

Abstract
Knowledge of the molecular networks controlling the proliferation and fate of hematopoietic stem cells (HSC) is essential to understand their function in maintaining blood cell production during normal hematopoiesis and upon clinical transplantation. Using highly purified stem and progenitor cell populations, we define the proliferation index and status of the cell cycle machinery at discrete stages of hematopoietic differentiation and during cytokine-mediated HSC mobilization. We identify distinct sets of cell cycle proteins that specifically associate with differentiation, self-renewal, and maintenance of quiescence in HSC and progenitor cells. Moreover, we describe a striking inequality of function among in vivo cycling and quiescent HSC by demonstrating that their long-term engraftment potential resides predominantly in the G0 fraction. These data provide a direct link between HSC proliferation and function and identify discrete molecular targets in regulating HSC cell fate decisions that could have implications for both the therapeutic use of HSC and the understanding of leukemic transformation.
Hematopoiesis is initiated by a rare population of BM multipotent hematopoietic stem cells (HSC) that is faced at each cell division with the decision to self-renew, differentiate, migrate, or die (1). During steady-state hematopoiesis, the HSC population is relatively quiescent (2, 3), but does give rise, upon cell cycle entry, to a hierarchy of differentiating progenitor populations that undergo the massive proliferative expansion required to replenish the blood system every day (1). The blood-forming system is also able to respond efficiently to hematological stresses such as blood loss, infection, or exposure to cytotoxic agents by expanding the HSC population, mainly through increased numbers of self-renewing divisions (4, 5). Thus, HSC proliferation must be highly adaptive to ensure both durable production of progenitor populations during steady-state hematopoiesis and extensive, stress-induced, self-renewing proliferation, without depletion of the stem cell pool. Such precise control of blood cell production requires appropriate regulation of cell cycle progression, which therefore plays a fundamental role in determining HSC fate decisions.
Although the molecular events controlling HSC proliferation are still poorly understood, they are likely determined, at least in part, by regulated expression and/or function of components and regulators of the cell cycle machinery. In mammals, the rate of cell proliferation is largely determined during the G1 phase of the cell cycle (6). During this period, G1 cyclins bind to and activate specific cyclin-dependent kinases (Cdks), thereby dictating the timing and substrate specificity of Cdk-mediated phosphorylation of critical cellular targets, such as the Rb family of proteins (Rb, p107, and p130). Two families of Cdk inhibitors (Ckis) regulate the activity of G1-type cyclin–Cdk complexes: the Ink4 family (p16, p15, p18, and p19), which blocks the activity of cyclin D–Cdk4-6 complexes, and the Cip/Kip family (p21, p27, and p57), which preferentially inhibits cyclin E–Cdk2 complexes and also acts as a scaffold for the catalytically active cyclin D–Cdk4-6 complexes. In quiescent cells, Ckis are present in excess of cyclin–Cdk complexes, which, together with the transcriptional repression of E2F target genes ensured by hypophosphorylated Rb proteins (mostly p130) (7), maintain the cells in a nonproliferating state. Mitogenic stimulation leads to induction of D-type cyclins, which initiate Rb phosphorylation and also titrate away Cip/Kip Ckis from cyclin E and cyclin A–Cdk2 complexes, thereby relieving their inhibition and allowing them to complete the hyperphosphorylation and inactivation of Rb proteins (mostly Rb and p107). The consecutive release of E2F transcription factors then enables transcriptional activation of genes required for S phase entry and progression through mitosis, where proteolytic degradation of the mitotic cyclins finally resets the system for the next G1 phase.
Several positive and negative regulators of the cell cycle machinery have already been shown to be critical for various aspects of HSC and/or progenitor proliferation, including the Ckis p21, p27, p16, and p18 (8–11) and the D-type cyclins and their catalytic partners Cdk4-6 (12, 13). Studies have also analyzed the proliferation rates and distribution of cell cycle components in various subsets of hematopoietic cells (14), including investigation by gene array of the global status of the cell cycle machinery in HSC (15). However, an integrated description of proliferation both at the cellular and molecular levels, which would allow us to understand how different sets of cell cycle proteins may be involved in controlling the different cellular outcomes and fate decisions in HSC and primitive progenitor cell populations, has yet to be provided. Previous studies have also demonstrated that among HSC, cells with 2N DNA content (in G0 or G1 phases) exhibit enhanced capacity for radioprotection and for long-term multilineage reconstitution when compared with cells with >2N DNA content (in S or G2/M phases) (16, 17), leading to the suggestion that HSC quiescence may promote engraftment function (15, 18), although this hypothesis has yet to be formally tested.
In this paper, we have used our ability to isolate nearly pure populations of mouse stem and progenitor cells to describe the molecular networks regulating their proliferation during both normal hematopoietic differentiation and cytokine-mediated HSC mobilization and to identify distinct molecular targets that specifically associate with these processes. Furthermore, we functionally assess the impact of HSC proliferation on hematopoietic repopulating potential and demonstrate that long-term engraftment capacity of transplanted HSC critically depends on maintenance of the quiescent state. Together, these findings provide a direct link between HSC proliferation and cell fate decisions that could have implications for both the therapeutic use of HSC and the understanding of leukemic transformation.
Results
Proliferation in the pool of multipotent BM cells
BM HSC can be divided into a long-term reconstituting Flk-2− HSC (LT-HSC) subset (Lin−/c-Kit+/Sca-1+/Thy1.1int/Flk2−) capable of extensive self-renewal, a short-term reconstituting Flk-2int HSC (ST-HSCF) subset (Lin−/c-Kit+/Sca-1+/Thy1.1int/Flk2int), which self-renews for a limited interval and then gives rise to a nonself-renewing Flk-2+ multipotent progenitor (MPPF) subset (Lin−/c-Kit+/Sca-1+/Thy1.1−/Flk2+; Fig. 1 A) (19). To determine the cell cycle profile of these populations in steady-state conditions, each subset was first purified by double FACS sorting and then analyzed both by HOECHST 33342/Pyronin Y (H/PY) vital staining for DNA/RNA content and intracellular propidium iodide (PI) staining of fixed cells for DNA content (Fig. 1 B). An average of two thirds (69.2 ± 2.4%; n = 3) of the LT-HSC population was found in the quiescent G0 phase (H2N/PY−) of the cell cycle, whereas the remaining one third was divided between the G1 phase (H2N/PY+; 20.2 ± 2.5%) and the S-G2/M phase (H>2N-4N/PY+; 9.7 ± 0.8%). Differentiating ST-HSCF and MPPF displayed a progressive decrease in the frequency of quiescent G0 cells (58.8 ± 1.4% and 48.3 ± 2.5%, respectively; n = 3), correlating with an increase in the frequency of cycling G1 (25.3 ± 0.9% and 37 ± 3.5%) and S-G2/M (15.3 ± 1% and 14.5 ± 0.9%) cells, respectively. Intracellular PI staining also indicated that each subset contained a substantial proportion (12–16%) of cells with a sub-2N DNA apoptotic profile (Fig. 1 B).
Figure 1.
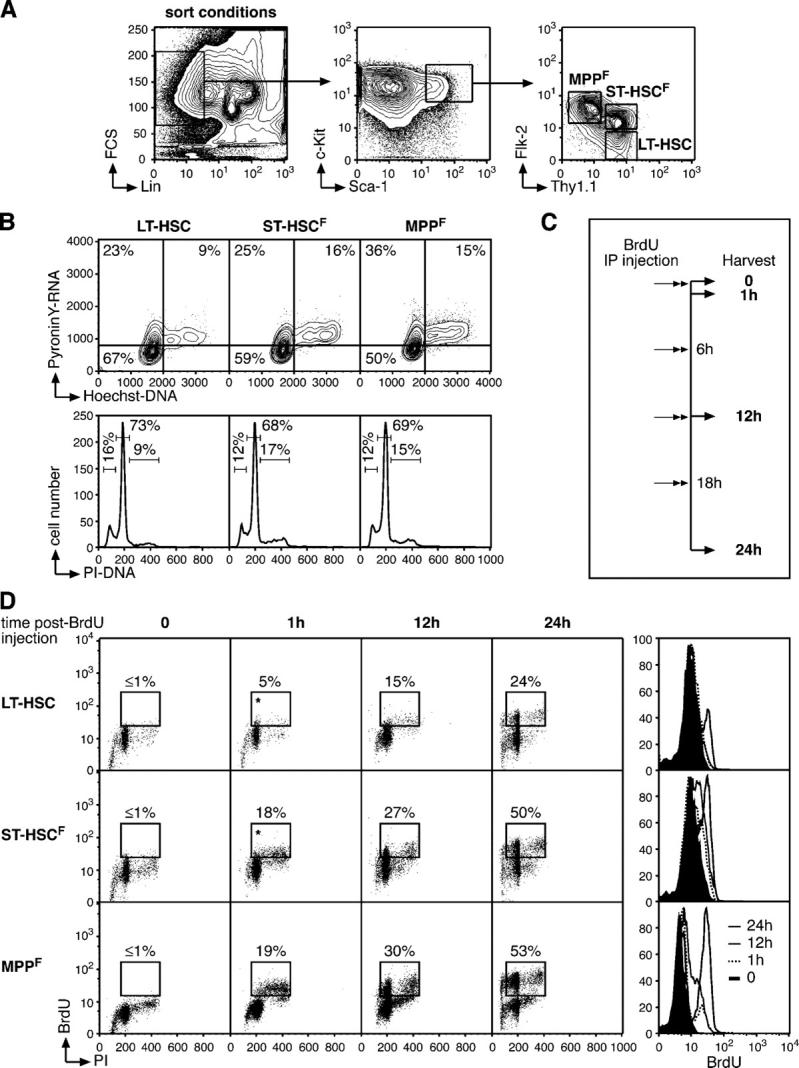
Cell cycle status in the pool of multipotent bone marrow cells. (A) FACS sorting conditions used to purify LT-HSC, ST-HSCF, and MPPF. (B) Cell cycle analyses of purified populations by H/PY (top, contour plot) and PI (bottom, histogram) staining. The percentages of cells in G0 (H2N/PY−), G1 (H2N/PY+), or S-G2/M (H>2N-4N/PY+) and in sub2N (<2N), G0/G1 (=2N), S (>2N and <4N), and/or G2/M (=4N) are indicated. (C) Scheme of the BrdU incorporation experiment. 1 mg BrdU (n = 20 mice/time point) was injected intraperitoneally at the start of the kinetic (t = 0) and every 6 h thereafter, and BM cells were harvested after 1 h (t = 1 h; one injection of BrdU), 12 h (t = 12 h; two injections of BrdU), and 24 h (t = 24 h; four injections of BrdU). (D) Short-term kinetics of BrdU incorporation. At the indicated time after BrdU injection, each population was purified by double FACS sorting, and analyzed by flow cytometry for BrdU/PI staining (dot plots). The percentage of BrdU+ cells is indicated (*, potential background). Histograms (right) indicate the changes over time in BrdU fluorescence intensity.
To analyze the proliferation changes that occur when LT-HSC lose their self-renewal potential and differentiate into ST-HSCF and MPPF, we performed a short-term kinetic analysis of bromodeoxyuridine (BrdU) incorporation (Fig. 1 C). Mice were injected intraperitoneally with BrdU at the start of the experiment and every 6 h thereafter to ensure that constant levels of BrdU remained available in vivo, and mice were killed at the indicated times after BrdU injection. Each subset was first purified by double FACS sorting and then analyzed for BrdU incorporation by flow cytometry (Fig. 1 D). As expected, LT-HSC showed the slowest kinetics of BrdU incorporation with only ∼5% BrdU+ cells after 1 h and approximately one fourth (∼24%) of the population BrdU+ after 24 h. Although this percentage is considerably higher than the ∼8-10%/day described in a previous report (3), we believe that it provides a better approximation of the number of cycling LT-HSC per day because the data were obtained using a more efficient BrdU delivery system (repeated intraperitoneal injection vs. drinking water), a more sensitive BrdU detection system (FACS vs. immunofluorescence), and a more refined combination of cell surface markers to isolate the LT-HSC population (3, 19). Differentiating ST-HSCF and MPPF displayed even faster kinetics of BrdU incorporation, with 18–19% BrdU+ cells after 1 h and more than half of each population having incorporated BrdU after 24 h. These results indicate that although quiescence is the numerically dominant state of the LT-HSC, a substantial fraction of this rare population of cells is actively cycling and differentiating to give rise to the faster proliferating ST-HSCF and MPPF.
HSC long-term engraftment potential and cell cycle status
We and others have previously demonstrated that cycling or recently divided HSC have substantially reduced engraftment capacity upon transplantation into lethally irradiated hosts (16, 17). To study further the impact of cell cycle status on HSC function in hematopoietic reconstitution, double FACS–sorted LT-HSC and Lin−/c-Kit+/Sca-1+/Thy1.1int (KTLS HSC) (20) were stained with H/PY, resorted into G0, G1 or S-G2/M subpopulations, and subsequently tested for their engraftment capacity in limited dilution competitive reconstitution assays (Table I). At doses of 10–50 HSC per recipient, sustained multilineage reconstitution was observed only in animals transplanted with triple-sorted G0 LT-HSC or KTLS HSC (11/24 recipients of 10 G0 HSC and 8/9 recipients of 50 G0 HSC). Furthermore, transplanted G0 HSC generally showed increasing contributions to hematopoiesis over time, as indicated by increasing peripheral blood chimerism, whereas animals transplanted with 10 or 50 LT-HSC or KTLS HSC in either G1 or S-G2/M phases showed little or no contribution to mature peripheral blood lineages, and none exhibited multilineage reconstitution for >12 wk after transplant. However, G0, G1 and S-G2/M phase LT-HSC subpopulations readily proliferated and differentiated in liquid culture (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20050967/DC1), suggesting that the engraftment defect of cycling HSC does not result from an increased susceptibility to apoptosis or from an intrinsically impaired capacity to proliferate and/or to differentiate. These results indicate that the ability of HSC to functionally engraft irradiated recipients dramatically decreases as soon as they exit quiescence and enter G1.
Table I.
HSC engraftment capacity as a function of cell cycle status
| 3–4 wk
|
9–12 wk
|
>18 wk
|
|||||
|---|---|---|---|---|---|---|---|
| Phase | Cell # | Frequencyreconstitution(#/total) | Average chimerism(range)engraftment type(#/reconstituted) | Frequencyreconstitution(#/total) | Average chimerism(range)engraftment type(#/reconstituted) | Frequencyreconstitution(#/total) | Average chimerism(range)engraftment type(#/reconstituted) |
| LT-HSC | |||||||
| G0 | 10 | 57% (8/14) | 1.1% (0.1–3.9%) M + L (8/8) |
57% (8/14) | 4.7% (0.3–15.2%) M + L (8/8) |
54% (7/13) | 7.5% (0.6–23.7%) M + L (7/7) |
| 50 | 100% (5/5) | 1.5% (0.1–5.8%) M + L (5/5) |
100% (5/5) | 5.7% (0.5–14.3%) M + L (5/5) |
100% (5/5) | 11.8% (0.3–35.3%) M + L (4/5), L only (1/5) |
|
| G1 | 10 | 8% (1/12) | 0.2% M only (1/1) |
0% (0/11) | n/a | 0% (0/11) | n/a |
| 50 | 50% (3/6) | 4.2% (0.1–12.5%) M only (3/3) |
0% (0/5) | n/a | 0% (0/5) | n/a | |
| S-G2/M | 10 | 0% (0/9) | n/a | 0% (0/9) | n/a | 0% (0/9) | n/a |
| 50 | 50% (3/6) | 0.4% (0.2–0.7%) M + L (3/3) |
33% (2/6) | 0.35% (0.3–0.4%) M + L (1/2), L only (1/2) |
33% (2/6) | 0.15% (0.1–0.2%) L only (2/2) |
|
| KTLS HSC | |||||||
| G0 | 10 | 50% (6/12) | 0.7% (0.1–1.2%) M + L (5/6), M only (1/6) |
42% (5/12) | 3% (0.2–10.6%) M + L (5/5) |
42% (5/12) | 3.2% (0.3–8.3%) M + L (4/5), L only (1/5) |
| 50 | 100% (4/4) | 0.7% (0.1–1.2%) M + L (4/4) |
100% (4/4) | 2.9% (0.4–8.2%) M + L (4/4) |
100% (4/4) | 3.0% (0.2–6.2%) M + L (4/4) |
|
| G1 | 10 | 0% (0/10) | n/a | 0% (0/10) | n/a | 0% (0/10) | n/a |
| 50 | 25% (1/4) | 0.3% M (1/1) |
0% (0/4) | n/a | 0% (0/4) | n/a | |
| S-G2/M | 10 | 0% (0/10) | n/a | 0% (0/10) | n/a | 0% (0/10) | n/a |
| 50 | 20% (1/5) | 0.6% M + L (1/1) |
20% (1/5) | 0.3% L only (1/1) |
0% (0/5) | n/a | |
LT-HSC and KTLS HSC were first double sorted from C57BL/6-Ly5.1 donor mice, stained with H/PY, resorted for cell cycle G0 (H2N/PY−), G1 (H2N/PY+), and S-G2/M (H>2N-4N/PY+) subpopulations and tested for their engraftment capacities by competitive reconstitution assays. 10 or 50 cells were transplanted into lethally irradiated congenic C57BL/6-Ly5.2 recipient mice, together with 3 × 105 Ly5.2+ helper BM cells. Recipient mice were considered to be reconstituted by Ly5.1+ donor cells if the frequencies of Ly5.1+ donor-derived myeloid (M, Gr-1+, or Mac-1+) or lymphoid (L, B220+, or CD3+) cells detected in the peripheral blood at the indicated times after transplantation were greater than background levels (0.1% as determined by parallel analysis of untransplanted control C57BL/6-Ly5.2 mice). M+L indicates multilineage reconstitution. n/a, not applicable.
Proliferation status in differentiating progenitor populations
Seeded by differentiating HSC, hematopoiesis progresses with the production of both myeloid and lymphoid lineages through the generation of common myeloid progenitors (CMP; Lin−/IL-7R−/c-Kit+/Sca-1−/CD34+/FcγRlow) (21) and common lymphoid progenitors (CLP; Lin−/IL-7R+/Thy1.1−/c-Kitint/Sca-1int) (22). Oligolineage-committed CLP ultimately produce mature lymphocytes, including splenic B cells (B220+/CD19+), whereas CMP differentiate into megakaryocytic/erythroid progenitors (MEP; Lin−/IL-7R−/c-Kit+/Sca-1−/CD34−/FcγRlow) and myelomonocytic progenitors (GMP; Lin−/IL-7R−/c-Kit+/Sca-1−/CD34+/FcγRhigh) (23), which finally produce mature myeloid cells, including granulocytes (Gr; Gr-1+/Mac-1+; Fig. 2, A and B).
Figure 2.

Cell cycle status in progenitor and mature cell populations. (A and B) FACS sorting conditions used to purify myeloid progenitors (CMP, GMP, and MEP), mature Gr, lymphoid progenitors (CLP), and mature splenic B cells (B). (C and D) Short-term kinetics of BrdU incorporation (n = 5 mice/time point). At the indicated time after BrdU injection, each population was first purified by double FACS sorting and then analyzed by flow cytometry for BrdU/PI staining (dot plots). The percentages of BrdU+ cells are indicated. Histograms on the left represent intracellular PI staining (with the percentage of cells in G0/G1, S, and/or G2/M indicated), and histograms on the right represent the changes over time in BrdU fluorescence intensity.
To analyze the changes in proliferation occurring during lineage commitment and maturation, we performed similar H/PY double staining analysis (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20050967/DC1) and short-term kinetic analysis of BrdU incorporation, with intraperitoneal BrdU injection every 6 h (Fig. 2, C and D). Within the myeloid lineage, CMP and MEP displayed similar profiles of DNA content by PI staining, with ∼20% of the population in S, G2, or M phase, whereas H/PY staining indicated a twofold higher frequency of cells in G1 phase within the CMP compartment. Consistently, CMP showed faster kinetics of BrdU incorporation than MEP, with 92 versus 77% of these populations BrdU+ after 24 h. GMP displayed an even higher proliferation index, with almost no quiescent G0 subpopulation, approximately one third of the population in S, G2, or M phase, and every cell BrdU+ after 24 h. Interestingly, approximately one third of the noncycling Gr population became BrdU+ by 24 h, suggesting that it requires ∼18–24 h for a proliferative granulocyte progenitor (perhaps a GMP) to generate growth-arrested mature granulocytes (Fig. 2 C). Within the lymphoid lineage, CLP displayed a low proliferation index, with only 35% of cells BrdU+ after 24 h (Fig. 2 D). Resting mature B lymphocytes did not show any BrdU incorporation within the time frame of the experiment, thereby confirming that de novo production of mature B lymphocytes from cycling lymphoid progenitors requires >24 h (23).
Collectively, these results indicate that the process of hematopoietic differentiation is accompanied by a progressive reduction in the percentage of G0 cells (LT-HSC, 67%; ST-HSC, 59%; MPP, 50%; CMP, 49%; and GMP, 4%) and a correlative increase in the intrinsic proliferation rate of each progenitor population, indicating that cell cycle distribution is of critical importance for compartment dynamics and production of the various lineages of mature blood cells. Moreover, these results can be used to correlate the intrinsic proliferation rates of multipotent and oligopotent precursor cells with their population size in vivo (Fig. 3, A and B). Hence, the rare population of LT-HSC slowly and asynchronously differentiates into ST-HSCF and then MPPF, both of which proliferate at approximately three times the LT-HSC rate despite the fact that MPPF accumulate in vivo at about twice the frequency of ST-HSCF. Differentiation of oligolineage-committed progenitors is also associated with a marked proliferative expansion whose magnitude, at least for the myelomonocytic lineage, directly correlates with the size of the corresponding populations. GMP exhibit twice the proliferation capacity of CMP and accumulate at approximately five times their frequency. In contrast, neither MEP nor CLP show a direct correlation between their proliferation rates and population size in vivo because both exhibit a relatively large pool size given their restricted proliferation capacities. One explanation for these expanded pool sizes could be that these cell populations have a survival advantage, as suggested by the increased expression of at least one key mediator of cell survival, Bcl2 (24), in MPPF and CLP compared with LT-HSC (unpublished data).
Figure 3.
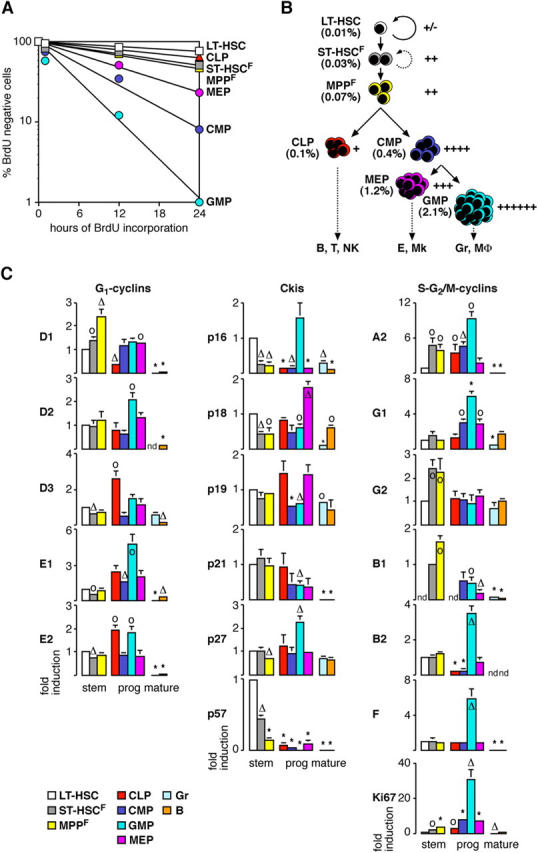
Proliferation index and status of the cell cycle machinery during hematopoietic differentiation. (A) Semilogarithmic plot giving the proportion of BrdU-negative stem and progenitor cells. (B) Schematic and color coded representation correlating the proliferation index (right) of each stem and progenitor compartment with their frequency in vivo (left). The different lineages of mature cells produced by each progenitor population are also indicated. B, B lymphocytes; T, T lymphocytes; NK: natural killer cells; E: erythrocytes; Mk: megakaryocytes; MΦ: macrophages. (C) Analysis by qRT-PCR of the expression levels of various components and regulators of the cell cycle machinery during hematopoietic differentiation. Each value has been standardized for β-actin expression levels. For each gene, the relative expression levels in stem, progenitor (prog), and mature cell (mature) populations are expressed as fold induction compared with the levels (set to 1) detected in LT-HSC (except for cyclin B1, in which ST-HSCF was used instead). Results (means ± SD) are obtained from three independently sorted sets of populations. *, P ≤ 0.001; Δ, P ≤ 0.01; o, P ≤ 0.05. nd, not detectable.
Molecular regulation of the cell cycle machinery in stem and progenitor cells
To gain insights into the molecular control of proliferation during steady-state hematopoiesis, we analyzed the expression levels of a select number of components and regulators of the cell cycle machinery using quantitative real-time RT-PCR (qRT-PCR; Fig. 3 C and Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20050967/DC1). Within the pool of multipotent BM cells, LT-HSC expressed the highest levels of most of the Ckis and the lowest levels of most of the cyclins. Strikingly, differentiation of LT-HSC into ST-HSCF and then MPPF specifically correlated with increased expression of cyclin D1, whereas the expression of the other D cyclins did not change. Differentiation toward MPPF was also accompanied by decreased expression of at least four Ckis (p16, p18, p27, and p57), which together correlated with a partial relief of Rb-mediated transcriptional repression (as evidenced by induction of cyclin A2 but not E cyclins) and with a specific increase in the expression levels of two mitotic cyclins, cyclin G2 and cyclin B1.
In the pool of myeloid progenitors, differentiation toward GMP was specifically associated with induction of cyclin D2, which presumably overcame the inhibitory threshold imposed by relatively high levels of Ckis (mostly p16 and p27), as evidenced by the complete relief of Rb-mediated transcriptional repression (both E cyclins and cyclin A2 induction) and the induction of numerous mitotic cyclins in this compartment (cyclins G1, B1, B2, and F). In contrast, CMP and MEP did not show major induction of any D cyclins, expressed high levels of Ckis (mainly p18), and had strong Rb-mediated repression of E2F target genes (almost no E cyclins or cyclin A2 induction), but still displayed accumulation of a limited number of mitotic cyclins (mostly cyclin G1) that likely contribute to their intrinsic proliferation index. Previous reports have shown that myeloid progenitors express higher levels of p27 and lower levels of p21 than LT-HSC (8, 9). Strikingly, our analysis indicated that among all myeloid progenitors, only GMP displayed this inverted ratio of p21 and p27 (Fig. 3 C and Fig. S3).
Interestingly, lymphoid differentiation also correlated with increased expression of one specific D cyclin, cyclin D3, which is functionally important for lymphocyte development (25) and was presumably able to overcome the Ckis' inhibitory threshold and relieve Rb-mediated transcriptional repression (cyclins E2 and A2 induction). However, CLP did not show substantial induction of any mitotic cyclins, suggesting that other as yet unidentified regulators may limit their proliferation during the S or G2/M phases of the cell cycle. As expected, mature hematopoietic cell populations generally displayed low levels of cyclins and of the proliferation marker Ki67, as well as rather high levels of Ckis, confirming the results of the BrdU incorporation studies (Fig. 2). Together, these results provide a detailed description of the cell cycle machinery controlling HSC and progenitors proliferation during steady-state hematopoiesis.
Proliferation changes during HSC mobilization
Treatment with a combination of cyclophosphamide (Cy) plus granulocyte-CSF (G) is commonly used in human clinical transplantation to increase the numbers of HSC available in the peripheral blood. In mice, as in humans, such treatment drives HSC self-renewing proliferation and induces HSC migration from the BM into the bloodstream (Fig. 4 A) (4, 5, 26). After administration of Cy plus two daily doses of G (D2 Cy/G treatment), the BM HSC population dramatically expands, reaching ∼10–12 times the size of the HSC compartment in untreated animals. After two additional daily doses of G (D4 Cy/G treatment), BM HSC frequency declines, and HSC begin to appear in increased numbers in mobilized peripheral blood (mpb; 10–fold increase) and in spleen (100-fold increase). Previous experiments have indicated that all HSC divide in response to Cy/G treatment (4) and that mpb HSC derive from recently divided BM HSC that transit through the bloodstream as they move from the BM to the spleen (5).
Figure 4.

Cell cycle status in mobilized HSC. (A) Schematic of the Cy/G treatment used to mobilize HSC. Mice were injected intraperitoneally with a single dose of Cy (4 mg/mouse) and injected subcutaneously with recombinant human G (5 μg/mouse) for two successive days (D2 Cy/G treatment) or 4 successive days (D4 Cy/G treatment). For the BrdU experiments, mice were injected intraperitoneally with BrdU (n = 15 normal mice, 3 D2 mice, or 7 D4 mice, respectively, per time point) 1 h (t = 1 h) or 12 h (t = 12 h, with repeated injection at 6 h) before killing them at the end of the D2 or D4 Cy/G treatment. Histograms illustrate the increased numbers of KTLS HSC found in BM, spleen (spl), blood (bld), or mpb at D2 and D4 Cy/G treatment compared with untreated mice (reference 4). (B) Intracellular PI staining analysis of DNA content. The percentages of cells in G0/G1, S, and/or G2/M are indicated. (C) Kinetics of BrdU incorporation. At the indicated times after BrdU injection, each population of HSC was first purified by double FACS sorting and then analyzed by flow cytometry for BrdU/PI staining (dot plots). The percentages of BrdU+ cells are indicated. Histograms at the bottom indicate the changes over time in BrdU fluorescence intensity.
To study further the mechanisms by which cell cycle progression is regulated in mobilized HSC, we analyzed KTLS HSC for cell cycle profile (Fig. 4 B) and short-term kinetic analysis of BrdU incorporation (Fig. 4 C) at D2 and D4 Cy/G treatment. In comparison to untreated BM HSC, D2 BM HSC exhibited a substantial increase in the frequency of cells in S-G2/M phase (35.4 ± 5.1% vs. 9.1 ± 0.5%; n = 4) and very rapid kinetics of BrdU incorporation, with ∼85% of the cells having incorporated BrdU after only 12 h, suggesting a rapid recruitment of self-renewing HSC into cycle. Interestingly, D4 BM HSC displayed an almost normal cell cycle profile, with only 10.9 ± 1.5% of cells in S-G2/M phase and levels of BrdU incorporation very similar to those of untreated BM HSC. In contrast, D4 spleen HSC showed increased proliferation, with 20.5 ± 2.3% cells in S-G2/M phase, and increased levels of BrdU incorporation, with ∼40% BrdU+ cells after 12 h. At D4, mpb HSC displayed DNA content very similar to untreated BM HSC (8.5 ± 1.2% cells in S-G2/M phase), although they are found exclusively in the G0/G1 fraction after another day of G administration (5). However, D4 mpb HSC displayed intermediate levels of BrdU incorporation between those of D4 BM and spleen HSC, suggesting that HSC found in the bloodstream may derive from both recently divided BM and spleen HSC. To determine whether a subset of noncycling HSC could also be released into the bloodstream during HSC mobilization, we analyzed BrdU incorporation into mpb HSC in D4 Cy/G-treated mice exposed to BrdU during the final 36 h of the mobilization protocol (Fig. S4, available at http://www.jem.org/cgi/content/full/jem.20050967/DC1). Remarkably, every D4 mpb HSC was BrdU positive after only 36 h of labeling, clearly demonstrating that mobilized HSC are specifically released into the bloodstream only after having recently divided.
To investigate the impact of mobilization on HSC function in hematopoietic reconstitution, we also tested double FACS–sorted LT-HSC isolated from BM of untreated and D2 Cy/G-treated mice for their engraftment capacity in limited dilution competitive reconstitution assays (Table II). Sustained multilineage reconstitution was observed in every animal transplanted with 50 untreated BM LT-HSC. In contrast, less than half of the animals transplanted with 50 D2 BM LT-HSC displayed enduring, multilineage hematopoietic engraftment. Moreover, those mice receiving D2 BM LT-HSC that did show donor cell engraftment exhibited substantially lower (approximately sixfold) contributions to overall blood cell chimerism as compared with those receiving untreated BM LT-HSC. These findings clearly demonstrate that the 10-fold expanded population of D2 BM HSC has considerably reduced engraftment capacity, in accordance with its increased proliferation rate.
Table II.
Engraftment capacity of mobilized BM LT-HSC
| 4 wk
|
8 wk
|
12–18 wk
|
||||
|---|---|---|---|---|---|---|
| Cell # | Frequencyreconstitution(#/total) | Average chimerism(range)engraftment type(#/reconstituted) | Frequencyreconstitution(#/total) | Average chimerism(range)engraftment type(#/reconstituted) | Frequencyreconstitution(#/total) | Average chimerism(range)engraftment type(#/reconstituted) |
| Untreated BM LT-HSC |
||||||
| 50 | 100% (8/8) | 11.07% (2.3–22.7%) M + L (8/8) |
100% (7/7) | 29.2% (6.7–56.9%) M + L (7/7) |
100% (7/7) | 30.2% (6.2–56.2%) M + L (7/7) |
| D2 Cy/G mobilized BM LT-HSC |
||||||
| 50 | 46% (7/15) | 2.9% (0.2–12.5%) M + L (7/7) |
40% (6/15) | 6.1% (0.4–26.8%) M + L (4/6), L only (2/6) |
40% (6/15) | 4.6% (0.1–18.1%) M + L (4/6), L only (2/6) |
LT-HSC (Lin−/c-Kit+/Sca-1+/Thy1.1int/Flk-2−) were double FACS–sorted from BM of untreated C57BL/6-Ly5.1 donor mice or from mice subjected to the D2 Cy/G mobilization treatment and tested for their engraftment capacities by limited dilution competitive reconstitution assays. 50 cells were transplanted into lethally irradiated congenic C57BL/6-Ly5.2 recipient mice together with 3 × 105 Ly5.2+ helper BM cells. Recipient mice were considered to be reconstituted by Ly5.1+ donor cells if the frequencies of Ly5.1+ donor-derived myeloid (M, Gr-1+, or Mac-1+) or lymphoid (L, B220+, or CD3+) cells detected in the peripheral blood at the indicated times after transplantation were greater than background levels (≥0.1% as determined by parallel analysis of untransplanted control C57BL/6-Ly5.2 mice). M + L indicates multilineage reconstitution.
Collectively, these results indicate that the self-renewing proliferative response of BM HSC induced by Cy/G treatment is only transient and that when HSC migrate to the periphery, starting at D3 and continuing at D4, the HSC that remain in the BM revert to normal proliferation rates (Fig. 5, A and B). In contrast, HSC migrating to the spleen remain actively cycling even though they may be more likely to differentiate to repopulate the injured hematopoietic system than to self-renew, as suggested by the reduction in spleen HSC frequency typically observed after cessation of the Cy/G treatment (4). These results further clarify the ongoing debate about the origin of mobilized HSC by demonstrating that HSC are rapidly released into the bloodstream only after progressing through M phase.
Figure 5.

Proliferation index and status of cell cycle machinery during HSC mobilization. (A) Semilogarithmic plot giving the proportion of BrdU-negative HSC in untreated mice and mice that received D2 and D4 Cy/G treatment. (B) Schematic and color-coded representation correlating the proliferation index of each HSC population following D2 and D4 Cy/G treatment with their pool size in vivo. (C) Analysis by qRT-PCR of the expression levels of various components and regulators of the cell cycle machinery following D2 and D4 Cy/G treatment. Results represent means ± SD of duplicate measurement performed on three independently sorted sets of HSC populations. Each value has been standardized for β-actin expression levels and is expressed as fold induction compared with the levels (set to 1) detected in untreated BM HSC. *, P ≤ 0.001; Δ, P ≤ 0.01; o, P ≤ 0.05.
Molecular regulation of the cell cycle machinery during HSC mobilization
Next, we investigated by qRT-PCR the mobilization-induced changes in expression levels of key components and regulators of the cell cycle machinery (Fig. 5 C). Self-renewing proliferation in D2 BM specifically correlated with a modest induction of cyclin D3 expression, whereas cyclin D1 stayed unchanged and cyclin D2 levels actually decreased. D2 BM HSC also displayed reduced levels of all Ckis except for p21, which was increased possibly because of its requirement as a scaffold protein for the active cyclin D–Cdk4-6 complexes. Together, these molecular events contributed to the relief of Rb-mediated transcriptional repression (cyclins E1 and A2 induction) and to the induction of all the mitotic cyclins tested except for cyclin G2. The importance of these changes is further attested by their later reversion in D4 BM HSC, which were indistinguishable from untreated BM HSC in terms of proliferation kinetics and population size. Hence, D4 BM HSC showed repression of all D cyclins, reinduction of all Ckis, and restoration of Rb-mediated transcriptional repression, which together provide a strong proliferation brake leading to an almost complete absence of mitotic cyclin expression. In contrast, mobilized D4 spleen HSC exhibited activated cell cycle machinery, though they did not display induction of any D cyclins. This result suggests that D4 spleen HSC are either becoming refractory to mitogenic stimulation or that alternative pathways to cyclin D–Cdk4-6 might sense such mitogens (6). In fact, mobilized D4 spleen HSC showed an easing of Rb-mediated transcriptional repression (cyclin A2 induction), reduced expression of most of the Ckis (except for p21 and p27), and a striking and unique induction of cyclin B1 rather than the broad induction of all mitotic cyclins seen in self-renewing D2 BM HSC. As expected, proliferating D2 BM HSC and D4 spleen HSC also harbored increased expression levels of Ki67 and of cdc-20/APC, which mediates proteolytic degradation of A and B cyclins in late G2 phase. Altogether, these results provide a comprehensive description of the molecular networks controlling HSC proliferation during the various phases of cytokine-mediated mobilization.
Molecular regulation of quiescence in HSC
To gain insights into how exit from quiescence and cell cycle entry is regulated in LT-HSC, we also used qRT-PCR to investigate potential changes in the expression levels of genes known to play a role in establishing and/or maintaining quiescence or in regulating the G0-to-G1 transition (27) that occurs when LT-HSC differentiate toward MPPF or when KTLS HSC expand in response to Cy/G treatment (Fig. 6). Differentiation of LT-HSC into ST-HSCF was associated with a striking increase in the expression level of the G0/G1 transition kinase cyclin C, although cyclin C was again down-regulated at the MPPF stage. As expected, LT-HSC predominantly expressed p130, which is mainly found in quiescent G0 cells (7), over p107 and Rb (Fig. S3). Rb expression did not change during LT-HSC differentiation into ST-HSCF and MPPF, whereas p107 levels increased from the ST-HSCF stage and stayed elevated in MPPF, and p130 levels decreased at the MPPF stage. In contrast, the expression levels of E2F4, the partner of p130 for transcriptional repression of E2F target genes in G0 cells and of Rb and p107 in proliferative cells, did not change, nor did expression of E2F6, another E2F family member associated with quiescence. The proliferative response associated with mobilization did not induce changes in the expression levels of most G0 and G0/G1 transition genes other than p130, which was down-regulated in both proliferative D2 BM HSC and D4 spleen HSC and conversely reexpressed to levels found in untreated, resting BM HSC in D4 BM HSC (Fig. 6). Collectively, these results suggest a switch from p130–E2F4 complexes to p107- and Rb-associated complexes as cells are recruited into cycle. Most importantly, they indicate that quiescence in LT-HSC is actively established and/or maintained by specific molecular mechanisms that support HSC function in hematopoietic reconstitution.
Figure 6.
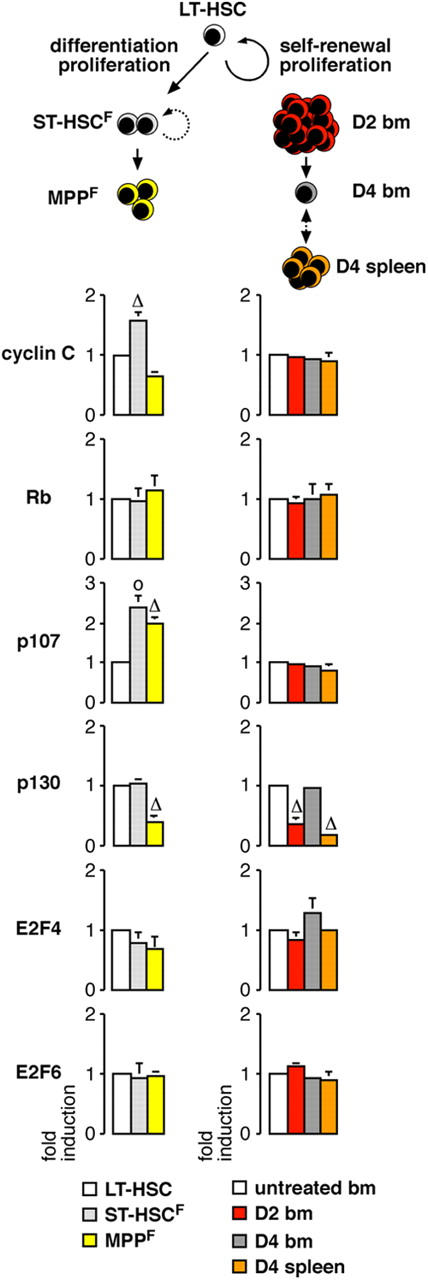
Molecular control of quiescence. Analysis by qRT-PCR of the expression levels of various regulators of G0 or G0/G1 phase transition in homeostatic proliferation during LT-HSC differentiation toward ST-HSCF and MPPF and stress-induced HSC self-renewing proliferation following D2 and D4 Cy/G treatment. The results represent the means ± SD of duplicate measurements performed on three independently sorted sets of populations. Each value has been standardized for β-actin expression levels and is expressed as fold induction compared with the levels (set to 1) detected in LT-HSC or untreated BM HSC. *, P ≤ 0.001; Δ, P ≤ 0.01; o, P ≤ 0.05.
Discussion
In this paper, we demonstrate that the rare population of adult BM LT-HSC exists in two distinct states that are both equally important for their in vivo functions as stem cells: a numerically dominant quiescent state, which is critical for LT-HSC function in hematopoietic reconstitution, and a proliferative state, which represents almost one fourth of this population and is essential for LT-HSC functions in differentiation and self-renewal. We show that when LT-HSC exit quiescence and enter G1 as a prelude to cell division, at least two critical events occur: first, during the G1 and subsequent S-G2/M phases, they temporarily lose efficient in vivo engraftment activity while retaining in vitro differentiation potential, and second, they select the particular cell cycle proteins that are associated with specific developmental outcomes (self-renewal vs. differentiation) and developmental fates (myeloid vs. lymphoid). A growing body of published literature already exists demonstrating the functional involvement of many of these cell cycle proteins in maintaining normal numbers of hematopoietic cells (9–15). In this paper, we provide a global description of the precise stages of hematopoietic differentiation in which such regulators likely play a role and a comprehensive description of their relative expression levels that can directly be used to further understand these published studies and to design future experiments targeting the relevant cell populations.
Our results demonstrate that LT-HSC function in hematopoietic reconstitution after transplantation critically depends on maintenance of the quiescent state. Hence, actively cycling G1 and S-G2/M phase LT-HSC display dramatically impaired long-term engraftment and multilineage reconstitution capabilities compared with G0 LT-HSC. Because purified G1 and S-G2/M LT-HSC readily proliferate and differentiate in vitro, their functional deficiencies in vivo may result in part from impaired or altered homing after intravenous transplant. Such an explanation has some support from observations in human CD34+ BM progenitor cells, which in G0/G1 phase show increased adhesiveness to stromal cells (28) and appear to migrate to the BM of conditioned recipients with increased frequency over S-G2/M cells (17). The recent microarray analysis of quiescent and proliferative mouse HSC (15) also suggests the importance of migratory molecules for establishing HSC quiescence and supports the hypothesis that at least a component of the enhanced engraftment capacity of G0 LT-HSC may relate to a superior BM homing efficiency. The demonstration that HSC cell cycle status directly and dramatically affects HSC engraftment function has important implications for human therapy because cycling HSC derived from mpb or from ex vivo–expanded HSC cultures are commonly used, or proposed for use, in clinical transplantation. It suggests that strategies designed to reestablish quiescence in HSC before transplantation should dramatically improve their efficiency. A detailed investigation of cycling and resting HSC is now essential to address completely the functional relationship between their proliferation, homing, and engraftment capacity. In this context, the analysis of HSC cell cycle progression and regulation during homeostatic HSC maintenance, induced HSC expansion, and HSC differentiation (Fig. 7) presented here provides a molecular foothold from which such functional relationships can be further examined.
Figure 7.

Molecular networks linking proliferation and cell fate decision in HSC and progenitor cells. Summary of the proliferation index, phase of the cell cycle, and status of the cell cycle machinery associated with differentiation, self-renewal, or long-term engraftment capacity in HSC and progenitor cells. Several regulators of the cell cycle found to be specifically associated with maintenance of quiescence, differentiation, self-renewal, expansion, and mobilization are identified (underlined and bold). Such genes represent potential targets to encourage self-renewal, prevent differentiation, and reestablish quiescence during ex vivo expansion of HSC for clinical transplantation. They also represent potential targets for deregulation during leukemic transformation.
Because cycling LT-HSC display impaired function in hematopoietic reconstitution, one may wonder whether non-G0 LT-HSC should be called LT-HSC at all, as the name stands for long-term reconstituting cells. Historically, LT-HSC have been identified and isolated based on their ability to reconstitute the blood system of an irradiated host. With the evolution of the stem cell field, LT-HSC are now more precisely defined by their unique ability to balance self-renewal and differentiation division without depletion of the stem cell pool. This definition implies that LT-HSC must contain a proliferative component to fulfill both self-renewal and differentiation divisions. In fact, it has been shown that the only precursors for LT-HSC in mouse BM are the LT-HSC themselves (29), and as all LT-HSC enter cell cycle at least once every 15–30 d (3), the products of LT-HSC division must include G0 phase LT-HSC. It is also important to note that although our analysis does demonstrate a substantially more robust capacity for hematopoietic reconstitution in the G0 fraction of LT-HSC, these data do not exclude the existence of less potent engraftment potential within other cell cycle stages. In previous experiments (16, 30), though reduced compared with G0/G1 cells, long-term multilineage engraftment function could be detected within the S-G2/M population when large numbers of these cells were transplanted. Thus, LT-HSC at all cell cycle stages do retain some engraftment capacity and, for these reasons, we feel it is appropriate to continue to refer to the entire population as LT-HSC.
Our results indicate that LT-HSC are maintained in G0 phase of the cell cycle through the concerted action of high levels of Ckis, and predominant expression of p130–E2F4 complexes, which are thought to transcriptionally repress most of the E2F target genes involved in S phase progression (7). Together, these factors preclude accumulation of high levels of cyclins (mostly mitotic cyclins) and limit LT-HSC proliferation rates. Although every member of the Ink4 and Cip/Kip families of Ckis is highly expressed in LT-HSC, they are not functionally equivalent in preserving HSC quiescence. Analyses of p21 and p18 knockout mice have demonstrated their critical functions in maintaining normal numbers of resting LT-HSC capable of long-term engraftment and multilineage reconstitution (8, 10). In contrast, p27-null mice have a normal HSC compartment but show increased numbers of progenitor cells (9), whereas p16-null mice specifically develop thymic hyperplasia (31) and p19- or p57-deficient mice do not show overt hematopoietic abnormalities (32, 33). These results suggest that these Ckis are either dispensable for HSC function or are highly specialized in controlling proliferation in subsets of differentiating hematopoietic cells. However, further investigation may uncover additional functions in controlling HSC proliferation. Hence, the recent study of bmi-1 knockout mice demonstrated a novel and critical function for p16 in regulating adult HSC self-renewing proliferation (11), which was not previously appreciated in p16-null mice (31). Mice deficient for single Rb proteins or for E2F4 also show defects in various hematopoietic lineages (34) but have not yet been analyzed specifically for changes in the stem cell compartment. Future investigations will therefore be required to assay their importance in controlling HSC quiescence.
Although analyses of genetically modified mice are usually performed to demonstrate the functional involvement of specific cell cycle components in regulating stem and progenitor cell proliferation, these in vivo approaches have also their limitations. A good example comes from analyses of the D cyclin–Cdk4-6 complexes, whose critical function during hematopoietic development has only recently been appreciated through the generation of D1/D2/D3 triple knockout (12) and Cdk4/6 double knockout (13) mice. Both mice have complete hematopoietic failure starting at the HSC level and die during late embryonic development due, in part, to severe anemia. However, none of the single or double D cyclins knockout mice previously analyzed display major hematopoietic defects or impaired HSC function. This result emphasizes the functional redundancy existing in vivo between the three D cyclins that limits the understanding of their specific function during hematopoiesis. Our analysis sheds new light on the involvement of each particular D cyclin in controlling stem and progenitor fates (Fig. 7). Hence, HSC differentiation specifically associates with cyclin D1 induction, whereas self-renewing HSC do not show induction of cyclin D1. Furthermore, myeloid differentiation only correlates with cyclin D2 induction, whereas lymphoid differentiation specifically associates with cyclin D3 induction. Together, these findings suggest that specific cellular outcomes may be effected by the induction of a particular D cyclins. In addition, differentiating HSC show specific induction of cyclins G2 and B1, whereas self-renewing HSC display specific induction of cyclin G1 and B2 and no change in cyclin G2 expression (Fig. 7). Hence, cyclin D1, cyclin G2, and, to a lower extent, cyclin B1 may be privileged mediators of HSC differentiation-associated proliferation, whereas cyclin G1 and cyclin B2 could be privileged mediators of HSC self-renewal–associated proliferation. HSC fate determination is controlled by many intrinsic and extrinsic signals, which act through regulation of different signaling pathways. Because all these pathways must eventually converge on cell cycle regulation, it is conceivable that one might drive HSC fate determination by directly manipulating the expression levels of specific components and regulators of the cell cycle machinery, as suggested by the recent demonstration that ex vivo targeting of the Cki p21 permits a relative expansion of human HSC (35). Our results provide a molecular basis for the rational design of strategies to test whether inactivation or activation of specific sets of cyclins may prevent differentiation and favor self-renewal proliferation during ex vivo expansion of HSC.
In conclusion, understanding the particular transcriptional programs used by HSC as they exit quiescence and transit through the cell cycle are likely to reveal how HSC fate decisions are regulated at the molecular level and to provide ways to allow their manipulation for clinical applications. Moreover, because leukemia (and other cancer) stem cells also undergo similar self-renewal versus differentiation cell fate decisions (36), identifying the key entities involved in these decisions could reveal new molecular targets for human therapies.
Materials and Methods
Mice
4–6-wk-old C57BL/6-Thy1.1 mice were used as donors for all stem, progenitor, and mature cell populations. BrdU (Sigma-Aldrich) was administrated by intraperitoneal injection (1 mg/mouse) as a single dose or as repeated doses every 6 h. Cytokine mobilization of HSC by combined administration of Cy/G was performed as previously described (4). For transplantation experiments, cells were purified from C57BL/6-Thy1.1-Ly5.1 mice and transplanted via retroorbital injection into lethally irradiated (950 rad) C57/B6-Ly5.2 congenic recipient mice. Peripheral blood was obtained from the tail vein of each mouse and analyzed by flow cytometry. Donor-derived cells were distinguished from host cells by the expression of different Ly5 antigens (Ly5.1 vs. Ly5.2). All mice were maintained in Stanford University's Research Animal Facility in accordance with institutional guidelines.
Cell sorting
Cell staining and enrichment procedures for sorting of multipotent BM cells (LT-HSC, ST-HSCF, and MPPF), KTLS HSC, myeloid progenitors (CMP, GMP, and MEP), and lymphoid progenitors (CLP) were performed as previously described (19–22). Cells were finally resuspended in HBSS with 2% heat-inactivated FCS and 1 μg/ml PI and sorted on a FACSVantage (Becton Dickinson). Dead cells were gated out by high PI staining and forward light scatter.
BrdU staining
Double FACS–sorted stem, progenitor, and mature cell populations were fixed in 70% ethanol overnight at −20°C, denatured in 2N HCL/0.5% Triton X-100 for 20 min at room temperature, neutralized with 0.1 M sodium borate for 5 min, and stained with anti-BrdU antibody (BD Biosciences) for 30 min at room temperature in PBS/0.5% BSA/0.5% Tween 20. Cells were finally resuspended in 500 μl PBS containing 10 μg RNase A and 5 μg PI, incubated for 30 min, and immediately analyzed on a FACScan (Becton Dickinson).
H/PY staining
Double FACS–sorted stem, progenitor, and mature cell populations were incubated for 45 min at 37°C with 20 μg/ml HOECHST 33342 (Invitrogen) in HBSS medium containing 10% FCS, 20 mM Hepes, 1 g/liter glucose, and 50 μg/ml Verapamil as described previously (5). Pyronin Y (Sigma-Aldrich) was then added at 1 μg/ml, and the cells were incubated for another 15 min at 37°C, washed, and immediately analyzed or sorted by flow cytometry on a UV laser–equipped Flasher II MoFlo (DakoCytomation).
qRT-PCR
Total RNA was isolated using Trizol reagent (Invitrogen) from equivalent numbers of double FACS–sorted stem, progenitor, and mature cell populations (1,000–8,000 cells), digested with DNase I to remove DNA contamination, and used for RT according to the manufacturer's instructions (SuperScript II kit; Invitrogen). qRT-PCR primers were designed using Primer Express software (Applied Biosystems; Table S1, available at http://www.jem.org/cgi/content/full/jem.20050967/DC1). All reactions were performed in an ABI-7000 sequence detection system using SYBR green PCR Core reagents according to the manufacturer's instructions (Applied Biosystems). PCR amplification was performed in a 10-μl final volume containing 1× SYBR Green PCR buffer, 2 mM magnesium chloride, 0.5 mM dNTP mix with dUTP, 8 ng of each primer, 0.1 U AmpErase UNG, 0.25 U AmpliTaq Gold, and 1 μl of cDNA templates (cDNA equivalent of 50 cells per reaction) using 50°C for 2 min and 95°C for 10 min, followed by 45 cycles at 95°C for 15 s and 60°C for 1 min. For each sample, expression of the β-actin gene was used to normalize the amount of the investigated transcript. Statistical analyses were performed using Student's t test for two samples.
Online supplemental material
Fig. S1 depicts the proliferation capacity of LT-HSC cell cycle subpopulations. Fig. S2 shows the cell cycle distribution in progenitor and mature cell populations. Fig. S3 depicts the differential expression of components and regulators of the cell cycle machinery in stem, progenitor, and mature cell populations. Fig. S4 shows that mobilized D4 mpb HSC originate from recently divided HSC. Table S1 provides sequences of qRT-PCR primers. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20050967/DC1.
Acknowledgments
We thank J. Sage, T. Serwold, D. Bhattacharya, D. Rossi, and C. Forsberg for critical readings of the manuscript, L. Jerabek for excellent lab management, S. Smith for antibody production, and L. Hidalgo and B. Lavarro for animal care.
E. Passegué is a fellow of the Jose Carreras International Leukemia Foundation, and S. Giuriato is a fellow of the Lymphoma Research Foundation. A.J. Wagers was supported by a Frederick Frank/Lehman Brothers, Inc. Irvington Institute Fellowship and a Burroughs Wellcome Fund Career Award. This work was supported by grants from the National Institutes of Health (CA55209 and CA86017) and a De Villier Award from the Leukemia and Lymphoma Society to I.L. Weissman.
I.L. Weissman owns stock in Amgen and is a co-founder and member of the Board of Director of Cellerant Inc. The authors have no other conflicting financial interests.
Abbreviations used: BrdU, bromodeoxyuridine; Cdk, cyclin-dependent kinase; Cki, Cdk inhibitor; CLP, common lymphoid progenitors; CMP, common myeloid progenitors; Cy, cyclophosphamide; G, granulocyte-CSF; GMP, myelomonocytic progenitors; Gr, granulocytes; H/PY, HOECHST 33342/Pyronin Y; HSC, hematopoietic stem cells; KTLS HSC, Lin−/c-Kit+/Sca-1+/Thy1.1int HSC; LT-HSC, long-term reconstituting Flk-2− HSC; MEP, megakaryocytic/erythroid progenitors; mpb, mobilized peripheral blood; MPPF, nonself- renewing Flk-2+ multipotent progenitor; PI, propidium iodide; qRT-PCR, quantitative real-time RT-PCR; ST-HSCF, short-term reconstituting Flk-2int HSC.
E. Passegué and A.J. Wagers contributed equally to this work.
A.J. Wagers's present address is Section on Developmental and Stem Cell Biology, Joslin Diabetes Center, Boston, MA 02215.
References
- 1.Weissman, I.L. 2000. Stem cells: units of development, units of regeneration, and units of evolution. Cell. 100:157–168. [DOI] [PubMed] [Google Scholar]
- 2.Bradford, G.B., B. Williams, R. Rossi, and I. Bertoncello. 1997. Quiescence, cycling, and turnover in the primitive hematopoietic stem cell compartment. Exp. Hematol. 25:445–453. [PubMed] [Google Scholar]
- 3.Cheshier, S.H., S.J. Morrison, X. Liao, and I.L. Weissman. 1999. In vivo proliferation and cell cycle kinetics of long-term hematopoietic stem cells. Proc. Natl. Acad. Sci. USA. 96:3120–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morrison, S.J., D.E. Wright, and I.L. Weissman. 1997. Cyclophosphamide/granulocyte colony-stimulating factor induces hematopoietic stem cells to proliferate prior to mobilization. Proc. Natl. Acad. Sci. USA. 94:1908–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wright, D.E., S.H. Cheshier, A.J. Wagers, T.D. Randall, J.L. Christensen, and I.L. Weissman. 2001. Cyclophosphamide/granulocyte colony-stimulating factor causes selective mobilization of bone marrow hematopoietic stem cells into the blood after M phase of the cell cycle. Blood. 97:2278–2285. [DOI] [PubMed] [Google Scholar]
- 6.Sherr, C.J., and J.M. Roberts. 2004. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 18:2699–2711. [DOI] [PubMed] [Google Scholar]
- 7.Smith, E.J., G. Leone, J. DeGregori, L. Jakoi, and J.R. Nevins. 1996. The accumulation of an E2F-p130 transcriptional repressor distinguishes a G0 cell state from a G1 cell state. Mol. Cell. Biol. 16:6965–6976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng, T., N. Rodrigues, H. Shen, Y. Yang, D. Dombkowski, M. Sykes, and D.T. Scadden. 2000. Hematopoietic stem cell quiescence maintained by p21CIP1/WAF1. Science. 287:1804–1808. [DOI] [PubMed] [Google Scholar]
- 9.Cheng, T., N. Rodrigues, D. Dombkowski, S. Stier, and D.T. Scadden. 2000. Stem cell repopulation efficiency but not pool size is governed by p27KIP1. Nat. Med. 6:1235–1240. [DOI] [PubMed] [Google Scholar]
- 10.Yuan, Y., H. Shen, D.S. Franklin, D.T. Scadden, and T. Cheng. 2004. In vivo self-renewing division of haematopoietic stem cells are increased in the absence of the early G1-phase inhibitor, p18INK4c. Nat. Cell Biol. 6:436–442. [DOI] [PubMed] [Google Scholar]
- 11.Park, I.-K., D. Qian, M. Kiel, M.W. Becker, M. Pihalja, I.L. Weissman, S.J. Morrison, and M.F. Clarke. 2003. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 423:302–305. [DOI] [PubMed] [Google Scholar]
- 12.Kozar, K., M.A. Ciemerych, V.I. Rebel, H. Shigematsu, A. Zagozdzon, E. Sicinska, Y. Geng, Q. Yu, S. Bhattacharya, R.T. Bronson, et al. 2004. Mouse development and cell proliferation in the absence of d-cyclins. Cell. 118:477–491. [DOI] [PubMed] [Google Scholar]
- 13.Malumbres, M., R. Sotillo, D. Santamaria, J. Galan, A. Cerezo, S. Ortega, P. Dubus, and M. Barbacid. 2004. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell. 118:493–504. [DOI] [PubMed] [Google Scholar]
- 14.Steinman, R.A. 2002. Cell cycle regulator and hematopoiesis. Oncogene. 21:3403–3413. [DOI] [PubMed] [Google Scholar]
- 15.Venezia, T.A., A.A. Merchant, C.A. Ramos, N.L. Whitehouse, A.S. Young, C.A. Shaw, and M.A. Goodell. 2004. Molecular signatures of proliferation and quiescence in hematopoietic stem cells. PLoS Biol. 2:e301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fleming, W.H., E.J. Alpern, N. Uchida, K. Ikuta, G.J. Spangrude, and I.L. Weissman. 1993. Functional heterogeneity is associated with the cell cycle status of murine hematopoietic stem cells. J. Cell Biol. 122:897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glimm, H., J.H. Oh, and C.J. Eaves. 2000. Human hematopoietic stem cells stimulated to proliferate in vitro lose engraftment potential during their S/G(2)/M transit and do not reenter G(0). Blood. 96:4185–4193. [PubMed] [Google Scholar]
- 18.Cashman, J., B. Dykstra, I. Clark-Lewis, A. Eaves, and C. Eaves. 2002. Changes in the proliferative activity of human hematopoietic stem cells in NOD/SCID mice and enhancement of their transplantability after in vivo treatment with cell cycle inhibitors. J. Exp. Med. 196:1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Christensen, J.L., and I.L. Weissman. 2001. Flk-2 is a marker in hematopoietic stem cell differentiation: a simple method to isolate long-term stem cells. Proc. Natl. Acad. Sci. USA. 98:14541–14546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morrison, S.J., and I.L. Weissman. 1994. The long-term repopulating subset of hematopoietic stem cells is deterministic and isolatable by phenotype. Immunity. 1:661–673. [DOI] [PubMed] [Google Scholar]
- 21.Akashi, K., D. Traver, T. Miyamoto, and I.L. Weissman. 2000. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 404:193–197. [DOI] [PubMed] [Google Scholar]
- 22.Kondo, M., I.L. Weissman, and K. Akashi. 1997. Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell. 91:661–672. [DOI] [PubMed] [Google Scholar]
- 23.Forster, I., P. Vieira, and K. Rajewsky. 1989. Flow cytometric analysis of cell proliferation dynamics in the B cell compartment of the mouse. Int. Immunol. 1:321–331. [DOI] [PubMed] [Google Scholar]
- 24.Domen, J. 2000. The role of apoptosis in regulating hematopoiesis and hematopoietic stem cells. Immunol. Res. 22:83–94. [DOI] [PubMed] [Google Scholar]
- 25.Sicinska, E., I. Aifantis, L. Le Cam, W. Swat, C. Borowski, Q. Yu, A.A. Ferrando, S.D. Levin, Y. Geng, H. von Boehmer, and P. Sicinski. 2003. Requirement for cyclin D3 in lymphocyte development and T cell leukemias. Cancer Cell. 4:451–461. [DOI] [PubMed] [Google Scholar]
- 26.Neben, S., K. Marcus, and P. Mauch. 1993. Mobilization of hematopoietic stem and progenitor cell subpopulations from the marrow to the blood of mice following cyclophosphamide and/or granulocyte colony-stimulating factor. Blood. 81:1960–1967. [PubMed] [Google Scholar]
- 27.Sage, J. 2004. Cyclin C makes an entry into the cell cycle. Dev. Cell. 6:607–616. [DOI] [PubMed] [Google Scholar]
- 28.Yamaguchi, M., K. Ikebuchi, F. Hirayama, N. Sato, Y. Mogi, J. Ohkawara, Y. Yoshikawa, K. Sawada, T. Koike, and S. Sekiguchi. 1998. Different adhesive characteristics and VLA-4 expression of CD34(+) progenitors in G0/G1 versus S+G2/M phases of the cell cycle. Blood. 92:842–848. [PubMed] [Google Scholar]
- 29.Uchida, N., and I.L. Weissman. 1992. Searching for hematopoietic stem cells: evidence that Thy-1.1loLin−Sca-1+ cells are the only stem cells in C57BL/Ka-Thy-1.1 bone marrow. J. Exp. Med. 175:175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uchida, N., A.M. Friera, D. He, M.J. Reitsma, A.S. Tsukamoto, and I.L. Weissman. 1997. Hydroxyurea can be used to increase mouse c-kit+Thy-1.1(lo)Lin-/loSca-1(+) hematopoietic cell number and frequency in cell cycle in vivo. Blood. 90:4354–4362. [PubMed] [Google Scholar]
- 31.Sharpless, N.E., N. Bardeesy, K.H. Lee, D. Carrasco, D.H. Castrillon, A.J. Aguirre, E.A. Wu, J.W. Horner, and R.A. DePinho. 2001. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature. 413:86–91. [DOI] [PubMed] [Google Scholar]
- 32.Zindy, F., J. van Deursen, G. Grosveld, C.J. Sherr, and M.F. Roussel. 2000. INK4d-deficient mice are fertile despite testicular atrophy. Mol. Cell. Biol. 20:372–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang, P., N.J. Liegeois, C. Wong, M. Finegold, H. Hou, J.C. Thompson, A. Silverman, J.W. Harper, R.A. DePinho, and S.J. Elledge. 1997. Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith-Wiedemann syndrome. Nature. 387:151–158. [DOI] [PubMed] [Google Scholar]
- 34.Lipinski, M.M., and T. Jacks. 1999. The retinoblastoma gene family in differentiation and development. Oncogene. 18:7873–7882. [DOI] [PubMed] [Google Scholar]
- 35.Stier, S., T. Cheng, R. Forkert, C. Lutz, D.M. Dombkowski, J.L. Zhang, and D.T. Scadden. 2003. Ex vivo targeting of p21Cip1/Waf1 permits relative expansion of human hematopoietic stem cells. Blood. 102:1260–1266. [DOI] [PubMed] [Google Scholar]
- 36.Reya, T., S.J. Morrison, M.F. Clarke, and I.L. Weissman. 2001. Stem cells, cancer, and cancer stem cells. Nature. 414:105–111. [DOI] [PubMed] [Google Scholar]