

Abstract
Extracellular adenosine (Ado) has been implicated as central signaling molecule during conditions of limited oxygen availability (hypoxia), regulating physiologic outcomes as diverse as vascular leak, leukocyte activation, and accumulation. Presently, the molecular mechanisms that elevate extracellular Ado during hypoxia are unclear. In the present study, we pursued the hypothesis that diminished uptake of Ado effectively enhances extracellular Ado signaling. Initial studies indicated that the half-life of Ado was increased by as much as fivefold after exposure of endothelia to hypoxia. Examination of expressional levels of the equilibrative nucleoside transporter (ENT)1 and ENT2 revealed a transcriptionally dependent decrease in mRNA, protein, and function in endothelia and epithelia. Examination of the ENT1 promoter identified a hypoxia inducible factor 1 (HIF-1)–dependent repression of ENT1 during hypoxia. Using in vitro and in vivo models of Ado signaling, we revealed that decreased Ado uptake promotes vascular barrier and dampens neutrophil tissue accumulation during hypoxia. Moreover, epithelial Hif1 α mutant animals displayed increased epithelial ENT1 expression. Together, these results identify transcriptional repression of ENT as an innate mechanism to elevate extracellular Ado during hypoxia.
Limited oxygen availability (hypoxia) is central to the pathogenesis of many cardiovascular, infectious, and inflammatory diseases. Pathophysiologic changes related to hypoxia include changes in tissue permeability, accumulation of inflammatory cells, and transcriptional induction of proinflammatory cytokines (1, 2). Recent studies have also identified hypoxia-elicited factors that counterregulate such proinflammatory circuit, thereby functioning as endogenous antiinflammatories (3). Central to this latter pathway is extracellular adensoine (Ado), which through activation of surface-expressed receptors on a variety of cells, dampens ongoing inflammation (2, 4–6) and promotes wound healing (7). At present, the exact metabolic steps for generation of extracellular Ado in hypoxia are not well characterized, but likely involve increased enzymatic phosphohydrolysis from precursor adenine nucleotides (ATP, ADP, and AMP). For instance, we recently demonstrated that hypoxia coordinates both transcriptional and metabolic control of the surface ectonucleotidases CD39 and CD73 (8–10), and thereby amplifies extracellular accumulation of Ado. Additional mechanisms also exist to amplify Ado signaling during hypoxia, including coordinate changes at the Ado receptor level. For instance, the vascular endothelial Ado receptor subtype AdoRA2B is selectively induced by hypoxia and such increases in receptor density are associated with increased vascular barrier responses to Ado (8).
Once generated into the extracellular milieu, Ado is rapidly cleared through passive or active uptake by nucleoside transporters, termed equilibrative nucleoside transporters (ENTs) and concentrative nucleoside transporters, respectively, expressed on a variety of cell types (11, 12). Recent studies suggest that the predominant functional nucleoside transporters in the vascular endothelium are ENT1 and ENT2, with a minimal contribution by centrative nucleoside transporters (13), wherein ENT1 and 2 are bidirectional transporters functioning as diffusion-limited channels for transmembrane nucleoside flux. Previous studies have suggested that vascular Ado transport during hypoxia is predominantly inward (14), thereby terminating extracellular Ado signaling. However, more recent studies show that the expression of ENT1 may be transcriptionally regulated by hypoxia (15, 16), thereby functioning to fine tune extracellular levels of Ado.
Therefore, we examined the influence of hypoxia on endothelial and epithelial Ado transport. Results from these studies revealed that ENT1 and ENT2 gene expression and function are attenuated by hypoxia, and that this regulatory circuit maps to hypoxia inducible factor 1 (HIF-1)–mediated repression of ENT expression. These studies provide new molecular insight into endogenous mechanisms of tissue protection during hypoxia.
Results
Increased extracellular Ado half-life in posthypoxic endothelia
Recent studies suggest that hypoxia promotes a cellular phenotype that supports high capacity for rapid nucleotide phosphohydrolysis and enhanced Ado signaling (8, 9). As an extension of these studies, we addressed whether mechanisms of extracellular Ado clearance were different in posthypoxic endothelial cells. Initial studies indicated that hypoxia significantly decreased endothelial uptake of exogenous Ado (Fig. 1 a), thereby increasing Ado half-life. Indeed, these studies indicated that human microvascular endothelial cells (HMEC)-1 subjected to hypoxia (pO2 20 torr, 48 h) and examined for their capacity to transport extracellular Ado (AdoEC, 50 μM final concentration), had significantly attenuated ability to transport Ado (AdoEC 19.2 ± 0.6 μM at 2 h) compared with normoxic controls (AdoEC 1.3 ± 1.1μM at 2 h, P < 0.01). Control experiments performed at 4°C or in the presence of the ENT-inhibitor dipyridamole (10 μM) revealed significant attenuation of Ado uptake (AdoEC 40 ± 1.6 and 33 ± 2.1 μM concentrations, respectively; P < 0.001), indicating that endothelial Ado uptake requires metabolism and depends on dipyridamole-sensitive carriers. Subsequent studies addressing the hypoxia time course (Fig. 1 b) and dose response (Fig. 1 c) revealed a time-dependent (P < 0.01) and dose-dependent (P < 0.05) decrease in transport of AdoEC.
Figure 1.

Influence of hypoxia on endothelial Ado uptake. (a) HMEC-1 cells were subjected to normoxia (N, pO2 147 torr, 48 h) or hypoxia (H, pO2 20 torr, 48 h) and Ado transport (50 μM at t = 0) was quantified. Control experiments were performed in the presence of dipyridamole (10 μM) (D), or at 4°C (cold). (b) HMEC-1 were exposed to indicated periods to hypoxia (pO2 20 torr) and Ado transport (50 μM at t = 0) was quantified. (c) Measurement of Ado half life in the supernatant of HMEC-1 cells cultured under hypoxic conditions with indicated oxygen concentration (*P < 0.01, different from normoxia). (d) To measure intracellular Ado uptake, HMEC-1 cells were subjected to normoxia (N) or hypoxia (H) over 48 h, and were exposed to 50 μM Ado containing 10 nCi of 8-14C-Ado. Cells were washed and lysed after indicated time periods, and radioactivity was measured.
To confirm that the observed decreases in AdoEC reflects transport and to rule out other potential mechanisms of Ado loss (e.g., metabolism by the ecto-Ado deaminase) (17), we determined the kinetics of 14C-AdoEC transmembrane transport (Fig. 1 d). These studies confirmed our studies with native Ado and revealed that 14C-AdoEC transport was 63 ± 6% decreased in posthypoxic endothelia relative to normoxia (P < 0.001). In parallel, 14C-AdoEC transport at 4°C or in the presence of dipyridamole revealed significant attenuation of endothelial uptake (P < 0.001). Together, these data suggest an additional mechanism to elevate AdoEC involves diminished active uptake through endothelial membrane transporters.
Hypoxia rapidly represses expression of endothelial and epithelial ENT1 and ENT2 mRNA
Prompted by these results, recent findings that ENT1/2 predominate in the vascular endothelium (13) and that hypoxia down-regulates dipyridamole-sensitive Ado transporters (15, 16), we examined the influence of hypoxia on expression of ENT1 and ENT2 mRNA. Initial insight gained by microarray analysis revealed an 85 ± 5 and 78 ± 4% inhibition of ENT1 and ENT2 mRNA expression, respectively (18 h hypoxia, pO2 20 torr). As shown in Fig. 2 a, real-time PCR analysis confirmed our microarray results and revealed a time-dependent loss of both ENT1 (95 ± 8% loss at 24 h, P < 0.01) and ENT2 (81 ± 7% loss at 24 h of hypoxia, P < 0.01). In addition, we found that expression levels of ENT1 mRNA in HMEC-1 were approximately fourfold higher compared with ENT2 mRNA.
Figure 2.

Repression of ENT1 and ENT2 mRNA and protein expression by hypoxia. (a) Confluent HMEC-1 monolayers were exposed to normoxia or hypoxia for indicated time periods. Total RNA was isolated, and ENT1/ENT2 mRNA levels were determined by real-time PCR. Data were calculated relative to β-actin and expressed as fold change relative to normoxia ± SD. Results are derived from three experiments (*P < 0.01, different from normoxia and ENT2; #P < 0.01, different from normoxia). (b) Human saphenous vein was obtained from patients undergoing coronary bypass surgery and exposed ex vivo to normoxia (24 h) or hypoxia (2, 8, or 24 h). Real-time PCR was used to define ENT1 and ENT2 mRNA levels. Data were calculated relative to β-actin and are expressed as fold change compared with normoxia ± SD. Results are derived from three experiments (*P < 0.01, different from normoxia and ENT2; #P < 0.01, different from normoxia). (c) Confluent T84 epithelial cell monolayers were exposed to normoxia (24 h) or hypoxia for indicated time periods. Total RNA was isolated and ENT1/ENT2 mRNA levels were determined by real-time PCR. Results are derived from three experiments (*P < 0.01, different from normoxia and ENT2; #P < 0.01 different from normoxia). (d) HMEC-1 cells were grown to confluence and exposed to indicated periods of hypoxia. Shown here is a representative Western blot of ENT-1. The same blot was probed for β-actin expression as a control for protein loading.
Similar results of ENT1 and ENT2 repression were found when human saphenous vein tissues were subjected to hypoxia, indicating that such findings are not limited to cultured endothelium (Fig. 2 b, P < 0.001). Additionally, ENT1 and ENT2 expression were decreased in T84 cells and intestinal epithelial cell line (Fig. 2 c, P < 0.001), further suggesting that these findings are not limited to endothelial cells.
We extended these mRNA findings to examine ENT1 protein expression in cultured HMEC-1. As shown in Fig. 2 d, Western blot analysis of lysates derived from HMEC-1 subjected to hypoxia (pO2 20 torr for 6–36 h) revealed a dramatic loss of ENT1 with increasing time in hypoxia. Protein repression was maximal at 36 h time periods of hypoxia (no additional loss was evident at 48 h).
ENT1 repression mediates attenuated endothelial Ado uptake during hypoxia
We next determined whether inhibition of ENT gene expression was required for the observed functional differences in Ado uptake between posthypoxic and postnormoxic endothelia. Here, we used siRNA-mediated repression in HMEC-1 of ENT1 or ENT2 for gaining insight into relative contributions of these individual gene products on AdoEC transport. As a first step, we generated siRNA double-stranded oligoribonucleotides specific for ENT1 and ENT2 and profiled ENT expression by PCR (Fig. 3, a and b). As control siRNA, we used a single-stranded forward oligoribonucleotide, which did not influence ENT1 or ENT2 mRNA expression (Fig. 3 c), suggesting that the aforementioned findings of decreased ENT1 or ENT2 expression (Fig. 3, a and b) were specific for targeted gene knock-down by the chosen siRNA molecules. As additional control, we used double-stranded siRNA directed against the 5′-ectonucleotidase (CD73), an endothelial surface enzyme that facilitates conversion of Ado monophosphate to Ado. As shown in Fig. 3 (d and e), siRNA repression of CD73 did not influence expressional levels of ENT1 or ENT2. After showing that siRNA-mediated repression of ENT1 and ENT2 was specific and highly efficient using the aforementioned conditions (81 ± 8% inhibition of ENT1 mRNA expression, P < 0.01, and 79 ± 10% inhibition of ENT2, P < 0.01), we measured endothelial Ado uptake using 14C-Ado. As shown in Fig. 3 f, whereas 14C-Ado uptake was not influenced by siRNA repression with control siRNA (5 ± 1% decrease compared with mock-loaded cells, p-values were not significant) or siRNA directed against ENT2 (14 ± 7% decrease compared with mock-loaded cells, p-values were not significant), Ado uptake was decreased after siRNA-mediated repression of ENT1 (76 ± 7% decrease compared with mock-loaded cells, P < 0.01) or simultaneous ENT1 and ENT2 repression (81 ± 5% decrease compared with mock-loaded cells, P < 0.01). Together, these results implicate ENT1 as the prevailing mechanism for the observed changes in endothelial Ado.
Figure 3.
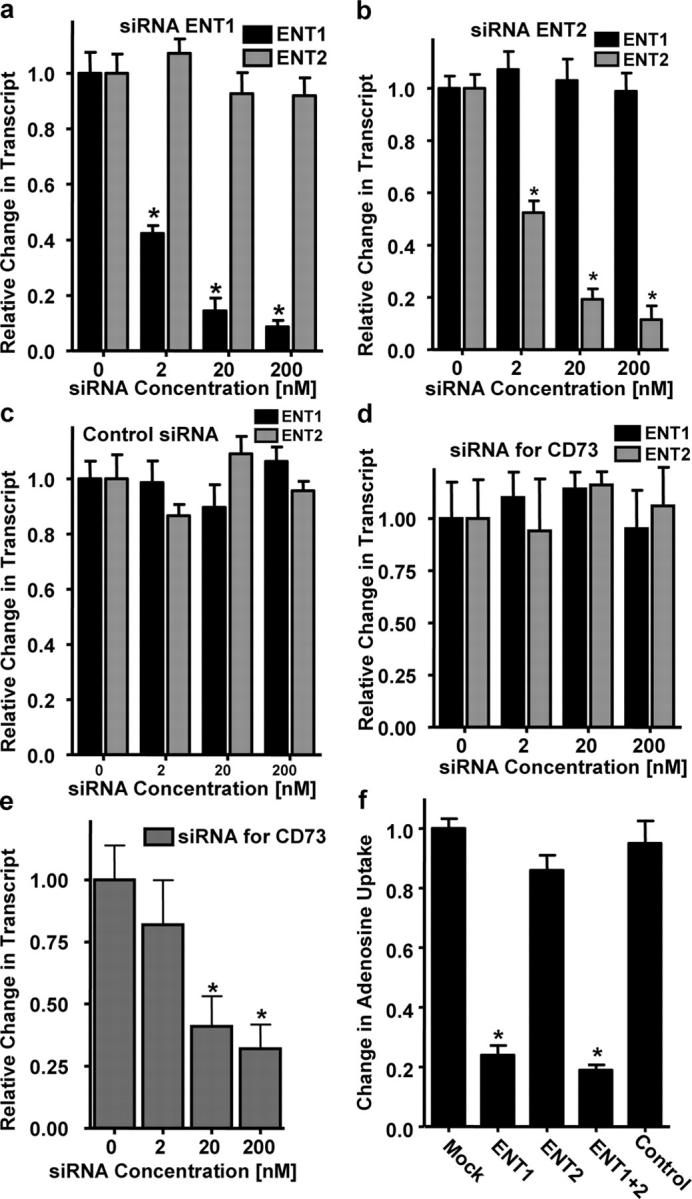
Influence of siRNA knockdown of ENT1 and ENT2 on endothelial Ado uptake. Real-time PCR was used to confirm down-regulation of ENT1 and ENT2 mRNA by siRNA loading in cultured endothelial cells. HMEC-1 cells were loaded with siRNA for ENT1 (a), ENT2 (b), control siRNA (c), or siRNA specific for CD73 (d and e). Data were calculated relative to β-actin and are expressed as fold decrease over control ± SD at each indicated siRNA concentration. Results are derived from three experiments (*P < 0.01, different from control). (d) siRNA knockdown with siRNA directed against CD73 did not influence expression of ENT1 or ENT2. (e) Repression of CD73 dose response. (f) Measurement of intracellular Ado uptake after siRNA loading. HMEC-1 cells were transfected with 200-nM concentrations of siRNA for ENT1, ENT2, ENT1 and ENT2, or control siRNA for 48 h. Intracellular Ado uptake was measured by exposing cell to 50 μM Ado containing 10 nCi of 8-14C-Ado. Cells were lysed after 30 min and radioactivity was measured (*P < 0.01, different from control).
Decreased endothelial Ado uptake promotes endothelial barrier function in vitro
We next determined whether hypoxia-mediated repression of ENT1 might enhance signaling mediated by AdoEC. Here, we compared Ado responses of normoxic and posthypoxic endothelia using a previously described in vitro model of endothelial barrier (8, 18). As reported previously (8), we observed that barrier function (i.e., flux 70-kD FITC-dextran) in response to Ado was enhanced to a greater extent in posthypoxic HMEC-1 than in normoxic HMEC-1 (Ado EC50's of ∼30 μM and ∼1 μM for normoxia and posthypoxia, respectively; Fig. 4 a). To define the contribution of hypoxia-mediated repression of ENT1/2, we repeated this experiment in the presence of the ENT inhibitor dipyridamole (10 μM). As shown in Fig. 4 b, in the presence of dipyridamole, the dose response to Ado became indistinguishable between hypoxia and normoxia (Ado EC50 of ∼0.1 μM for both), implicating hypoxia-mediated repression of ENT1/2 in functional Ado responses.
Figure 4.

Influence of Ado uptake inhibition on endothelial barrier function in vitro. (a) Indicated concentrations of Ado were added to the apical surface of normoxic (48 h, pO2 147) or posthypoxic (48 h, pO2 20 torr) HMEC-1 and permeability to FITC-dextran (70kD) was quantified. Data are derived from six monolayers in each condition and expressed as mean ± SD of percent control flux. *, significant differences from baseline (P < 0.05); #, differences from baseline and from normoxia (P < 0.05). (b) Influence of 10 μM dipyridamole on Ado barrier responses in normoxic and posthypoxic endothelia. (c) Influence of ENT1 and ENT2 (d) suppression by siRNA on Ado elicited endothelial barrier responses. HMEC-1 cells were loaded with ENT1- or ENT2-specific siRNA or control ribonucleotide (200 nM) and, after 48 h, permeability to 70 kD FITC was measured in the presence of indicated Ado concentrations. Data are derived from six monolayers in each condition and expressed as mean ± SD of percent control flux.
We next defined the contribution of ENT1 or ENT2 to such changes in Ado-elicited barrier responses of the endothelium. To do this, we used the aforementioned siRNA repression approach. As shown in Fig. 4 c, siRNA repression of ENT1 was associated with an increase of Ado elicited barrier responses (P < 0.01). In contrast, loss of ENT2 was not associated with a measurable change in endothelial Ado response (Fig. 4 d). These data reveal that inhibition of Ado uptake, either via hypoxia inhibition of ENT gene expression, pharmacological intervention, or siRNA repression of ENT1, represents an adaptive mechanism to elevate extracellular Ado levels, and thereby prolong signaling mediated by this nucleoside.
HIF-1α–dependent repression of ENT1 promoter
In an attempt to gain specific insight into the molecular mechanisms of ENT1 repression by hypoxia, we profiled promoter activity in HMEC-1 transfected with luciferase promoter constructs expressing varying length 5′-truncations (225, 282, 344, and 756 bp relative to the major transcription start site). Highest basal expression (i.e., normoxia) was observed in the 756-bp construct (15 ± 2.1-fold over background) and graded decreases in baseline activity were observed with increasing 5′-truncation (6.8 ± 1.7, 3 ± 1.1, and 5 ± 0.6-fold over background for 344-, 282-, and 225-bp truncations, respectively). Comparison of promoter activity in HMEC-1 exposed to hypoxia or normoxia revealed that, whereas other constructs were expressed at or near the level of the normoxia control, expression of the 756-bp fragment was repressed by 59 ± 6% compared with normoxia controls (Fig. 5 a, P < 0.01). Such analysis indicates that the region spanning −344 to −756 of the ENT1 promoter confers hypoxia-repressive activity.
Figure 5.

Influence of hypoxia on ENT1 promoter activity. (a) Indicated serial truncations of the ENT1 promoter reporter were transfected in HMEC-1 cells along with Renilla reporter plasmids and assayed for firefly and Renilla luciferase activity, respectively, after exposure to hypoxia for 48 h. Results depict the fold change in relative luminescence in hypoxia relative to normoxic controls. An HRE plasmid containing four tandem hypoxia-responsive elements is shown as a positive control (HRE). Data shown are a pooled experiments from n = 4 and are normalized for background vector (empty PGL3) and Renilla luciferase to control for transfection efficiency, and presented as mean ± SD, where * indicates significance between individual plasmids and empty PGL3 plasmid (P < 0.025) and # indicates significance between HRE and ENT1-756 (P < 0.025). (b) HMEC-1 cells were grown to confluence and exposed to indicated periods of hypoxia. Nuclear lysates were isolated and resolved by SDS-PAGE. Shown here is a representative Western blot of HIF-1α under these conditions. The same blot was probed for β-actin expression as control for protein loading. (c) The putative HIF sites located at −721 and −657 bp were mutated in the −756 construct by site-directed mutagenesis. The resulting constructs (ENT1Δ721) and (ENT1Δ657) was cotransfected along with Renilla luciferase vector into HMEC1 cells followed by 48 h of hypoxia. An HRE plasmid containing four tandem hypoxia-responsive elements is shown as a hypoxia positive control. Data shown are pooled from n = 4 and are normalized for background vector (empty PGL3) and Renilla luciferase to control for transfection efficiency and presented as mean ± SD, where * indicates significance between ENT1-756 and ENT1Δ721 (P < 0.025) and # indicates significance between HRE and ENT1-756 (P < 0.025). (d) Chromatin immunoprecipitation was used to examine HIF-1α binding to the ENT1 promoter in normoxic and hypoxic HMEC-1. Reaction controls included PCR performed using whole HMEC-1 genomic DNA (input) and samples precipitated by protein G sepharose beads alone (Cntl IP).
In the course of our experiments, we identified two potential HIF binding sites oriented on the antisense strand of the ENT1 gene promoter (DNA consensus motif 3′-CCGTG-5′ and 3′-CCGTG-5′ located at positions from −653 to −657 and −717 to −721, respectively, relative to the major transcription start site). Because HIF can mediate transcriptional induction or repression (19, 20), we determined if either of these putative HIF-1–binding sites conferred hypoxia repression of ENT1 in HMEC-1. First, we examined the kinetics of HIF-1α expression in our HMEC-1 cultures. As shown in Fig. 5 b, Western blot analysis of HIF-1α revealed minimal protein in hypoxia, and time-dependent induction over a 24-h period of hypoxia. Such kinetics are consistent with the loss of ENT1 protein during hypoxia (Fig. 2 d). Second, HIF-1α–binding site mutations (Δ657 and Δ721) were introduced in the 756-bp construct, and as shown in Fig. 5 c, mutation of the −717 to −721 site, but not the −653 to −657 site, reversed hypoxia repression of this promoter construct, suggesting that this region of the promoter, at least in part, confers hypoxia repressor activity. Third, we determined whether this region of the ENT1 promoter binds HIF-1α. For these purposes, we used chromatin immunoprecipitation (ChIP) to analyze HIF-1α binding in live cells. As shown in Fig. 5 d, ChIP analysis of nuclei derived from HMEC-1 cells revealed a prominent band of 192 bp in hypoxic but not normoxic samples. No bands were evident in control IgG immunoprecipitates, and input samples (preimmunoprecipitation) revealed the predictable 192-bp band under conditions of both hypoxia and normoxia. Such results indicate that hypoxia induces HIF-1α binding to the distal 192-bp region of the ENT1 promoter. Together, these results provide strong evidence for a functional hypoxia repressor activity, mediated by HIF-1, in the distal 5′-region the ENT1 promoter.
Role of HIF-1α in epithelial ENT1 regulation
We next pursued the finding that epithelial ENT expression, like that in endothelia, is repressed by hypoxia (Fig. 2 c). To probe the role of HIF-1α in an epithelial model, we generated a T84 cell line expressing oxygen-stable HIF-1α (T84ΔODD) (21) via lentiviral transduction (Fig. 6). As shown in Fig. 6 a, using this lentiviral vector-expressing GFP, >90% of T84 cells were stably transduced by this method. Analysis of the T84ΔODD by Western blot (Fig. 6 b) and by hypoxia-responsive element (HRE) luciferase (Fig. 6 c) revealed increased HIF function and activity in both normoxia and hypoxia, releative to the control T84-GFP cell line. The further increase of HIF function and activity with hypoxia most likely reflects the presence of oxygen-stable and oxygen-unstable HIF-1α.
Figure 6.
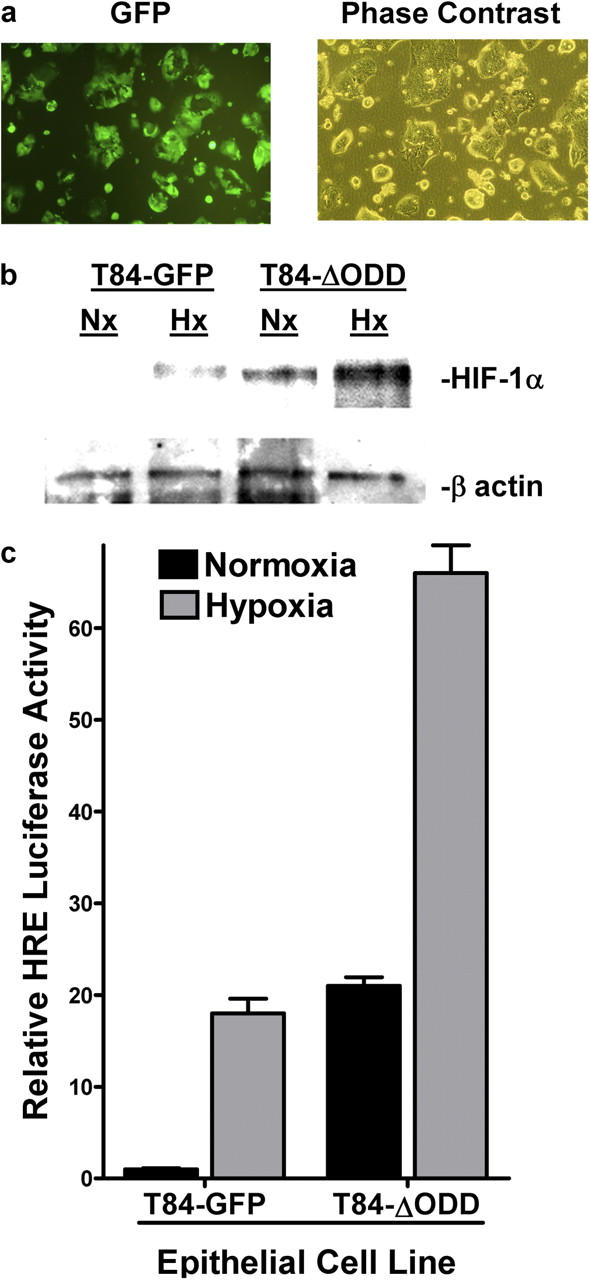
Characterization of epithelial cell line expressing oxygen-stable HIF-1α. (a) Imaging of T84 cells stably transduced with a control lentivirus encoding GFP. For comparison, the phase contrast image is also shown. (b) Cells were transduced with control GFP lentivirus (T84-GFP) or ΔODD lentivirus (T84-ΔODD), exposed to normoxia or hypoxia (6 h), and nuclear HIF-1α expression was examined by Western blot (β-actin controls are also shown). (c) T84-GFP and T84-ΔODD cells were transiently transfected with the HIF reporter HRE-luciferase plasmid, and exposed to hypoxia or normoxia (24 h). Relative HIF activity was assessed by luciferase relative to empty vector (pGL3). Results are presented as fold above PGL3 background (relative to control plasmids expressing Renilla in each condition).
Using the T84ΔODD cell line, we addressed whether ENT1 expression was differentially regulated. As shown in Fig. 7 a, ENT1 mRNA expression in normoxia was decreased by >80% in the T84ΔODD relative to the control T84-GFP cell line (P < 0.01). Likewise, enhanced repression of ENT1 was observed in T84ΔODD compared with control T84-GFP cells at 6, 12, and 24 h of hypoxia (P < 0.025 for each). Additionally, comparison of promoter activity in T84ΔODD and T84-GFP cell lines exposed to hypoxia or normoxia revealed enhanced repression of the 756-bp fragment in cells expressing oxygen-stable HIF-1α (Fig. 7 b). Such findings support the hypothesis that ENT1 repression is proportional to HIF-1α activity.
Figure 7.

Functional regulation of ENT1 expression in epithelial cells expressing oxygen-stable HIF-1α. (a) T84-GFP and T84-ΔODD cells were exposed to indicated periods of hypoxia and examined for ENT1 expression by real-time PCR. Data were calculated relative to β-actin and are expressed as relative change ± SD at each indicated time. Results are derived from three experiments in each condition (*P < 0.01, different from normoxia and ENT2; #P < 0.01, different from normoxia). (b) T84-GFP and T84-ΔODD cells were transiently transfected with the wild-type ENT promoter construct (ENT1-756) and exposed to hypoxia or normoxia (24 h). Relative activity was assessed by luciferase relative to empty vector (pGL3). Results are presented as relative change in activity above PGL3 background (relative to control plasmids expressing Renilla in each condition).
In vivo analysis of ENT function on the vasculature
As proof of principle for these in vitro and ex vivo findings of hypoxia-induced ENT repression, we extended these findings to an in vivo model. We and others have shown that hypoxia promotes vascular leak and tissue PMN accumulation in vivo (1, 8, 10, 22–24). Thus, we pursued pharmacological inhibition of Ado uptake in a previously described murine hypoxia model (8, 10, 22).
As first step, we administered the ENT inhibitor dipyridamole (10 mg/kg i.p. and 10 mg/kg s.c.) or PBS and subjected animals to either normoxia or hypoxia (8% O2 and 92% N2) for 4 h. Plasma levels of dipyridamole (determined by HPLC) ranged between 25- and 110-nm concentrations during the course of the experiment (Fig. 8 a), consistent with previous dosing studies with dipyridamole and dipyridamole analogues (25). In previous in vivo hypoxia experiments, we observed the lungs as particularly prone to developing hypoxia-induced vascular leak and pulmonary edema (10). Therefore, we assessed pulmonary edema by determining lung water content (wet/dry ratio). As shown in Fig. 8 b, hypoxia increased lung water content from 3.81± 0.42 mg/mg dry tissue to 5.83 ± 0.54 (P < 0.05) in PBS treated mice. In contrast, lung water was decreased in dipyridamole-treated animals by 16 ± 2% under normoxic conditions, and by 21 ± 3% (P < 0.025) under hypoxic conditions (P < 0.05). These results suggest that pharmacological inhibition of Ado uptake is associated with a decrease in lung water.
Figure 8.
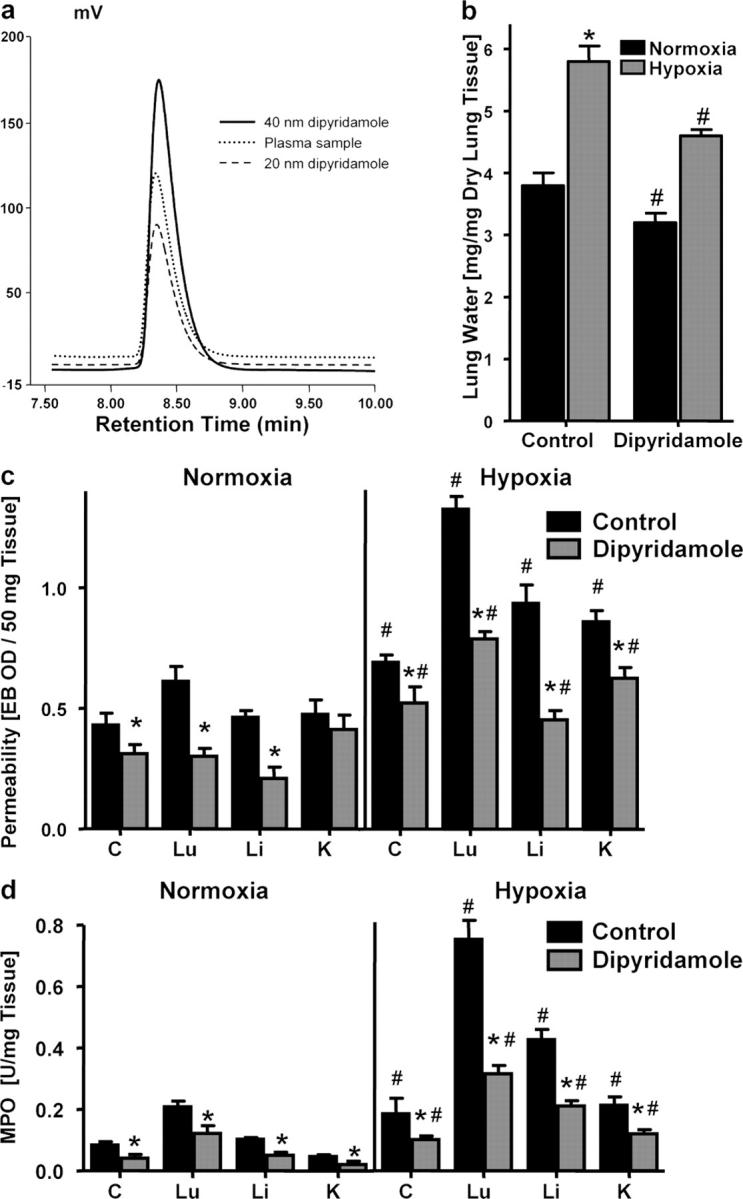
Influence of dipyridamole on pulmonary edema, vascular permeability, and PMN accumulation in vivo. BL/6/129 mice were injected with dipyridamole (10 mg/kg i.p. and 10 mg/kg s.c.) or with PBS, and exposed to normoxia (room air) or normobaric hypoxia (8% O2 and 92% N2) for 4 h. (a) Dipyridamole plasma levels were measured by HPLC and compared with standards of known dipyridamole concentration. Mean plasma dipyridamole concentrations were 33.7 ± 10.2 nM with no differences between treatment groups (P > 0.05). (b) Assessment of lung water content in normoxia (black bars) and hypoxia (gray bars) after dipyridamole or PBS treatment. Data are expressed as mean ± SD mg H2O/mg dry tissue, and are pooled from four animals per condition. *, difference between hypoxia and normoxia (P < 0.025); #, difference between dipyridamole treatment and vehicle control (P < 0.025). (c) Evan's blue vascular permeability. Animals were killed and indicated organs were harvested. Evan's blue concentrations were quantified as described in Materials and methods. Data are expressed as mean ± SD Evan's blue OD/50mg wet tissue, and are pooled from four to six animals per condition, where * indicates differences between dipyridamole/PBS treatment groups (P < 0.025) and # indicates differences between normoxia/hypoxia exposure (P < 0.05). (d) Organ assessment of PMN accumulation by MPO measurements in the indicated organs after 4 h of normoxia/hypoxia exposure (*P < 0.025 compared with PBS, #P < 0.025 compared with normoxia).
To assess vascular barrier function of other organs, we administered the albumin marker Evan's blue before hypoxia. Comparative analysis in normoxia revealed decreased Evan's blue concentrations in all examined organs (colon, lung, liver, and kidney) of dipyridamole-treated mice compared with PBS controls. Comparison of vascular permeability in dipyridamole or PBS-treated mice subjected to normobaric hypoxia (8% O2, 92% N2 for 4 h) revealed significantly increased vascular leak in all animals, however to smaller degree in dipyridamole-treated animals (P < 0.05 for all organs; Fig. 8 c). These findings support our hypothesis that decreased Ado uptake by the endothelium is a protective cellular strategy to dampen changes in vascular barrier function, both at baseline and during hypoxia-induced barrier dysfunction.
We next examined whether a reduction of Ado uptake at the vascular surface may also function as antiinflammatory response during hypoxia, particularly given the antiinflammatory properties of Ado (4, 6). To test this hypothesis, we screened multiple mucosal organs (colon, lung, liver, and kidney) for PMN accumulation after administration of dipyridamole (Fig. 8 d). This analysis revealed decreased myeloperoxidase (MPO) activity in all organs of dipyridamole-treated animals. Consistent with previous studies (26), MPO levels were significantly increased after hypoxia exposure in all animals, however to a much lesser extend in all organs of dipyridamole-treated animals compared with PBS controls (P < 0.05 for all organs). These in vivo findings support the hypothesis that Ado uptake via dipyridamole-sensitive carriers is a critical control point in the regulation of PMN accumulation within normoxic and hypoxic tissues and suggest ENT down-regulation as innate antiinflammatory adaptation.
It is known that the two nucleoside transporters ENT1 and ENT2 differ in their sensitivity to inhibition by the inosine analogue NBTI (27, 28). NBTI is a highly potent inhibitor of ENT1 (IC50 ∼0.4–8 nM), whereas it is only a moderate inhibitor of ENT2 (IC50 ∼3 μM). Based on pharmacokinetic studies of NBTI in mice, we used an NBTI dose that would result in estimated plasma levels between 1 and 10 nM (29). To pursue this, we administered NBTI (1 mg/kg i.p.) or vehicle (DMSO) and subjected animals to either normoxia or normobaric hypoxia. As shown in Fig. 9 a, hypoxia significantly increased lung water content in vehicle-treated animals (P < 0.05). In contrast, lung water was decreased in NBTI-treated animals under normoxic or hypoxic conditions. Measurement of vascular barrier function revealed significantly increased vascular leak with hypoxia in all animals, however to a smaller degree in NBTI treated animals (P < 0.05 for all organs; Fig. 9 b). Finally, we also screened multiple mucosal organs for PMN accumulation (Fig. 9 c). Consistent with the aforementioned results with dipyridamole, we found decreased MPO activity in all organs of NBTI-treated animals after hypoxia exposure (P < 0.05 for all organs). These in vivo results suggest that inhibition of ENT1 contributes to the in vivo resuscitation of vascular leak syndrome by nucleoside transport inhibitors.
Figure 9.
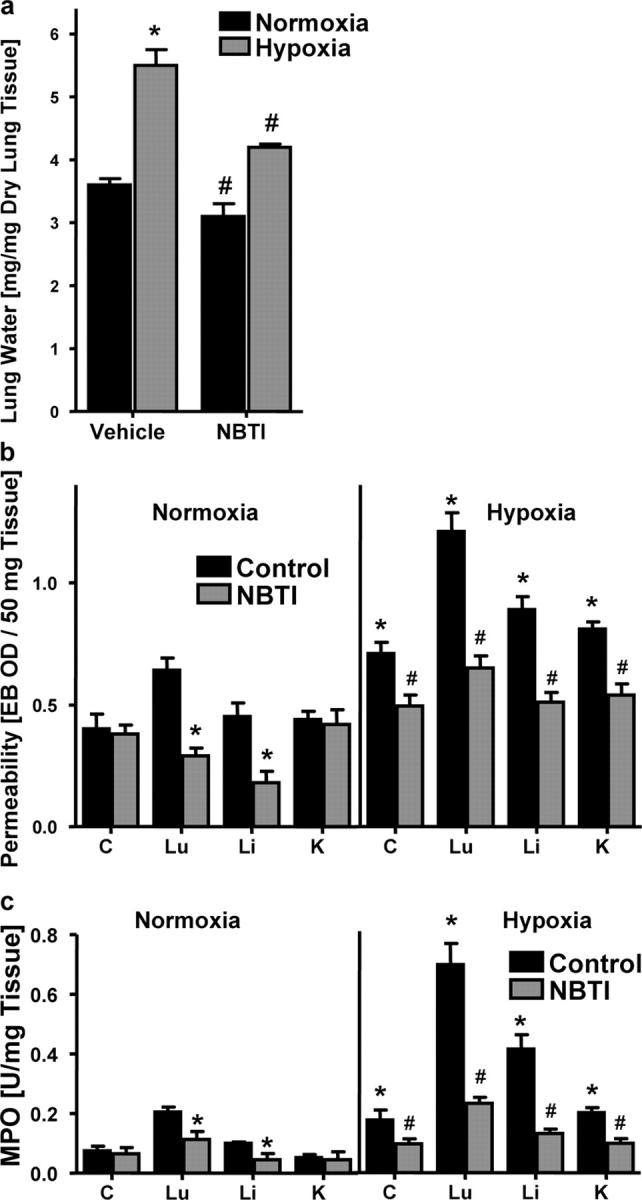
Influence of S-(4-Nitrobenzyl)-6-thioinosine (NBTI) on pulmonary edema, vascular permeability, and PMN accumulation in vivo. BL/6/129 mice were administered NBTI (1 mg/kg i.p. in DMSO) or vehicle alone, and exposed to normoxia or hypoxia for 4 h. (a) Assessment of lung water content in normoxia (black bars) and hypoxia (gray bars) after dipyridamole or vehicle treatment. Data are expressed as mean ± SD mg H2O/mg dry tissue, and are pooled from four animals per condition. *, difference between hypoxia and normoxia (P < 0.025); #, difference between dipyridamole treatment and vehicle control (P < 0.025). (b) To assess vascular barrier function, animals were administered intravenous Evan's blue solution (0.2 ml of 0.5% in PBS) before normoxia/hypoxia exposure. Animals were killed and indicated organs were harvested. Evan's blue concentrations were quantified as described in Materials and methods. Data are expressed as mean ± SD Evan's blue OD/50 mg wet tissue and are pooled from four to six animals per condition. *, differences between NBTI/vehicle treatment groups (P < 0.025); #, differences between normoxia/hypoxia exposure (P < 0.05). (c) Organ assessment of PMN accumulation by MPO measurements in the indicated organs after 4 h of normoxia/hypoxia exposure (*P < 0.025 compared with vehicle; #P < 0.025 compared with normoxia).
Role of HIF in epithelial ENT1 expression in vivo
We extended these findings of HIF-1–mediated repression of ENT1 expression into a genetic in vivo model. Here, we examined baseline expression and the influence of hypoxia on mENT1 mRNA levels in intestinal epithelia derived from conditional Hif1α knockout mice, in which intestinal epithelia from these mice lack detectable Hif1α expression in >70% of cells (3). As shown in Fig. 10, ENT1 levels in mice expressing wild-type Hif1α showed a normal pattern of hypoxia-associated ENT repression (68 ± 3% decrease after hypoxia for 6, P < 0.025). Consistent with our hypothesis that HIF-1 transcriptionally represses ENT1, real-time PCR analysis revealed a 37 ± 6.8-fold increase in intestinal epithelial ENT1 expression in Hif1α mutant animals (Fig. 10), relative to their littermate controls (P < 0.01). Exposure of HIF1α mutants to hypoxia decreased ENT1 expression (P < 0.05), but to a far lesser extent than in wild-type animals. These latter findings likely reflect the remaining ∼30% wild-type Hif1α expression in these mice (3). Together, such findings support our in vitro findings and indicate the likelihood that HIF-1 directly regulates murine ENT1 expression.
Figure 10.
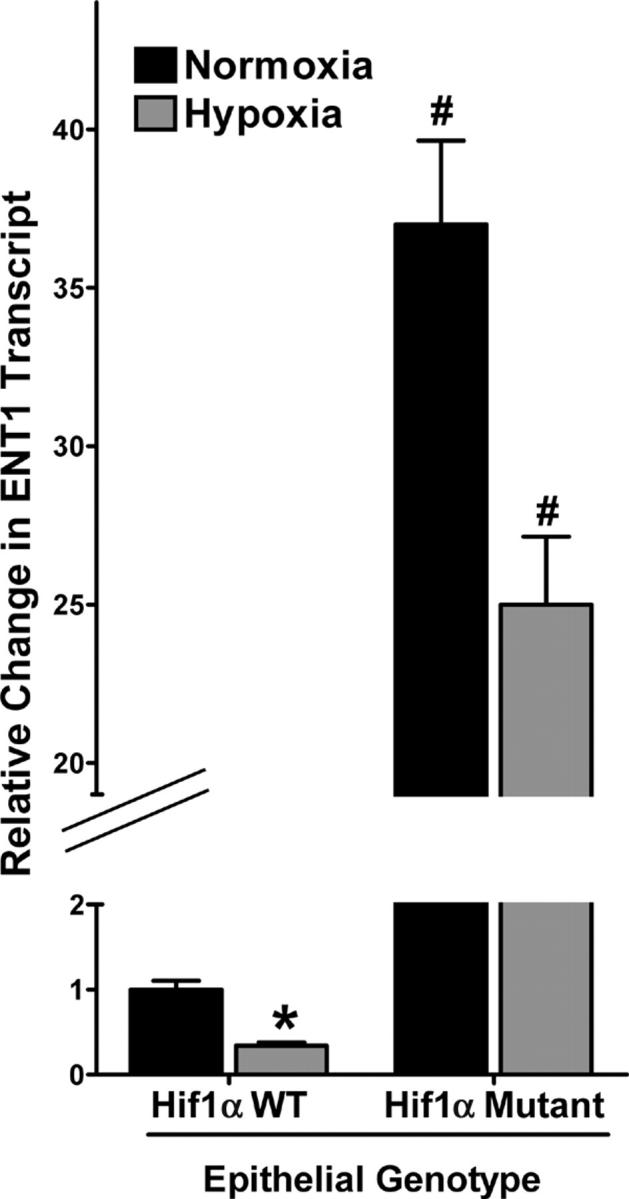
ENT1 expression patterns in conditional Hif1α mutant tissue. Real-time PCR analysis of murine epithelial ENT1 transcript in conditional Hif1α mutant and littermate control animals subjected to normoxia or hypoxia. Data were calculated relative to β-actin and are expressed as relative ENT1 mRNA ± SD, where transcript levels in control animals were normalized to 1. Results are derived from three animals in each condition.
Discussion
Ado exerts paracrine and autocrine functions on most cell types. Pathophysiologic conditions of hypoxia/ischemia result in numerous adenine nucleotide metabolic changes, and targets for extracellular Ado signaling is now an area of much interest. In the present studies, we explored the mechanisms of extracellular Ado accumulation during hypoxia. For these studies, we used vascular endothelial and mucosal epithelial cells, which both lie anatomically positioned to function as determinants of the inflammatory response. Our results revealed that hypoxia increases extracellular Ado half-life. This response is mechanistically determined, at least in part, by HIF-1–regulated ENT1 repression.
The results from the present study are consistent with previous findings that hypoxia inhibits intracellular metabolism of Ado to AMP. Decking et al. showed, by measuring Ado and AMP concentrations in the coronary sinus and coronary arteries of isolated guinea pig hearts, that hypoxia is associated with a functional inhibition of the Ado kinase (30). The authors propose that hypoxia-associated inhibition of the Ado kinase leads to increased intracellular Ado levels. It is known that during hypoxia, Ado flux is predominantly directed from the extracellular to the intracellular space (14). Therefore, two cellular strategies with regard to Ado transport may play a role to elevate extracellular Ado levels during hypoxia. First, as described by Decking et al., increased intracellular Ado levels due to inhibition of Ado kinase–dependent metabolism of Ado to AMP would decrease the transcellular Ado gradient, thereby decreasing flux through bidirectional ENTs and, thus, elevating extracellular Ado levels during hypoxia (30). Second, transcriptional repression of ENTs, as we demonstrate in the present work, decreases overall equilibrative Ado transport capacities, thereby decreasing intracellularly directed Ado transport. It should be noted that we have not observed a direct change in ENT function by hypoxia per se. Rather, our studies addressed function as it related to expression levels of ENT. From this standpoint, it is likely that inhibition of adenine nucleotide metabolism (e.g., inhibition of Ado kinase) could serve as an immediate response to hypoxia (within minutes), whereas our proposed transcriptional mechanism serves as a more adaptive phenotype (within hours). Therefore it is reasonable that both mechanisms could function during hypoxia and, from this perspective, contribute synergistically to elevate extracellular Ado during hypoxia.
Consistent with recent work in cardiac myocytes (16), the present studies in vascular endothelia and mucosal epithelia identified transcriptional repression of ENT as a mechanism for such decreased Ado uptake during hypoxia. Given the temporal and robust hypoxia response observed in the repression of ENT1, a candidate regulator was HIF-1, a heterodimeric transcription factor whose activation is dependent on stabilization of an O2-dependent degradation domain (31). It is recently appreciated that HIF can function as both a transcriptional activator as well as a repressor (19). Therefore, a search of the cloned ENT1 gene promoter identified two potential HIF-1–binding DNA consensus motifs located in the distal 5′ region of the ENT1 promoter. However, the existence of a HIF-1α–binding consensus is not evidence for HIF-1α–mediated response; instead, the HRE is defined as a cis-acting transcriptional regulatory sequence located within 5′-flanking, 3′-flanking, or intervening sequences of target genes (32, 33). Therefore, two strategies were used to define the role of HIF-1 in repression of ENT1. First, mutational analysis of ENT1 promoter constructs defined a functional HRE located in the distal 5′region of the ENT1 promoter (−717 to −721 relative to the major transcription start site). Second, ChIP analysis confirmed HIF-1α binding to the ENT1 promoter. At present, we do not know if this binding is direct or indirect, and we do not know the nature of HIF-mediated transcription. While previous work with peroxisome proliferator-activated receptor α (PPAR-α) gene implicated potential repressor activity with HIF-1α binding sites oriented on the antisense strand (20), implying some degree of transcriptional directionality, there is not direct evidence for such a mechanism. Moreover, while a recent study comparing transcriptional responses between hypoxia and constitutively active HIF1-1α identified a large cohort of transcriptionally repressed genes (19), no unique patterns of HRE expression were noted. Thus, more work will be necessary to define the nature of HIF-mediated repression.
The present study also identified ENT1 as a potential physiologic target in vivo. Indeed, using the ENT inhibitor dipyridamole, we demonstrated that blockade of ENT1 prevents vascular leakage associated with hypoxia, presumably through accumulation of vascular Ado. Several lines of evidence support the presumption that elevated Ado attenuates vascular leakage. First, Nozik-Grayck determined that dipyridamole effectively attenuates edema in an isolated perfused lung model of hyperoxia (34). Second, in murine models of hypoxia, decreased vascular Ado is associated with substantially increased vascular leakage (8, 10, 22). In particular, these observations have been made in animals lacking the ability to metabolize adenine nucleotide precursors into Ado, including cd39 and cd73-null animals (8, 10, 22). Third, the administration of Ado analogues in the form of Ado receptor A2a or A2b agonists, as well as the nonspecific analogue 5′-(N-ethylcarboxamido)-Ado (NECA), protect the vasculature during hypoxia-induced leakage (10). This hypothesis was also tested in a conditional Hif1α mutant mice, a mouse line that has revealed a protective role for murine HIF-1α in a mucosal inflammation model (3). Using this same mouse line, we confirmed our hypothesis that HIF transcriptionally represses ENT1. Indeed, our findings revealed that colonic epithelial cells lacking Hif1α express increased ENT1 transcript. Of particular interest, analysis of unmanipulated Hif1α mutant mice (i.e., normoxia condition) revealed substantial increases in ENT1. Such findings suggest, as previously speculated (3, 9), that HIF may regulate homeostatic gene expression (i.e., “physiologic hypoxia”) in tissues with normally low pO2 values (e.g., colon) (35).
These results identify ENT repression as a mechanism for attenuating both inflammatory responses and vascular leakage during hypoxia. Interaction of HIF-1 with the ENT1 promoter region is central to the observed inhibition of ENT1 gene expression. Extensions of these findings will determine whether ENT regulation via HIF-1 might function as a pathway for development of therapies for disorders involving vascular leak syndromes or excessive inflammatory responses.
Materials and Methods
Cell culture
HMEC-1 were a gift from F. Candal (Centers for Disease Control, Atlanta, GA) (36) and were cultured as described previously (37). For preparation of experimental HMEC-1 monolayers, confluent endothelial cells were seeded at ∼105 cells/cm2 onto either permeable polycarbonate inserts or 100 mm Petri dishes. Endothelial cell purity was assessed by phase microscopic “cobblestone” appearance and uptake of fluorescent acetylated low-density lipoprotein. Where indicated, T84 cells were cultured as described previously (9, 38).
Measurement of Ado in supernatants
HMEC-1 were grown to full confluency and exposed as indicated to normoxia or different degrees of hypoxia. To develop a model for kinetics of endothelial Ado uptake, we first measured Ado uptake in the supernatant of HMEC-1 monolayers with different Ado concentrations (1–500 μM) and found optimal resolution of Ado uptake when using 50 μM Ado concentrations. These conditions were used throughout for further measurements. After washing in HBSS and addition of 50 μM Ado, samples from the supernatant were collected at indicated time points, and Ado concentrations were resolved by HPLC with a pump P680 and an UVD 170 detector on a reverse-phase column (Grom-Sil 120-ODS-ST-5μ; 150 × 3 mm Grom) using a mobile phase gradient from 0 to 25% acetonitrile/0.3 mM KH2PO4 (pH 5) in 5 min. Ultraviolet absorption spectra were measured at 260 nm with a retention time for Ado of 2.8 min.
Measurement of Ado uptake.
HMEC-1 were grown to full confluency and exposed as indicated to normoxia or hypoxia. To measure endothelial Ado uptake, 10 nCi of 8-14C-Ado (Sigma-Aldrich) was added to the supernatant, in addition to a 50 μM concentration of “cold” Ado. After indicated time periods, cells were washed with HBSS and lysed in cold H2O. Assessment of intracellular Ado uptake was done by measuring the radioactivity within the lysates by directly diluting the lysate into the scintillation mixture (Ultima Gold HFL-LSC-Cocktail; Packard) and counting decays per minute (Liquid Scintillation Counter Packard 1600).
Transcriptional analysis
Conventional PCR was used to verify endothelial ENT1 and ENT2 mRNA regulation, as described previously (38). The PCR reaction contained 1 μM each of the sense primer 5′-AAGGCACCTGGTTTCTGTC-3′ and the antisense primer 5′-CTTGGGCTTGGAGAACAC-3′ for ENT1, and 5′-CTTCCATACCCACTCTCTCACC-3′ and 5′-GAGAGAGAGGGGATTGGGTC-3′ for ENT2, respectively. The primer set was amplified using increasing numbers of cycles of 94°C for 1 min, 58°C for 2 min, 72°C for 4 min, and a final extension of 72°C for 7 min. The PCR transcripts were visualized on a 1.5% agarose gel containing 5 μg/ml of ethidium bromide. Human β-actin (sense primer, 5′-TGACGGGGTCACCCACACTGTGCCCATCTA-3′ and antisense primer, 5′-CTAGAAGCATTTGCGGTGGACGATGGAGGG-3′) in identical reactions was used to control for the starting template.
In subsets of experiments, the transcriptional profile of endothelial cells subjected to normobaric hypoxia (12 h) was compared in RNA derived from control or hypoxic endothelia using quantitative genechip expression arrays (Affymetrix, Inc.) as described before (9). Where indicated, mRNA was also quantified by real-time PCR (iCycler; Bio-Rad Laboratories Inc.) as described previously (9). The primer sets contained 1 μM sense and 1 μM antisense containing SYBR green I (GE Healthcare) in the reaction mixture. Transcript levels and fold change in mRNA were determined as described previously (39). In addition, real-time PCR was performed from RNA isolations of human saphenous vein after ex vivo exposure to hypoxia. After approval by the institutional review board, saphenous vein material was obtained from patients undergoing aorta-coronary bypass surgery. Equal portions of dissected vein tissue were subjected to hypoxia (pO2 20 torr) immediately after the operation for different time points (0, 2, 8, and 24 h). After hypoxia exposure, total RNA was isolated from the tissues and real-time PCR was performed as described before.
Macromolecule paracellular permeability assay
Using a modification of methods previously described (18), HMEC-1 were grown on polycarbonate permeable supports (0.4-μm pore, 6.5-mm diam; Costar Corp.) and studied 7–10 d after seeding (2–5 d after confluency). Inserts were placed in HBSS-containing wells (0.9 ml), and HBSS (alone or with indicated concentrations of Ado, with and without 10 μM concentration of dipyridamole) was added to inserts (100 μl). At the start of the assay (t = 0), FITC-labeled dextran 70 kD (concentration 3.5 μM) was added to fluid within the insert. The size of FITC-dextran, 70 kD, approximates that of human albumin, both of which have been used in similar endothelial paracellular permeability models (18). Fluid from opposing well (reservoir) was sampled (50 μl) over 60 min (t = 20, 40, and 60 min). Fluorescence intensity of each sample was measured (excitation, 485 nm; emission, 530 nm; Cytofluor 2300; Millipore Corp., Waters Chromatography) and FITC-dextran concentrations were determined from standard curves generated by serial dilution of FITC-dextran. Paracellular flux was calculated by linear regression of sample fluorescence.
Immunoblotting experiments
HMEC-1 were grown to confluency on 100-mm dishes and exposed to indicated experimental conditions. The monolayers were lysed for 10 min in 300 μl lysis buffer (150 mM NaCl, 25 mM Tris, pH 8.0, 5 mM EDTA, 2% Triton X-100, and 10% mammalian tissue protease inhibitor cocktail; Sigma-Aldrich), scraped and collected into microfuge tubes. After spinning at 14,000 g to remove cell debris, the pellet was discarded. Proteins were solublized in nonreducing Laemmli sample buffer and heated to 90°C for 5 min. Samples were resolved on a 12% polyacrylamide gel and transferred to nitrocellulose membranes. The membranes were blocked for 1 h at room temperature in PBS supplemented with 0.2% Tween 20 (PBS-T) and 4% BSA. The membranes were incubated in 5 μg/ml polyclonal rabbit anti-ENT1 in PBS-T for 1 h at room temperature, followed by 10 min washes in PBS-T. The membranes were incubated in 1:3,000 goat anti–rabbit IgG (ICN Biomedicals/Cappel), and conjugated to horseradish peroxidase for 1 h at room temperature. The wash was repeated and proteins were detected by enhanced chemiluminescence. To control for protein loading, blots were stripped and reprobed for β-actin using a mouse monoclonal anti–human β-actin antibody (Abcam Inc.). In subsets of experiments, identical conditions were used to examine HIF-1α protein levels in nuclear lysates derived from HMEC-1. For these purposes, 1 μg/ml of monoclonal mouse anti–HIF-1α (Novus Biologicals) was used.
ENT1 and ENT2 suppression with RNA interference.
HMEC-1 were either grown on inserts or in 60-mm Petri dishes. Different sets of siRNA directed against human ENT1 and ENT2 were designed using standard molecular tools, synthesized by Dharmacon, and tested with RT-PCR for their efficiency to suppress ENT1 and ENT2 expression at different concentrations. Highest efficiencies were found with 5′-CGGCCACUCAGUAUUUCACdTdT-3′ (sense strand) and 5′-GUGAAAUACUGAGUGGCCGdTdT-3′ (antisense strand) for ENT1 and 5′-CUCUCUCACCGAAGCCUAAdTdT-3′ (sense strand) and 5′-UUAGGCUUCGGUGAGAGAGdTdT-3′ (antisense strand) for ENT2. As nonspecific control, the sense strand of the ENT2 oligoribonucleotide was used under identical conditions. As additional control, knockdown with a set of siRNA directed against human 5′-ectonucleotidase (CD73) was performed using the sense sequence 5′-CUAUCUGGUUCACCGUGUAdTdT-3′ (sense strand) and 5′-UACACGGUGAACCAGAUAGdTdT-3′ (antisense strand). HMEC-1 loading was accomplished using standard conditions of Fugene6 (Roche Diagnostics Corp.), when cells had reached 40–60% confluence. After 48 h of loading, RNA was isolated as described before or functional assays (flux assays, Ado uptake) were performed.
ENT1 reporter assays
Plasmids expressing sequence corresponding to full-length ENT1 (−756 to +1), or the following 5′ truncations: ENT1-344 (bp −334 to +1), ENT1-282 (bp −282 to +1), ENT1-225 (bp −225 to +1), have been previously characterized (unpublished data). As a control for hypoxia, cells were transfected with a PGL3-based HRE plasmid containing four tandem HIF-1 enhancer sequences from the 3′-region of the erythropoietin gene (40). HMEC-1 were cotransfected with constitutively expressed Renilla plasmids using standard methods of overnight transfection using polyfect transfection reagent (QIAGEN). T84 cells were transfected using solution T and a modified form of electroporation (Amaxa Inc.). After transfection, cells were subjected to hypoxia or normoxia for 48 h. Luciferase activity was assessed (Turner Designs) using a luciferase assay kit (Promega). All firefly luciferase activity was normalized with respect to the constitutively expressed Renilla luciferase reporter gene.
In subsets of experiments, HIF-1–binding site mutations were introduced in ENT1-756 plasmids using the GeneEditor in vitro site-directed mutagenesis system (Promega). In brief, mutations encoding two nucleotide mutations ENT1 HIF-1 binding sites at positions −721 and −657, respectively, as described previously. All mutations were confirmed by sequencing using pGL3-basic primers. Hypoxia inducibility in transient transfectants using such mutated luciferase constructs was exactly as described in the previous paragraph.
ChIP assay
ChIP assays were performed using HMEC-1 subjected to normoxia or hypoxia. In brief, 2 × 107 cells were fixed with 1% paraformaldehyde for 10 min. Cross-linking was stopped by the addition of 125 mM glycine, and chromatin derived from isolated nuclei was sheared using a F550 micro-tip cell sonicator (Fisher Scientific). After centrifugation, supernatants containing sheared chromatin were incubated for 4 h with 5 μg of anti–HIF-1α antibody (Novus Biologics) or control IgG. Protein A sepharose was added and the incubation continued overnight at 4°C. Immune complexes were washed extensively and eluted from the protein A sepharose. The supernatants were transferred to a new tube, and 1 μg/μl of RNase was added and incubated for 5 h at 67°C. Samples were frozen at −80°C and 60 μg/μl proteinase K was added and incubated for 2 h at 45°C. Next, samples were diluted with TE containing 10 μg of tRNA followed by one extraction with phenol/chloroform and one extraction with chloroform. DNA was precipitated from the samples, washed, dried, and resuspended in 30 μl of autoclaved water. 2 μl of sample was used for each PCR reaction. The sequences of the ENT1 promoter-specific primers spanning the putative HIF-1–binding region were as follows: sense, 5′-GTGTCAGTGCACATCTGCCTGGC-3′ and antisense, 5′-CCTGTCCGCTTCCCCTTTCTAAG-3′. The size of the amplified product resulting from the use of this primer pair was 192 bp.
Lentiviral vector design, production, and transduction in T84 cells.
The HIV-1 lentiviral vector used was based on a vector previously described in detail (41). In short, the ΔODD variant of HIF-1α (containing a proline→alanine substitution at position 564; the ODD mutant HIF-1α plasmid was provided by H. Franklin Bunn [Harvard Medical School, Boston, MA]) (21) was cloned into the Bam HI/ClaI sites of the lentiviral vector. Virus was produced by first transfecting 293T cells with 3.2 μg of vector, 4.0 μg of the packaging plasmid, and 0.4 μg of REV, TAT, and VSV-G and collecting and filtering the cell supernatant through a 0.45-μm filter. Viral titers were determined by FACS analysis and/or quantitative Southern blot analysis. T84 cells were plated at a density of 2 × 105 cells/10cm2 dish 24 h before transduction. These cells were transduced using 2 × 106 infectious U/ml (MOI = 10) of viral supernatant along with 5 μg/ml protamine sulfate for 3 h at 37°C. Cells were fed and propagated in standard T84 media.
In vivo hypoxia model.
C57BL/6/129 svj mice were matched according to sex, age, and weight. Total organ vascular permeability was quantified by intravascular administration of Evan's blue as described previously (42). Animals were injected with dipyridamole (10 mg/kg i.p. and 10 mg/kg s.c) or of S-(4-nitrobenzyl)-6-thioinosine (NBTI, 1 mg/kg i.p.[Sigma-Aldrich]) or vehicle (DMSO). For the purpose of quantifying vascular permeability, 0.2 c.c. of Evan's blue (0.5% in PBS) were injected intravenously. Animals were exposed to normobaric hypoxia (8% O2, 92% N2) or room temperature air for 4 h (n = 6 animals per condition). After hypoxia/normoxia exposure, the animals were killed and the colon, kidney, liver, and lungs were harvested. Organ Evan's blue concentrations were quantified after formamide extraction (55°C for 2 h) by measuring absorbances at 610 nm with subtraction of reference absorbance at 450 nm. Pulmonary edema was assessed in additional experiments. For this purpose, lungs of the animals (n = 6) were collected, weighed, and dried by speed-vac (Eppendorf Vacufuge). Weight differences before and after drying were used to calculate lung water content. For the purpose of quantifying PMN tissue concentrations, the animals were exposed to normobaric hypoxia (8% O2, 92% N2) or room temperature air for 4 h (n = 4–6 animals per condition). After hypoxia/normoxia exposure, the animals were killed, organs were harvested, and the PMN marker MPO was quantified as previously described (43). Plasma levels of dipyridamole were determined by HPLC with a pump P680 and a Hitachi Fluorescence Detector L-7480 on a reverse-phase column (Grom-Sil 120-ODS-ST-5μ; 150 × 3 mm Grom) using a mobile phase gradient from 0 to 33% acetonitrile/0.3 mM KH2PO4 (pH 5) in 10 min. Fluorescence was measured at 404 nm excitation and 480 nm emission wavelength, with a retention time for dipyridamole of 8 min.
In subsets of experiments, colonic mucosal scrapings (enriched in epithelial cells) were obtained from 6–8-wk-old conditional Hif1α mutant mice or littermate controls, as described before (3). Scrapings were homogenized in RNAlater (QIAGEN) using a 22-gauge syringe (Becton Dickinson) and Qiashredder column (QIAGEN). RNA extraction including DNase digestion was performed using the RNeasy kit (QIAGEN). Reverse-transcription was done using the iScript cDNA Synthesis Kit (BioRad Laboratories). Amplification was performed on an i-Cycler IQ real-time PCR detection system (BioRad Laboratories) using the gene-specific primers for ENT1 as described before. These protocols were in accordance with National Institutes of Health guidelines for use of live animals and were approved by the Institutional Animal Care and Use Committee at Brigham and Women's Hospital.
Data analysis.
Data were compared by two-factor analysis of variance, or by Student's t test where appropriate. Values are expressed as the mean ± SD from at least three separate experiments.
Acknowledgments
The authors wish to thank Dr. P. Allen for advice on lentiviral construction and A. Mager, S. Zug, M. Faigle, and M. Hochgutbrot for technical assistance.
This work was supported by Fortune grant no. 1319-0-0 and DFG grant no. EL274/2-2 (to H.K. Eltzschig), National Institutes of Health grant nos. HL60569 and DK50189 (to S.P. Colgan), and by grants from the NSERC and PREA (to I.R. Coe).
The authors have no conflicting financial interests.
Abbreviations used: Ado, adenosine; ChIP, chromatin immunoprecipitation; ENT, equilibrative nucleoside transporter; HIF-1, hypoxia inducible factor 1; HMEC, human microvascular endothelial cells; HRE, hypoxia-responsive element; MPO, myeloperoxidase.
References
- 1.Kong, T., H.K. Eltzschig, J. Karhausen, S.P. Colgan, and C.S. Shelley. 2004. Leukocyte adhesion during hypoxia is mediated by HIF-1-dependent induction of β2 integrin gene expression. Proc. Natl. Acad. Sci. USA. 101:10440–10445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eltzschig, H.K., and C.D. Collard. 2004. Vascular ischaemia and reperfusion injury. Br. Med. Bull. 70:71–86. [DOI] [PubMed] [Google Scholar]
- 3.Karhausen, J., G.T. Furuta, J.E. Tomaszewski, R.S. Johnson, S.P. Colgan, and V.H. Haase. 2004. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J. Clin. Invest. 114:1098–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ohta, A., and M. Sitkovsky. 2001. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 414:916–920. [DOI] [PubMed] [Google Scholar]
- 5.Linden, J. 2001. Molecular approach to adenosine receptors: receptor-mediated mechanisms of tissue protection. Annu. Rev. Pharmacol. Toxicol. 41:775–787. [DOI] [PubMed] [Google Scholar]
- 6.Sitkovsky, M.V., D. Lukashev, S. Apasov, H. Kojima, M. Koshiba, C. Caldwell, A. Ohta, and M. Thiel. 2004. Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annu. Rev. Immunol. 22:657–682. [DOI] [PubMed] [Google Scholar]
- 7.Desai, A., C. Victor-Vega, S. Gadangi, M.C. Montesinos, C.C. Chu, and B.N. Cronstein. 2005. Adenosine A2A receptor stimulation increases angiogenesis by down-regulating production of the antiangiogenic matrix protein thrombospondin 1. Mol. Pharmacol. 67:1406–1413. [DOI] [PubMed] [Google Scholar]
- 8.Eltzschig, H.K., J.C. Ibla, G.T. Furuta, M.O. Leonard, K.A. Jacobson, K. Enjyoji, S.C. Robson, and S.P. Colgan. 2003. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J. Exp. Med. 198:783–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Synnestvedt, K., G.T. Furuta, K.M. Comerford, N. Louis, J. Karhausen, H.K. Eltzschig, K.R. Hansen, L.F. Thompson, and S.P. Colgan. 2002. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J. Clin. Invest. 110:993–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thompson, L.F., H.K. Eltzschig, J.C. Ibla, C.J. Van De Wiele, R. Resta, J.C. Morote-Garcia, and S.P. Colgan. 2004. Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J. Exp. Med. 200:1395–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baldwin, S.A., P.R. Beal, S.Y. Yao, A.E. King, C.E. Cass, and J.D. Young. 2004. The equilibrative nucleoside transporter family, SLC29. Pflugers Arch. 447:735–743. [DOI] [PubMed] [Google Scholar]
- 12.Kong, W., K. Engel, and J. Wang. 2004. Mammalian nucleoside transporters. Curr. Drug Metab. 5:63–84. [DOI] [PubMed] [Google Scholar]
- 13.Archer, R.G.E., V. Pitelka, and J.R. Hammond. 2004. Nucleoside transporter subtype expression and function in rat skeletal muscle microvascular endothelial cells. Br. J. Pharmacol. 143:202–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saito, H., M. Nishimura, H. Shinano, H. Makita, I. Tsujino, E. Shibuya, F. Sato, K. Miyamoto, and Y. Kawakami. 1999. Plasma concentration of adenosine during normoxia and moderate hypoxia in humans. Am. J. Respir. Crit. Care Med. 159:1014–1018. [DOI] [PubMed] [Google Scholar]
- 15.Chaudary, N., Z. Naydenova, I. Shuralyova, and I.R. Coe. 2004. The adenosine transporter, mENT1, is a target for adenosine receptor signaling and PKCε in hypoxic and pharmacological preconditioning in the mouse cardiomyocyte cell line, HL-1. J. Pharmacol. Exp. Ther. jpet.104.067157. [DOI] [PubMed]
- 16.Chaudary, N., Z. Naydenova, I. Shuralyova, and I.R. Coe. 2004. Hypoxia regulates the adenosine transporter, mENT1, in the murine cardiomyocyte cell line, HL-1. Cardiovasc. Res. 61:780–788. [DOI] [PubMed] [Google Scholar]
- 17.Hashikawa, T., S.W. Hooker, J.G. Maj, C.J. Knott-Craig, M. Takedachi, S. Murakami, and L.F. Thompson. 2004. Regulation of adenosine receptor engagement by ecto-adenosine deaminase. FASEB J. 18:131–133. [DOI] [PubMed] [Google Scholar]
- 18.Lennon, P.F., C.T. Taylor, G.L. Stahl, and S.P. Colgan. 1998. Neutrophil-derived 5′-adenosine monophosphate promotes endothelial barrier function via CD73-mediated conversion to adenosine and endothelial A2B receptor activation. J. Exp. Med. 188:1433–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manalo, D.J., A. Rowan, T. Lavoie, L. Natarajan, B.D. Kelly, S.Q. Ye, J.G.N. Garcia, and G.L. Semenza. 2005. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 105:659–669. [DOI] [PubMed] [Google Scholar]
- 20.Narravula, S., and S.P. Colgan. 2001. Hypoxia-inducible factor 1-mediated inhibition of peroxisome proliferator-activated receptor α expression during hypoxia. J. Immunol. 166:7543–7548. [DOI] [PubMed] [Google Scholar]
- 21.Huang, L.E., J. Gu, M. Schau, and H.F. Bunn. 1998. Regulation of hypoxia-inducible factor 1α is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA. 95:7987–7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eltzschig, H.K., L.F. Thompson, J. Karhausen, R.J. Cotta, J.C. Ibla, S.C. Robson, and S.P. Colgan. 2004. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: coordination by extracellular nucleotide metabolism. Blood. 104:3986–3992. [DOI] [PubMed] [Google Scholar]
- 23.Pinsky, D.J., Y. Naka, H. Liao, M.C. Oz, D.D. Wagner, T.N. Mayadas, R.C. Johnson, R.O. Hynes, M. Heath, C.A. Lawson, and D.M. Stern. 1996. Hypoxia-induced exocytosis of endothelial cell Weibel-Palade bodies. A mechanism for rapid neutrophil recruitment after cardiac preservation. J. Clin. Invest. 97:493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shreeniwas, R., S. Koga, M. Karakurum, D. Pinsky, E. Kaiser, J. Brett, B.A. Wolitzky, C. Norton, J. Plocinski, W. Benjamin, et al. 1992. Hypoxia-mediated induction of endothelial cell interleukin-1 α. An autocrine mechanism promoting expression of leukocyte adhesion molecules on the vessel surface. J. Clin. Invest. 90:2333–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith, P.G., H.D. Thomas, H.C. Barlow, R.J. Griffin, B.T. Golding, A.H. Calvert, D.R. Newell, and N.J. Curtin. 2001. In vitro and in vivo properties of novel nucleoside transport inhibitors with improved pharmacological properties that potentiate antifolate activity. Clin. Cancer Res. 7:2105–2113. [PubMed] [Google Scholar]
- 26.Collard, C.D., K.A. Park, M.C. Montalto, S. Alapati, J.A. Buras, G.L. Stahl, and S.P. Colgan. 2002. Neutrophil-derived glutamate regulates vascular endothelial barrier function. J. Biol. Chem. 277:14801–14811. [DOI] [PubMed] [Google Scholar]
- 27.Griffiths, M., N. Beaumont, S.Y. Yao, M. Sundaram, C.E. Boumah, A. Davies, F.Y. Kwong, I. Coe, C.E. Cass, J.D. Young, and S.A. Baldwin. 1997. Cloning of a human nucleoside transporter implicated in the cellular uptake of adenosine and chemotherapeutic drugs. Nat. Med. 3:89–93. [DOI] [PubMed] [Google Scholar]
- 28.SenGupta, D.J., and J.D. Unadkat. 2004. Glycine 154 of the equilibrative nucleoside transporter, hENT1, is important for nucleoside transport and for conferring sensitivity to the inhibitors nitrobenzylthioinosine, dipyridamole, and dilazep. Biochem. Pharmacol. 67:453–458. [DOI] [PubMed] [Google Scholar]
- 29.Gati, W.P., and A.R. Paterson. 1997. Measurement of nitrobenzylthioinosine in plasma and erythrocytes: a pharmacokinetic study in mice. Cancer Chemother. Pharmacol. 40:342–346. [DOI] [PubMed] [Google Scholar]
- 30.Decking, U.K.M., G. Schlieper, K. Kroll, and J. Schrader. 1997. Hypoxia-induced inhibition of adenosine kinase potentiates cardiac adenosine release. Circ. Res. 81:154–164. [DOI] [PubMed] [Google Scholar]
- 31.Schofield, C.J., and P.J. Ratcliffe. 2004. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 5:343–354. [DOI] [PubMed] [Google Scholar]
- 32.Semenza, G.L. 1999. Perspectives on oxygen sensing. Cell. 98:281–284. [DOI] [PubMed] [Google Scholar]
- 33.Hofer, T., H. Wenger, and M. Gassmann. 2002. Oxygen sensing, HIF-1α stabilization and potential therapeutic strategies. Pflugers Arch. 443:503–507. [DOI] [PubMed] [Google Scholar]
- 34.Nozik-Grayck, E., C.A. Piantadosi, J. van Adelsberg, S.L. Alper, and Y.C. Huang. 1997. Protection of perfused lung from oxidant injury by inhibitors of anion exchange. Am. J. Physiol. Lung Cell. Mol. Physiol. 273:L296–L304. [DOI] [PubMed] [Google Scholar]
- 35.Sheridan, W.G., R.H. Lowndes, and H.L. Young. 1990. Intraoperative tissue oximetry in the human gastrointestinal tract. Am. J. Surg. 159:314–319. [DOI] [PubMed] [Google Scholar]
- 36.Robinson, K.A., F.J. Candal, N.A. Scott, and E.W. Ades. 1995. Seeding of vascular grafts with an immortalized human dermal microvascular endothelial cell line. Angiology. 46:107–113. [DOI] [PubMed] [Google Scholar]
- 37.Ades, E.W., F.J. Candal, R.A. Swerlick, V.G. George, S. Summers, D.C. Bosse, and T.J. Lawley. 1992. HMEC-1: establishment of an immortalized human microvascular endothelial cell line. J. Invest. Dermatol. 99:683–690. [DOI] [PubMed] [Google Scholar]
- 38.Furuta, G.T., J.R. Turner, C.T. Taylor, R.M. Hershberg, K. Comerford, S. Narravula, D.K. Podolsky, and S.P. Colgan. 2001. Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J. Exp. Med. 193:1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pfaffl, M.W. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sheta, E.A., H. Trout, J.J. Gildea, M.A. Harding, and D. Theodorescu. 2001. Cell density mediated pericellular hypoxia leads to induction of HIF-1α via nitric oxide and Ras/MAP kinase mediated signaling pathways. Oncogene. 20:7624–7634. [DOI] [PubMed] [Google Scholar]
- 41.Pawliuk, R., K.A. Westerman, M.E. Fabry, E. Payen, R. Tighe, E.E. Bouhassira, S.A. Acharya, J. Ellis, I.M. London, C.J. Eaves, et al. 2001. Correction of sickle cell disease in transgenic mouse models by gene therapy. Science. 294:2368–2371. [DOI] [PubMed] [Google Scholar]
- 42.Barone, G.W., P.C. Farley, J.M. Conerly, T.L. Flanagan, and I.L. Kron. 1989. Morphological and functional techniques for assessing endothelial integrity: the use of Evans blue dye, silver stains, and endothelial derived relaxing factor. J. Card. Surg. 4:140–148. [DOI] [PubMed] [Google Scholar]
- 43.Parkos, C.A., C. Delp, M.A. Arnaout, and J.L. Madara. 1991. Neutrophil migration across a cultured intestinal epithelium. Dependence on a CD11b/CD18-mediated event and enhanced efficiency in physiological direction. J. Clin. Invest. 88:1605–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]