

Abstract
The established model for the mechanism of action of aspirin is the inhibition of prostaglandin synthesis. However, this has never fully explained aspirin's repertoire of antiinflammatory properties. We found in acute pleuritis that aspirin, but not salicylate, indomethacin, or piroxicam, increased plasma nitric oxide (NO), which correlated with a reduction in inflammation. Inhibiting aspirin-elicited NO pharmacologically in this model nullified the antiinflammatory effects of aspirin. Moreover, aspirin was not antiinflammatory in either constitutive (eNOS) or inducible NO synthase (iNOS) knockout mice with IL-1β–induced peritonitis. It transpires that aspirin generates NO through its unique ability to trigger the synthesis of 15-epi-lipoxin A4. Aspirin and 15-epi-lipoxin A4 were shown to inhibit leukocyte trafficking in an NO-dependent manner using intravital microscopy on IL-1β–stimulated mouse mesentery. Not only did aspirin inhibit leukocyte–endothelial interaction in a manner similar to NO in wild-type mice but both aspirin and 15-epi-lipoxin A4 had markedly reduced effects on leukocyte–endothelial cell adherence in eNOS- and iNOS-deficient mice compared with wild type. Collectively, these data suggest that aspirin triggers the synthesis of 15-epi-lipoxin A4, which increases NO synthesis through eNOS and iNOS. This aspirin-elicited NO exerts antiinflammatory effects in the microcirculation by inhibiting leukocyte–endothelium interactions.
Keywords: leukocyte trafficking, resolution, nonsteroidal antiinflammatory drugs, epi-lipoxins, microvascular endothelium
Introduction
Aspirin has been used for over a century to treat the cardinal signs of inflammation (heat, redness, swelling, and pain), whereas recently it has been shown to prevent intravascular thrombosis and cancer as well as to slow Alzheimer's disease. Since the classic work of Vane, it has been widely accepted that the pharmacological action of nonsteroidal antiinflammatory drugs (NSAIDs) is mediated by inhibiting the activity of cyclooxygenase (COX), a key enzyme in the biosynthesis of proinflammatory PGs (1). However, not all of aspirin's biological effects can be explained by the inhibition of PGs alone. For example, the doses of aspirin needed to exert an antiinflammatory effect are higher than that which is required to inhibit PG synthesis (2), whereas aspirin itself was found to trigger the synthesis of novel lipid metabolites that directly halt leukocyte trafficking (3). Moreover, the original description of PG inhibition does not explain how aspirin inhibits cell accumulation during inflammation, as it is generally accepted that the COX pathway is not involved in the recruitment of inflammatory leukocytes (4). However, this is not exclusive to aspirin as other NSAIDs also inhibit leukocyte accumulation, suggesting additional mechanisms of action to that of PG inhibition. In an attempt to understand how aspirin inhibits acute inflammation, we found serendipitously that it increased plasma nitrite levels in inflamed animals through the actions of both constitutive (eNOS) and inducible nitric oxide (NO) synthase (iNOS). This was of importance to acute inflammation as NO plays a central role in regulating leukocyte trafficking. Here, we report that the inhibition of acute inflammation by aspirin is dependent on the induction of plasma NO, which negatively regulates leukocyte–endothelial interaction in the microcirculation. In doing so, we contend that this is the principal mechanism by which aspirin inhibits acute inflammation.
Materials and Methods
Animals
eNOS and neuronal NO synthase (nNOS) knockout mice were obtained from P.L. Huang (Harvard Medical School, Boston, MA) and I. MacIntyre (The Royal London School of Medicine and Dentistry, London, England, UK), and iNOS knockout mice were obtained from A.J. Hobbs (University College London, London, England, UK).
Animal Models of Inflammation
Carrageenin-induced Pleurisy.
Male Wistar rats (150 ± 20 g; Tuck and Sons) were used. A 1% (wt/vol) carrageenin (Sigma-Aldrich) solution in saline (0.15 ml) was injected into the pleural cavity as described previously (5).
Peritonitis.
Male SV129 or C57BL/6 wild type (15–20 g; Harlan) or male eNOS−/−, iNOS−/−, or nNOS−/− mice were injected into the peritoneal cavity with 10 ng IL-1β (R&D Systems) in sterile phosphate-buffered saline as described previously (6). For both pleurisy and peritonitis models, exudates were collected 4 h after stimulus injection by lavaging the pleural or peritoneal cavity with 1 ml 3.15% (wt/vol) sodium citrate in physiological saline. For the pleurisy, edema formation was assessed by weighing the collected inflammatory exudates and cells were counted with a Coulter® counter (model DN; Beckman Coulter). For the mouse peritonitis, lavage fluids were stained with Turk's solution (0.01% crystal violet in 3% acetic acid), and differential cell counts were performed by light microscopy (model B061; Olympus) using a Neubauer hemocytometer. All animal experiments were done in accordance with the UK Home Office regulations for the care and use of animals.
Measurement of Plasma Nitrite
Nitrite formation was measured by a modification of the Griess reaction (7). In brief, 10 μl of NADPH (10 μM), 5 mM glucose-6-phosphate, 0.16 U glucose-6-phosphate dehydrogenase, and 10 mM PBS was added to 50 μl of cell and protein (using 35% wt/vol sulfosalicylic acid as the precipitating agent) free lavage fluids or in plasma samples in a 96-well plate. Nitrate was converted to nitrite by addition of 10 μl (0.08 U) of nitrate reductase and incubated for 45 min; 200 μl of Griess reagent (equal volumes of 1% wt/vol sulfanilamide in 5% H3PO4 and 0.1% wt/vol [1-napthyl]ethylenediamine) was added and incubated for a further 10 min. Nitrite concentrations were measured at 570 nm with a reference filter at 620 nm and results expressed as micrometers of nitrite in cell-free exudates.
Eicosanoid Measurements and 15-epi-lipoxin A4 Analogue
Aspirin-triggered 15-epi-lipoxin A4 was measured by ELISA (Neogen Corp.; reference 8). 15-epi-16-(para-fluoro)-phenoxy-LXA4-methyl ester, the stable analogue of 15-epi-lipoxin A4 was a gift from K. Gotlinger (Harvard Medical School, Boston, MA).
Intravital Microscopy of the Mouse Mesentery Microcirculation
Male C57BL/6 mice were used. All animals were injected i.p. with 10 ng IL-1β 1 h after drug treatment. 2 h after IL-1β injection, the mouse mesentery was prepared for intravital microscopy as described previously (6).
Drug Dosing
100–400 mg/kg aspirin, 100–400 mg/kg salicylate, 10 m/kg piroxicam, 3 mg/kg indomethacin, or 0.5 mg/kg dexamethasone was dosed orally in vehicle (gum tragacanth) 1 h before initiation of either pleurisy or peritonitis. NOS inhibitors (10 mg/kg) were given into the pleural cavity concomitant with carrageenin. For intravital microscopy experiments on mice, 200 mg/kg aspirin was given orally and 3 μg 15-epi-16-(para-fluoro)-phenoxy-LXA4-methyl ester was given i.v. 1 h before injection of IL-1β.
Statistics
Statistical analysis was performed using analysis of variance (ANOVA) followed by a Bonferroni-T test. Data are expressed as mean ± standard error of the mean.
Results
Aspirin, But Not Other NSAIDs, Increases Plasma NO.
The injection of carrageenin into the pleural cavity of rats initiates an acute, self-resolving inflammatory response that has proven useful for screening the antiinflammatory properties of novel drugs. Dosing rats orally with aspirin 1 h before intrapleural carrageenin injection resulted in a decrease in total inflammatory cell numbers and edema accumulation within the pleural cavity after 4 h (Fig. 1 A). This predictable reduction in local inflammation correlated inversely with a dose-dependent and significant increase in plasma NO levels as measured by nitrite using the Griess reaction. In contrast, the major aspirin metabolite, salicylate (Fig. 1 B) as well as other NSAIDs such as piroxicam (Fig. 1 C) and indomethacin (Fig. 1 D), reduced pleural inflammation to varying degrees, but did not alter peripheral blood nitrite compared with controls, suggesting that the ability to trigger NO synthesis may be exclusive to aspirin.
Figure 1.
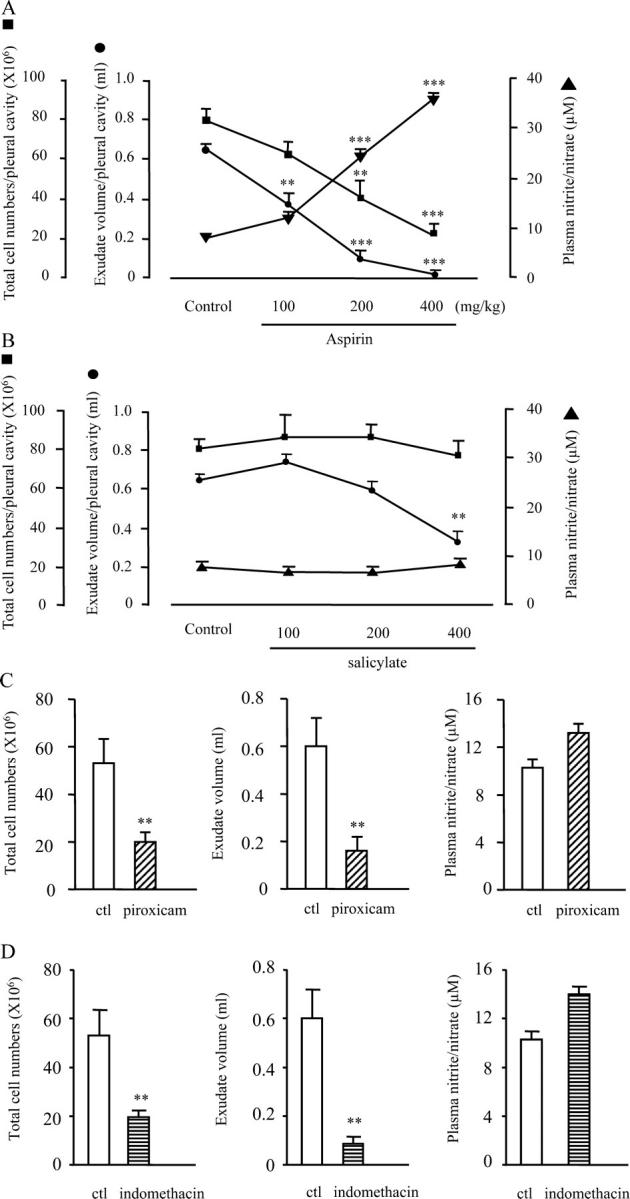
Aspirin dose-dependently increases peripheral blood nitrite levels while ameliorating acute pleural inflammation. Rats were dosed orally with (A) aspirin and compared with other NSAIDs including (B) salicylate, (C) piroxicam (10 mg/kg) or (D) indomethacin (3 mg/kg) 1 h before intrapleural carrageenin injection. 4 h later, peripheral blood nitrite levels were measured by the Griess reaction and compared with pleural cavity inflammatory cell numbers and exudate volume. n = 8–10 per group. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001, as determined by ANOVA followed by a Bonferroni-T test.
Inhibiting Aspirin-elicited NO Pharmacologically Abrogates Aspirin's Antiinflammatory Effects.
This increase in plasma NO by aspirin is of enormous potential impact on inflammatory diseases as some of the critical steps that result in the extravascular accumulation of leukocytes during acute inflammation (rolling, adhesion, and migration through the microvascular endothelium) are controlled by NO through its negative regulatory effect on cell adhesion molecule expression in the microcirculation (9, 10). As a result, we questioned whether part of aspirin's antiinflammatory properties lay in its ability to increase NO and prevent leukocyte trafficking across the endothelium. To address this, first we took a pharmacological approach. Despite the role NO plays in inhibiting leukocyte trafficking, NOS inhibitors are generally antiinflammatory (11–13) when given systemically, possibly due to their inhibition of eNOS remote from the inflammatory locus, resulting in vasoconstriction and reduced blood delivery to the inflamed site. Certainly, the antiinflammatory effects of systemically administered L-arginine analogues, for instance, are reversed by vasodilators (14, 15). Thus, to investigate the possible role of aspirin-generated NO in regulating leukocyte trafficking in the rat pleurisy, the NOS inhibitor, AE-ITU (10 mg/kg), was given locally to inhibit eNOS in the microvascular endothelium surrounding the pleural cavity and alter leukocyte–endothelial interaction in this area. 200 mg/kg aspirin was dosed orally 1 h before the concomitant administration of carrageenin and AE-ITU into the pleural cavity. Aspirin inhibition of cell accumulation (Fig. 2 A, P ≤ 0.05) was associated with an increase in pleural nitrite levels (Fig. 2 B, P ≤ 0.05) 4 h after stimulus injection. This inhibition of cell trafficking by aspirin was reversed by AE-ITU (Fig. 2 A, P ≤ 0.05) at concentrations that reduced aspirin-increased pleural nitrite (Fig. 2 B, P ≤ 0.05). Although AE-ITU has a fourfold selectivity for iNOS over eNOS (16), selectivity at the doses used here would not be anticipated with similar effects also obtained with 1,400 W and L-NIO (unpublished data). The results of these experiments suggest that in rat pleuritis, aspirin-generated NO controls leukocyte migration to the inflamed site.
Figure 2.

Pharmacological inhibition of aspirin-elicited NO abrogates aspirin's antiinflammatory effects. To determine whether NO mediates the antiinflammatory effects of aspirin in this pleuritis model, the NOS inhibitor AE-ITU (10 mg/kg) was injected into the pleural cavity concomitant with carrageenin (aspirin was dosed orally 1 h previously at 200 mg/kg) and found to (A) reverse the antiinflammatory effects of aspirin at doses that (B) inhibited aspirin-generated NO, 4 h after carrageenin/AE-ITU injection. n = 8–10 per group.*, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001, as determined by ANOVA followed by a Bonferroni-T test.
Aspirin Is Not Antiinflammatory in NOS Knockout Mice.
To take these experiments further, we examined the antiinflammatory effects of aspirin in NOS knockout mice. Although eNOS is well known to negatively regulate leukocyte–endothelial cell interaction, iNOS and nNOS have also been suggested to play a role in this aspect of leukocyte trafficking (17, 18). From the aforementioned pharmacological studies in rats, it is reasonable to suggest that aspirin would be less effective at inhibiting inflammation in NO-deficient animals than in wild-type controls. To test this hypothesis, we dosed 200 mg/kg aspirin to eNOS−/− (19) and iNOS−/− mice (20) bearing an IL-1β–induced peritonitis and found that it was ineffective at inhibiting inflammation in these animals, as total inflammatory cell numbers in aspirin-treated knockouts were the same as untreated knockouts (Fig. 3, A and B). Wild-type controls for these animals responded normally to aspirin, which reduced inflammation by ∼60%. 0.5 mg/kg dexamethasone was used as a positive control and reduced peritonitis in both wild type and knockouts to the same extent. In contrast, nNOS−/− (21) responded to aspirin as normal with a 60% reduction in peritoneal PMN numbers compared with controls (Fig. 3 C). Together with the results of the aforementioned pharmacological experiments, these data imply that aspirin has a significant dependence on NO to inhibit acute inflammatory reactions.
Figure 3.

Aspirin is not antiinflammatory in eNOS−/− and iNOS−/− bearing an IL-1β–induced peritonitis. 200 mg/kg aspirin or 0.5 mg/kg dexamethasone (positive control) were dosed orally to (A) eNOS−/−, (B) iNOS−/−, and (C) nNOS−/− mice 1 h before intraperitoneal IL-1β injection. 4 h after IL-1β administration, local PMNs numbers and (D) plasma nitrite levels were determined. n = 6–8 per group. ***, P ≤ 0.001, as determined by ANOVA followed by a Bonferroni-T test.
Measuring nitrite in the plasma of these animals provided some insight into how NO is generated in response to aspirin (Fig. 3 D). Aspirin increased NO in mice bearing an IL-1β–induced peritonitis (P ≤ 0.001) to a similar level found in rats with pleuritis (Fig. 1 A). Interestingly, aspirin also increased NO in wild-type naive (without inflammation) mice (P ≤ 0.001). In inflamed eNOS−/− controls, nitrite levels were similar to those in untreated wild type and increased only marginally after aspirin treatment. Equally, in iNOS−/−, nitrite levels in controls were comparable to that in wild type and only slightly augmented by aspirin. These results show that aspirin cannot increase plasma NO levels in either eNOS−/− or iNOS−/− mice much above controls, helping to explain the lack of antiinflammatory effects of aspirin in these animals (i.e., failure to reach a critical level of antiadhesive NO). These data further suggest that there is a degree of cross-talk between eNOS and iNOS for the generation of NO to negatively regulate leukocyte–endothelium interaction.
Aspirin Mediates Its Effects through 15-epi-lipoxin A4.
The next objective was to discern the mechanism by which aspirin increases plasma NO. It was recently shown that 15-lipoxygenases and constitutive COX (COX 1) catalytically consume NO through peroxidase-dependent mechanisms implicating COX/lipoxygenase as an elimination route for NO. However, as neither indomethacin nor piroxicam (both COX 1 inhibitors) significantly altered plasma NO (Fig. 1, C and D), it is unlikely that inhibition of COX 1 enzyme activity is the cause of increased NO by aspirin. However, of all the NSAIDs, only aspirin possesses the unique ability to trigger lipoxin synthesis (so-called aspirin-triggered epi-lipoxins) as a result of acetylating the active site of COX 2 in endothelial or epithelial cells, a property not shared with other NSAIDs (3). This results in the synthesis of (15 R) hydroxyeicosatetraenoic acid (15R-HETE), which is rapidly metabolized in a transcellular manner by leukocyte 5-lipoxygenase to 15-epi-lipoxin A4 or B4. In addition, given that aspirin but not salicylate or other NSAIDs increased NO in the present work and that native lipoxins can generate NO in PMN/endothelial cells (22) and rabbit tracheal segments (23), we questioned whether 15-epi-lipoxin A4 mediates the NO-inducing properties of aspirin. To test this, mice with an IL-1β–induced peritonitis were predosed for 1 h with 200 mg/kg aspirin. 4 h after induction of peritonitis, peripheral blood 15-epi-lipoxin A4 levels were measured and found to increase significantly with aspirin but not indomethacin (Fig. 4 A). These results reflect the recent observation that aspirin increases urinary levels of 15-epi-lipoxin A4 in humans (24). Mice with peritonitis were dosed with the nonselective COX inhibitor, indomethacin (3 mg/kg), 1 h before dosing with aspirin to block the active site of COX 2 and blunt aspirin's ability to generate 15-epi-lipoxin A4. Administering indomethacin with aspirin was originally used to show that COX 2 is the source of aspirin-triggered 15R-HETE generation from human recombinant COX 2 (25) and tracheal epithelial cells (26) and that COX 2 was the source of aspirin-generated 15-epi-lipoxin A4 synthesis in IL-1β–stimulated coincubations of PMN/endothelial cells (3) and, as such, is a well-characterized strategy for inhibiting aspirin-triggered 15-epi-lipoxin A4 synthesis. This approach significantly reduced aspirin-triggered 15-epi-lipoxin A4 (Fig. 4 A) as well as plasma nitrite levels (Fig. 4 B) in murine IL-1β–induced peritonitis. When 15-epi-16-(para-fluoro)-phenoxy-LXA4-methyl ester (3 μg), a stable analogue of 15-epi-lipoxin A4 (27), was injected i.v. into either naive mice (uninflamed) or mice with a peritonitis, peripheral blood nitrite levels were increased (Fig. 4 C) at dosing levels that significantly inhibited inflammation (Fig. 4 D). As equivalent experiments found the same in a rat carrageenin–induced pleurisy (unpublished data), these experiments suggest that aspirin-generated NO is mediated, at least in part, through the effects of aspirin-triggered 15-epi-lipoxin A4.
Figure 4.

Aspirin increases systemic nitrite levels through the induction of aspirin-triggered 15-epi-lipoxin A4. In a mouse with IL-1β–induced peritonitis (A), 200 mg/kg aspirin was dosed orally to mice 1 h before IL-1β injection and found to increase plasma levels of 15-epi-lipoxin A4 4 h later, a known consequence of the acetylation of the active site of COX 2. Separate groups of animals were predosed with 3 mg/kg indomethacin 1 h before aspirin to block the active site of COX 2, thereby preventing access to aspirin and diminishing its ability to elicit 15-epi-lipoxin A4. (B) This increase in 15-epi-lipoxin A4 by aspirin and its partial blockade with indomethacin was associated with an increase and reduction, respectively, in plasma nitrite levels. In the final set of experiments, (C) 3 μg of the stable 15-epi-lipoxin A4 analogue, 15-epi-16-(para-fluoro)-phenoxy-LxA4-methyl ester, was injected i.v. into both naive as well as mice bearing an IL-1β–induced peritonitis and was found to increase plasma nitrite levels at concentrations that (D) ameliorated the severity of IL-1β–induced peritoneal inflammation. n = 6–8 per group. **, P ≤ 0.01; ***, P ≤ 0.001, as determined by ANOVA followed by a Bonferroni-T test.
Aspirin Prevents Leukocyte–Endothelium Interaction in a NO-dependent Manner.
Finally, we examined the effects of aspirin on leukocyte–endothelial cell interaction using intravital microscopy, a technique that allows visualization of leukocyte movement in the microcirculation in response to an inflammatory stimulus. In the mouse mesentery model, 200 mg/kg aspirin significantly reduced IL-1β–stimulated leukocyte flux (Fig. 5 A), which defines the number of leukocytes rolling along a predefined length of venule per minute, back to unstimulated control levels (P ≤ 0.01). IL-1β–mediated reduction in leukocyte rolling velocity, which facilitates adherence to the endothelium, was also returned to unstimulated control levels by aspirin (Fig. 5 B, P ≤ 0.01). In addition, leukocyte adherence to (P ≤ 0.01) and extravasation through the endothelium (P ≤ 0.001) was inhibited by aspirin (Fig. 5, C and D, respectively). Thus, aspirin inhibited leukocyte–endothelial interaction in a manner similar to NO. Importantly, aspirin and the 15-epi-lipoxin A4 analogue (3 μg, i.v.) did not significantly alter leukocyte adhesion to microvascular endothelial cells in eNOS−/− (Fig. 5 E) and had a reduced effect on this inflammatory parameter in iNOS−/− mice (Fig. 5 F) when compared with drug-treated wild-type controls (Fig. 5 C). These results suggest a central role for NO in mediating aspirin and 15-epi-lipoxin A4 inhibition of leukocyte trafficking.
Figure 5.

Aspirin and 15-epi-lipoxin A4 alter leukocyte–endothelium interaction in an NO-dependent manner. Mice were dosed orally with 200 mg/kg aspirin 1 h before intraperitoneal injection of 10 ng IL-1β, which elicits localized cell adhesion and emigration within the mouse mesentery. 2 h after IL-1β administration, mice were anesthetized, and a loop of the ileal mesentery was exposed to observe leukocyte dynamics in microcirculation by intravital microscopy. The effects of aspirin on leukocyte (A) flux, (B) velocity, (C) adherence, and (D) extravasation were determined off-line during playback of videotape images. In this model, the effects of 200 mg/kg aspirin as well as the stable 15-epi-lipoxin A4 analogue, 15-epi-16-(para-fluoro)-phenoxy-LXA4-methylester (3 μg i.v.) were examined for their effects on leukocyte adherence in (E) eNOS−/− and (F) iNOS−/− mice, where aspirin and the epi-lipoxin analogue were given 1 h before IL-1β, and their effects on cell trafficking were assessed 2 h after cytokine injection. n = 4–6 per group. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001, as determined by ANOVA followed by a Bonferroni-T test.
Discussion
The data presented here suggest that, once ingested, aspirin is exposed to the vasculature where it acetylates COX 2 within the endothelium or circulating leukocytes to trigger 15-epi-lipoxin A4, which, in turn, elicits NO synthesis from both eNOS and iNOS. Ultimately, aspirin-triggered NO mediates the antiinflammatory effects of aspirin in the microcirculation by negatively regulating leukocyte–endothelium interaction. What was initially a study to search for alternative modes of action of aspirin has raised further questions, namely how do aspirin and the 15-epi-lipoxin A4 increase NO synthesis and what cross-talk exists between eNOS and iNOS for the regulation of leukocyte trafficking in aspirin-treated inflammation? Regarding the latter, NO has a biphasic effect on NF-κB activity in murine macrophages, being able to up- and down-regulate the expression of several proinflammatory proteins, including iNOS (28). It appears that low levels of NO as produced from eNOS activate NF-κB, whereas higher levels of NO, typical of iNOS, inhibit NF-κB. In support of this, it was shown recently that lipopolysaccharide-stimulated, bone marrow–derived macrophages from eNOS−/− mice show greatly reduced NF-κB activity and iNOS expression compared with wild-type cells (29). These authors demonstrated that eNOS triggered iNOS expression, in part, through soluble guanylate cyclase. A critical role for this NO–guanylate cyclase pathway in cytokine signaling of iNOS expression was also shown in human mesangial cells (30). Thus, inhibiting the first wave of NO stimulated by IL-1β or tumor necrosis factor with NG-nitro-l-arginine methyl ester (L-NAME) resulted in a subsequent reduction in iNOS expression and activity, which was reversed using an NO donor (30). Collectively, these in vitro studies suggest that if the initial NO-mediated response to stimulation is absent, the magnitude of subsequent iNOS expression is diminished, which may explain why plasma NO levels in aspirin-treated eNOS−/− mice are much lower than aspirin-treated wild-type animals (Fig. 3 D). Similarly, in iNOS−/−, though eNOS activity may be enhanced slightly by aspirin, levels of NO from this constitutive isoform will also be low, explaining why plasma NO levels in aspirin-treated iNOS−/− mice are lower than in aspirin-treated wild-type animals (Fig. 3 D). These results also strongly suggest that there is a requirement for both eNOS and iNOS for the endogenous control of leukocyte–endothelial cell interaction during acute inflammation in response to aspirin. This is particularly important as eNOS has received the most attention regarding cell trafficking with comparatively less work reported on iNOS and nNOS in this setting (10). Whether both eNOS and iNOS are also essential during untreated (without aspirin) inflammatory reactions is unclear at this time.
One of the unexpected findings in this analysis was that, in addition to animals bearing a localized inflammation (Figs. 1 A and 3 D), aspirin and 15-epi-lipoxin A4 also elicited NO synthesis in naive animals (Fig. 3 D). This suggests that the biochemical machinery required to manufacture NO in response to aspirin is constitutively expressed. That 15-epi-lipoxin A4 mediates aspirin-generated NO identifies COX 2/5-lipoxygenase as this constitutively expressed pathway. COX 2 is not classically thought of as a constitutively expressed enzyme. However, given the size of the vasculature, it has been proposed that the vascular endothelium may contain focal regions or “hot spots” under stress that express COX 2 and can generate substantial amounts of COX 2–derived epi-lipoxins with aspirin treatment (31). Certainly, it was found that a physiological level of steady laminar sheer stress, an in vitro model that mimics the average wall shear encountered in the vasculature, caused a sustained expression of endothelial COX 2 (32). This constitutive expression of COX 2 helps to explain why we found increased plasma NO in naive, uninflamed animals (Fig. 3 D). COX 2 could also be constitutively expressed in circulating leukocytes. Indeed, we found that ∼10% of circulating PMNs from naive rats constitutively express COX 2 (33). However, the exact cellular source of this constitutively expressed COX 2 remains to be identified.
Aspirin's reduced effect on leukocyte–endothelial cell interaction in iNOS−/− mice (Fig. 5 F) is not consistent with its complete lack of effect on cell accumulation in these animals bearing an IL-1β–induced peritonitis (Fig. 3 B). However, a closer look at the kinetics of cell trafficking in acute pleuritis (34, 35), for instance, may provide an explanation for this discrepancy. This model shows two waves of cellular influx: the first within 1–3 h representing moderate cell migration, and the second between 4–6 h marking a period of enhanced cell trafficking. This two-phase accumulation mirrors NO synthesis: an early phase within the first hour or so that is dexamethasone insensitive and most likely from eNOS, and a second peak at 4–6 h that may be from iNOS as it is dexamethasone sensitive and mirrors iNOS protein expression (33). Given the antiadhesive effects of NO, this possibly represents an endogenous mechanism controlling cell trafficking during the early onset phase of acute inflammation. In which case, this two-phase concept of NO-regulated cell trafficking suggests that aspirin and 15-epi-lipoxin A4 would be antiinflammatory in iNOS−/− mice during the very early eNOS-dependent time frame of 1–2 h, as eNOS protein will still be expressed in these animals. This hypothesis supports our findings using intravital microscopy, which was performed 2 h after IL-1β injection (Fig. 5, this is our general laboratory practice as it is easier to tabulate cell trafficking during this earlier phase), whereas the peritonitis experiments were performed at 4 h after stimulus injection (Fig. 3).
Lipoxins and aspirin-triggered epi-lipoxins regulate the extravascular accumulation of leukocytes during acute inflammation by altering leukocyte–endothelial interaction (36) as well as having a direct effect on leukocyte diapedesis (37) and chemotaxis (38). For instance, lipoxin A4 stable analogues modulate the expression of both L-selectin and CD11/CD18 on resting and stimulated leukocytes and inhibit neutrophil adhesion to human coronary artery endothelial cells by reducing CD11/CD18 expression (39). Similarly, in a model of L-NAME–enhanced leukocyte–endothelial cell interaction in the rat mesentery, lipoxin analogues markedly attenuated L-NAME–induced leukocyte rolling and adherence concomitant with an inhibition of endothelial P-selectin expression (39), whereas aspirin-triggered 15-epi-lipoxin A4 has been implicated in the antiadhesive effects of aspirin on adhesion of PMN to endotoxin-stimulated human umbilical vein endothelial cells (40, 41). Whether lipoxins mediate these effects on cell adhesion molecule expression directly or indirectly through NO is unclear. Certainly, NO is known to regulate P-selectin (42) and CD11/CD18 expression (43). Also, inhibiting aspirin-triggered 15-epi-lipoxin A4 synthesis using a selective COX 2 inhibitor or blocking the effects of 15-epi-lipoxin A4 with a lipoxin receptor antagonist (lipoxins and epi-lipoxins share the same receptor (44) markedly attenuated aspirin's inhibition of PMN–endothelial cell interaction. In these papers, this strategy of COX 2 inhibition and lipoxin receptor antagonism had only a minimal effect on antiadhesion by NCX-4016, a NO-releasing derivative of aspirin (24, 41). Further evidence that aspirin's protective properties may be mediated through a epi-lipoxin–NO pathway is apparent from a recent paper examining the effects of NCX-4016 on clinical (24) and experimental models of mucosal damage (41). Volunteers who received a selective COX 2 inhibitor (celecoxib) in combination with aspirin developed twice the mucosal damage compared with aspirin alone. This was not observed when celecoxib was administered with NO-releasing NCX-4016 (24). Similarly, in rats, celecoxib exacerbated aspirin-induced gastric damage and inflammation, but in combination with NCX-4016 was without effect (45). These studies provide evidence that NO may mediate, to a large extent, the antiinflammatory effects of aspirin and aspirin-generated epi-lipoxins. In addition, the data presented here suggest that drugs that inhibit COX 2 may nullify the antiinflammatory effects of aspirin, which supports the cautionary note sounded by others for patients taking aspirin in combination with COX 2 inhibitors (24, 45). Although lipoxins are known to enhance prostacyclin synthesis (46), which also attenuates leukocyte–endothelial cell interaction, we suspect that the role of prostacyclin in this setting is negligible given the striking dependence of aspirin and 15-epi-lipoxin A4 on NO to inhibit leukocyte trafficking shown in this paper.
The results presented here raise several additional issues pertaining to the endogenous control of acute inflammation. For instance, we contend that COX 2 mediates the antiinflammatory effects of aspirin (through the epi-lipoxin/NO circuit), implying that the efficacy of aspirin will increase in parallel with the severity of an inflammatory response (i.e., a more aggressive inflammation will have greater COX 2 expression), thereby facilitating the generation of more antiinflammatory epi-lipoxin–NO. This confers on aspirin a type of “smart-drug” status, working only when and where it is needed. One caveat to this idea is that too much COX 2 could generate NO at high levels leading to local tissue damage. Moreover, NO can enhance COX 2 expression (47) and enzyme activity (48), suggesting that aspirin may worsen matters by creating an unstable feed-forward system, leading to sustained COX 2–15-epi-lipoxin A4–NO synthesis. However, such an unstable feed-forward system may be interrupted by high levels of NO derived from iNOS, which, as aforementioned, can inhibit NF-κB, a well-known regulator of COX 2 transcription (49), resulting in an inhibition of COX 2 expression. This may represent a potential control loop for prolonged COX–NOS synthesis, failure of which could contribute to tissue damage and an inability of acute inflammation to resolve normally.
In summary, the discovery by Vane in 1971 that aspirin exerts its antiinflammatory effect by inhibiting PG synthesis (1) has helped to explain many, but not all, of the properties of this enigmatic drug. Indeed, the inhibition of PGs is insufficient in itself to adequately explain how aspirin controls inflammation. As a result, many elegant experiments have been performed to help address this shortfall in our understanding of how aspirin works. To this end, aspirin has been shown to inhibit NF-κB activation (50), mediate the release of adenosine (51), activate heat shock proteins (52), and down-regulate inducible COX expression (53), to name but a few. However, these bio-effects of aspirin are also shared with salicylate, indicating that many of the antiinflammatory properties of aspirin may, in fact, be due to salicylate. But even the reported bio-effects of salicylate do not correlate with antiinflammation. For instance, salicylate was shown to inhibit COX 2 mRNA expression in LPS-elicited alveolar macrophages at 10–30 mg/kg (53), dosing levels that are far below that which are antiinflammatory in mice, leading us to speculate that under some circumstances the diverse effects of salicylate may be secondary to those of aspirin. In an attempt to identify its primary mechanism of action, we show that the induction of NO seems to be exclusive to aspirin. We propose that once ingested, aspirin is exposed to the vasculature where it acetylates COX 2 within the endothelium or circulating leukocytes to trigger 15-epi-lipoxin A4, which, in turn, elicits NO synthesis from both eNOS and iNOS. Ultimately, aspirin-triggered NO mediates the antiinflammatory effects of aspirin in the microcirculation by negatively regulating leukocyte–endothelium interaction. As aspirin is without antiinflammatory effect in NO-deficient animals, we believe this is the primary mode of action of aspirin and that the leukocyte–endothelial interface is its primary site of action. We believe these findings explain how aspirin inhibits acute inflammation. In addition, these data support the development of drugs that controllably elevate plasma NO to levels that inhibit leukocyte attachment to the endothelium but do not overload the system with potentially cytotoxic NO. Given the side effects of aspirin itself, perhaps the usage of NO-inducing agents maybe a further refinement on this theme and an advancement for the treatment of inflammatory arthropathies.
Acknowledgments
The authors would like to thank A.J. Hobbs for iNOS knockout mice and P.L. Huang and I. MacIntyre for nNOS knockout mice. We would also like to acknowledge the laboratory assistance of S. Yona.
D.W. Gilroy is funded by The Joint Research Board of St. Batholomew's Hospital and the Royal London School of Medicine and Dentistry.
Abbreviations used in this paper: ANOVA, analysis of variance; COX, cyclooxygenase; eNOS, constitutive NO synthase; iNOS, inducible NO synthase; L-NAME, NG-nitro-l-arginine methyl ester; nNOS, neuronal NO synthase; NO, nitric oxide; NSAID, nonsteroidal antiinflammatory drug.
References
- 1.Vane, J.R. 1971. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat. New Biol. 231:232–235. [DOI] [PubMed] [Google Scholar]
- 2.Abramson, S.B., and G. Weissmann. 1989. The mechanisms of action of nonsteroidal antiinflammatory drugs. Arthritis Rheum. 32:1–9. [DOI] [PubMed] [Google Scholar]
- 3.Claria, J., and C.N. Serhan. 1995. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc. Natl. Acad. Sci. USA. 92:9475–9479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williams, K.I., and G.A. Higgs. 1988. Eicosanoids and inflammation. J. Pathol. 156:101–110. [DOI] [PubMed] [Google Scholar]
- 5.Gilroy, D.W., P.R. Colville-Nash, D. Willis, J. Chivers, M.J. Paul-Clark, and D.A. Willoughby. 1999. Inducible cyclooxygenase may have anti-inflammatory properties. Nat. Med. 5:698–701. [DOI] [PubMed] [Google Scholar]
- 6.Fiorucci, S., E. Antonelli, E. Distrutti, P. Del Soldato, R.J. Flower, M.J. Clark, A. Morelli, M. Perretti, and L.J. Ignarro. 2002. NCX-1015, a nitric-oxide derivative of prednisolone, enhances regulatory T cells in the lamina propria and protects against 2,4,6-rinitrobenzene sulfonic acid-induced colitis in mice. Proc. Natl. Acad. Sci. USA. 99:15770–15775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verdon, C.P., B.A. Burton, and R.L. Prior. 1995. Sample pretreatment with nitrate reductase and glucose-6-phosphate dehydrogenase quantitatively reduces nitrate while avoiding interference by NADP+ when the Griess reaction is used to assay for nitrite. Anal. Biochem. 224:502–508. [DOI] [PubMed] [Google Scholar]
- 8.Chiang, N., T. Takano, C.B. Clish, N.A. Petasis, H.H. Tai, and C.N. Serhan. 1998. Aspirin-triggered 15-epi-lipoxin A4 (ATL) generation by human leukocytes and murine peritonitis exudates: development of a specific 15-epi-LXA4 ELISA. J. Pharmacol. Exp. Ther. 287:779–790. [PubMed] [Google Scholar]
- 9.Granger, D.N., and P. Kubes. 1994. The microcirculation and inflammation: modulation of leukocyte-endothelial cell adhesion. J. Leukoc. Biol. 55:662–675. [PubMed] [Google Scholar]
- 10.Hickey, M.J., D.N. Granger, and P. Kubes. 2001. Inducible nitric oxide synthase (iNOS) and regulation of leucocyte/endothelial cell interactions: studies in iNOS-deficient mice. Acta Physiol. Scand. 173:119–126. [DOI] [PubMed] [Google Scholar]
- 11.Ialenti, A., A. Ianaro, S. Moncada, and M. Di Rosa. 1992. Modulation of acute inflammation by endogenous nitric oxide. Eur. J. Pharmacol. 211:177–182. [DOI] [PubMed] [Google Scholar]
- 12.Tracey, W.R., M. Nakane, J. Kuk, G. Budzik, V. Klinghofer, R. Harris, and G. Carter. 1995. The nitric oxide synthase inhibitor, L-NG-monomethylarginine, reduces carrageenan-induced pleurisy in the rat. J. Pharmacol. Exp. Ther. 273:1295–1299. [PubMed] [Google Scholar]
- 13.Salvemini, D., Z.Q. Wang, P.S. Wyatt, D.M. Bourdon, M.H. Marino, P.T. Manning, and M.G. Currie. 1996. Nitric oxide: a key mediator in the early and late phase of carrageenan-induced rat paw inflammation. Br. J. Pharmacol. 118:829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Najafipour, H., and W.R. Ferrell. 1993. Nitric oxide modulates sympathetic vasoconstriction and basal blood flow in normal and acutely inflamed rabbit knee joints. Exp. Physiol. 78:615–624. [DOI] [PubMed] [Google Scholar]
- 15.Ridger, V.C., E.R. Pettipher, C.E. Bryant, and S.D. Brain. 1997. Effect of the inducible nitric oxide synthase inhibitors aminoguanidine and L-N6-(1-iminoethyl)lysine on zymosan-induced plasma extravasation in rat skin. J. Immunol. 159:383–390. [PubMed] [Google Scholar]
- 16.Garvey, E.P., J.A. Oplinger, G.J. Tanoury, P.A. Sherman, M. Fowler, S. Marshall, M.F. Harmon, J.E. Paith, and E.S. Furfine. 1994. Potent and selective inhibition of human nitric oxide synthases. Inhibition by non-amino acid isothioureas. J. Biol. Chem. 269:26669–26676. [PubMed] [Google Scholar]
- 17.Hickey, M.J., K.A. Sharkey, and E.G. Sihota. 1997. Inducible nitric oxide synthase (iNOS)-deficient mice have enhanced leukocyte-endothelium interactions in endotoxemia. FASEB J. 11:955–964. [DOI] [PubMed] [Google Scholar]
- 18.Lefer, D.J., S.P. Jones, W.G. Girod, A. Baines, M.B. Grisham, A.S. Cockrell, P.L. Huang, and R. Scalia. 1999. Leukocyte-endothelial cell interactions in nitric oxide synthase-deficient mice. Am. J. Physiol. 276:H1943–H1950. [DOI] [PubMed] [Google Scholar]
- 19.Huang, P.L., Z. Huang, H. Mashimo, K.D. Bloch, M.A. Moskowitz, J.A. Bevan, and M.C. Fishman. 1995. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 377:239–242. [DOI] [PubMed] [Google Scholar]
- 20.Laubach, V.E., E.G. Shesely, O. Smithies, and P.A. Sherman. 1995. Mice lacking inducible nitric oxide synthase are not resistant to lipopolysaccharide-induced death. Proc. Natl. Acad. Sci. USA. 92:10688–10692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang, P.L., T.M. Dawson, D.S. Bredt, S.H. Snyder, and M.C. Fishman. 1993. Targeted disruption of the neuronal nitric oxide synthase gene. Cell. 75:1273–1286. [DOI] [PubMed] [Google Scholar]
- 22.Bratt, J., and H. Gyllenhammar. 1995. The role of nitric oxide in lipoxin A4-induced polymorphonuclear neutrophil-dependent cytotoxicity to human vascular endothelium in vitro. Arthritis Rheum. 38:768–776. [DOI] [PubMed] [Google Scholar]
- 23.Tamaoki, J., E. Tagaya, I. Yamawaki, and K. Konno. 1995. Lipoxin A4 inhibits cholinergic neurotransmission through nitric oxide generation in the rabbit trachea. Eur. J. Pharmacol. 287:233–238. [DOI] [PubMed] [Google Scholar]
- 24.Fiorucci, S., E. Distrutti, A. Mencarelli, A. Morelli, S.A. Laufor, G. Cirino, and J.L. Wallace. 2003. Evidence that 5-lipoxygenase and acetylated cyclooxygenase 2-derived eicosanoids regulate leukocyte-endothelial asherence in response to aspirin. Br. J. Pharmacol. 139:1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O'Neill, G.P., J.A. Mancini, S. Kargman, J. Yergey, M.Y. Kwan, J.P. Falgueyret, M. Abramovitz, B.P. Kennedy, M. Ouellet, W. Cromlish, et al. 1994. Overexpression of human prostaglandin G/H synthase-1 and -2 by recombinant vaccinia virus: inhibition by nonsteroidal anti-inflammatory drugs and biosynthesis of 15-hydroxyeicosatetraenoic acid. Mol. Pharmacol. 45:245–254. [PubMed] [Google Scholar]
- 26.Holtzman, M.J., J. Turk, and L.P. Shornick. 1994. Identification of a pharmacologically distinct prostaglandin H synthase in cultured epithelial cells. J. Biol. Chem. 267:21438–21445. [PubMed] [Google Scholar]
- 27.Takano, T., S. Fiore, J.F. Maddox, H.R. Brady, N.A. Petasis, and C.N. Serhan. 1997. Aspirin-triggered 15-epi-lipoxin A4 (LXA4) and LXA4 stable analogues are potent inhibitors of acute inflammation: evidence for anti-inflammatory receptors. J. Exp. Med. 185:1693–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Connelly, L., M. Palacios-Callender, C. Ameixa, S. Moncada, and A.J. Hobbs. 2001. Biphasic regulation of NF-kB activity underlies the pro- and anti-inflammatory actions of nitric oxide. J. Immunol. 166:3873–3881. [DOI] [PubMed] [Google Scholar]
- 29.Connelly, L., A.T. Jacobs, M. Palacios-Callender, S. Moncada, and A.J. Hobbs. 2003. Macrophage endothelial nitric oxide synthase auto-regulates cellular activation and pro-inflammatory protein expression. J. Biol. Chem. 278:26480–26487. [DOI] [PubMed] [Google Scholar]
- 30.Pérez-Sala, D., E. Cernuda-Morollón, M. Díaz-Cazorla, F. Rodríguez-Pascual, and S. Lamas. 2001. Posttranscriptional regulation of human iNOS by the NO/cGMP pathway. Am. J. Physiol. Renal Physiol. 280:F466–F473. [DOI] [PubMed] [Google Scholar]
- 31.Serhan, C.N., and E. Oliw. 2001. Unorthodox routes to prostanoid formation: new twists in cyclooxygenase-initiated pathways. J. Clin. Invest. 107:1481–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Topper, J.N., J. Cai, D. Falb, and M.A.J. Gimbrone. 1996. Identification of vascular endothelial genes differentially responsive to fluid mechanical stimuli: cyclooxygenase-2, manganese superoxide dismutase, and endothelial cell nitric oxide synthase are selectively up-regulated by steady laminar shear stress. Proc. Natl. Acad. Sci. USA. 93:10417–10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tomlinson, A., I. Appleton, A.R. Moore, D.W. Gilroy, D. Willis, J.A. Mitchell, and D.A. Willoughby. 1994. Cyclo-oxygenase and nitric oxide synthase isoforms in rat carrageenin-induced pleurisy. Br. J. Pharmacol. 113:693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paul-Clark, M.J., D.W. Gilroy, D. Willis, D.A. Willoughby, and A. Tomlinson. 2001. Nitric synthase inhibitors have opposite effects on acute inflammation depending on their route of administration. J. Immunol. 166:1169–1177. [DOI] [PubMed] [Google Scholar]
- 35.Regnault, C., M. Roch-Arveiller, I. Florentin, J.P. Giroud, E. Postaire, and M. Delaforge. 1996. Kinetic evaluation of nitric oxide production in pleural exudate after induction of two inflammatory reactions in the rat. Inflammation. 20:613–622. [DOI] [PubMed] [Google Scholar]
- 36.Filep, J.G., C. Zouki, N.A. Petasis, M. Hachicha, and C.N. Serhan. 2002. Lipoxin A4 and aspirin-triggered 15-epi-lipoxin A4 modulate adhesion molecule expression on human leukocytes in whole blood and inhibit neutrophil-endothelial cell adhesion. Adv. Exp. Med. Biol. 507:223–228. [DOI] [PubMed] [Google Scholar]
- 37.Perretti, M., N. Chiang, M. La, I.M. Fierro, S. Marullo, S.J. Getting, E. Solito, and C.N. Serhan. 2002. Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A4 receptor. Nat. Med. 8:1296–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Colgan, S.P., C.N. Serhan, C.A. Parkos, C. Delp-Archer, and J.L. Madara. 1993. Lipoxin A4 modulates transmigration of human neutrophils across intestinal epithelial monolayers. J. Clin. Invest. 92:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Filep, J.G., C. Zouki, N.A. Petasis, M. Hachicha, and C.N. Serhan. 1999. Anti-inflammatory actions of lipoxin A(4) stable analogs are demonstrable in human whole blood: modulation of leukocyte adhesion molecules and inhibition of neutrophil-endothelial interactions. Blood. 94:4132–4142. [PubMed] [Google Scholar]
- 40.Fiorucci, S., L. Santucci, J.L. Wallace, M. Sardina, M. Romano, P. del Soldato, and A. Morelli. 2003. Interaction of a selective cyclooxygenase-2 inhibitor with aspirin and NO-releasing aspirin in the human gastric mucosa. Proc. Natl. Acad. Sci. USA. 100:10937–10941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fiorucci, S., E. Distrutti, A. Mencarelli, G. Rizzo, A.R. Di Lorenzo, M. Baldoni, P. Del Soldato, A. Morelli, and J. Wallace. 2004. Cooperation between aspirin-triggered lipoxin and nitric oxide (NO) mediates anti-adhesive properties of NCX-4016 (NO-aspirin) on neutrophil-endothelial cell adherence. J. Pharmacol. Exp. Ther. 309:1174–1182. [DOI] [PubMed] [Google Scholar]
- 42.Gauthier, T.W., K.L. Davenpeck, and A.M. Lefer. 1994. Nitric oxide attenuates leukocyte-endothelial interaction via P-selectin in splanchnic ischemia-reperfusion. Am. J. Physiol. 267:G562–G568. [DOI] [PubMed] [Google Scholar]
- 43.Kubes, P., M. Suzuki, and D.N. Granger. 1991. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc. Natl. Acad. Sci. USA. 88:4651–4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brink, C., S.E. Dahlen, J. Drazen, J.F. Evans, D.W. Hay, S. Nicosia, C.N. Serhan, T. Shimizu, and T. Yokomizo. 2003. International union of pharmacology XXXVII. Nomenclature for leukotriene and lipoxin receptors. Pharmacol. Rev. 55:195–227. [DOI] [PubMed] [Google Scholar]
- 45.Wallace, J.L., S.R. Zamuner, W. McKnight, M. Dicay, A. Mencarelli, P. del Soldato, and S. Fiorucci. 2004. Aspirin, but not NO-releasing aspirin (NCX-4016), interacts with selective COX-2 inhibitors to aggravate gastric damage and inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 286:G76–G81. [DOI] [PubMed] [Google Scholar]
- 46.Brezinski, M.E., M.A. Gimbrone, Jr., K.C. Nicolaou, and C.N. Serhan. 1989. Lipoxins stimulate prostacyclin generation by human endothelial cells. FEBS Lett. 245:167–172. [DOI] [PubMed] [Google Scholar]
- 47.Tetsuka, T., D. Daphna-Iken, B.W. Miller, Z. Guan, L.D. Baier, and A.R. Morrison. 1996. Nitric oxide amplifies interleukin 1-induced cyclooxygenase-2 expression in rat mesangial cells. J. Clin. Invest. 97:2051–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Salvemini, D., T.P. Misko, J.L. Masferrer, K. Seibert, M.G. Currie, and P. Needleman. 1993. Nitric oxide activates cyclooxygenase enzymes. Proc. Natl. Acad. Sci. USA. 90:7240–7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamamoto, K., T. Arakawa, N. Ueda, and S. Yamamoto. 1995. Transcriptional roles of nuclear factor kappa B and nuclear factor-interleukin-6 in the tumor necrosis factor alpha-dependent induction of cyclooxygenase-2 in MC3T3-E1 cells. J. Biol. Chem. 270:31315–31320. [DOI] [PubMed] [Google Scholar]
- 50.Kopp, E., and S. Ghosh. 1994. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science. 265:956–959. [DOI] [PubMed] [Google Scholar]
- 51.Cronstein, B.N., M.C. Montesinos, and G. Weissmann. 1999. Sites of action for future therapy: an adenosine-dependent mechanism by which aspirin retains its antiinflammatory activity in cyclooxygenase-2 and NFkappaB knockout mice. Osteoarthritis Cartilage. 7:361–363. [DOI] [PubMed] [Google Scholar]
- 52.Fawcett, T.W., Q. Xu, and N.J. Holbrook. 1997. Potentiation of heat stress-induced hsp70 expression in vivo by aspirin. Cell Stress Chaperones. 2:104–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu, K.K., R. Sanduja, A.L. Tsai, B. Ferhanoglu, and D.S. Loose-Mitchell. 1991. Aspirin inhibits interleukin 1-induced prostaglandin H synthase expression in cultured endothelial cells. Proc. Natl. Acad. Sci. USA. 88:2384–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]