not as much as B*4402 (6, 21). This HLA variation suggests that as well as diversifying epitope specificities class I polymorphism presents a tantalizing evolutionary choice for the host. On the one hand, class I allotypes that are incorporated into the PLC undergo efficient ligand optimization but are at greater risk of viral interference, whereas class I allotypes that can still load without incorporation into the PLC may optimize their cargo inefficiently but are less susceptible to viral evasion. The ancient nature of tapasin (64), and its role in the refinement of peptide loading by class I molecules (13), implies that this gene function has been highly selected through evolution. Therefore, complete independence from the PLC in the case of B*4405 may be unusual, since it comes at a significant cost to the host, perhaps being selected by rare pathogen determinants (7) or specific viral evasion mechanisms such as the tapasin inhibitors CMV US3 (24) or adenovirus E3/19Kd protein (65). This might explain the “schizophrenia” of known tapasin-independent class I allotypes that are actually incorporated into the PLC and undergo peptide optimization in the presence of tapasin but can still load peptides in its absence (6, 7, 43). The evolutionary cost of a high degree of independence from the PLC might be tolerated in hosts expressing a functional mixture of class I allotypes (1).
Acknowledgments
We would like to thank Frank Carbone for critical reading of the manuscript and Claerwen Jones for technical help with virus infections. Rajiv Khanna and Scott Burrows (both from Queensland Institute of Medical Research, Brisbane, Queensland, Australia) generously provided reagents. We thank the staff at BioCARS and the Australian Synchrotron Research Program for assistance.
J. Rossjohn is supported by a Wellcome Trust Senior Research Fellowship in Biomedical Science in Australia; A.W. Purcell is a C.R. Roper Fellow of the University of Melbourne; A.G. Brooks is supported by a R.D. Wright Fellowship. This work was supported by the National Health and Medical Research Council, Australian Research Council, the Australian Kidney Foundation, the Roche Organ Transplantation Research Foundation, The Wellcome Trust (UK), and the Juvenile Diabetes Research Foundation.
J. McCluskey and J. Rossjohn are senior authors on this paper.
D. Zernich and A.W. Purcell contributed equally to this work.
The online version of this article contains supplemental material.
Abbreviations used in this paper: Endo H, endoglycosidase H; PLC, peptide loading complex; TAP, transporter associated with Ag presentation.
References
- 1.Parham, P., and T. Ohta. 1996. Population biology of antigen presentation by MHC class I molecules. Science. 272:67–74. [DOI] [PubMed] [Google Scholar]
- 2.Messaoudi, I., J.A. Patino, R. Dyall, J. LeMaoult, and J. Nikolich-Zugich. 2002. Direct link between MHC polymorphism, T cell avidity, and diversity in immune defense. Science. 298:1797–1800. [DOI] [PubMed] [Google Scholar]
- 3.Potts, W.K., and P.R. Slev. 1995. Pathogen-based models favoring MHC genetic diversity. Immunol. Rev. 143:181–197. [DOI] [PubMed] [Google Scholar]
- 4.Greenwood, R., Y. Shimizu, G.S. Sekhon, and R. DeMars. 1994. Novel allele-specific, post-translational reduction in HLA class I surface expression in a mutant human B cell line. J. Immunol. 153:5525–5536. [PubMed] [Google Scholar]
- 5.Neisig, A., R. Wubbolts, X. Zang, C. Melief, and J. Neefjes. 1996. Allele-specific differences in the interaction of MHC class I molecules with transporters associated with antigen processing. J. Immunol. 156:3196–3206. [PubMed] [Google Scholar]
- 6.Peh, C.A., S.R. Burrows, M. Barnden, R. Khanna, P. Cresswell, D.J. Moss, and J. McCluskey. 1998. HLA-B27-restricted antigen presentation in the absence of tapasin reveals polymorphism in mechanisms of HLA class I peptide loading. Immunity. 8:531–542. [DOI] [PubMed] [Google Scholar]
- 7.Knittler, M.R., K. Gulow, A. Seelig, and J.C. Howard. 1998. MHC class I molecules compete in the endoplasmic reticulum for access to transporter associated with antigen processing. J. Immunol. 161:5967–5977. [PubMed] [Google Scholar]
- 8.Turnquist, H.R., H.J. Thomas, K.R. Prilliman, C.T. Lutz, W.H. Hildebrand, and J.C. Solheim. 2000. HLA-B polymorphism affects interactions with multiple endoplasmic reticulum proteins. Eur. J. Immunol. 30:3021–3028. [DOI] [PubMed] [Google Scholar]
- 9.Turnquist, H.R., E.L. Schenk, M.M. McIlhaney, H.D. Hickman, W.H. Hildebrand, and J.C. Solheim. 2002. Disparate binding of chaperone proteins by HLA-A subtypes. Immunogenetics. 53:830–834. [DOI] [PubMed] [Google Scholar]
- 10.Hildebrand, W.H., H.R. Turnquist, K.R. Prilliman, H.D. Hickman, E.L. Schenk, M.M. McIlhaney, and J.C. Solheim. 2002. HLA class I polymorphism has a dual impact on ligand binding and chaperone interaction. Hum. Immunol. 63:248–255. [DOI] [PubMed] [Google Scholar]
- 11.Park, B., S. Lee, E. Kim, and K. Ahn. 2003. A single polymorphic residue within the peptide-binding cleft of mhc class I molecules determines spectrum of tapasin dependence. J. Immunol. 170:961–968. [DOI] [PubMed] [Google Scholar]
- 12.Park, B., S. Lee, E. Kim, and K. Ahn. 2003. Correction: a single polymorphic residue within the peptide-binding cleft of MHC class I molecules determines spectrum of tapasin dependence. J. Immunol. 170:4869. [DOI] [PubMed] [Google Scholar]
- 13.Cresswell, P., N. Bangia, T. Dick, and G. Diedrich. 1999. The nature of the MHC class I peptide loading complex. Immunol. Rev. 172:21–28. [DOI] [PubMed] [Google Scholar]
- 14.Sadasivan, B., P.J. Lehner, B. Ortmann, T. Spies, and P. Cresswell. 1996. Roles for calreticulin and a novel glycoprotein, tapasin, in the interaction of MHC class I molecules with TAP. Immunity. 5:103–114. [DOI] [PubMed] [Google Scholar]
- 15.Williams, A., C.A. Peh, and T. Elliott. 2002. The cell biology of MHC class I antigen presentation. Tissue Antigens. 59:3–17. [DOI] [PubMed] [Google Scholar]
- 16.Lehner, P.J., M.J. Surman, and P. Cresswell. 1998. Soluble tapasin restores MHC class I expression and function in the tapasin-negative cell line .220. Immunity. 8:221–231. [DOI] [PubMed] [Google Scholar]
- 17.Peh, C.A., N. Laham, S.R. Burrows, Y. Zhu, and J. McCluskey. 2000. Distinct functions of tapasin revealed by polymorphism in MHC class I peptide loading. J. Immunol. 164:292–299. [DOI] [PubMed] [Google Scholar]
- 18.Williams, A.P., C.A. Peh, A.W. Purcell, J. McCluskey, and T. Elliott. 2002. Optimization of the MHC class I peptide cargo is dependent on tapasin. Immunity. 16:509–520. [DOI] [PubMed] [Google Scholar]
- 19.Dick, T.P., N. Bangia, D.R. Peaper, and P. Cresswell. 2002. Disulfide bond isomerization and the assembly of MHC class I-peptide complexes. Immunity. 16:87–98. [DOI] [PubMed] [Google Scholar]
- 20.Park, B., and K. Ahn. 2003. An essential function of tapasin in quality control of HLA-G molecules. J. Biol. Chem. 278:14337–14345. [DOI] [PubMed] [Google Scholar]
- 21.Ortmann, B., J. Copeman, P.J. Lehner, B. Sadasivan, J.A. Herberg, A.G. Grandea, S.R. Riddell, R. Tampe, T. Spies, J. Trowsdale, and P. Cresswell. 1997. A critical role for tapasin in the assembly and function of multimeric MHC class I-TAP complexes. Science. 277:1306–1309. [DOI] [PubMed] [Google Scholar]
- 22.Powis, S.J., L.L. Young, E. Joly, P.J. Barker, L. Richardson, R.P. Brandt, C.J. Melief, J.C. Howard, and G.W. Butcher. 1996. The rat cim effect: TAP allele-dependent changes in a class I MHC anchor motif and evidence against C-terminal trimming of peptides in the ER. Immunity. 4:159–165. [DOI] [PubMed] [Google Scholar]
- 23.Purcell, A.W., J.J. Gorman, M. Garcia-Peydro, A. Paradela, S.R. Burrows, G.H. Talbo, N. Laham, C.A. Peh, E.C. Reynolds, J.A. Lopez De Castro, et al. 2001. Quantitative and qualitative influences of tapasin on the class I peptide repertoire. J. Immunol. 166:1016–1027. [DOI] [PubMed] [Google Scholar]
- 24.Park, B., Y. Kim, J. Shin, S. Lee, K. Cho, K. Fruh, and K. Ahn. 2004. Human cytomegalovirus inhibits tapasin-dependent peptide loading and optimization of the MHC class I peptide cargo for immune evasion. Immunity. 20:71–85. [DOI] [PubMed] [Google Scholar]
- 25.Raffoux, C., R.C. Williams, C. Gorodezky, E.D. Albert, E. Gyodi, M.G. Hammond, Z. Layrisse, M. Mariani, G. de la Rosa, J.M. Tiercy, et al. 1997. AHS#7: HLA-B12, B13, B21, B37, B40, B41, B81, B4005. Genetic Diversity of HLA Functional and Medical Implication. D. Charron, editor. EDK Medical and Scientific International Publisher, Paris. 69–72.
- 26.Fleischhauer, K., N.A. Kernan, B. Dupont, and S.Y. Yang. 1991. The two major subtypes of HLA-B44 differ for a single amino acid in codon 156. Tissue Antigens. 37:133–137. [DOI] [PubMed] [Google Scholar]
- 27.Vilches, C. 2003. MHC class I peptide binding and tapasin. J. Immunol. 171:3. [DOI] [PubMed] [Google Scholar]
- 28.Copeman, J., N. Bangia, J.C. Cross, and P. Cresswell. 1998. Elucidation of the genetic basis of the antigen presentation defects in the mutant cell line .220 reveals polymorphism and alternative splicing of the tapasin gene. Eur. J. Immunol. 28:3783–3791. [DOI] [PubMed] [Google Scholar]
- 29.Zemmour, J., A.M. Little, D.J. Schendel, and P. Parham. 1992. The HLA-A,B “negative” mutant cell line C1R expresses a novel HLA-B35 allele, which also has a point mutation in the translation initiation codon. J. Immunol. 148:1941–1948. [PubMed] [Google Scholar]
- 30.Hill, A., P. Jugovic, I. York, G. Russ, J. Bennink, J. Yewdell, H. Ploegh, and D. Johnson. 1995. Herpes simplex virus turns off the TAP to evade host immunity. Nature. 375:411–415. [DOI] [PubMed] [Google Scholar]
- 31.Stam, N.J., H. Spits, and H.L. Ploegh. 1986. Monoclonal antibodies raised against denatured HLA-B locus heavy chains permit biochemical characterization of certain HLA-C locus products. J. Immunol. 137:2299–2306. [PubMed] [Google Scholar]
- 32.Carreno, B.M., J.C. Solheim, M. Harris, I. Stroynowski, J.M. Connolly, and T.H. Hansen. 1995. TAP associates with a unique class I conformation, whereas calnexin associates with multiple class I forms in mouse and man. J. Immunol. 155:4726–4733. [PubMed] [Google Scholar]
- 33.van Endert, P.M., R. Tampe, T.H. Meyer, R. Tisch, J.F. Bach, and H.O. McDevitt. 1994. A sequential model for peptide binding and transport by the transporters associated with antigen processing. Immunity. 1:491–500. [DOI] [PubMed] [Google Scholar]
- 34.Androlewicz, M.J., B. Ortmann, P.M. van Endert, T. Spies, and P. Cresswell. 1994. Characteristics of peptide and major histocompatibility complex class I/beta 2-microglobulin binding to the transporters associated with antigen processing (TAP1 and TAP2). Proc. Natl. Acad. Sci. USA. 91:12716–12720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Macdonald, W.A., A.W. Purcell, N. Mifsud, L.K. Ely, D.S. Williams, L. Chang, J.J. Gorman, C.S. Clements, L. Kjer-Nielsen, D.M. Koelle, et al. 2003. A naturally selected dimorphism within the HLA-B44 supertype alters class I structure, peptide repertoire and T cell recognition. J. Exp. Med. 198:679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Macdonald, W., D.S. Williams, C.S. Clements, J.J. Gorman, L. Kjer-Nielsen, A.G. Brooks, J. McCluskey, J. Rossjohn, and A.W. Purcell. 2002. Identification of a dominant self-ligand bound to three HLA B44 alleles and the preliminary crystallographic analysis of recombinant forms of each complex. FEBS Lett. 527:27–32. [DOI] [PubMed] [Google Scholar]
- 37.Ottwinowski, Z. 1993. Oscillation data reduction program. Data Collection and Processing. L. Sawyer, N. Issacs, and S. Bailey, editors. SERC Daresbury Laboratory, U.K., Warrington, U.K. 56–62.
- 38.Brunger, A.T., P.D. Adams, G.M. Clore, W.L. DeLano, P. Gros, R.W. Grosse-Kunstleve, J.S. Jiang, J. Kuszewski, M. Nilges, N.S. Pannu, et al. 1998. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54:905–921. [DOI] [PubMed] [Google Scholar]
- 39.Nicholls, A., K.A. Sharp, and B. Honig. 1991. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 11:281–296. [DOI] [PubMed] [Google Scholar]
- 40.Fruh, K., A. Gruhler, R.M. Krishna, and G.J. Schoenhals. 1999. A comparison of viral immune escape strategies targeting the MHC class I assembly pathway. Immunol. Rev. 168:157–166. [DOI] [PubMed] [Google Scholar]
- 41.Khanna, R., S.R. Burrows, A. Neisig, J. Neefjes, D.J. Moss, and S.L. Silins. 1997. Hierarchy of Epstein-Barr virus-specific cytotoxic T-cell responses in individuals carrying different subtypes of an HLA allele: implications for epitope-based antiviral vaccines. J. Virol. 71:7429–7435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neisig, A., C.J. Melief, and J. Neefjes. 1998. Reduced cell surface expression of HLA-C molecules correlates with restricted peptide binding and stable TAP interaction. J. Immunol. 160:171–179. [PubMed] [Google Scholar]
- 43.Ford, S., A.N. Antoniou, G.W. Butcher, and S.J. Powis. 2004. Competition for access to the rat MHC class I peptide loading complex reveals optimisation of peptide cargo in the absence of TAP association. J. Biol. Chem. 279:16077–16082. First published on February 4, 2004; 10.1074/jbc. M400456200. [DOI] [PubMed]
- 44.Garcia-Peydro, M., M. Marti, and J. Lopez de Castro. 1999. High T cell epitope sharing between two HLA-B27 subtypes (B*2705 and B*2709) differentially associated to ankylosing spondylitis. J. Immunol. 163:2299–2305. [PubMed] [Google Scholar]
- 45.Ramos, M., A. Paradela, M. Vazquez, A. Marina, J. Vazquez, and J.A. Lopez de Castro. 2002. Differential association of HLA-B*2705 and B*2709 to ankylosing spondylitis correlates with limited peptide subsets but not with altered cell surface stability. J. Biol. Chem. 277:28749–28756. [DOI] [PubMed] [Google Scholar]
- 46.Reits, E., A. Griekspoor, J. Neijssen, T. Groothuis, K. Jalink, P. van Veelen, H. Janssen, J. Calafat, J.W. Drijfhout, and J. Neefjes. 2003. Peptide diffusion, protection, and degradation in nuclear and cytoplasmic compartments before antigen presentation by MHC class I. Immunity. 18:97–108. [DOI] [PubMed] [Google Scholar]
- 47.Lybarger, L., X. Wang, M.R. Harris, H.W.t. Virgin, and T.H. Hansen. 2003. Virus subversion of the MHC class I peptide-loading complex. Immunity 18:121-130. [DOI] [PubMed] [Google Scholar]
- 48.Fruh, K., K. Ahn, and P.A. Peterson. 1997. Inhibition of MHC class I antigen presentation by viral proteins. J. Mol. Med. 75:18–27. [DOI] [PubMed] [Google Scholar]
- 49.Hughes, E.A., C. Hammond, and P. Cresswell. 1997. Misfolded major histocompatibility complex class I heavy chains are translocated into the cytoplasm and degraded by the proteasome. Proc. Natl. Acad. Sci. USA. 94:1896–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schoenhals, G.J., R.M. Krishna, A.G. Grandea III, T. Spies, P.A. Peterson, Y. Yang, and K. Fruh. 1999. Retention of empty MHC class I molecules by tapasin is essential to reconstitute antigen presentation in invertebrate cells. EMBO J. 18:743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barnden, M.J., A.W. Purcell, J.J. Gorman, and J. McCluskey. 2000. Tapasin-mediated retention and optimization of peptide ligands during the assembly of class I molecules. J. Immunol. 165:322–330. [DOI] [PubMed] [Google Scholar]
- 52.Froloff, N., A. Windemuth, and B. Honig. 1997. On the calculation of binding free energies using continuum methods: application to MHC class I protein-peptide interactions. Protein Sci. 6:1293–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Matthews, B.W. 1993. Structural and genetic analysis of protein stability. Annu. Rev. Biochem. 62:139–160. [DOI] [PubMed] [Google Scholar]
- 54.Serrano, L., M. Bycroft, and A.R. Fersht. 1991. Aromatic-aromatic interactions and protein stability. Investigation by double-mutant cycles. J. Mol. Biol. 218:465–475. [DOI] [PubMed] [Google Scholar]
- 55.Fruci, D., G. Niedermann, R.H. Butler, and P.M. van Endert. 2001. Efficient MHC class I-independent amino-terminal trimming of epitope precursor peptides in the endoplasmic reticulum. Immunity. 15:467–476. [DOI] [PubMed] [Google Scholar]
- 56.Brouwenstijn, N., T. Serwold, and N. Shastri. 2001. MHC class I molecules can direct proteolytic cleavage of antigenic precursors in the endoplasmic reticulum. Immunity. 15:95–104. [DOI] [PubMed] [Google Scholar]
- 57.Lorenzo, M.E., H.L. Ploegh, and R.S. Tirabassi. 2001. Viral immune evasion strategies and the underlying cell biology. Semin. Immunol. 13:1–9. [DOI] [PubMed] [Google Scholar]
- 58.Burgert, H.G., Z. Ruzsics, S. Obermeier, A. Hilgendorf, M. Windheim, and A. Elsing. 2002. Subversion of host defense mechanisms by adenoviruses. Curr. Top. Microbiol. Immunol. 269:273–318. [DOI] [PubMed] [Google Scholar]
- 59.Boname, J.M., B.D. De Lima, P.J. Lehner, and P.G. Stevenson. 2004. Viral degradation of the MHC class I peptide loading complex. Immunity. 20:305–317. [DOI] [PubMed] [Google Scholar]
- 60.Reddehase, M.J. 2002. Antigens and immunoevasins: opponents in cytomegalovirus immune surveillance. Nat. Rev. Immunol. 2:831–844. [DOI] [PubMed] [Google Scholar]
- 61.Benjamin, R.J., J.A. Madrigal, and P. Parham. 1991. Peptide binding to empty HLA-B27 molecules of viable human cells. Nature. 351:74–77. [DOI] [PubMed] [Google Scholar]
- 62.Carreno, B.M., and T.H. Hansen. 1994. Exogenous peptide ligand influences the expression and half-life of free HLA class I heavy chains ubiquitously detected at the cell surface. Eur. J. Immunol. 24:1285–1292. [DOI] [PubMed] [Google Scholar]
- 63.Purcell, A.W., A.J. Kelly, C.A. Peh, N.L. Dudek, and J. McCluskey. 2000. Endogenous and exogenous factors contributing to the surface expression of HLA B27 on mutant APC. Hum. Immunol. 61:120–130. [DOI] [PubMed] [Google Scholar]
- 64.Kaufman, J., S. Milne, T.W. Gobel, B.A. Walker, J.P. Jacob, C. Auffray, R. Zoorob, and S. Beck. 1999. The chicken B locus is a minimal essential major histocompatibility complex. Nature. 401:923–925. [DOI] [PubMed] [Google Scholar]
- 65.Beier, D.C., J.H. Cox, D.R. Vining, P. Cresswell, and V.H. Engelhard. 1994. Association of human class I MHC alleles with the adenovirus E3/19K protein. J. Immunol. 152:3862–3872. [PubMed] [Google Scholar]