

Abstract
Streptococcus pneumoniae is the most common cause of bacterial meningitis of high mortality and morbidity. Neurological sequelae include paralysis, mental retardation, and learning disorders. In humans, neurons of the hippocampus undergo apoptosis as a result of meningitis. Phosphatidylcholine (PtdCho) is an essential component of mammalian cell membranes and PtdCho deficiency, either due to chemicals or altered nutrition, leads to apoptosis, especially in hippocampal neurons. We show that apoptosis of a variety of brain cells after pneumococcal infection arises from inhibition of PtdCho biosynthesis, the first such activity described for a bacterium. Apoptosis inhibitors did not prevent the bacterial-dependent inhibition of PtdCho biosynthesis. Supplementation with exogenous lyso-phosphatidylcholine prevents cell death and treatment of mice with cytidine diphosphocholine attenuates hippocampal damage during meningitis, even after the onset of infection. We conclude that bacterial inhibition of PtdCho biosynthesis activates an apoptotic cascade that is a causative event in pathogenesis and amenable to therapeutic intervention.
Keywords: meningitis, Streptococcus pneumoniae, lyso-phosphatidylcholine, CDP-choline, pneumolysin
Introduction
Streptococcus pneumoniae remains one of the most common causes of bacterial meningitis and despite effective antimicrobial therapy, continues to be associated with high mortality and morbidity (1). About one third of the survivors, especially children, experience neurological sequelae including deafness, mental retardation, focal neurologic deficits, and cognitive impairment (2–4). Defects in learning and memory result from neuronal apoptosis in the hippocampus (5), a pathological process arising from bacterial induction of two equally important mechanisms, each with a different trigger and different apoptotic pathway. Caspase-dependent apoptosis accounts for about half of the neuronal loss in the hippocampus during meningitis as evidenced by partial rescue in animals treated with the broad spectrum caspase inhibitor z-VAD-fmk (6). Initiation of caspase-dependent neuronal death critically depends on leukocyte-derived products because damage to neurons can be attenuated by anti-CD18 antibody and recapitulated in vitro by inflamed, leukocyte-containing cerebrospinal fluid, but not cell-free fluid. In the second apoptotic process, the bacterium itself induces hippocampal neuronal death directly (7, 8). This process is not associated with the activation of caspases 1–10 and is not inhibited by z-VAD-fmk in vitro or in vivo. Rather, apoptosis is attributable to damage to mitochondria by two pneumococcal toxins, pneumolysin and H2O2, leading to the release of apoptosis-inducing factor (AIF) from mitochondria and large-scale DNA fragmentation. Injection of cells with anti-AIF antiserum or treatment with a calcium chelator blocks AIF release and neuronal cell death (7). In experimental pneumococcal meningitis, pneumolysin colocalizes with apoptotic neurons of the hippocampus, and infection with pneumococci unable to produce pneumolysin and H2O2 causes about half as much damage as infection with wild-type strains. Recent evidence in support of the coexistence of two apoptotic processes during pneumococcal infection comes from studies on dendritic cells (9), which demonstrate bacterial toxin–induced, caspase-independent apoptosis and host response–derived, caspase-dependent apoptosis. Such a multiplicity of apoptotic triggers complicates efforts to deploy apoptosis inhibitors to improve neuronal survival after meningitis, a setting where a single or at least dominant salvage treatment would be highly desirable.
Phosphatidylcholine (PtdCho) is a major component of eukaryotic cell membranes. Its biosynthesis occurs mainly via the cytidine diphosphocholine (CDP-choline) pathway and involves three enzymatic reactions (Fig. 1; references 10 and 11). The rate-limiting step in this pathway is the second step, the CTP:phosphocholine cytidylyltransferase (CCT)-catalyzed reaction, and the activity of this enzyme is highly regulated (12). Induction of apoptosis by inhibition of the enzymes CCT or choline phosphotransferase (CPT) is a mechanism of action of the anticancer drugs ET-18-OCH3 (13), HexPC (14), camptothecin and etoposide (15), the cytotoxic agents farnesol and chelerythrine (15), and the short-chain ceramides (16, 17). Choline deprivation, leading to PtdCho deficiency, is also known to trigger apoptosis in neurons (18). Here we report that the apoptosis of neurons and microglia triggered during pneumococcal infection appears to involve the inhibition of PtdCho biosynthesis as a determining factor, the first example of this type of toxicity induced by a bacterium.
Figure 1.

The Kennedy pathway. The CDP-choline pathway is catalyzed by choline kinase (CK), CTP:phosphocholine cytidylyltransferase (CCT), and CDP-choline:1,2-diacylglycerol cholinephosphotransferase (CPT; reference 10).
Materials and Methods
Bacterial Cell Culture.
The following isogenic strains of pneumococci were used: D39 capsular type 2 and R6, its nonencapsulated derivative (provided by A. Tomasz, Rockefeller University, New York, NY), the pneumolysin− mutant plnA − (provided by D. Briles, University of Alabama, Birmingham, AL), the pyruvate oxidase mutant spxB − deficient in H2O2 production (19), the plnA −/spxB− double mutant (6), and the adherence-defective cbpA − mutant (20). T4 capsular serotype 4 and its nonencapsulated derivative T4R were also tested. Bacteria were grown overnight in a casein hydrolysate medium supplemented with yeast extract and appropriate antibiotics for maintaining mutant selection. The bacterial inoculum (107 bacteria/ml) was adjusted photometrically (OD, 620 nm), centrifuged, washed with PBS, and then resuspended in cell culture media. For some experiments, bacteria were replaced by 1 μg/ml recombinant pneumolysin (provided by T. Mitchell, University of Glasgow, Glasgow, UK) or 1 mM hydrogen peroxide (Sigma-Aldrich).
Cell Culture.
A human microglial cell line (HMC; provided by C.A. Colton, Georgetown University, Washington, DC) that expresses most markers common to primary human microglia was cultured as described previously (7). Primary human microglia, primary rat hippocampal neurons (21), A549 human lung epithelial cell line (American Type Culture Collection [ATCC]), RBEC6 rat brain endothelial cell line (22), or PC12 rat pheochromocytoma cell line (ATCC) were seeded in 60-mm culture dishes (4–5 × 105 cells) and cultured in DMEM medium containing 5% fetal calf serum at 37°C in a humidified atmosphere of 5% CO2. Cells were infected 48 h later at a confluency of >80% with different strains of S. pneumoniae for 3–4 h as indicated. Cell number and viability were assessed by counting on a hemocytometer, trypan blue exclusion, lactate dehydrogenase (LDH) release, and staining by acridine orange and ethidium bromide.
Metabolic Labeling.
12 h before infection, 4–5 × 105 cells were cultured in choline-free media. At the time of infection, [methyl- 3H]choline chloride (1 μCi/ml, 80 Ci/mmol; American Radiolabel Chemicals Inc.) and unlabeled choline (4 μM final concentration/dish) were added without change of media during the 4 h of infection. After the infection, cells were scraped, washed twice with ice-cold PBS, and centrifuged, and the cell pellet was stored at −20°C. Extraction of lipids was performed according to Bligh and Dyer (23). Incorporation of [methyl- 3H]choline into PtdCho was measured by quantification of total radioactivity in the lipid extract using scintillation counting because >95% of the radiolabel was identified as PtdCho. Separation of the water soluble [methyl-3H]choline metabolites was performed by spotting aliquots of the upper phase onto preadsorbent silica gel thin layer chromatography plates, which were developed in 95% ethanol/2% NH4OH (1:1, vol/vol). Identification of radiolabeled choline, phosphocholine, and CDP-choline was made by comigration with authentic standards. After chromatography, the samples were scraped from the plates and dissolved in water, and radioactivity was quantified by scintillation counting.
Apoptosis Assays.
Morphological changes were documented by light microscopy and uptake of acridine orange and ethidium bromide. Apoptotic cells were detected by flow cytometric analysis (FACSCalibur™; Becton Dickinson) of cells stained for phosphatidylserine exposed on the plasma membrane. Cells were treated with fluorescein-conjugated annexin V and propidium iodide according to the manufacturer's instructions (Becton Dickinson), and then analyzed by FACS® using a 488-nm argon ion laser. For analyzing mitochondrial transmembrane potential (ΔΨm), cells were incubated with 5,5′,6,6′-tetrachloro-1′1,3,3′-tetrahylbenzimidazolcarbocyanine-iodide (JC-1) according to the manufacturer's instructions (Molecular Probes). Emitted fluorescence was measured by FACS® analysis.
Antibodies to activated caspase 3 and AIF (BD Biosciences), cytochrome C (Santa Cruz Biotechnology, Inc.), and Lamin B (Oncogene Research Products) were used in Western blot analyses to characterize apoptotic cells. 0.5–107 cells were harvested at 80% confluence and extraction of the nucleus was performed as described elsewhere (24).
For inhibition of apoptosis, cells were treated 30 min before infection with either 80 μM l-α-lysophosphatidylcholine from egg (Avanti Polar Lipids, Inc.), 60 μM caspase inhibitor Z-Val_Ala_ASP(Ome)-FMK (ZVAD-fmk; Enzyme Systems Products), 25 μM calpain inhibitor 1 N-acetyl-Leu-Leu-Nle-CHO (ALLN; Enzyme Systems Products), 50 μM fumonisin B1 (Sigma-Aldrich), or 5 μM Ca2-chelator BAPTA-AM (Molecular Probes). For some experiments, 100 μM citicoline (BIOMOL Research Laboratories, Inc.) was applied to the cells for 24 h before infection.
Inhibition of Apoptosis In Vivo.
4–5-wk-old C57BL/6 mice (The Jackson Laboratory) were anesthetized and infected via lumbar puncture with either saline (n = 6) or 104 strain D39 pneumococci (n = 13) in 20 μl as described previously (25). Six mice received 500 mg/kg citicoline intraperitoneally 24 h before infection and via lumbar puncture at the time of infection. A second set of five mice per group received citicoline via lumbar puncture only at 6 h after infection. Mice receiving citicoline alone showed no hippocampal apoptosis and citicoline did not affect bacterial growth. Mice were killed at 24 h and perfused transcardially with 3% paraformaldehyde and 0.1% glutaraldehyde (Sigma-Aldrich) in 0.1 M cacodylate buffer (Ted Pella, Inc.), pH 7.4, adjusted with sucrose to 300 mOsm. Brains were removed, postfixed overnight in 10% formalin, embedded in paraffin, sectioned (5 μm), deparaffinized, rehydrated, and stained. Hematoxylin and eosin stain was used to visualize and measure the area of dentate gyrus of the hippocampus using the NIH Scion Image Beta 4.02 software. To quantitate apoptosis, TUNEL staining (five sections per mouse) was performed (Apop Tag Plus Peroxidase In Situ Apoptosis Detection Kit; Serologicals Corp.). All experiments were performed in compliance with National Institutes of Health (NIH) and institutional guidelines.
Statistical Analysis.
Statistical analysis was performed using SigmaStat software version 2.03. Data are presented as mean ± SD or ± SEM as indicated in the figure legends. Differences between groups were evaluated using the Student's t test or the Mann-Whitney rank sum test unless otherwise stated. P < 0.05 was considered an indication of statistical significance.
Results
Infection with S. pneumoniae Inhibits PtdCho Biosynthesis.
Microglia are an effective model cell type to study brain cell injury because they proliferate well in vitro and their metabolism is tightly coupled to neuronal activity (26). Exposure of a human microglial cell line (7) to S. pneumoniae D39 or its unencapsulated derivative R6 for 4 h (multiplicity of infection, 30 bacteria/human cell) induced apoptosis as measured by markers of cell death such as translocation of phosphatidylserine and loss of mitochondrial membrane potential (Fig. 2 A), cleavage of lamin B, and release of cytochrome C and AIF into the cytoplasm (not depicted).
Figure 2.
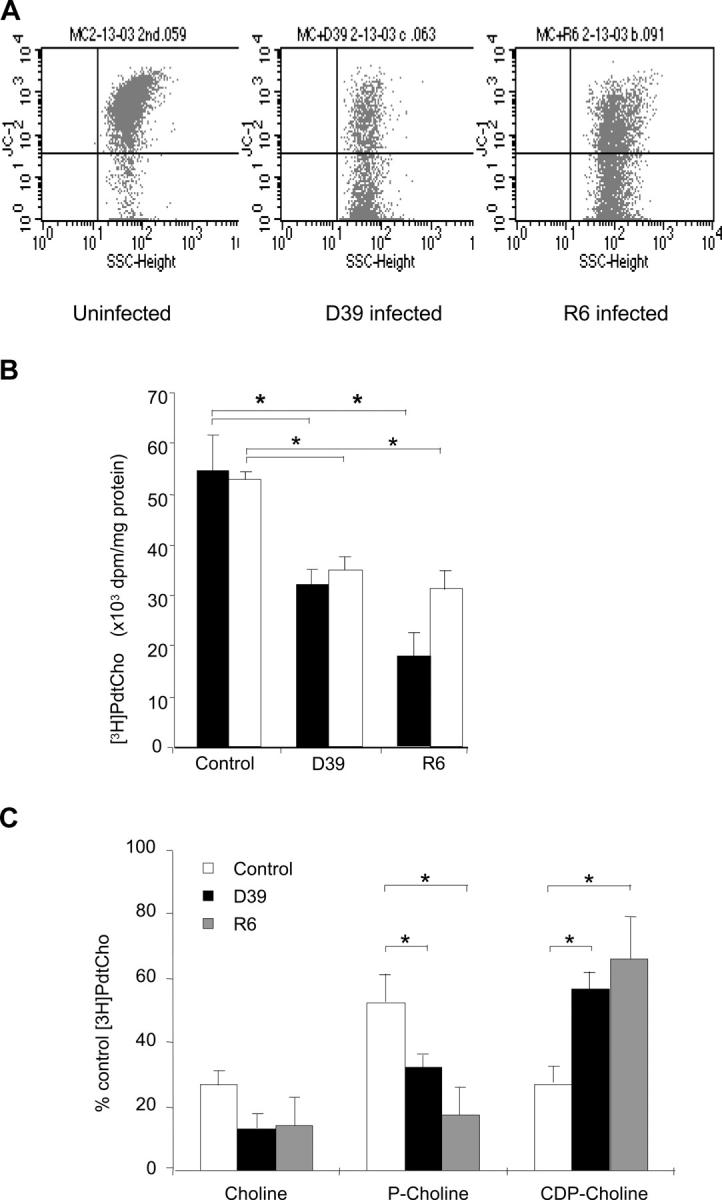
Inhibition of PtdCho biosynthesis by S. pneumoniae induces apoptosis. (A) FACS® analysis of mitochondrial membrane potential of control (left) and infected (middle and right) microglia. Dot plots show side scatter signals (x axis) versus fluorescence intensity (y axis) for microglia cells. Shift of fluorescence of JC-1 from 590 (FL2) to 530 nm (FL1) indicates membrane depolarization. Analysis representative of six independent experiments. (B) Rate of PtdCho biosynthesis. [methyl-3H]choline–labeled human primary microglia (black bars) or cultured cells (white bars) were infected with 2 × 107 cells of strain D39 or R6 pneumococci for 4 h. PtdCho synthesis was measured by scintillation counting and values were normalized to the protein concentration of the sample. Values are mean ± SE of 10 duplicate experiments. *, P < 0.05. (C) Distribution of [methyl-3H]choline incorporation into water-soluble choline intermediates in microglia. *, P < 0.05. Total cpm incorporated were 292,536 ± 68,812. Bacteria alone incorporated 13,410 ± 4,797 (4.5%).
To determine if infection with S. pneumoniae influenced PtdCho biosynthesis, the cell line and primary microglia were labeled with [methyl- 3H]choline for 4 h and challenged with S. pneumoniae D39 or R6. Chromatographic separation of the phospholipids showed that the majority of the label was incorporated into PtdCho (unpublished data). The ratio of phosphocholine to CDP-choline was 5:1, in agreement with previous studies in neurons after short-term labeling (27). Significant inhibition of [methyl- 3H]choline incorporation into PtdCho occurred upon infection with D39 (41.2 ± 6% inhibition) or R6 (67.4 ± 8% inhibition) as compared with the uninfected cells (Fig. 2 B). S. pneumoniae T4 and its unencapsulated derivative T4R yielded similar results of 56.4 ± 9 and 55.4 ± 8% inhibition, respectively. This effect was not the result of depletion of nutritional choline because [methyl- 3H]choline was used in 40-fold excess over that which is needed by cells and pneumococci together. Furthermore, bacteria incorporated only 2.4% of the total label into CDP-choline and this amount was rendered negligible in additional analyses by washing away the bacteria from adherent microglia before quantitation of [3H]choline incorporation into host cellular products.
Exposure of other cell types to R6, including a neuronal-type cell line (PC12), an epithelial cell line (A-549), and a brain endothelial cell line (RBE6), also showed an inhibition of incorporation of label into PtdCho, but to a lesser degree than for microglia (22.2 ± 4.8, 29.6 ± 3.3, and 42 ± 11.8% inhibition, respectively). To determine which step of the CDP-choline pathway was blocked by pneumococci, the distribution of water-soluble choline intermediates was assessed by thin layer chromatography and quantified by scintillation counting. Pneumococcal-infected cells accumulated CDP-choline (Fig. 2 C), suggesting that the CPT, the final step in the pathway, was inhibited.
Lyso-Phosphatidylcholine (lyso-PC) Prevents Pneumococcal Apoptosis in Microglia and Neurons.
To determine whether the inhibition of PtdCho synthesis was causative or secondary to induction of a known apoptotic pathway, lyso-PC was added to the infected cells to bypass the defect in the CDP-choline pathway (28) by direct acylation to PtdCho (29). Bacterial growth was unaffected by treatment with lyso-PC under tissue culture conditions and concentrations of lyso-PC up to 80 μM showed no toxicity to human cells (Fig. 3 B). The addition of 80 μM lyso-PC to the culture medium before infection significantly prevented apoptosis as compared with the untreated infected cells (Fig. 3 A). Lower doses were less effective. AIF was released from mitochondria in pneumococcal-infected cells and this effect was blocked by lyso-PC or the calcium chelator, BAPTA-AM (Fig. 3 B).
Figure 3.

Rescue of microglia from apoptosis by lyso-PC. (A) LDH release and trypan blue uptake of microglia incubated with different concentrations of lyso-PC. Microglia were exposed to different concentrations of lyso-PC for 4 h or to 0.5% Triton X-100 for 30 min and release of LDH (white bars; reference 49) and uptake of trypan blue (gray bars) were measured. Values are mean ± SEM of 12 experiments (LDH assay) and 6 experiments (trypan blue assay), and the mean of the cells treated with 80 μM lyso-PC was compared with the cells treated with 120 and 160 μM lyso-PC, or 0.5% Triton X-100, respectively. * and #, P < 0.05. (B) 5 × 105–106 microglia were exposed to 80 μM lyso-PC 1 h before infection with strain D39 or R6 for 4 h. Cells were considered viable according to their scatter characteristics and their lack of annexin V and/or annexin V/propidium iodide staining. Values are mean ± SE of six duplicate experiments counting 10,000 events. *, P < 0.05 with versus without treatment. Inset: Morphology by light microscopy (magnification, 400). (C) Immunoblot of AIF in mitochondrial and cytosolic cell fractions. Microglia were either uninfected (lane 1) or infected with R6 ± pretreatment with 80 μM Lyso-PC (lanes 2 and 3) or 5 μM BAPTA (lane 4).
Role of Bacterial Factors in Inhibition of PtdCho Biosynthesis.
Inhibition of PtdCho biosynthesis was lost upon challenge of host cells with heat-killed or antibiotic-treated bacteria (Fig. 4 A). Bacterial adherence to the target cells mediated by the major adhesin CbpA was not required (Fig. 4 A). Exposure to a filtered pneumococcal lysate from 108 bacteria/ml strongly inhibited PtdCho incorporation, suggesting a product of active bacterial metabolism. This was supported by testing pure H2O2 and pneumolysin, a pore-forming toxin, both of which are secreted by pneumococci and are known to induce apoptosis in host cells (7, 8). Attenuation of H2O2 production by mutation of pyruvate oxidase, spxB, or deletion of pneumolysin, plnA, significantly reduced the ability of pneumococci to cause cell death and inhibit host PtdCho synthesis (Fig. 4 B). Infection with plnA, spxB, or the spxB/plnA double mutant strains caused less inhibition of PtdCho synthesis, with [methyl-3H]choline incorporation into PtdCho increasing to 57.8, 77.9, and 92.4%, respectively, compared with uninfected control cells (Fig. 4, A and B). Addition of either H2O2 or pneumolysin in concentrations comparable to 108 bacteria to the host cell culture significantly inhibited PtdCho synthesis (Fig. 4 A) and induced apoptosis. The precise mechanism of inhibition of PtdCho biosynthesis by pneumolysin and H2O2 is not known. Cells treated for 1 h with lyso-PC before exposure to either toxin were protected from apoptosis (75 ± 17% apoptotic cells with pneumolysin alone vs. 32 ± 5% pneumolysin plus lyso-PC; 95 ± 3% apoptotic cells with H2O2 alone vs. 40 ± 4% H2O2 plus lyso-PC; baseline 4 ± 1% with no treatment or 5 ± 2% lyso-PC alone).
Figure 4.

Effect of loss of function mutations in S. pneumoniae on ability to induce apoptosis and inhibit PtdCho synthesis. Microglia were exposed for 4 h to 107 CFU/ml of each bacterial mutant indicated or to 1 μg purified pneumolysin or 1 mM hydrogen peroxide. [methyl-3H]choline incorporation was quantitated by scintillation counting. Values are mean ± SE of six experiments and were compared with mean of control, untreated cells (50,764 cpm/mg protein = 100%). Apoptosis was determined as described in Fig 2 A. *, P < 0.05.
Role of Host-derived Factors in Inhibition of PtdCho Biosynthesis.
Several pathways have been suggested to mediate apoptosis in response to inhibition of PtdCho biosynthesis. To rule out accumulation of ceramides as an underlying mechanism (14), we treated microglia with fumonisin B1, a natural inhibitor of ceramide synthase that blocks ceramide-induced apoptosis (Fig. 5; reference 30). Pretreating the cells with fumonisin B1 failed to restore PtdCho biosynthesis (34.8 ± 11.8% inhibition) or prevent apoptosis in pneumococcal-challenged cells. Caspase activation is also known to occur in apoptosis induced by choline starvation (31), farnesol (32), and in pneumococcal-induced apoptosis of hippocampal neurons (6). Yet, inhibition of caspase activity by ZVAD, which partially decreased apoptosis (from 55 ± 3 to 42 ± 1%), did not prevent inhibition of PtdCho biosynthesis (55.3 ± 8.3% inhibition). Release of mitochondrial AIF accompanied by increased intracellular Ca2+ represents another pathway of pneumococcal-induced apoptosis (8), but incorporation of label into PtdCho was still inhibited in infected cells treated with BAPTA-AM (53.6 ± 2.1% inhibition). Ca2+-dependent activation of calpains can cause proteolytic inactivation of the CCT (33). However, exposure to the calpain inhibitor ALLN before the pneumococcal infection did not rescue PtdCho biosynthesis (62.1 ± 4.8% inhibition). These data place the inhibition of PtdCho synthesis independent of the indicated inciting elements of the apoptotic cascade.
Figure 5.

Effect of inhibition of apoptotic pathways on PtdCho synthesis inhibition by S. pneumoniae. Microglia were treated with the indicated inhibitor and then exposed to bacteria for 4 h. [methyl-3H]choline incorporation was quantified by scintillation counting and apoptosis was determined by annexin V labeling. Values were compared with control, untreated cells (29,221 dpm/mg protein = 100%). ZVAD, Z-Val_Ala_Asp(Ome)-Fmk; ALLN, N-acetyl-Leu-Leu-NLE-CHO; BAPTA, Ca2- chelator. *, P < 0.05 comparing inhibition of labeling in infected cells plus inhibitor to control cells plus inhibitor. There is no significant difference between values shown and infected cells without inhibitor.
Citicoline Treatment Protects from Pneumococcal-induced Apoptosis in the Hippocampus In Vivo.
CDP-choline provides protection against neuronal death caused by decreased phospholipid biosynthesis after cerebral ischemia (34, 35). Administration of CDP-choline increases cytidine (or uridine) and choline levels in rodent and human tissues (36, 37), rescues CA1 hippocampal neurons from ischemic death (38), and increases PtdCho formation in neuronal tissues in vitro and in vivo (39). Our in vitro results suggested that application of excess CDP-choline might also rescue PtdCho production in cells dying during pneumococcal meningitis by overcoming the partial inhibition of CPT. Treatment of microglia or primary neurons with 100 μM citicoline (CDP-choline) 24 h before exposure to strain R6 rescued the cells from apoptosis. Although media alone or with citicoline caused little cell death (<10%), microglia exposed to R6 for 2 h showed 52 ± 15% apoptosis and this value was reduced to 16 ± 3% in the presence of citicoline (n = 4; P < 0.05). Similarly for primary neurons, exposure to R6 for 3 h caused 40 ± 12% apoptosis and this value was reduced to 28 ± 16% in the presence of citicoline (control baseline 22 ± 7% apoptosis; n = 4–8; P < 0.05).
Citicoline prevention of pneumococcal-induced apoptosis was extended to the in vivo setting using a mouse model of meningitis (21). Animals were treated with citicoline and 24 h later, they were treated again and challenged with strain D39 pneumococci. The amount of apoptosis in the hippocampus was quantitated 24 h after infection. Animals receiving citicoline had a dramatic reduction in neuronal cell death (Fig. 6) as compared with untreated, infected controls (P < 0.05). The damage was reduced to levels found in uninfected controls. This degree of protection has not been achieved by other apoptosis inhibitors directed at either the caspase-dependent (6) or -independent pathways of pneumococcal-induced apoptosis (7). To determine if apoptosis could be prevented after the onset of infection, as would be applicable to the clinical situation, mice were challenged with bacteria and then treated with citicoline 6 h later. Brain histopathology was examined at 24 h. Although all infected mice harbored 3–9 × 107 bacteria/ml cerebrospinal fluid, cells in the hippocampus of mice receiving citicoline were protected against apoptosis to levels approximating uninfected controls (apoptotic cells/mm2 dentate gyrus: saline controls: 37 ± 18; infected: 71 ± 29; infected plus citicoline: 39 ± 8; P < 0.04).
Figure 6.

Protection from pneumococcal-induced apoptosis in the hippocampus in vivo. Three groups of six mice were injected via lumbar puncture with saline or pneumococci as indicated. One group also received citicoline intraperitoneally 24 h before infection and via lumbar puncture at the time of infection. Mean bacterial number in cerebrospinal fluid for infected animals was 107 bacteria/ml for pneumococci alone and 3 × 107 bacterial/ml for pneumococci with citicoline. Histopathology of the dentate gyrus: (A) normal control, (B) infected with pneumococcus, and (C) infected with pneumococcus and treated with citicoline. (D) Number of apoptotic neurons per mm2 in the hippocampus ascertained at 24 h of infection. The data were statistically analyzed by a One Way Analysis of Variance (P < 0.05) followed by a Student-Newman-Keuls test. #, the difference between controls and meningitis (P = 0.01). *, the difference between untreated meningitis and citicoline treatment (P = 0.02).
Discussion
PtdCho is a major component of eukaryotic but not prokaryotic membranes. We demonstrate for the first time that a bacterium can inhibit the de novo synthesis of PtdCho in human cells. Significantly, this inhibition triggers apoptosis. Although inhibition of PtdCho biosynthesis initiates apoptosis in cells exposed to xenobiotics (13–15, 17, 28), in mutant cells with genetic CCT dysfunction (40), or in nutritional choline deprivation (18, 31), the response of human cells to pneumococcus is the first case where this mechanism plays a critical role in pathogenesis.
In the brain, PtdCho has an additional role as a precursor for the biosynthesis of the important neurotransmitter acetylcholine. It is unlikely that acetylcholine deprivation accounts for the observed pneumococcal-induced neuronal cell death because a low level of acetylcholine is not known to be a death signal and PtdCho serves as a reservoir of choline for acetylcholine biosynthesis under conditions of nutritional choline deprivation (41). Furthermore, inhibition of PtdCho biosynthesis and apoptosis were observed in nonneuronal cells that do not synthesize or secrete acetylcholine.
The membranes of most bacteria are comprised of phosphatidylethanolamine and phosphatidylglycerol rather than the PtdCho that dominates eukaryotic cell membranes (42). Thus, although respiratory pathogens add choline to a variety of constituents on their surfaces, they do not incorporate choline into PtdCho (43). Rather, the pneumococcal licD genes (44, 45), the bacterial equivalents of the mammalian CPT, transfer CDP-choline to teichoic acid. There is no homology between the CPT enzymes of mammals and of S. pneumoniae, thus explaining the selective inhibition of the mammalian enzyme by the bacterial toxin(s). The apoptosis detected in pneumococcal-infected human cells was not caused by consumption of PtdCho or precursors by pneumococci with resultant starvation of human cells because a pneumococcal lysate and purified pneumococcal toxins also induced inhibition of PtdCho biosynthesis and apoptosis.
Interference with caspase activation, calpain activation, or AIF translocation did not restore synthesis of PtdCho in pneumococcal-infected cells. This suggests that inhibition of PtdCho synthesis in this setting is triggered independently of the well-known biochemical and cellular events associated with apoptosis. Purified pneumolysin and H2O2 strongly inhibited PtdCho synthesis. Pneumococcus is unique among meningeal pathogens for its lack of catalase and thus, it produces substantial amounts of H2O2. The mode of H2O2 inhibition of PtdCho synthesis needs to be further elucidated to determine whether lipid peroxidation or inhibition of CPT by intracellular acidification (15) occurs. Pneumolysin is well-known to bind to cholesterol in membranes, oligomerize into ring-shaped assemblies, and form pores within the lipid bilayer (46). One can hypothesize that pneumolysin-induced destabilization of the cell membrane inhibits the enzymes CPT or CCT because their substrate binding and enzymatic activities are dependent on the biophysical properties of the bilayer (12).
Exogenous lyso-PC is taken up into cells in a linear fashion for 24 h and converted to PtdCho almost immediately after cell association (13). 70–80% of the exogenous lyso-PC in the medium is incorporated into cells. The newly formed PtdCho is converted to phosphatidylethanolamine until a final equilibrium ratio of ∼50% PtdCho and 50% phosphatidylethanolamine is reached. A significant portion of the newly formed PtdCho is degraded to either glycerophosphocholine or phosphocholine. As the amount of exogenous lyso-PC increases, the proportion of glycerophosphocholine also increases in normal cultured cells and is released to the medium. Lyso-PC must be added at 24-h intervals to replenish the medium (27), an effect consistent with repeated dosing to achieve protection in this study.
PtdCho biosynthesis plays a crucial role in proliferating and resting neuronal cells of the hippocampus (31), a fact that may explain, in part, the very high sensitivity of this region of the brain to injury during meningitis. Treatment with excess of the pathway intermediate CDP-choline (citicoline) has shown beneficial effects in a number of central nervous system injury models and pathological conditions of the brain (35, 38, 39), although the results of human clinical trials in stroke remain mixed (34). Our data show that neuronal apoptosis after experimental S. pneumoniae meningitis is amenable to medical intervention using CDP-choline and/or its metabolic derivatives. This effect was seen even when citicoline was administered after the animals were symptomatic. The strength of the protective effect is significantly greater than that seen with apoptosis inhibitors and argues for further study of citicoline to define its clinical potential in meningitis. It is likely that the mechanism of the beneficial effect of CDP-choline is to increase the supply of pathway intermediates and thereby overcome the bacterial-induced partial inhibition of CPT, the final, nonrate limiting enzyme in the pathway. However, in theory, the effect might also be due to reduced phospholipid destruction through inhibition of phospholipase activation (47) or arachidonic acid release (39). It is also possible that clinical benefit might be aided by replenishing acetylcholine in neurons, although this pool is very small compared with membrane PtdCho (48).
Pneumococci are known to induce caspase- and AIF-dependent apoptosis. Yet the process studied here was not reversible with inhibitors of known apoptotic mediators, but rather complete rescue from apoptosis was achieved by the addition of lyso-PC or CDP-choline. These findings would be consistent with a final common pathway of damage at the membrane level regardless of the original stimulus and thus, a dominant salvage effect of agents preserving phospholipid integration into membranes. The field of meningitis research has long sought an agent capable of protecting neurons from damage during antibiotic treatment of disease. This goal appeared to become less realistic with the discovery of the multiple apoptotic pathways triggered by pneumococci in neurons. The strong response to lyso-PC or CDP-choline justifies optimism that despite a diversity of proapoptotic signals during infection, significant neuronal rescue might be achieved by a single therapeutic intervention for meningitis in vivo.
Acknowledgments
We are grateful to G. Gao and J. Wang for assisting in Western blot analyses and in the radiolabeling assays. We thank Dr. M. Bastholm Bille for performing experiments with inhibition of calpains and D. Freyer for culturing primary neurons.
This work was supported by NIH (grant AI27913 to E.I. Tuomanen and grant GM45737 to S. Jackowski), the Cancer Center CORE grant CA21765, and the American Lebanese and Syrian Associated Charities.
J. Zweigner's present address is Institute for Microbiology and Hygiene, Charité-Universitaetsmedizin Berlin, Schumannstrasse 20/21, D-10117 Berlin, Germany.
Abbreviations used in this paper: AIF, apoptosis-inducing factor; ALLN, N-acetyl-Leu-Leu-Nle-CHO; CCT, CTP:phosphocholine cytidylyltransferase; CDP-choline, cytidine diphosphocholine; CPT, choline phosphotransferase; LDH, lactate dehydrogenase; lyso-PC, lyso-phosphatidylcholine; PtdCho, phosphatidylcholine.
References
- 1.Schuchat, A., K. Robinson, J.D. Wenger, L.H. Harrison, M. Farley, A.L. Reingold, L. Lefkowitz, and B.A. Perkins. 1997. Bacterial meningitis in the United States in 1995. Active Surveillance Team. N. Engl. J. Med. 337:970–976. [DOI] [PubMed] [Google Scholar]
- 2.Bohr, V., O.B. Paulson, and N. Rasmussen. 1984. Pneumococcal meningitis. Late neurologic sequelae and features of prognostic impact. Arch. Neurol. 41:1045–1049. [DOI] [PubMed] [Google Scholar]
- 3.Arditi, M., E.O. Mason, Jr., J.S. Bradley, T.Q. Tan, W.J. Barson, G.E. Schutze, E.R. Wald, L.B. Givner, K.S. Kim, R. Yogev, et al. 1998. Three-year multicenter surveillance of pneumococcal meningitis in children: clinical characteristics, and outcome related to penicillin susceptibility and dexamethasone use. Pediatrics. 102:1087–1097. [DOI] [PubMed] [Google Scholar]
- 4.van de Beek, D., B. Schmand, J. de Gans, M. Weisfelt, H. Vaessen, J. Dankert, and M. Vermeulen. 2002. Cognitive impairment in adults with good recovery after bacterial meningitis. J. Infect. Dis. 186:1047–1052. [DOI] [PubMed] [Google Scholar]
- 5.Nau, R., A. Soto, and W. Bruck. 1999. Apoptosis of neurons in the dentate gyrus in humans suffering from bacterial meningitis. J. Neuropathol. Exp. Neurol. 58:265–274. [DOI] [PubMed] [Google Scholar]
- 6.Braun, J.S., R. Novak, K.H. Herzog, S.M. Bodner, J.L. Cleveland, and E.I. Tuomanen. 1999. Neuroprotection by a caspase inhibitor in acute bacterial meningitis. Nat. Med. 5:298–302. [DOI] [PubMed] [Google Scholar]
- 7.Braun, J.S., R. Novak, P.J. Murray, C.M. Eischen, S.A. Susin, G. Kroemer, A. Halle, J.R. Weber, E.I. Tuomanen, and J.L. Cleveland. 2001. Apoptosis-inducing factor mediates microglial and neuronal apoptosis caused by pneumococcus. J. Infect. Dis. 184:1300–1309. [DOI] [PubMed] [Google Scholar]
- 8.Braun, J.S., J.E. Sublett, D. Freyer, T.J. Mitchell, J.L. Cleveland, E.I. Tuomanen, and J.R. Weber. 2002. Pneumococcal pneumolysin and H(2)O(2) mediate brain cell apoptosis during meningitis. J. Clin. Invest. 109:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Colino, J., and C.M. Snapper. 2003. Two distinct mechanisms for induction of dendritic cell apoptosis in response to intact Streptococcus pneumoniae. J. Immunol. 171:2354–2365. [DOI] [PubMed] [Google Scholar]
- 10.Kennedy, E.P., and S.B. Weiss. 1956. The function of cytidine coenzyme in the biosynthesis of phospholipids. J. Biol. Chem. 222:193–214. [PubMed] [Google Scholar]
- 11.Lykidis, A., J. Wang, M.A. Karim, and S. Jackowski. 2001. Overexpression of a mammalian ethanolamine-specific kinase accelerates the CDP-ethanolamine pathway. J. Biol. Chem. 276:2174–2179. [DOI] [PubMed] [Google Scholar]
- 12.Attard, G.S., R.H. Templer, W.S. Smith, A.N. Hunt, and S. Jackowski. 2000. Modulation of CTP:phosphocholine cytidylyltransferase by membrane curvature elastic stress. Proc. Natl. Acad. Sci. USA. 97:9032–9036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baburina, I., and S. Jackowski. 1998. Apoptosis triggered by 1-O-octadecyl-2-O-methyl-rac-glycero-3-phosphocholine is prevented by increased expression of CTP:phosphocholine cytidylyltransferase. J. Biol. Chem. 273:2169–2173. [DOI] [PubMed] [Google Scholar]
- 14.Wieder, T., C.E. Orfanos, and C.C. Geilen. 1998. Induction of ceramide-mediated apoptosis by the anticancer phospholipid analog, hexadecylphosphocholine. J. Biol. Chem. 273:11025–11031. [DOI] [PubMed] [Google Scholar]
- 15.Anthony, M.L., M. Zhao, and K.M. Brindle. 1999. Inhibition of phosphatidylcholine biosynthesis following induction of apoptosis in HL-60 cells. J. Biol. Chem. 274:19686–19692. [DOI] [PubMed] [Google Scholar]
- 16.Ramos, B., G.M. Salido, M.L. Campo, and E. Claro. 2000. Inhibition of phosphatidylcholine synthesis precedes apoptosis induced by C2-ceramide: protection by exogenous phosphatidylcholine. Neuroreport. 11:3103–3108. [DOI] [PubMed] [Google Scholar]
- 17.Ramos, B., M. El Mouedden, E. Claro, and S. Jackowski. 2002. Inhibition of CTP:phosphocholine cytidylyltransferase by C(2)-ceramide and its relationship to apoptosis. Mol. Pharmacol. 62:1068–1075. [DOI] [PubMed] [Google Scholar]
- 18.Holmes-McNary, M.Q., A.S. Baldwin, Jr., and S.H. Zeisel. 2001. Opposing regulation of choline deficiency-induced apoptosis by p53 and nuclear factor kappaB. J. Biol. Chem. 276:41197–41204. [DOI] [PubMed] [Google Scholar]
- 19.Spellerberg, B., D.R. Cundell, J. Sandros, B.J. Pearce, I. Idanpaan-Heikkila, C. Rosenow, and H.R. Masure. 1996. Pyruvate oxidase, as a determinant of virulence in Streptococcus pneumoniae. Mol. Microbiol. 19:803–813. [DOI] [PubMed] [Google Scholar]
- 20.Rosenow, C., P. Ryan, J.N. Weiser, S. Johnson, P. Fontan, A. Ortqvist, and H.R. Masure. 1997. Contribution of novel choline-binding proteins to adherence, colonization and immunogenicity of Streptococcus pneumoniae. Mol. Microbiol. 25:819–829. [DOI] [PubMed] [Google Scholar]
- 21.Hoffmann, O., N. Keilwerth, M.B. Bille, U. Reuter, K. Angstwurm, R.R. Schumann, U. Dirnagel, and J.R. Weber. 2002. Triptans reduce the inflammatory response in bacterial meningitis. J. Cereb. Blood Flow Metab. 2002:988–996. [DOI] [PubMed] [Google Scholar]
- 22.Blasig, I.E., H. Giese, M.L. Schroeter, A. Sporbert, D.I. Utepbergenov, I.B. Buchwalow, K. Neubert, G. Schonfelder, D. Freyer, and I. Schimke. 2001. *NO and oxyradical metabolism in new cell lines of rat brain capillary endothelial cells forming the blood-brain barrier. Microvasc. Res. 62:114–127. [DOI] [PubMed] [Google Scholar]
- 23.Bligh, E.G., and W.J. Dyer. 1959. A rapid method of total lipid extraction and purification. Can. J. Med. Sci. 37:911–917. [DOI] [PubMed] [Google Scholar]
- 24.Lee, K.A.W., K. Zerivitz, and G. Akusjärvi. 1994. Small-scale preparation of nuclear extracts from mammalian cells. Cell Biology: A Laboratory Handbook, Vol. 3. J.E. Celis, editor. Elsevier Science & Technology Books, New York. 668–673.
- 25.Hoffmann, O., N. Keilwerth, M. Bastholm Bille, U. Reuter, K. Angstwurm, R. Schumann, U. Dirnagl, and J.R. Weber. 2002. Triptans reduce the inflammatory response in bacterial meningitis. J. Cereb. Blood Flow Metab. 22:988–996. [DOI] [PubMed] [Google Scholar]
- 26.Banati, R.B. 2003. Neuropathological imaging: in vivo detection of glial activation as a measure of disease and adaptive change in the brain. Br. Med. Bull. 65:121–131. [DOI] [PubMed] [Google Scholar]
- 27.Ramos, B., J.M. Lahti, E. Claro, and S. Jackowski. 2003. Prevalence of necrosis in C2-ceramide-induced cytotoxicity in NB16 neuroblastoma cells. Mol. Pharmacol. 64:502–511. [DOI] [PubMed] [Google Scholar]
- 28.Boggs, K.P., C.O. Rock, and S. Jackowski. 1995. Lysophosphatidylcholine attenuates the cytotoxic effects of the antineoplastic phospholipid 1-O-octadecyl-2-O-methyl-rac-glycero-3-phosphocholine. J. Biol. Chem. 270:11612–11618. [DOI] [PubMed] [Google Scholar]
- 29.Baburina, I., and S. Jackowski. 1999. Cellular responses to excess phospholipid. J. Biol. Chem. 274:9400–9408. [DOI] [PubMed] [Google Scholar]
- 30.Merrill, A.H., D.C. Liotta, and R.I. Riley. 1996. Fumonisins: fungal toxins that shed light on sphingolipid function. Trends Cell Biol. 6:218–223. [DOI] [PubMed] [Google Scholar]
- 31.Yen, C.L., M.H. Mar, R.B. Meeker, A. Fernandes, and S.H. Zeisel. 2001. Choline deficiency induces apoptosis in primary cultures of fetal neurons. FASEB J. 15:1704–1710. [DOI] [PubMed] [Google Scholar]
- 32.Lagace, T.A., J.R. Miller, and N.D. Ridgway. 2002. Caspase processing and nuclear export of CTP:phosphocholine cytidyltransferase α during farnesol-induced apoptosis. Mol. Cell. Biol. 22:4851–4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mallampalli, R.K., A.J. Ryan, R.G. Salome, and S. Jackowski. 2000. Tumor necrosis factor-alpha inhibits expression of CTP:phosphocholine cytidylyltransferase. J. Biol. Chem. 275:9699–9708. [DOI] [PubMed] [Google Scholar]
- 34.Adibhatla, R.M., and J.F. Hatcher. 2002. Citicoline mechanisms and clinical efficacy in cerebral ischemia. J. Neurosci. Res. 70:133–139. [DOI] [PubMed] [Google Scholar]
- 35.Clark, W., L. Gunion-Rinker, N. Lessov, and K. Hazel. 1998. Citicoline treatment for experimental intracerebral hemorrhage in mice. Stroke. 29:2136–2140. [DOI] [PubMed] [Google Scholar]
- 36.Lopez-Coviella, I., J. Agut, V. Savci, J.A. Ortiz, and R.J. Wurtman. 1995. Evidence that 5′-cytidinediphosphocholine can affect brain phospholipid composition by increasing choline and cytidine plasma levels. J. Neurochem. 65:889–894. [DOI] [PubMed] [Google Scholar]
- 37.Wurtman, R.J., M. Regan, I. Ulus, and L. Yu. 2000. Effect of oral CDP-choline on plasma choline and uridine levels in humans. Biochem. Pharmacol. 60:989–992. [DOI] [PubMed] [Google Scholar]
- 38.Grieb, P., R. Gadamski, R. Wojda, and M. Janisz. 2001. CDP-choline, but not cytidine, protects hippocampal CA1 neurones in the gerbil following transient forebrain ischemia. Folia Neuropathol. 39:141–145. [PubMed] [Google Scholar]
- 39.Rao, A.M., J.F. Hatcher, and R.J. Dempsey. 2000. Lipid alterations in transient forebrain ischemia: possible new mechanisms of CDP-choline neuroprotection. J. Neurochem. 75:2528–2535. [DOI] [PubMed] [Google Scholar]
- 40.Cui, Z., M. Houweling, M.H. Chen, M. Record, H. Chap, D.E. Vance, and F. Terce. 1996. A genetic defect in phosphatidylcholine biosynthesis triggers apoptosis in chinese hamster ovary cells. J. Biol. Chem. 271:14668–14671. [DOI] [PubMed] [Google Scholar]
- 41.Blusztajn, J.K., M. Liscovitch, and U.I. Richardson. 1987. Synthesis of acetylcholine from choline derived from phosphatidylcholine in a human neuronal cell line. Proc. Natl. Acad. Sci. USA. 84:5474–5477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sohlenkamp, C., I.M. Lopez-Lara, and O. Geiger. 2003. Biosynthesis of phosphatidylcholine in bacteria. Prog. Lipid Res. 42:115–162. [DOI] [PubMed] [Google Scholar]
- 43.Badger, E. 1944. The structural specificity of choline for the growth of type III pneumococcus. J. Biol. Chem. 153:183–191. [Google Scholar]
- 44.Weiser, J.N., J.M. Love, and E.R. Moxon. 1989. The molecular mechanism of phase variation of H. influenza lipopolysaccharide. Cell. 59:657–665. [DOI] [PubMed] [Google Scholar]
- 45.Zhang, J.-R., I. Idanpaan-Heikkila, W. Fischer, and E.I. Tuomanen. 1999. Pneumococcal licD2 gene is involved in phosphorylcholine metabolism. Mol. Microbiol. 31:1477–1488. [DOI] [PubMed] [Google Scholar]
- 46.Bonev, B.B., R.J.C. Gilbert, P.W. Andrew, O. Byron, and A. Watts. 2001. Structural analysis of the protein/lipid complexes associated with pore formation by the bacterial toxin pneumolysin. J. Biol. Chem. 276:5714–5719. [DOI] [PubMed] [Google Scholar]
- 47.Arrigoni, E., N. Averet, and F. Cohadon. 1987. Effects of CDP-choline on phospholipase A2 and cholinephosphotransferase activities following a cyrogenic brain injury in the rabbit. Biochem. Pharmacol. 36:3697–3700. [DOI] [PubMed] [Google Scholar]
- 48.Bussiere, M., J.E. Vance, R.B. Campenot, and D.E. Vance. 2001. Compartmentalization of choline and acetylcholine metabolism in cultured sympathetic neurons. J. Biochem. 130:561–568. [DOI] [PubMed] [Google Scholar]
- 49.Koh, J.Y., and D.W. Choi. 1987. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J. Neurosci. Methods. 20:83–90. [DOI] [PubMed] [Google Scholar]