

Abstract
B cell terminal differentiation involves development into an antibody-secreting plasma cell, reflecting the concerted activation of proplasma cell transcriptional regulators, such as Blimp-1, IRF-4, and Xbp-1. Here, we show that the microphthalmia-associated transcription factor (Mitf) is highly expressed in naive B cells, where it antagonizes the process of terminal differentiation through the repression of IRF-4. Defective Mitf activity results in spontaneous B cell activation, antibody secretion, and autoantibody production. Conversely, ectopic Mitf expression suppresses the expression of IRF-4, the plasma cell marker CD138, and antibody secretion. Thus, Mitf regulates B cell homeostasis by suppressing the antibody-secreting fate.
Keywords: autoimmunity, antibody secretion, cellular differentiation, immunoglobulin, IRF-4
Introduction
Terminal differentiation of B cells into plasma cells requires at least three transcription factors (Blimp-1, IRF-4 and Xbp-1) and involves the simultaneous acquisition of a capacity for antibody secretion as well as an inhibition of class switching, somatic hypermutation, and cell cycle progression (1). During the germinal center reaction, other transcription factors, including bcl-6 or Pax5, antagonize this process, likely to allow for affinity maturation and isotype switching before terminal differentiation. However, whether or not these developmental processes are regulated in naive B cells remains largely undetermined.
The microphthalmia-associated transcription factor (Mitf) is a basic-helix-loop-helix-leucine-zipper protein that has been primarily studied for its critical role in melanocyte function, where it regulates the expression of essential pigment enzymes (2). Its importance in cellular differentiation likely further extends to multiple areas of cellular proliferation and viability because it also regulates expression of antiapoptotic proteins such as bcl-2, at least in melanocytes (3), and plays a role in the development and function of mast cells, natural killer cells, and osteoclasts (4, 5). Previous studies in mi/mi mice, which are functionally Mitf-deficient due to a functionally disabling ΔArg mutation in their DNA binding domain (6, 7), have suggested that dysfunctional Mitf activity results in defective B cell development (4, 5). However, given the osteopetrotic phenotype and multiple other anatomical abnormalities of mi/mi animals, it is difficult to discern whether or not their apparently diminished B cell numbers in the marrow and spleen indeed reflects a B cell–intrinsic role for Mitf. Nonetheless, it is tempting to speculate on a role for Mitf in B cells because a review of microarray data in resting and activated B cells suggests that it is highly expressed in resting cells and rapidly diminished in activated cells, suggesting a role in the regulation of B cell activation (references 8, 9, and unpublished data).
Here, we demonstrate that Mitf plays a critical role in the maintenance of the mature, resting B cell state. Dysfunction of Mitf appears to result in spontaneous differentiation of B cells into plasma cells, as well as autoantibody production. Enforced expression of Mitf in activated B cells represses the plasmacytoid phenotype, suppressing Ig secretion and correlating with the ability of Mitf to repress IRF-4. Thus, Mitf suppresses the antibody-secreting cell fate.
Materials and Methods
Mice.
B6C3Fe a/a-Mitf Mi/J, Rag-2-deficient, and wild-type C57BL/6J mice were obtained from The Jackson Laboratory. To generate wild-type versus mi/mi animals, Mitf Mi/+ animals were intercrossed and genotyped by PCR on tail DNA using primers 5′-GTCCTGGCTGGTGACGTCAGTACG-3′ and 5′-ACCTAGCTCCTTAATGCGGTCGTTTATG-3′, which span the ΔArg of the mi mutation (6); sequencing of the product unequivocally distinguished genotype (see Fig. 1 F). For chimeras, bone marrow cells from day 0 newborn wild type versus mi/mi animals were adoptively transferred intravenously to six Gy-irradiated Rag-2–deficient recipients. Animals were analyzed 6–12 wk after reconstitution. Immunizations and antihapten responses were performed and determined as described previously (10). All experiments were performed in compliance with the relevant laws and institutional guidelines of the Washington University School of Medicine.
Figure 1.

Expression characteristics of lymphoid Mitf (Mitf-L). (A) Genomic structure of the murine Mitf locus. Alternative use of the 1a and 1b exons, in combination with the common exons 2–9, generates the Mitf-L isoform. Exon combinations for the melanocyte, heart, and mast cell–specific isoforms consist of 1m (depicted), 1h–1b, and 1mc–1b, respectively, followed by exons 2–9. The location of the ΔArg mutation in mi mice in exon 7 is indicated. (B) RT-PCR analysis of Mitf expression in B cells. 100 ng cDNA from naive B cells treated with the indicated stimuli in vitro for 24 h was assessed for Mitf expression using primers specific for the heart, melanocyte, and mast cell isoforms, or “common” primers that detect all Mitf isoforms (exons 2–3). Control cDNA was generated from whole E14 murine embryo. White line indicates that intervening lanes have been spliced out. (C) Western analysis of Mitf expression in B cells. 107 naive B cells were treated with the indicated stimuli in vitro for 24 h, and the whole cell lysate (equivalent to 107 originally incubated cells) was subjected to Western blotting with anti-Mitf and antiactin antisera. (D) Real-time PCR analysis of Mitf in B cells. cDNA from naive B cells treated with the indicated stimuli in vitro for 24 h was assessed for Mitf expression by real-time PCR. Error bars indicate standard deviations for individually tested cells from three animals. (E) RACE product for the lymphoid isoform of Mitf. RACE was performed on wild-type B cell cDNA using a reverse primer in Mitf exon 2. (F) Genotyping of mi animals. Tail DNA of representative animals was subjected to PCR with primers spanning exon 7. Subsequent sequencing revealed unambiguous changes corresponding to the ΔAGA mutation of mi mice. Results are representative of at least three experiments for each figure.
Mitf Plasmids and Retroviruses.
To isolate murine Mitf-M, cDNA from a C57BL/6 day 14 embryo was amplified by PCR using primers 5′-CTTGGGGCTGCCTGAAACCTTG-3′ and 5′-TGAATGAAAACGGACAGACACTTACTTC-3′. A single ∼1.6-kb fragment was obtained that was ligated to pCR2.1-TOPO (Invitrogen), generating plasmid pMitf-M. To generate Mitf-A, cDNA from a C57BL/6 day 14 embryo was amplified by PCR using primers 5′-CCCCCATCTTTCTCAGGTGCCCG-3′ and 5′-GCCGGATCCATCAAGCCCAAAAT-3′. The 812-bp product was ligated to pCR2.1-TOPO, and an XhoI–XhoI fragment from this vector, corresponding to some vector sequence, exon 1a, exon 1b, and part of exon 2 of the deduced Mitf-A isoform, was used to replace the corresponding XhoI–XhoI fragment of pMitf-M, generating pMitf-A. Identities of both clones were confirmed by sequencing. To generate Mitf expression vectors, the EcoRI–EcoRI fragments of pMitf-A and pMitf-M containing their respective coding sequences were ligated to the EcoRI site of pcDNA3 (Invitrogen), generating pcDNA3-Mitf-A and pcDNA3-Mitf-M, respectively. For retroviral constructs, the same EcoRI–EcoRI fragments were ligated into the EcoRI site of pMX-IRES–green fluorescence protein (GFP; reference 11), generating pMX-Mitf-A-IRES-GFP and pMX-Mitf-M-IRES-GFP, respectively. Retroviral supernatants were generated in the PlatE packaging line (11), and transductions of murine B cells were performed as described previously (12).
Rapid Amplification of cDNA Ends (RACE).
To identify the 5′ sequence of Mitf-A, RNA was combined from purified naive T and B cells of wild-type C57BL/6 mice and was subjected to the SMART RACE cDNA amplification kit (CLONTECH Laboratories, Inc.) with the reverse primer 5′-AGGCCCTGGTTGCTGTAGAG-3′, which is located in exon 2 (see Fig. 1 A). Sequencing of the PCR product and comparison with the National Center for Biotechnology Information murine genomic sequence allowed the determination of the expressed exons.
Lymphocyte Purification, Culture, and Analyses.
Naive-enriched B cells were purified from spleens by negative selection against CD43 (12). Results comparable to those shown in this paper were obtained with B cells purified by negative selection against both CD43 and CD5 (unpublished data). For in vitro differentiation assays, B cells were further purified over a discontinuous Percoll gradient (70/66/60/50%) with resting B cells isolated from the 66–70 interface. Cells were cultured in RPMI 1640 medium supplemented with 10% FCS (BioWhittaker) and 100 U penicillin/streptomycin (Sigma-Aldrich) in the presence or absence of 25 μg/ml LPS (Sigma-Aldrich), 2 μg/ml anti-CD40 antibody (BD Biosciences), 5–10 μg/ml anti-IgM (Jackson Immunologicals), 3 mM CpG-1 stimulatory phosphorothioate oligonucleotide 5′-TCCATGACGTTCCTGACGTT-3′, 10,000 U/ml IFN-A/D (R&D Systems), 2 ng/ml APRIL, 100 ng/ml IFN-γ, 5 ng/ml TGF-β, 100 U/ml hIL-2, 10 ng/ml IL-4, 500 ng/ml IL-5, 100 ng/ml IL-6, 5 ng/ml IL-7, 5 ng/ml IL-9, 10 ng/ml IL-10, 10 ng/ml IL-12, 5 ng/ml IL-13, 20 ng/ml IL-15, and/or 25 ng/ml IL-18 (PeproTech). Where indicated, cells were supplemented with 10 μM antisense IRF-4, 5′-GCTGCCCGTCTCCAAGTTCAT-3′, or missense oligonucleotide, 5′-CTCTTCTCCTCGTTTTTTTAC-3′ (Integrated DNA Technologies). This strategy was derived from an antisense strategy used to inhibit chicken IRF-4 and resulted in a 75–80% reduction in IRF-4 mRNA under the indicated culture conditions (reference 13 and unpublished data). Flow cytometric assays, as well as assessments for Ig secretion, Ig titers, autoantibodies, and cellular proliferation were performed as described previously (12, 14). Viability/cellular ATP content was assessed using the Cell Titer-Glo assay (Promega).
Expression Analysis.
Isoform-specific RT-PCR for Mitf was performed using primers and conditions described previously (15). For real-time PCR detection of Mitf-A, primers 5′-GGCCTTGCAAATGGCAAA-3′ and 5′-AGGCCCTGGTTGCTGTAGAG-3′ were used on samples analyzed by an ABI PRISM® 7000 Sequence Detection System (Applied Biosystems) under standard conditions with specificity reinforced via the dissociation protocol. Other gene-specific primers, including those for IRF-4 (16), have been described previously. Relative mRNA abundance of each transcript was normalized against tubulin as described previously (10). Western blots used a polyclonal goat anti-Mitf (C-17; Santa Cruz Biotechnology, Inc.).
Luciferase Assays.
An IRF-4 promoter–luciferase construct was generated by PCR from C57BL/6 tail DNA using primers 5′-GGGGTACCAATTCGTCGGTTTCATTCACCCAACATG-3′ and 5′-GAAGATCTACACTCCTCCTTCTGCCCGACTACAG-3′, which created a KpnI–BglII fragment containing basepairs −633 to +71 (relative to the transcriptional start site) of the IRF-4 promoter (17) that could be ligated into the KpnI–BglII sites of TK-luc (14). Sequences were confirmed by routine sequencing. Derivatives with mutant putative Mitf binding sites (CANNTG) were generated by site-directed mutagenesis (QuikChange®; Stratagene). For luciferase activity assessment, 107 M12 murine B cell lymphoma cells (12) in 400 μl complete RPMI 1640 medium were electroporated in a 0.4-cm cuvette at 280 mV, 975 μF in the presence of 10 μg of luciferase reporter, 40 ng pRL-CMV, and 10 μg pcDNA3 (Invitrogen), pcDNA3-Mif-A, pcDNA3-Mitf-A(AS), or pcDNA3-Mitf-M expression plasmids and returned to cell culture medium. At the indicated times, reporter activity was determined by the Dual-Luciferase® Reporter Assay System (Promega); relative activity was determined after normalization for Renilla luciferase.
ELISPOT Analyses.
For quantification of antibody-secreting cells, microtiter plates were coated with 10 μg/ml anti–mouse Ig (Southern Biotechnology Associates, Inc.) in PBS and blocked with 1% BSA. Single cell suspensions of splenocytes or bone marrow cells were incubated in the plates with RPMI 1640 medium supplemented with 10% FCS, using limiting dilutions, overnight at 37°C, 5% CO2. Bound antibody was detected with IgM-, IgG1-, or IgG2b-specific alkaline phosphatase–conjugated antibodies (Southern Biotechnology Associates, Inc.) followed by development with 5-bromo,4-chloro,3-indolylphosphate (Sigma-Aldrich) in 3% low-melt agarose. Spots were quantified upon examination under a dissecting microscope.
Online Supplemental Material.
Table S1 describes B cell populations in native and chimeric mi mice. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20040612/DC1.
Results and Discussion
Identification and Characterization of Lymphoid Mitf (Mitf-L/Mitf-A).
Ongoing microarray studies to identify novel regulatory transcription factors in B lymphocytes revealed a significant signal from the 101159_at Affymetrix probe set by resting but not activated B cell cDNA, corresponding to Mitf (references 8, 9, and unpublished data). The expression pattern of Mitf is tissue specific, with specific isoforms formed by alternative promoter activity before distinct first exons to produce melanocyte- (Mitf-M), mast cell– (Mitf-MC), and heart-specific (Mitf-H) transcripts, among others (Fig. 1 A); however, functionally relevant Mitf expression has not been described previously in the lymphoid system.
Standard RT-PCR and Western blot analyses confirmed expression of Mitf in B cells, particularly that its expression is highest in resting, but not activated, cells (Fig. 1, B and C). Interestingly, its expression could not be detected with primers specific for the predominantly studied isoforms, but only by primers within the common exons shared by all known Mitf isoforms. Real-time PCR studies confirmed these observations, demonstrating a suppression of Mitf expression in response to several stimuli, but most prominently in response to polyclonal B cell activators such as LPS or CpG oligonucleotides (Fig. 1 D).
Because the Mitf isoforms are only known to differ at the 5′ end due to alternative promoter use, 5′ RACE was performed to identify the lymphocyte form (Mitf-L), producing a dominant ∼600-bp product (Fig. 1 E). Sequencing revealed that the cDNA reflected the use of two Mitf exons previously termed 1a and 1b (Fig. 1 A). Thus, Mitf-L uses exons 1a, 1b, and the common exons 2–9; because this structure corresponds to a previously described A isoform of Mitf (18), we will use Mitf-A to refer to this species henceforth.
Mature B Cell Development Does Not Require Functional Mitf.
To determine the role of Mitf in B cells, we examined mi/mi mice (Fig. 1 A and references 6, 7). Here, mi/mi T cell development and function were largely normal, but mi/mi animals possessed substantially fewer peripheral B cells (reference 4, Table S1, available at http://www.jem.org/cgi/content/full/jem.20040612/DC1, and unpublished data). However, because the impaired B cell development could have resulted from the animals' generalized developmental delay (6, 7), we performed bone marrow chimerization from wt/wt versus mi/mi animals into Rag-deficient recipients, confirming genotype by direct sequencing of the mutant region (Fig. 1 F). mi/mi bone marrow cells were clearly capable, surprisingly so, of fully repopulating the B cell populations, both conventional and B1 cells, as evidenced by largely normal flow cytometric profiles for the various B cell subpopulations 8–12 wk after reconstitution (Fig. 2 A and Table S1). Therefore, all subsequent B cell analyses were performed with bone marrow chimeric animals.
Figure 2.
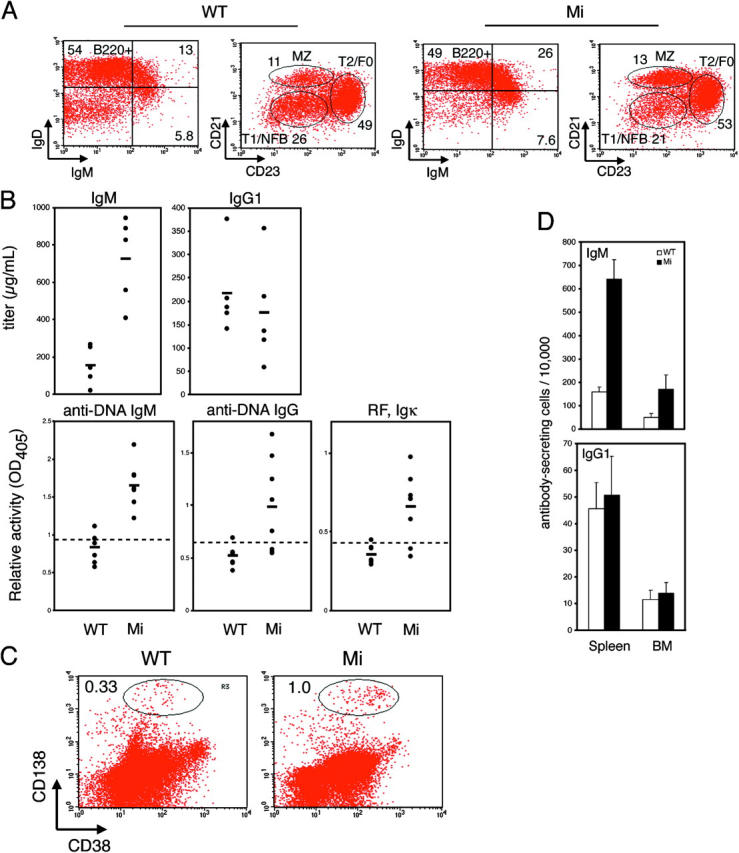
Hypergammaglobulinemia and plasma cell differentiation in the absence of functional Mitf. (A) Splenocytes from Rag-deficient animals reconstituted 12 wk previously by bone marrow derived from wild-type or mi animals were analyzed by flow cytometry and gated on B220+ or B220+IgM+ cells as indicated. Numbers indicate percentages of gated cells, and are representative of four to six animals of each genotype. Similar results were seen when CD19 was used instead of B220 (not depicted). NFB/T1, new follicular B/T1 transitional; T2/F0, T2 transitional/follicular; MZ, marginal zone. (B) Sera from wild-type or mi Rag-deficient chimeras (n = 5–7 in each group) were assessed for total Ig isotype concentrations, as well as anti-DNA and rheumatoid factor autoantibodies. Horizontal bars indicate means. Dashed line indicates the threshold for positivity, as indicated by three standard deviations above the mean OD of simultaneously assessed sera from wild-type C57BL/6 animals. (C) Spleens from wild-type or mi Rag-deficient chimeras were analyzed by flow cytometry for plasma cells, CD38midCD138+CD5−B220−. CD138 versus CD38 plots after gating on CD5−B220− double negative cells are shown. Numbers indicate percentages of the total splenocyte population, representative of three animals of each genotype. (D) Antibody-secreting cells of the IgM and IgG1 isotypes were quantified in the spleen and bone marrow of wild-type or mi Rag-deficient chimeras by ELISPOT analysis. Error bars indicate standard deviations for three individually tested animals of each genotype.
Hyperactivation of mi/mi B Cells.
Chimeric mi/mi animals developed hypergammaglobulinemia of the IgM but not IgG1 isotype (Fig. 2 B, P < 0.0001). They also developed evidence of humoral autoimmunity, including anti-DNA and rheumatoid factor (anti-IgG) activities (P < 0.0001 for all indicated specificities). However, these likely represented largely nonpathogenic (e.g., anti-ssDNA) rather than pathogenic (e.g., anti-dsDNA) autoantibody responses because mi/mi sera were not reactive with the dsDNA-containing kinetoplast of Crithidia luciliae substrates, nor was there evidence of inflammation in the end-organs of mi/mi chimeric animals (unpublished data).
Interestingly, chimeric mi/mi animals could mount IgM and IgG antihapten responses comparable to their wild-type counterparts when immunized with T-dependent (NP-CGG or TNP-CGG) or T-independent (NP-Ficoll, TNP-Ficoll, or TNP-LPS) antigens, indicating that mi B cells were not hyperreactive per se (unpublished data). Such findings suggested that mi/mi B cells simply undergo spontaneous activation and/or differentiation into plasma cells in vivo, without antigen-driven specificity. Indeed, mi/mi chimeras contained increased numbers of CD138+B220− plasma cells in their spleen, as well as IgM antibody-secreting cells in their spleen and bone marrow (PFig. 2, C and D, P < 0.01). In vitro, naive mi/mi B cells spontaneously produced IgM in the absence of external stimuli (Fig. 3 A, P < 0.00001 for both isotypes), albeit it was diminished in comparison to LPS-stimulated B cells, and they spontaneously up-regulated the plasma cell marker CD138 (syndecan-1) in culture (Fig. 3 B, P < 0.0001 comparing mean fluorescence intensities at 2 d). At the same time, mi B cells proliferated less well than their wild-type counterparts (Fig. 3 C, P < 0.01 comparing wild-type to mi at LPS concentrations of 10 and 25 μg/ml), but had increased intracellular metabolic activity, as judged by ATP activity (Fig. 3 D, P < 0.01). These findings, such as increased antibody secretory capacity, metabolic activity, and CD138 expression despite diminished proliferative capacity, are characteristic of plasma cell differentiation (1).
Figure 3.

Spontaneous Ig secretion by B cells in the absence of functional Mitf. (A) Naive B cells from wild-type or mi Rag-deficient chimeras were incubated in vitro in the presence or absence of LPS, IL-4, and/or IFN-γ. After 10 d, supernatants were assessed for Ig secretion by ELISA. Individual data points represent B cells obtained from individual mice (n = 3 for each genotype), with means indicated by horizontal bars. *, undetectable by assay (<20 pg/ml). (B) Syndecan-1 (CD138) expression was assessed on naive B cells from wild-type or mi Rag-deficient chimeras after 2 d of incubation in culture without mitogenic stimulation. (C) Proliferation of naive B cells from wild-type or mi Rag-deficient chimeras was assessed by BrdU incorporation on day 3 of stimulation with the indicated concentrations of LPS. (D) Metabolic activity of naive B cells from wild-type or mi Rag-deficient chimeras was assessed by the CellTiter Glo assay for ATP activity on the sixth day of treatment with the indicated stimuli. Error bars reflect standard deviations from cells derived from three individually tested animals. These data are representative of at least three separately performed experiments.
Suppression of B Cell Activation by Mitf via IRF-4.
These findings suggested that Mitf may enforce the naive B cell state by suppressing plasma cell differentiation. Retroviral transduction of Mitf-A into LPS-stimulated wild-type B cells suppressed Ig secretion and CD138 expression (Fig. 4, A and B): although control-transduced B cells secreted significant levels of IgM over 2 d in culture, Mitf-A–transduced B cells were largely unable to secrete Ig (P < 0.0001). Also, although ∼40–50% of control-transduced B cells up-regulated CD138, <10% of Mitf-transduced B cells did (P < 0.0001). In addition, MHC class II expression remained higher in Mitf-transduced B cells (Fig. 4 B; mean fluorescence intensity 662 ± 43 vs. 853 ± 23, comparing control- to Mitf-transduced cells, respectively; P < 0.001), which was consistent with a block in plasma cell differentiation (1).
Figure 4.

Mitf represses plasmacytoid differentiation via IRF-4. (A) B cells from wild-type C57BL/6 animals were stimulated with 25 μg/ml LPS for 24 h and infected with a control (pMX) or Mitf-A–expressing (pMX-Mitf) GFP retrovirus in the continued presence of LPS. (A) On day 2 after infection, GFP-positive cells were sorted and returned to medium containing LPS, and IgM secretion was assessed 1 and 2 d thereafter by ELISA. (B) On day 2 after infection, cells were assessed for syndecan-1 (CD138) and MHC class II (I-A) expression by flow cytometry. (C) GFP-sorted B cells infected with a control or Mitf-expressing retrovirus, as in A, were returned to medium containing LPS for an additional 24 h and assessed for the expression of IRF-4 by real-time PCR. (D) The ability of Mitf to regulate the IRF-4 promoter was assessed by transient transfection in M12 B cell lymphoma cells, using a control, Mitf-A, or Mitf-A antisense (AS) expression construct. Firefly light units were normalized against control Renilla luciferase activity. (E) Naive B cells from wild-type or mi Rag-deficient chimeras were incubated in vitro in the presence or absence of LPS for 24 h. The expression of IRF-4 was assessed by real-time PCR. (F) Naive B cells from mi Rag-deficient chimeras were incubated in vitro without mitogenic stimulation in the presence or absence of IRF-4 antisense (AS) or missense (MS) oligonucleotides. After 10 d, supernatants were assessed for IgM secretion by ELISA. Standard deviations reflect triplicate samples performed simultaneously, and are representative of at least three experiments.
To gain insight into the potential mechanism by which Mitf might regulate B cells, we sought to identify target genes regulated by Mitf and, therefore, examined its ability to regulate genes known to participate in plasma cell differentiation. Interestingly, ectopic Mitf significantly suppressed IRF-4 mRNA in LPS-stimulated B cells (Fig. 4 C, P < 0.0001), but not Blimp-1 or Xbp-1 (spliced or unspliced form; reference 19 and not depicted). Indeed, Mitf-A could repress the activity of an IRF-4 promoter construct in the M12 B cell lymphoma cell line (Fig. 4 D, P < 0.001 comparing pcDNA to pcDNA-Mitf-A), and mi B cells expressed significantly elevated levels of IRF-4 (Fig. 4 E, P < 0.001). To test the importance of IRF-4 to the mi B cell phenotype, we incubated B cells from mi chimeric animals in culture medium without mitogens, but in the presence or absence of an IRF-4 antisense oligonucleotide (Fig. 4 F). Strikingly, the antisense oligonucleotide, but not a missense control oligonuclotide, significantly reduced the spontaneous secretion of IgM (P < 0.001 comparing no or missense to antisense oligonucleotide). Interestingly, although the IRF-4 promoter contains two potential Mitf binding sites (CANNTG) at basepairs −449 to −444 and −10 to −4, mutagenesis of both sites failed to abrogate the ability of Mitf to suppress our IRF4 promoter luciferase reporter in M12 cells, and we have been unable to demonstrate binding of Mitf to any portion of the IRF4 promoter by electrophoretic mobility shift assays (unpublished data). Thus, the effects of Mitf on B cell activation clearly require IRF-4, but other genes likely cooperate with and/or are regulated by Mitf to modulate IRF-4 expression.
Mitf in B Cell Development.
As such, these data support a model in which Mitf suppresses spontaneous plasma cell differentiation in naive B cells by ultimately inhibiting the expression of IRF-4. As a result, defective Mitf function leads to IRF-4 overexpression and spontaneous plasma cell differentiation, and is interestingly associated with mild humoral autoimmunity. Therefore, defective Mitf activity may contribute to diseases of humoral immune dysregulation, such as autoimmune diseases (i.e., lupus). However, a Mitf defect as a sole cause of such diseases seems unlikely; instead, additional factors, such as T cell help (which is not accentuated in mi animals), are probably required for full-blown autoimmune disease (20). Nonetheless, a particularly intriguing finding in the present paper is the presence of IgG anti-DNA autoantibodies in mi chimeric mice (Fig. 2), even though Mitf primarily appears to affect IgM secretion (Figs. 2–4). Perhaps Mitf plays an additional role in the regulation of autoreactive B cells, suppressing their activation and/or subsequent differentiation, and/or enforcing tolerance.
In humans, mutations in Mitf result in the Waardenburg syndrome (WS) type 2a, consisting of hypopigmentation and deafness (for review see reference 2), but to our knowledge, a humoral immune phenotype has not been described in WS, and no mutations in Mitf have yet been found to be associated with antibody-related disorders. Nonetheless, Mitf resides on human chromosome 3 near a known susceptibility locus for lupus (21), and increased IRF-4 activity (22) and abnormal Wnt signaling (23), which modulates Mitf activity (at least in melanocytes; reference 2), have both been observed in multiple myeloma. Therefore, it will be of significant interest to perform a systematic study of antibody responses in WS patients, and to determine if defective Mitf or overactive IRF-4 activity contributes to the perturbed humoral immunity seen in plasma cell dyscrasias or autoimmune diseases.
Interestingly, most functions of Mitf have been attributed to its ability to transactivate target genes (24, 25), but it may act as a transcriptional repressor upon interacting with other transcription factors, such as Pax6 (26). Thus, it is possible that Mitf regulates IRF-4 expression by interacting with transcription factors, such as members of the NF-κB or NF-AT families, that can transactivate the IRF-4 promoter (17), becoming a transcriptional repressor of IRF-4 itself. However, the inability of Mitf to bind to the IRF-4 promoter (unpublished data) suggests that Mitf instead either represses the expression of a factor required to transactivate IRF-4, or more likely transactivates a transcriptional repressor of IRF-4. Regardless, our present findings strongly indicate that active transcriptional repression of IRF-4 by Mitf, via whatever means, is required to prevent spontaneous B cell activation, as well as tolerance loss. Therefore, continued investigation into the mechanisms by which Mitf regulates B cell activation and antibody production will undoubtedly shed insight into the mechanisms of immune tolerance and effector differentiation.
Acknowledgments
We thank B. Sleckman for critical commentary during the preparation of this paper, and J. Li and L. Robbins for technical assistance.
This work was supported in part by the Siteman Cancer, Rheumatic Diseases, Diabetes Research and Training, and the Digestive Diseases Research (DK52574) Core Centers of the Washington University School of Medicine, as well as grants from the National Institutes of Health (AI01803 and AI057471) and the Lupus Research Institute. S.L. Peng is supported in part by an Arthritis Investigator Award from the Arthritis Foundation.
The online version of this article contains supplemental material.
References
- 1.Calame, K.L., K.I. Lin, and C. Tunyaplin. 2003. Regulatory mechanisms that determine the development and function of plasma cells. Annu. Rev. Immunol. 21:205–230. [DOI] [PubMed] [Google Scholar]
- 2.Widlund, H.R., and D.E. Fisher. 2003. Microphthalamia-associated transcription factor: a critical regulator of pigment cell development and survival. Oncogene. 22:3035–3041. [DOI] [PubMed] [Google Scholar]
- 3.McGill, G.G., M. Horstmann, H.R. Widlund, J. Du, G. Motyckova, E.K. Nishimura, Y.L. Lin, S. Ramaswamy, W. Avery, H.F. Ding, et al. 2002. Bcl2 regulation by the melanocyte master regulator Mitf modulates lineage survival and melanoma cell viability. Cell. 109:707–718. [DOI] [PubMed] [Google Scholar]
- 4.Roundy, K., A. Kollhoff, E.J. Eichwald, J.J. Weis, and J.H. Weis. 1999. Microphthalmic mice display a B cell deficiency similar to that seen for mast and NK cells. J. Immunol. 163:6671–6678. [PubMed] [Google Scholar]
- 5.Roundy, K., R. Smith, J.J. Weis, and J.H. Weis. 2003. Overexpression of RANKL implicates IFN-beta-mediated elimination of B-cell precursors in the osteopetrotic bone of microphthalmic mice. J. Bone Miner. Res. 18:278–288. [DOI] [PubMed] [Google Scholar]
- 6.Hodgkinson, C.A., K.J. Moore, A. Nakayama, E. Steingrimsson, N.G. Copeland, N.A. Jenkins, and H. Arnheiter. 1993. Mutations at the mouse microphthalmia locus are associated with defects in a gene encoding a novel basic-helix-loop-helix-zipper protein. Cell. 74:395–404. [DOI] [PubMed] [Google Scholar]
- 7.Morii, E., K. Takebayashi, H. Motohashi, M. Yamamoto, S. Nomura, and Y. Kitamura. 1994. Loss of DNA binding ability of the transcription factor encoded by the mutant mi locus. Biochem. Biophys. Res. Commun. 205:1299–1304. [DOI] [PubMed] [Google Scholar]
- 8.Glynne, R., G. Ghandour, J. Rayner, D.H. Mack, and C.C. Goodnow. 2000. B-lymphocyte quiescence, tolerance and activation as viewed by global gene expression profiling on microarrays. Immunol. Rev. 176:216–246. [DOI] [PubMed] [Google Scholar]
- 9.Fruman, D.A., G.Z. Ferl, S.S. An, A.C. Donahue, A.B. Satterthwaite, and O.N. Witte. 2002. Phosphoinositide 3-kinase and Bruton's tyrosine kinase regulate overlapping sets of genes in B lymphocytes. Proc. Natl. Acad. Sci. USA. 99:359–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerth, A.J., L. Lin, and S.L. Peng. 2003. T-bet regulates T-independent IgG2a class switching. Int. Immunol. 15:937–944. [DOI] [PubMed] [Google Scholar]
- 11.Morita, S., T. Kojima, and T. Kitamura. 2000. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 7:1063–1066. [DOI] [PubMed] [Google Scholar]
- 12.Peng, S.L., S.J. Szabo, and L.H. Glimcher. 2002. T-bet regulates IgG class switching and autoantibody production. Proc. Natl. Acad. Sci. USA. 99:5545–5550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hrdlicková, R., J. Nehyba, and H.R. Bose, Jr. 2001. Interferon regulatory factor 4 contributes to transformation of v-Rel-expressing fibroblasts. Mol. Cell. Biol. 21:6369–6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin, L., M. Spoor, A.J. Gerth, S.L. Brody, and S.L. Peng. 2004. Modulation of Th1 activation and inflammation by the NF-κB repressor Foxj1. Science. 303:1017–1020. [DOI] [PubMed] [Google Scholar]
- 15.Takemoto, C.M., Y.J. Yoon, and D.E. Fisher. 2002. The identification and functional characterization of a novel mast cell isoform of the microphthalmia-associated transcription factor. J. Biol. Chem. 277:30244–30252. [DOI] [PubMed] [Google Scholar]
- 16.Marecki, S., M.L. Atchison, and M.J. Fenton. 1999. Differential expression and distinct functions of IFN regulatory factor 4 and IFN consensus sequence binding protein in macrophages. J. Immunol. 163:2713–2722. [PubMed] [Google Scholar]
- 17.Sharma, S., N. Grandvaux, Y. Mamane, P. Genin, N. Azimi, T. Waldmann, and J. Hiscott. 2002. Regulation of IFN regulatory factor 4 expression in human T cell leukemia virus-I-transformed T cells. J. Immunol. 169:3120–3130. [DOI] [PubMed] [Google Scholar]
- 18.Amae, S., N. Fuse, K. Yasumoto, S. Sato, I. Yajima, H. Yamamoto, T. Udono, Y.K. Durlu, M. Tamai, K. Takahashi, and S. Shibahara. 1998. Identification of a novel isoform of microphthalmia-associated transcription factor that is enriched in retinal pigment epithelium. Biochem. Biophys. Res. Commun. 247:710–715. [DOI] [PubMed] [Google Scholar]
- 19.Iwakoshi, N.N., A.H. Lee, and L.H. Glimcher. 2003. The X-box binding protein-1 transcription factor is required for plasma cell differentiation and the unfolded protein response. Immunol. Rev. 194:29–38. [DOI] [PubMed] [Google Scholar]
- 20.Shlomchik, M.J., J.E. Craft, and M.J. Mamula. 2001. From T to B and back again: positive feedback in systemic autoimmune disease. Nat. Rev. Immunol. 1:147–153. [DOI] [PubMed] [Google Scholar]
- 21.Kelly, J.A., K.L. Moser, and J.B. Harley. 2002. The genetics of systemic lupus erythematosus: putting the pieces together. Genes Immun. 3:S71–S85. [DOI] [PubMed] [Google Scholar]
- 22.Iida, S., P.H. Rao, M. Butler, P. Corradini, M. Boccadoro, B. Klein, R.S. Chaganti, and R. Dalla-Favera. 1997. Deregulation of MUM1/IRF4 by chromosomal translocation in multiple myeloma. Nat. Genet. 17:226–230. [DOI] [PubMed] [Google Scholar]
- 23.Derksen, P.W.B., E. Tjin, H.P. Meijer, M.D. Klok, H.D. MacGillavry, M.H.J. van Oers, H.M. Lokhorst, A.C. Bloem, H. Clevers, R. Nusse, et al. 2004. Illegitimate WNT signaling promotes proliferation of multiple myeloma cells. Proc. Natl. Acad. Sci. USA. 101:6122–6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yasumoto, K., K. Yokoyama, K. Takahashi, Y. Tomita, and S. Shibahara. 1997. Functional analysis of microphthalmia-associated transcription factor in pigment cell-specific transcription of the human tyrosinase family genes. J. Biol. Chem. 272:503–509. [DOI] [PubMed] [Google Scholar]
- 25.Adachi, S., E. Morii, D. Kim, H. Ogihara, T. Jippo, A. Ito, Y.M. Lee, and Y. Kitamura. 2000. Involvement of mi-transcription factor in expression of alpha-melanocyte-stimulating hormone receptor in cultured mast cells of mice. J. Immunol. 164:855–860. [DOI] [PubMed] [Google Scholar]
- 26.Planque, N., L. Leconte, F.M. Coquelle, P. Martin, and S. Saule. 2001. Specific Pax-6/microphthalmia transcription factor interactions involve their DNA-binding domains and inhibit transcriptional properties of both proteins. J. Biol. Chem. 276:29330–29337. [DOI] [PubMed] [Google Scholar]