

Abstract
MEG2, a protein tyrosine phosphatase with a unique NH2-terminal lipid-binding domain, binds to and is modulated by the polyphosphoinositides PI(4,5)P2 and PI(3,4,5)P3. Recent data implicate MEG2 in vesicle fusion events in leukocytes. Through the genesis of Meg2-deficient mice, we demonstrate that Meg2−/−embryos manifest hemorrhages, neural tube defects including exencephaly and meningomyeloceles, cerebral infarctions, abnormal bone development, and >90% late embryonic lethality. T lymphocytes and platelets isolated from recombination activating gene 2−/− mice transplanted with Meg2−/− embryonic liver–derived hematopoietic progenitor cells showed profound defects in activation that, in T lymphocytes, was attributable to impaired interleukin 2 secretion. Ultrastructural analysis of these lymphocytes revealed near complete absence of mature secretory vesicles. Taken together, these observations suggest that MEG2-mediated modulation of secretory vesicle genesis and function plays an essential role in neural tube, vascular, and bone development as well as activation of mature platelets and lymphocytes.
Tyrosine phosphorylation is pivotal in diverse physiological processes in eukaryotes, including cell proliferation and differentiation, cell–cell communication, and regulation of transmembrane and intracellular signaling pathways (1, 2). Protein tyrosine phosphorylation reflects the balance between the activities of protein tyrosine kinases and protein tyrosine phosphatases (PTPs). The latter selectively remove phosphate from tyrosine residues, an outcome that can negatively or positively regulate signaling pathways. Recent estimates suggest that the human genome contains at least 107 PTP genes, remarkably similar to the number of tyrosine kinases (3, 4). However, by comparison with protein tyrosine kinases, relatively little is known about the specific functions of the majority of PTPs.
MEG2, also known as PTPN9, was originally cloned from a megakaryocytic cell line and is distinguished from other mammalian PTPs by virtue of an NH2-terminal lipid-binding domain with homology to yeast Sec14p that binds to and is activated by polyphosphoinositides PIP2 and PIP3 as well as by phosphatidyl serine (5–10). We have recently reported that MEG2 resides on internal membranes, including secretory vesicles and granules in neutrophils and lymphocytes, and regulates secretory vesicle size and fusion via dephosphorylation of a key vesicle fusion protein, N-ethylmaleimide sensitive factor (NSF), which is involved in the disassembly of cis complexes of soluble NSF attachment protein receptor (SNARE) proteins under ATP hydrolysis (6, 7, 11).
To investigate the functional importance of MEG2 in development and in hematopoietic cell function, we generated mice deficient in MEG2 by homologous recombination. Meg2−/− embryos displayed multiple neurodevelopmental defects and hemorrhages as well as >90% late embryonic lethality. Lymphocytes and platelets from mice transplanted with Meg2−/− embryonic liver–derived hematopoietic progenitor cells demonstrated severe functional defects reflecting anomalous agonist-induced secretion.
Results
To characterize the genomic organization of murine Meg2, we isolated a BAC clone containing the full-length Meg2 gene and sequenced it. By comparison of the genomic sequence with the cDNA sequence, we deduced that Meg2 comprises 13 exons, with the catalytic domain encoded by exons 8–13 (Fig. 1 A). During embryogenesis, Meg2 mRNA expression was detected as early as embryonic day 9 (E9) and persisted throughout the embryonic period as assessed by northern analysis (Fig. 1 B). In adult mice, Meg2 mRNA was widely expressed with the highest concentrations in the brain, lung, heart, liver, kidneys, and testes and lower levels of expression in the spleen and bone marrow (Fig. 1 B). The predominant mRNA species was a 3.9-kb transcript present in all tissues examined. However, alternative transcripts of lower abundance were noted in several tissues, including the liver, brain, and testes, that were attributable to alternative mRNA splicing.
Figure 1.

Confirmation of null Meg2 allele in Meg2−/−mice. (A) Genomic organization of murine Meg2 illustrating introns and exons. H, HindIII. The long dashed lines represent HindIII restriction sites. The short lines and black boxes represent exons. Exon1′ is an exon that exists only in an alternatively spliced transcript characterized by RT-PCR and RACE. The size and location of the exons were determined by comparison of the sequences of full-length cDNA and genomic cDNA. The genomic size of Meg2 is ∼67 kb. The bar represents 10 kb. (B) Northern blots from embryos and adult mice. Embryo and tissue northern analysis demonstrating Meg2 mRNA expression in different tissues and during embryogenesis. FirstChoice Northern Blot Mouse Blot I was hybridized with a mouse Meg2 full-length cDNA probe and reprobed with human β-actin. He, heart; Br, brain; Li, liver; Sp, spleen; Ki, kidney; E, embryo; Lu, lung; Te, testis. (C) Gene targeting strategy. The wild-type Meg2 locus and the targeted allele after ES cell homologous recombination are illustrated. The deleted region of genomic DNA in the targeted allele contains exons 10, 11, 12, and part of exon 13. R, RcaI; P, PstI; B, BamHI; S, StuI; Neo, neomycin- resistance element cassette. Bar, 1 kb. The black rectangles represent the probes used for the genotyping of ES cells and mice by Southern hybridization. (D) Genotyping of Meg2+/+, Meg2+/−, and Meg2−/− mice by multiplex PCR. The 2.0- and 1.7-kb bands correspond to the wild-type and targeted alleles, respectively. (E) Southern analysis of tail DNA from wild-type (+/+) mice and mice containing the targeted (null) allele (+/− heterozygote). Genomic DNA was digested with RcaI or StuI and probed with 5′ or 3′ probe individually. (F) Northern analysis of Meg2 +/+, Meg2+/−, and Meg2−/− embryos at day E15. PolyA+ RNA from embryos was probed with a murine Meg2 full-length cDNA probe. The blots were subsequently reprobed with a human β-actin RNA probe. The 3.9-kb band corresponds to the major Meg2 transcript. The uniform density of the actin bands confirm equal loading of PolyA+ RNA. (G) Western blots of tissues from the liver, lung, and brain of Meg2+/+, Meg2+/−, and Meg2−/− mice demonstrating the absence of detectable MEG2 protein in tissues from the Meg2−/− mice, confirming a null mutation.
To ascertain the functional importance of Meg2, we generated Meg2-deficient mice by homologous recombination in embryonic stem (ES) cells targeting exons 10–13 containing part of the catalytic domain (Fig. 1, C–E). Mice heterozygous for Meg2 (Meg2+/−) appeared healthy and were fertile. At day (E12), Mendelian ratios of Meg2+/+, Meg2+/−, and Meg2−/− (1:2:1, respectively) embryos were detected. However, there was a decrease in the fraction of Meg2−/− homozygous embryos between days E15 and birth, with <10% of Meg2−/− embryos surviving to birth and the immediate postnatal period. Of the live-born Meg2−/− pups, all were severely runted and very few survived to weaning. Meg2−/− embryos and pups did not express Meg2 RNA or protein as determined by northern and western analysis, respectively (Fig. 1, F and G), confirming that these mice carry a null Meg2 allele.
MEG2−/−mice display craniofacial and neurodevelopmental abnormalities and hemorrhages
As early as day E10, Meg2−/− embryos demonstrated growth retardation and evidence of neural tube defects, including craniofacial abnormalities (shortened snout and domed cranium), exencephaly, encephaloceles, and meningomyeloceles (Fig. 2 A) that were also apparent in the few live-born pups (Fig. 2 B). The brains of neonatal Meg2−/−mice were grossly smaller than Meg2+/− or Meg2+/+ littermates, and histological analysis revealed areas of colliquative necrosis containing foamy and debris-laden macrophages (Fig. 2, C and D). Within such areas, there was petechial hemorrhage containing macrophages with ingested erythrocytes (Fig. 2 D), indicating that the hemorrhage occurred before death. The areas of necrosis involved the cerebral cortex and subcortical white matter. The adjacent lateral ventricles were enlarged, and the white matter demonstrated rarefaction and microcystic changes. These alterations are consistent with either a primary endothelial defect or with impaired platelet function (vide infra) that may enhance a bleeding tendency by impairment of hemostatic plug formation. Of the few Meg2−/− mice surviving past weaning, several demonstrated a conspicuous tremor and paraparesis, the latter likely due to spinal meningomyeloceles.
Figure 2.
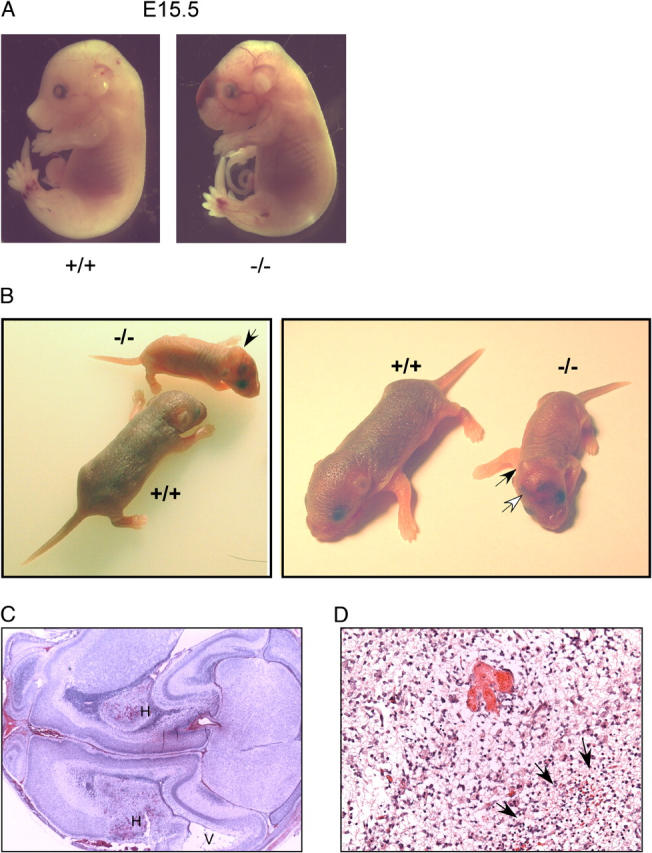
Neural tube developmental defects and cerebral infarctions in Meg2−/−mice. (A) Whole mount views of Meg2−/− embryos showing exencephaly, craniofacial maldevelopment, meningomyeloceles, and hemorrhages. (B) Photographs of wild-type (Meg2+/+) and Meg2−/− newborn pups showing domed heads, hemorrhages, and runting in the Meg2−/− pups. (C) Low power view (15×) of histology of brain tissue showing cerebral hemorrhagic infarction. H indicates the areas of hemorrhagic infarction, and V indicates dilation of the adjacent lateral ventricle. (D) Higher power view (200×) of histology of brain tissue showing cerebral hemorrhagic infarction. Arrows denote the area of infarction.
Most Meg2−/− embryos demonstrated large areas of hemorrhage in the craniofacial and cervical regions as well as dorsal aspects of the trunk (Fig. 2 A). Meg2−/− embryos also demonstrated abnormalities of calvarial and long bone formation. Meg2−/− embryos and newborn pups had absent calcification of the cranium as well as thinning and deficient calcification of the diaphyseal portion of the long bones (Fig. 3). Development of the other major organs, including the heart, lungs, kidneys, and liver, was grossly normal in Meg2−/− embryos, and histological analysis of these organs did not reveal any abnormalities.
Figure 3.
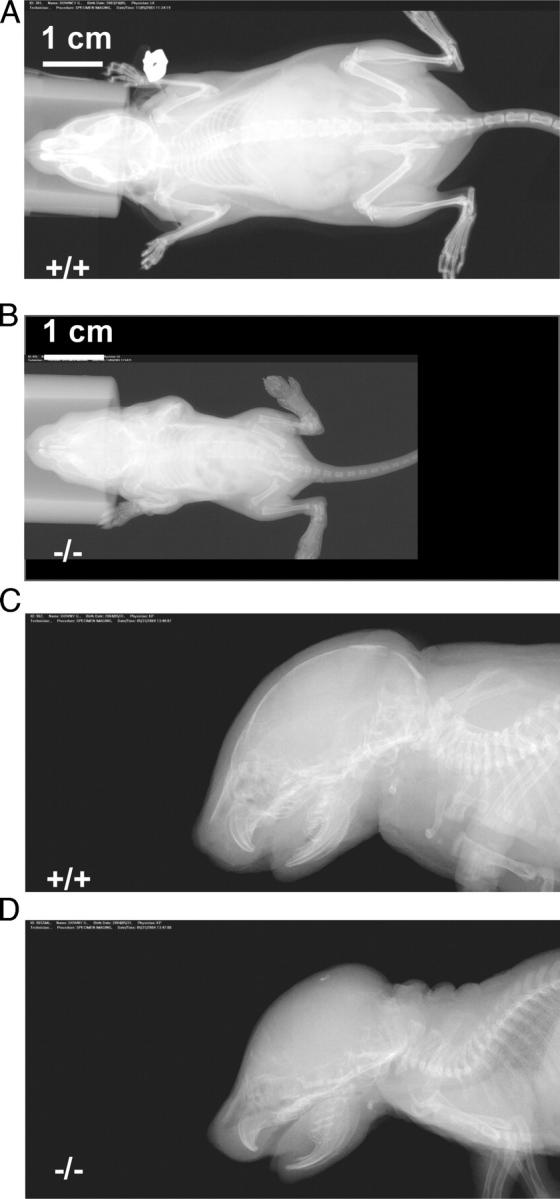
Abnormal formation of calvarial and long bones in Meg2−/−mice. X-ray (FAXetron) images showing calvarial and long bone abnormalities of young Meg2+/+ (A and C) Meg2−/− (B and D) pups.
Death of Meg2−/− embryos during late embryogenesis was likely attributable to hemorrhage into the cranial and dorsal areas and/or to severe neural tube closure defects. The vast majority of late embryonic and live-born Meg2−/− mice demonstrated obvious craniofacial malformations, including abnormal dentition and cleft palate, doming of the cranium, and shortened snout, and all were severely runted (Fig. 2 B). In the immediate perinatal period, serum electrolytes, glucose, calcium, creatinine, urea nitrogen, and albumin, and liver enzymes were within normal limits in surviving Meg2−/− mice. Most of these mice had difficulty feeding that was attributable to the facial, dental, and neurological abnormalities. Most did not survive until weaning despite attempts at liquid nutritional supplementation. Taken together, these observations indicate that MEG2 is required for normal embryonic development, including neural tube formation as well as bone and dental development.
Meg2 deficiency results in defective platelet and lymphocyte function
Next, we examined the role of MEG2 in hematopoietic cell function. The high degree of lethality in Meg2−/− mice and their sickly condition leading to premature death in the neonatal period precluded reliable analysis of hematopoietic cell function in these mice. Accordingly, we transplanted hematopoietic progenitor cells from the livers of day E15.5 Meg2−/− and Meg2+/+ embryos into lethally irradiated Rag2−/− mice (12). Mice transplanted with Meg2−/− hematopoietic progenitor cells demonstrated normal numbers and morphology of peripheral blood leukocytes, red blood cells, and platelets. Neutrophils from these mice were morphologically normal and displayed levels of GR-1 (Ly6G) comparable to their wild-type controls as assessed by flow cytometry. Functional analysis of Meg2−/− neutrophils, including surface expression of CD11b, adhesion to serum-coated tissue culture plastic, and activation of the NADPH oxidase, did not reveal any abnormalities (Table I).
Table I.
Analysis of neutrophils from Meg2+/+ and Meg2−/− hematopoietic progenitor cell–transplanted mice
| Parameter | Meg2+ /+ | Meg2− /− |
|---|---|---|
| GR-1 fluorescence (RFU) | 12.4 ± 1.4 | 12.4 ± 1.7 |
| Dihydrorhodamine fluorescence (RFU) (unstimulated cells) | 76.6 ± 5.88 | 71.9 ± 8.0 |
| Dihydrorhodamine fluorescence (PMA stimulation) | 140.9 ± 23.59 | 148.18 ± 10.30 |
| CD11b fluorescence (RFU) (unstimulated cells) | 93.3 ± 15.0 | 86.19 ± 6.2 |
| CD11b fluorescence (RFU) (PMA stimulation) | 269.3 ± 75.8 | 312.2 ±46.5 |
| % Adhesion (unstimulated cells) | 8.9 ± 1.6 | 10.4 ± 1.6 |
| % Adhesion (PMA stimulation) | 14.6 ± 1.6 | 14.3 ± 1.5 |
Data are presented as relative fluorescence units (RFU) as assessed by flow cytometry or as percent adhesion.
By contrast, Meg2−/− platelets and lymphocytes demonstrated significant functional abnormalities. Thrombin-induced platelet aggregation was considerably diminished in Meg2−/− compared with Meg2+/+ platelets (Fig. 4). By comparison, ADP-induced aggregation was only slightly impaired in Meg2−/− platelets. These observations are consistent with defective release of platelet granules in Meg2−/− platelets, a process that is involved in the second phase of platelet aggregation (13, 14).
Figure 4.

Abnormal platelet function in Meg2−/−mice. (A) Images of platelet aggregates in response to ADP and thrombin. (a and b) Platelet-rich plasma from Meg2+/+ (a) and Meg2−/− (b) hematopoietic progenitor cell–transplanted mice was treated with 20 μM ADP. (c and d) Washed platelets from Meg2+/+ (c) and Meg2−/− (d) hematopoietic progenitor cell–transplanted mice were treated with 1 U/ml thrombin. (d) Note that the platelet aggregates from wild-type hematopoietic progenitor cell–transplanted mice are substantially larger than those derived from Meg2−/− mice. (B) Platelet aggregometry traces in response to thrombin and ADP. Left panel: platelet-rich plasma from Meg2+/+ (black trace) and Meg2−/− (gray trace) hematopoietic progenitor cell–transplanted mice was treated with thrombin to induce platelet aggregation. Platelet aggregation was measured by aggregometry after stimulation with 1 U/ml thrombin. Note the decreased thrombin-induced platelet aggregation in Meg2−/− platelets. Right panel: Washed platelets from Meg2+/+ (black trace) and Meg2−/− (gray trace) hematopoietic progenitor cell–transplanted mice (black trace) were treated with 20 μM ADP. Meg2−/− platelets demonstrate diminished platelet aggregation compared with Meg2+/+ platelets.
Although lymphocyte development and maturation was normal in mice transplanted with Meg2−/− hematopoietic progenitor cells (Table II and Fig. 5), their function was severely compromised. Proliferation of Meg2−/− T lymphocytes (both from the thymus and peripheral lymph nodes) was deficient in response to receptor-dependent (anti-CD3, CD3 plus anti-CD28 antibodies, and Con A) or -independent (PMA plus ionomycin) stimulation (Fig. 6). The defect in T lymphocyte activation was likely related, in part, to suboptimal IL-2 secretion because the amount of secreted IL-2 was substantially diminished from Meg2−/− lymphocytes (Fig. 6 C). This defect could not be explained by alterations in the surface expression of IL-2 receptors as CD25 expression was normal in Meg2−/− lymphocytes (Fig. 6 D). Importantly, supplementation (“rescue”) of Meg2−/− lymphocytes with exogenous IL-2 restored their proliferative capacity to both receptor-dependent and -independent stimulation (Fig. 6 E), providing strong evidence that the primary defect in Meg2−/− lymphocytes is related to IL-2 secretion. In support of this notion, intracellular levels of IL-2 were normal (Fig. 6 F), indicating that the primary defect is attributable to abnormal agonist-regulated exocytosis (secretion) of IL-2. A defect in agonist-induced secretion was observed with other cytokines, such as GM-CSF, IFN-γ, and IL-6, but was not universal because secretion of TNF-α, IL-5, IL-10, and IL-12 were largely unaffected (Table III).
Table II.
Immune cell numbers in Meg2+/+ and Meg2−/− hematopoietic progenitor cell–transplanted mice
| Immune organ | Cell number | |
|---|---|---|
| Meg2+/+ | Meg2−/− | |
| Thymus | 1.98 × 108 | 2.23 × 108 |
| Lymph Node | 5.5 × 107 | 4.2 × 107 |
| Spleen | 9.57 × 108 | 10.12 × 108 |
Figure 5.

Normal development of lymphoid cell populations in Meg2−/−mice. Flow cytometric analysis of lymphocytes from the (A) spleen, (B) thymus, (C) bone marrow, and (D) lymph nodes from Rag2−/− mice transplanted with Meg2+/+ and Meg2−/− embryonic liver–derived hematopoietic progenitor cells.
Figure 6.

Defective activation in Meg2−/−T lymphocytes. (A) Thymus-derived T lymphocyte proliferation in response to agonist stimulation as assessed by [3H]thymidine uptake. Thymus-derived lymphocytes from Rag2−/− mice transplanted with Meg2+/+ (wild- type) and Meg2−/− hematopoietic progenitor cells were activated with either TCR-mediated stimuli (anti-CD3 or anti-CD3 plus anti-CD28 antibodies) or with a combination of PMA and ionomycin. [3H]thymidine incorporation was measured by β scintillation counting. *, significantly different than Meg2+/+ responses as determined by ANOVA with post hoc Sheffe test. (B) Lymph node– derived T lymphocyte proliferation in response to agonist stimulation as assessed by [3H]thymidine uptake. *, significantly different than Meg2+/+ responses as determined by ANOVA with post hoc Sheffe test. (C) IL-2 production in Meg2+/+ and Meg2−/− T lymphocytes as assessed by ELISA. *, significantly different than Meg2+/+ responses as determined by ANOVA. (D) Surface expression of IL-2 receptors (CD25) by Meg2+/+ and Meg2−/− T lymphocytes as assessed by flow cytometry. (E) IL-2 rescue of Meg2−/− lymphocytes. Proliferation of Meg2+/+ and Meg2−/− T lymphocytes in response to agonist stimulation without or with the addition of 50 U/ml exogenous IL-2 as assessed by [3H]thymidine uptake. *, significantly different than Meg2−/− lymphocytes without IL-2 rescue as determined by ANOVA. (F) Intracellular IL-2 production in Meg2+/+ and Meg2−/− T lymphocytes as assessed by flow cytometry.
Table III.
Cytokine production by Meg2+/+ and Meg2−/− peripheral lymphocytes
| Stimulus | Control | Anti-CD3 | Anti-CD3 + CD28 | Con A | PMA + Ionomycin | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Meg2 Genotype | +/+ | −/− | +/+ | −/− | +/+ | −/− | +/+ | −/− | +/+ | −/− |
| GM-CSF | 43 | 35 | 151 | 82 | 194 | 92 | 84 | 71 | 191 | 90 |
| IFN-γ | nd | nd | 356 | 87 | 451 | 121 | nd | 62 | 141 | 124 |
| TNF-α | nd | nd | 56 | 35 | 62 | 31 | 4 | 26 | 47 | 32 |
| IL-1β | nd | nd | nd | nd | nd | nd | nd | nd | nd | nd |
| IL-4 | nd | nd | 100 | 43 | 129 | 49 | nd | 19 | 31 | 37 |
| IL-5 | nd | nd | 89 | 41 | 117 | 30 | 10 | nd | 18 | 64 |
| IL-6 | 9 | 9 | 761 | 306 | 1,042 | 360 | 51 | 231 | 401 | 376 |
| IL-10 | 9 | 9 | 74 | 37 | 71 | 44 | 13 | 34 | 23 | 34 |
| IL-12 (p70) | 9 | 8 | 13 | 11 | 13 | 10 | 9 | 10 | 12 | 11 |
Data represent extracellular cytokine levels in pg/ml as assessed by a bead binding array. n.d., none detected (below limits of detection). These results are representative of two separate experiments performed in duplicate.
To determine the ultrastructural basis for this defect, T lymphocytes were examined by electron microscopy. These studies revealed that the number of secretory vesicles was markedly diminished in Meg2−/− compared with Meg2+/+ lymphocytes (Fig. 7). Moreover, the Meg2−/− lymphocytes displayed nonmembrane-bound collections of electron dense particles with a “cluster-of-grapes” appearance. These clusters of particles may represent failed attempts to fuse small transport vesicles into immature secretory vesicles, which normally is the first step in the biogenesis of mature secretory vesicles.
Figure 7.
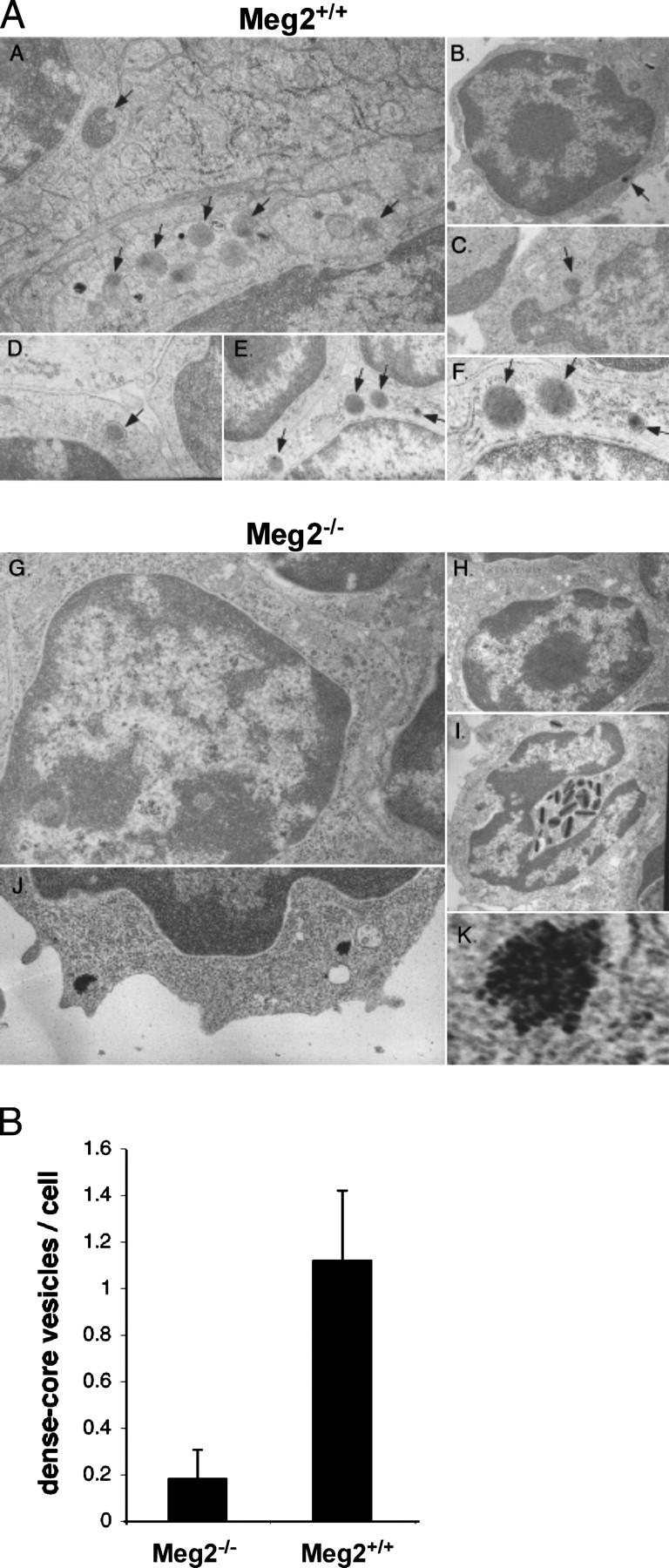
Absence of secretory vesicles in Meg2−/−lymphocytes. (A) Electron microscopic analysis of vesicles in Meg2+/+ and Meg2−/− splenocytes, thymocytes, and lymph node cells. Cells are from spleen (a and i), thymus (b–d, g, and h), and lymph node (e, f, j, and k). (a–f) Meg2+/+ lymphocytes. The arrows indicate typical dense core secretory vesicles that are present in normal numbers. (g and h–k) Meg2−/− lymphocytes. Note the near complete absence of dense core secretory vesicles and the presence of nonmembrane-bound collections of electron dense particles with a cluster-of-grapes appearance in Meg2−/− lymphocytes (i and k). (h) A Meg2−/− eosinophil with relatively normal appearance except that the vesicles are more elongated than normal. Magnifications are 15,000 (a, f, j, and k), 3,000 (b, c, h, and i), and 8,000 (d, e, and g). (k) The image is electronically magnified by a factor of 10. (B) Morphometric quantification of dense core vesicles from Meg2−/− and Meg2+/+ T lymphocytes.
Discussion
Our studies demonstrate the fundamental role played by MEG2 in diverse aspects of murine embryonic development and in the function of mature cells of hematopoietic origin. Meg2−/− T lymphocytes and platelets demonstrated profound defects in activation. Importantly, the common mechanism responsible for this anomalous function is faulty agonist-induced secretion (exocytosis). In T lymphocytes, this was manifest by diminished IL-2 secretion and correlated with the absence of mature secretory vesicles as assessed by electron microscopy. These vesicles are known to contain IL-2, and their fusion with the plasma membrane is required for agonist-induced extracellular secretion of the cytokine (7, 11). These data confirm and extend our previous observations in human Jurkat T lymphoma cells where overexpression of functional MEG2 resulted in enlarged vesicles and, conversely, expression of catalytically inactive MEG2 prevented the formation of mature vesicles and abrogated agonist-induced IL-2 secretion (11). The absence of mature secretory vesicles in Meg2−/− T lymphocytes underscores the importance of MEG2 in the biogenesis of these crucial components of the exocytic apparatus.
The functional defect in the second phase of thrombin-induced aggregation observed in Meg2−/− platelets is also likely attributable to defective agonist-induced secretion because this phase of aggregation is dependent on the exocytosis of granule-associated proteins, mediators, and adhesion molecules (13, 14). By comparison, the early phase of platelet aggregation that is dependent on integrin GPIIb/IIIa–fibrinogen interaction is preserved in the Meg2−/− platelets. Taken together, these observations reveal the importance of MEG2 in the function of cells of hematopoietic origin that involve regulated secretion.
Our observations point to a fundamental role for MEG2 in regulated secretion, a complex process involving biogenesis and fusion of the vesicle membranes with the plasma membrane followed by release of the vesicular contents. SNARE proteins are key participants in these events (15–17). A common feature of these proteins is the SNARE motif, which can self-assemble into a four-helix bundle (17). When this pairing occurs in trans, between v (or R)-SNAREs from a vesicle and t (or Q)-SNAREs from a target compartment, membrane fusion proceeds. Vesicle fusion involves a cascade of molecular events, beginning with a priming stage that is followed by docking with target membranes and the development of the fusion pore (18, 19). The key reaction in the priming stage is the dissociation of cis–SNARE complexes by NSF, a cytosolic protein that regulates membrane fusion by disassembling cis complexes of SNARE proteins under ATP hydrolysis. Importantly, we have recently reported that NSF is a substrate for MEG2 (11). Disassembly of cis complexes of SNARE proteins is critical for formation of a multimeric trans complex at the site of contact between fusion partners and for recycling of the proteins for subsequent rounds of fusion (20, 21). Null mutants of NSF are lethal in yeast and Drosophila, consistent with its obligatory role in general membrane fusion (22–24). The ATPase activity of NSF is enhanced by α-soluble NSF attachment protein (SNAP), which mediates NSF binding to SNARE complexes with other factors (25, 26). NSF is proposed to bind directly to syntaxin and SNAP-25 in a complex with vesicle-associated membrane protein (VAMP; references 21 and 23). Although NSF is required for virtually all membrane fusion events, its participation in agonist-stimulated secretion might be accounted for by changes in activity and/or subcellular distribution. In this regard, recent evidence from us and others indicates that posttranslational modification of NSF, via reversible phosphorylation of tyrosine 83 or nitrosylation, alters its capacity to facilitate membrane fusion (11, 27). This provides a plausible mechanism for involvement of NSF in regulated secretion and a role for MEG2 in these events.
Regulated (agonist-induced) secretion is pivotal for leukocyte function in immune responses (28, 29). For example, during T lymphocyte activation, IL-2 is secreted and binds to its receptor, triggering a cascade of intracellular events involved in cell activation and proliferation (30). To facilitate secretion, lymphocytes express a variety of SNARE proteins, such as syntaxin 11 and SNAP-23 (27, 28, 31). In addition to classical secretion (exocytosis), SNARE proteins are also involved in internalization and recycling of TCRs, events that are crucial during interaction with APCs. During this interaction, TCRs accumulate at the immunological synapse (the site of T cell–APC contact) by a mechanism that involves polarized recycling of the TCR in recycling endosomes mediated by exocytic SNARE proteins and presumably NSF (28). Interference with SNARE function reduces TCR accumulation at this site and impedes T cell activation (28).
Agonist-induced secretion in other leukocyte types also involves SNARE proteins. For example, platelets are known to express multiple SNARE proteins, including cellubrevin, SNAP-23 and SNAP-25, and syntaxin-2 and syntaxin-4, as well as VAMP-3 and VAMP-8, and interference with their function abolishes exocytosis from platelet α, dense, and lysosomal granules (32–37). Myeloid cells, such as neutrophils, also express a variety of SNAREs, of which SNAP-23, VAMP-2, and syntaxin-4 and syntaxin-6 appear to be involved in exocytosis of granule contents (38, 39). Unexpectedly, the function of Meg2−/− neutrophils and macrophages was relatively preserved, indicating that there must be cell-specific effects of the regulation of NSF by MEG2 that remain to be clarified. This is the subject of our current investigations.
Our studies clearly indicate the importance of MEG2 in neural tube and cranial-facial development, but the molecular mechanisms remain unknown. In murine models of neural tube closure defects, various genetic mutations result in an array of defects, including exencephaly, anecephaly, rachischsis, and spina bifida, each reflecting the failure of elevation of specific zones of the neural folds. The most common neural tube closure defect is isolated exencephaly, whereas the combination of exencephaly with a craniofacial defect, such as occurs in the Meg2−/− mice, is much less common (40–43). Meg2−/− embryos and pups demonstrate exencephaly, encephaloceles, foreshortened snout, and midline hemorrhages in the frontal and nasal regions, indicating that the most rostral part of the neural tube (zones A and B, closures 2 and 3) has failed to close (40, 44). Meningomyeloceles were also observed in some Meg2−/− embryos and pups, indicating failure of closure of the caudal part of the neural tube.
Distinct types of neural tube defects correlate with specific genetic mutations in mice, implying spatial and temporal heterogeneity of molecular mechanisms regulating the elevation of neural folds. These include convergent extension of the neural plate (45), elevation and apposition of the neural folds (46), and fusion of the neural tube folds (47). Discrete cellular events are required for the successful closure of cranial neural folds compared with spinal folds. For example, expansion of the cranial mesenchyme (48, 49), contraction of sub-apical actin microfilaments (50), and neural crest migration are considered to be important in the transition from biconvex to biconcave neural plate morphology (51, 52) and essential to cranial neural tube closure. Additionally, defects in apoptosis and proliferation of neuroepithelium may also contribute to failure of cranial neurulation (53). Investigation of the role of MEG2 in these specific events is a focus of our current investigations.
The cranial malformations and midline hemorrhages observed in Meg2−/− mice are reminiscent of the phenotype observed in mice with conditional deletion of PDGFRα (Wnt1 Cre+ PDGFRα neural crest cells) in cranial and cardiac neural crest cells (54). However, in contrast to Meg2−/− mice, the PDGFRα neural crest cell conditional mutant mice demonstrate cardiovascular defects likely related to the loss of essential PDGFRα functions in cardiac neural crest cells. During murine embryogenesis, the cranial neural crest cells differentiate into bone, cartilage, cranial ganglia, connective tissue of the head and neck, odontoblasts, and craniofacial mesenchyme (55). They also differentiate into pericytes and smooth muscle cells of all blood vessels of the face and forebrain (56, 57). Various genetic mutations affecting processes in cranial neural crest cells in mice result in facial malformations and exencephaly (51, 58). We speculate that the craniofacial malformations, midline hemorrhages, and bone defects in Meg2−/− mice are related to defective migration and/or differentiation of cranial neural crest cells.
In addition to the conspicuous developmental defects related to cranial neural crest cell function discussed above, we have preliminary evidence that there are defects in other neural crest cell populations in Meg2−/− mice. Pigmentation defects on the head, abdomen, and megacolon were observed infrequently in Meg2−/− mice. Because these abnormalities are associated with defects in trunk neural crest and enteric neural crest cells, respectively, it is possible that MEG2 regulates the development of these neural crest cells directly or indirectly. However, given the low frequency of these defects, it is possible that these are attributable to spontaneous mutations in other loci and not related to MEG2 per se.
Published studies provide insights into certain developmentally regulated pathways involved in neurulation in which MEG2 may play a role. Genetic mutations that result in cranial neural tube closure defects are related to cell proliferation, gap junction formation (59), organization of the actin cytoskeleton (60), transcription factors such as Pax3 (61), and DNA methylation (62). As a tyrosine phosphatase, MEG2 may affect neurulation and cranial structure formation through regulation of the phosphorylation state of signaling proteins involved in these pathways. Given the importance of MEG2 in secretory vesicle biogenesis and secretion, we speculate that defective secretion of growth factors or embryonic chemotaxins in the Meg2−/− cells might result in aberrant migration and function of neural crest cells. In this regard, signaling pathways involving PDGFRα, FGF8, Wnt1, ET-1, and TGF-β signaling (63–66) that are known to be important in the regulation of craniofacial development are potential pathways that might be regulated by MEG2.
Our observations that Meg2−/− mice display cranial and dorsal hemorrhages suggest a role for this PTP in certain aspects of vascular development. As MEG2 is expressed by endothelial cells (5), it is possible that the hemorrhages reflect abnormal embryonic vascular development and/or integrity related to faulty endothelial cell migration. This defective vascular development would be compounded by the defective platelet function observed in these mice. Additionally, the areas of hemorrhagic cerebral infarction might be accounted for by a combination of endothelial dysfunction leading to abnormal vascular development in association with defective platelet function.
In summary, our studies demonstrate that MEG2 plays a crucial role in neural tube, craniofacial, and bone development as well as in platelet and lymphocyte activation. One of the primary functions of MEG2 appears to be the control of agonist-induced secretory (exocytic) processes, including secretory vesicle genesis, maturation, and membrane fusion.
Materials and Methods
All antibodies were obtained from BD Biosciences unless otherwise specified. Monoclonal hamster anti–mouse CD3ɛ was produced by the 145-2C11 hybridoma (Ontario Cancer Institute). PMA, ionomycin, and murine IL-2 were from Sigma-Aldrich, and rabbit anti–hamster and anti–mouse IgG antibodies and Cy5-conjugated streptavidin were from Jackson ImmunoResearch Laboratories.
Targeted disruption of the murine Meg2 gene.
All animal studies were approved by the Institutional Review Boards of the University of Toronto. A mouse 129/SvJ BAC genomic library was screened for Meg2, and a clone containing the full-length Meg2 gene was isolated and sequenced using fluorescence dideoxy sequencing. In the targeting vector, a 4.2-kb DNA fragment encompassing exons 10, 11, 12, and part of exon 13 was deleted and replaced by a neomycin-resistance marker and cloned into a targeting vector (pPNTLoxP). The linearized targeting vector was electroporated into 129/SvJ and R1 ES cells. Two independent heterozygous ES cell clones were expanded and used to generate chimeric mice by blastocyst injection, and the mutant animals were bred to a C57BL/6J background for nine generations. Genotyping was performed by Southern blotting (Fig. 1 E) or by multiplex PCR (Fig. 1 D). For Southern analysis, genomic DNA was digested with RcaI or StuI and hybridized with 3′ and 5′ probes, respectively. The 5′ probe corresponded to a 194-bp fragment amplified by PCR with primer 5′ probe-5 and 5′ probe-6 (5′-AAAGACCTCCCCCCT-3′ and 5′-CCCAAATTCCAAACTGT-3′), and the 3′ probe corresponded to a DNA fragment amplified by PCR from the murine genomic DNA with primer PRM1 and PRM2 (5′-TAAACTAACATTGAGCCCTT-3′ and 5′-TTCCAGTACATCCAGCAGT-3′). Primers for multiplex PCR analysis were 5′-TAGAGCTTGCGGAACCCTTA-3′, 5′-CTGGACACTGCTGAGCCTAT-3′, and 5′-AGTCTCCAAAGCACCATTCC-3′. The PCR parameters are: denaturation at 95°C for 5 min, 35 cycles at 95°C for 30 s, 60°C for 30 s, and 72°C for 3 min.
RNA extraction and northern hybridization.
Mouse Northern Blot MTN1 and MTN2 (CLONTECH Laboratories, Inc.) and FirstChoice Northern Mouse Blot I (Ambion) were hybridized with a 33P random– or digoxigenin-labeled Meg2 cDNA probe. The latter probes were made with the StripEZ PCR Kit (Ambion) and digoxigenin-11-dUTP (Roche Applied Science). The blots were detected with a DIG luminescent detection Kit (Roche Applied Science). PolyA+ RNA was extracted from mouse embryos at day E15 with the Micro PolyA Pure Kit (Ambion). Equal amounts of PolyA+ RNA were loaded, separated by electrophoresis, transferred to a positively charged nylon membrane (Roche Applied Science), and hybridized with a digoxigenin-labeled mouse Meg2 cDNA probe and then reprobed with a human β-actin RNA probe (Roche Applied Science).
Southern hybridization.
Mouse genomic DNA was digested with the specified restriction enzymes, electrophoretically separated, and hybridized with digoxigenin-labeled DNA probes according to the manufacturer's instructions (Roche Applied Sciences).
SDS PAGE and immunoblotting.
SDS PAGE and immunoblotting were performed as described previously (6).
Electron microscopy.
Cells were fixed in 2% formaldehyde and 2% glutaraldehyde in 0.1 M cacodylate, pH 7.3, for 2 h at 4°C. Cells were washed three times in the same buffer before a secondary fixation in 2% OsO4 (in the same buffer) for 1 h at room temperature. Cells were washed an additional three times in the same buffer and then successively dehydrated in ethanol, infused with propylene oxide, and embedded in Epon 812. Ultrathin sections were obtained using the Reichert-Jung microtome, double stained with uranyl-acetate and lead citrate. Specimens were examined using a Hitachi-600 electron microscope at 75 kV with magnifications ranging from 8,000 to 100,000.
Hematopoietic cell transplantation.
Livers from day E15.5 Meg2+/+, Meg+/−, and Meg2−/− embryos were genotyped and used to repopulate lethally irradiated (9.5 Gy) syngeneic Rag2−/− recipients. Fetal livers were disaggregated in Opi-MEM-1 medium. Irradiated C57B/6 or Rag2−/− mice were injected via tail vein with a cell suspension containing 107 cells. The reconstituted mice were kept in a pathogen-free environment, fed with antibiotic-treated water, and used between 8 and 10 wk after transplantation.
Flow cytometry.
Flow cytometry was conducted as described previously (67) using a FACScan flow cytometer with CELLQuest software (Becton Dickinson).
Lymphocyte stimulation.
Lymphocyte proliferation was assessed using single cell suspensions prepared from thymuses, lymph nodes, and spleens of age-matched Meg2−/− and Meg2+/+ (C57BL/6) hematopoietic stem cell–reconstituted Rag2−/− mice. Thymocytes and lymph node cells were cultured for 48 h in culture medium alone or in the presence of either 0–25 μg/ml of plate-bound or 2 μg/ml of soluble anti-CD3ɛ antibody with or without 0.2 μg/ml anti-CD28 antibody or with 1 μg/ml of soluble anti-CD3 antibody plus 5 ng/ml PMA, 1 μg/ml Con A, or 5 μg/ml PMA plus 250 ng/ml ionomycin or 1 μg/ml of soluble anti-CD3 antibody. Cultured cells were pulsed with 1 μCi/well [3H]thymidine for 16 h before terminating the incubation. Incorporated radioactivity was measured using an automated β scintillation counter.
Cytokine analysis.
IL-2 levels in cell supernatants were quantified by ELISA (Genzyme). Levels of other cytokines were quantified using a multiplex fluorescent bead binding assay (LINCOplex; LINCO Research) and analyzed using a Luminex 100 analyzer. Intracellular IL-2 levels were assessed by flow cytometry on saponin-permeabilized cells as described previously (67).
Platelet isolation and aggregation.
Hematopoietic progenitor cell–transplanted mice were anesthetized and bled from the retro-orbital plexus using a heparin-coated glass capillary tube containing 3.8% sodium citrate. Platelet-rich plasma was washed twice with Pipes buffer (pH 7.0, 5 mM Pipes, 137 mM NaCl, 4 mM KCl, 0.1% glucose). Platelet aggregation was performed using a Chrono-Log aggregometer (Chrono-Log Corporation) as described previously (68). Aggregation was induced by 20 μM ADP (Sigma-Aldrich). The same experiments were also performed with washed platelets (2 × 108 platelets/ml) treated with 1 U/ml thrombin (Sigma-Aldrich) at 1,000 rpm. The washed aggregates from each sample were visualized with an inverted microscope (32X, 0.4NA; Zeiss Axiovert 135; Carl Zeiss MicroImaging, Inc.), and images were captured with a digital camera (DP70; Olympus).
Acknowledgments
We thank Drs. J. Rossant, M. Glogauer, and M. Post and their research groups for extensive comments and discussion, and R. Martin for technical assistance.
This research was supported by an operating grant from the Canadian Institutes of Health Research (MT-12255), a block term grant from the Ontario Thoracic Society (to G.P. Downey), and an operating grant from the National Institutes of Health (NIH AI55741 to T. Mustelin). G.P. Downey is the recipient of a Tier 1 Canada Research Chair in Respiration and is a scholar of the McLaughlin Center for Molecular Medicine, University of Toronto.
The authors have no conflicting financial interests.
Abbreviations used: ES, embryonic stem; NSF, N-ethylmaleimide sensitive factor; PTP, protein tyrosine phosphatase; SNAP, soluble NSF attachment protein; SNARE, soluble NSF attachment protein receptor; VAMP, vesicle-associated membrane protein.
References
- 1.Hunter, T. 1995. Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell. 80:225–236. [DOI] [PubMed] [Google Scholar]
- 2.Tonks, N.K., and B.G. Neel. 1996. From form to function: signaling by protein tyrosine phosphatases. Cell. 87:365–368. [DOI] [PubMed] [Google Scholar]
- 3.Manning, G., D.B. Whyte, R. Martinez, T. Hunter, and S. Sudarsanam. 2002. The protein kinase complement of the human genome. Science. 298:1912–1934. [DOI] [PubMed] [Google Scholar]
- 4.Alonso, A., J. Sasin, N. Bottini, I. Friedberg, A. Osterman, A. Godzik, T. Hunter, J. Dixon, and T. Mustelin. 2004. Protein tyrosine phosphatases in the human genome. Cell. 117:699–711. [DOI] [PubMed] [Google Scholar]
- 5.Gu, M., I. Warshawsky, and P.W. Majerus. 1992. Cloning and expression of a cytosolic megakaryocyte protein-tyrosine-phosphatase with sequence homology to retinaldehyde-binding protein and yeast SEC14p. Proc. Natl. Acad. Sci. USA. 89:2980–2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kruger, J.M., T. Fukushima, V. Cherepanov, N. Borregaard, C. Loeve, C. Shek, K. Sharma, A.K. Tanswell, C.W. Chow, and G.P. Downey. 2002. Protein-tyrosine phosphatase MEG2 is expressed by human neutrophils. Localization to the phagosome and activation by polyphosphoinositides. J. Biol. Chem. 277:2620–2628. [DOI] [PubMed] [Google Scholar]
- 7.Wang, X., H. Huynh, A. Gjorloff-Wingren, E. Monosov, M. Stridsberg, M. Fukuda, and T. Mustelin. 2002. Enlargement of secretory vesicles by protein tyrosine phosphatase PTP-MEG2 in rat basophilic leukemia mast cells and Jurkat T cells. J. Immunol. 168:4612–4619. [DOI] [PubMed] [Google Scholar]
- 8.Huynh, H., X. Wang, W. Li, N. Bottini, S. Williams, K. Nika, H. Ishihara, A. Godzik, and T. Mustelin. 2003. Homotypic secretory vesicle fusion induced by the protein tyrosine phosphatase MEG2 depends on polyphosphoinositides in T cells. J. Immunol. 171:6661–6671. [DOI] [PubMed] [Google Scholar]
- 9.Krugmann, S., K.E. Anderson, S.H. Ridley, N. Risso, A. McGregor, J. Coadwell, K. Davidson, A. Eguinoa, C.D. Ellson, P. Lipp, et al. 2002. Identification of ARAP3, a novel PI3K effector regulating both Arf and Rho GTPases, by selective capture on phosphoinositide affinity matrices. Mol. Cell. 9:95–108. [DOI] [PubMed] [Google Scholar]
- 10.Zhao, R., X. Fu, Q. Li, S.B. Krantz, and Z.J. Zhao. 2003. Specific interaction of protein tyrosine phosphatase-MEG2 with phosphatidylserine. J. Biol. Chem. 278:22609–22614. [DOI] [PubMed] [Google Scholar]
- 11.Huynh, H., N. Bottini, S. Williams, V. Cherepanov, L. Musumeci, K. Saito, S. Bruckner, E. Vachon, X. Wang, J. Kruger, et al. 2004. Control of vesicle fusion by a tyrosine phosphatase. Nat. Cell Biol. 6:831–839. [DOI] [PubMed] [Google Scholar]
- 12.Forrester, L.M., A.M. Bernstein, J. Rossant, and A. Nagy. 1991. Long-term reconstitution of the mouse hematopoietic system by embryonic stem cell-derived fetal liver. Proc. Natl. Acad. Sci. USA. 88:7514–7517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramasamy, I. 2004. Inherited bleeding disorders: disorders of platelet adhesion and aggregation. Crit. Rev. Oncol. Hematol. 49:1–35. [DOI] [PubMed] [Google Scholar]
- 14.Flaumenhaft, R. 2003. Molecular basis of platelet granule secretion. Arterioscler. Thromb. Vasc. Biol. 23:1152–1160. [DOI] [PubMed] [Google Scholar]
- 15.Jahn, R., T. Lang, and T.C. Sudhof. 2003. Membrane fusion. Cell. 112:519–533. [DOI] [PubMed] [Google Scholar]
- 16.Antonny, B. 2004. SNARE filtering by dynamin. Cell. 119:581–582. [DOI] [PubMed] [Google Scholar]
- 17.Ungar, D., and F.M. Hughson. 2003. SNARE protein structure and function. Annu. Rev. Cell Dev. Biol. 19:493–517. [DOI] [PubMed] [Google Scholar]
- 18.Conradt, B., J. Shaw, T. Vida, S. Emr, and W. Wickner. 1992. In vitro reactions of vacuole inheritance in Saccharomyces cerevisiae. J. Cell Biol. 119:1469–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang, C.W., P.E. Stromhaug, J. Shima, and D.J. Klionsky. 2002. The Ccz1-Mon1 protein complex is required for the late step of multiple vacuole delivery pathways. J. Biol. Chem. 277:47917–47927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jahn, R., and T.C. Sudhof. 1999. Membrane fusion and exocytosis. Annu. Rev. Biochem. 68:863–911. [DOI] [PubMed] [Google Scholar]
- 21.Hay, J.C., and R.H. Scheller. 1997. SNAREs and NSF in targeted membrane fusion. Curr. Opin. Cell Biol. 9:505–512. [DOI] [PubMed] [Google Scholar]
- 22.Thompson, C.R., and M.S. Bretscher. 2002. Cell polarity and locomotion, as well as endocytosis, depend on NSF. Development. 129:4185–4192. [DOI] [PubMed] [Google Scholar]
- 23.Littleton, J.T., R.J. Barnard, S.A. Titus, J. Slind, E.R. Chapman, and B. Ganetzky. 2001. SNARE-complex disassembly by NSF follows synaptic-vesicle fusion. Proc. Natl. Acad. Sci. USA. 98:12233–12238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaiser, C.A., and R. Schekman. 1990. Distinct sets of SEC genes govern transport vesicle formation and fusion early in the secretory pathway. Cell. 61:723–733. [DOI] [PubMed] [Google Scholar]
- 25.Sagiv, Y., A. Legesse-Miller, A. Porat, and Z. Elazar. 2000. GATE-16, a membrane transport modulator, interacts with NSF and the Golgi v-SNARE GOS-28. EMBO J. 19:1494–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han, S.Y., D.Y. Park, S.D. Park, and S.H. Hong. 2000. Identification of Rab6 as an N-ethylmaleimide-sensitive fusion protein-binding protein. Biochem. J. 352:165–173. [PMC free article] [PubMed] [Google Scholar]
- 27.Matsushita, K., C.N. Morrell, B. Cambien, S.X. Yang, M. Yamakuchi, C. Bao, M.R. Hara, R.A. Quick, W. Cao, B. O'Rourke, et al. 2003. Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide-sensitive factor. Cell. 115:139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Das, V., B. Nal, A. Dujeancourt, M.I. Thoulouze, T. Galli, P. Roux, A. Dautry-Varsat, and A. Alcover. 2004. Activation-induced polarized recycling targets T cell antigen receptors to the immunological synapse; involvement of SNARE complexes. Immunity. 20:577–588. [DOI] [PubMed] [Google Scholar]
- 29.Logan, M.R., P. Lacy, B. Bablitz, and R. Moqbel. 2002. Expression of eosinophil target SNAREs as potential cognate receptors for vesicle-associated membrane protein-2 in exocytosis. J. Allergy Clin. Immunol. 109:299–306. [DOI] [PubMed] [Google Scholar]
- 30.Berridge, M.J. 1997. Lymphocyte activation in health and disease. Crit. Rev. Immunol. 17:155–178. [DOI] [PubMed] [Google Scholar]
- 31.Valdez, A.C., J.P. Cabaniols, M.J. Brown, and P.A. Roche. 1999. Syntaxin 11 is associated with SNAP-23 on late endosomes and the trans-Golgi network. J. Cell Sci. 112:845–854. [DOI] [PubMed] [Google Scholar]
- 32.Lemons, P.P., D. Chen, A.M. Bernstein, M.K. Bennett, and S.W. Whiteheart. 1997. Regulated secretion in platelets: identification of elements of the platelet exocytosis machinery. Blood. 90:1490–1500. [PubMed] [Google Scholar]
- 33.Chen, D., P.P. Lemons, T. Schraw, and S.W. Whiteheart. 2000. Molecular mechanisms of platelet exocytosis: role of SNAP-23 and syntaxin 2 and 4 in lysosome release. Blood. 96:1782–1788. [PubMed] [Google Scholar]
- 34.Feng, D., K. Crane, N. Rozenvayn, A.M. Dvorak, and R. Flaumenhaft. 2002. Subcellular distribution of 3 functional platelet SNARE proteins: human cellubrevin, SNAP-23, and syntaxin 2. Blood. 99:4006–4014. [DOI] [PubMed] [Google Scholar]
- 35.Flaumenhaft, R., J.R. Dilks, N. Rozenvayn, R.A. Monahan-Earley, D. Feng, and A.M. Dvorak. 2005. The actin cytoskeleton differentially regulates platelet alpha-granule and dense-granule secretion. Blood. 105:3879–3887. [DOI] [PubMed] [Google Scholar]
- 36.Polgar, J., W.S. Lane, S.H. Chung, A.K. Houng, and G.L. Reed. 2003. Phosphorylation of SNAP-23 in activated human platelets. J. Biol. Chem. 278:44369–44376. [DOI] [PubMed] [Google Scholar]
- 37.Polgar, J., S.H. Chung, and G.L. Reed. 2002. Vesicle-associated membrane protein 3 (VAMP-3) and VAMP-8 are present in human platelets and are required for granule secretion. Blood. 100:1081–1083. [DOI] [PubMed] [Google Scholar]
- 38.Mollinedo, F., B. Martin-Martin, J. Calafat, S.M. Nabokina, and P.A. Lazo. 2003. Role of vesicle-associated membrane protein-2, through Q-soluble N-ethylmaleimide-sensitive factor attachment protein receptor/R-soluble N-ethylmaleimide-sensitive factor attachment protein receptor interaction, in the exocytosis of specific and tertiary granules of human neutrophils. J. Immunol. 170:1034–1042. [DOI] [PubMed] [Google Scholar]
- 39.Martin-Martin, B., S.M. Nabokina, J. Blasi, P.A. Lazo, and F. Mollinedo. 2000. Involvement of SNAP-23 and syntaxin 6 in human neutrophil exocytosis. Blood. 96:2574–2583. [PubMed] [Google Scholar]
- 40.Juriloff, D.M., and M.J. Harris. 2000. Mouse models for neural tube closure defects. Hum. Mol. Genet. 9:993–1000. [DOI] [PubMed] [Google Scholar]
- 41.Nozaki, M., K. Ohishi, N. Yamada, T. Kinoshita, A. Nagy, and J. Takeda. 1999. Developmental abnormalities of glycosylphosphatidylinositol-anchor-deficient embryos revealed by Cre/loxP system. Lab. Invest. 79:293–299. [PubMed] [Google Scholar]
- 42.Lohnes, D., M. Mark, C. Mendelsohn, P. Dolle, A. Dierich, P. Gorry, A. Gansmuller, and P. Chambon. 1994. Function of the retinoic acid receptors (RARs) during development (I). Craniofacial and skeletal abnormalities in RAR double mutants. Development. 120:2723–2748. [DOI] [PubMed] [Google Scholar]
- 43.Berk, M., S.Y. Desai, H.C. Heyman, and C. Colmenares. 1997. Mice lacking the ski proto-oncogene have defects in neurulation, craniofacial, patterning, and skeletal muscle development. Genes Dev. 11:2029–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Copp, A.J., N.D. Greene, and J.N. Murdoch. 2003. The genetic basis of mammalian neurulation. Nat. Rev. Genet. 4:784–793. [DOI] [PubMed] [Google Scholar]
- 45.Zohn, I.E., C.R. Chesnutt, and L. Niswander. 2003. Cell polarity pathways converge and extend to regulate neural tube closure. Trends Cell Biol. 13:451–454. [DOI] [PubMed] [Google Scholar]
- 46.Ybot-Gonzalez, P., P. Cogram, D. Gerrelli, and A.J. Copp. 2002. Sonic hedgehog and the molecular regulation of mouse neural tube closure. Development. 129:2507–2517. [DOI] [PubMed] [Google Scholar]
- 47.Holmberg, J., D.L. Clarke, and J. Frisen. 2000. Regulation of repulsion versus adhesion by different splice forms of an Eph receptor. Nature. 408:203–206. [DOI] [PubMed] [Google Scholar]
- 48.Chen, Z.F., and R.R. Behringer. 1995. twist is required in head mesenchyme for cranial neural tube morphogenesis. Genes Dev. 9:686–699. [DOI] [PubMed] [Google Scholar]
- 49.Zhao, Q., R.R. Behringer, and B. de Crombrugghe. 1996. Prenatal folic acid treatment suppresses acrania and meroanencephaly in mice mutant for the Cart1 homeobox gene. Nat. Genet. 13:275–283. [DOI] [PubMed] [Google Scholar]
- 50.Hildebrand, J.D., and P. Soriano. 1999. Shroom, a PDZ domain-containing actin-binding protein, is required for neural tube morphogenesis in mice. Cell. 99:485–497. [DOI] [PubMed] [Google Scholar]
- 51.Ewart, J.L., M.F. Cohen, R.A. Meyer, G.Y. Huang, A. Wessels, R.G. Gourdie, A.J. Chin, S.M. Park, B.O. Lazatin, S. Villabon, and C.W. Lo. 1997. Heart and neural tube defects in transgenic mice overexpressing the Cx43 gap junction gene. Development. 124:1281–1292. [DOI] [PubMed] [Google Scholar]
- 52.Morriss-Kay, G., and F. Tuckett. 1989. Immunohistochemical localisation of chondroitin sulphate proteoglycans and the effects of chondroitinase ABC in 9- to 11-day rat embryos. Development. 106:787–798. [DOI] [PubMed] [Google Scholar]
- 53.Franz, T. 1992. Neural tube defects without neural crest defects in splotch mice. Teratology. 46:599–604. [DOI] [PubMed] [Google Scholar]
- 54.Tallquist, M.D., and P. Soriano. 2003. Cell autonomous requirement for PDGFRalpha in populations of cranial and cardiac neural crest cells. Development. 130:507–518. [DOI] [PubMed] [Google Scholar]
- 55.Chai, Y., X. Jiang, Y. Ito, P. Bringas Jr., J. Han, D.H. Rowitch, P. Soriano, A.P. McMahon, and H.M. Sucov. 2000. Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development. 127:1671–1679. [DOI] [PubMed] [Google Scholar]
- 56.Etchevers, H.C., C. Vincent, N.M. Le Douarin, and G.F. Couly. 2001. The cephalic neural crest provides pericytes and smooth muscle cells to all blood vessels of the face and forebrain. Development. 128:1059–1068. [DOI] [PubMed] [Google Scholar]
- 57.Etchevers, H.C., G. Couly, C. Vincent, and N.M. Le Douarin. 1999. Anterior cephalic neural crest is required for forebrain viability. Development. 126:3533–3543. [DOI] [PubMed] [Google Scholar]
- 58.Xu, W., H. Baribault, and E.D. Adamson. 1998. Vinculin knockout results in heart and brain defects during embryonic development. Development. 125:327–337. [DOI] [PubMed] [Google Scholar]
- 59.Simon, A.M., A.R. McWhorter, J.A. Dones, C.L. Jackson, and H. Chen. 2004. Heart and head defects in mice lacking pairs of connexins. Dev. Biol. 265:369–383. [DOI] [PubMed] [Google Scholar]
- 60.Harris, M.J., and D.M. Juriloff. 1999. Mini-review: toward understanding mechanisms of genetic neural tube defects in mice. Teratology. 60:292–305. [DOI] [PubMed] [Google Scholar]
- 61.Conway, S.J., D.J. Henderson, M.L. Kirby, R.H. Anderson, and A.J. Copp. 1997. Development of a lethal congenital heart defect in the splotch (Pax3) mutant mouse. Cardiovasc. Res. 36:163–173. [DOI] [PubMed] [Google Scholar]
- 62.Martin, C.C., L. Laforest, M.A. Akimenko, and M. Ekker. 1999. A role for DNA methylation in gastrulation and somite patterning. Dev. Biol. 206:189–205. [DOI] [PubMed] [Google Scholar]
- 63.Chai, Y., Y. Ito, and J. Han. 2003. TGF-beta signaling and its functional significance in regulating the fate of cranial neural crest cells. Crit. Rev. Oral Biol. Med. 14:78–88. [DOI] [PubMed] [Google Scholar]
- 64.Clouthier, D.E., K. Hosoda, J.A. Richardson, S.C. Williams, H. Yanagisawa, T. Kuwaki, M. Kumada, R.E. Hammer, and M. Yanagisawa. 1998. Cranial and cardiac neural crest defects in endothelin-A receptor-deficient mice. Development. 125:813–824. [DOI] [PubMed] [Google Scholar]
- 65.Crump, J.G., L. Maves, N.D. Lawson, B.M. Weinstein, and C.B. Kimmel. 2004. An essential role for Fgfs in endodermal pouch formation influences later craniofacial skeletal patterning. Development. 131:5703–5716. [DOI] [PubMed] [Google Scholar]
- 66.Maschhoff, K.L., and H.S. Baldwin. 2000. Molecular determinants of neural crest migration. Am. J. Med. Genet. 97:280–288. [DOI] [PubMed] [Google Scholar]
- 67.Morales-Tirado, V., S. Johannson, E. Hanson, A. Howell, J. Zhang, K.A. Siminovitch, and D.J. Fowell. 2004. Cutting edge: selective requirement for the Wiskott-Aldrich syndrome protein in cytokine, but not chemokine, secretion by CD4+ T cells. J. Immunol. 173:726–730. [DOI] [PubMed] [Google Scholar]
- 68.Reheman, A., P. Gross, H. Yang, P. Chen, D. Allen, V. Leytin, J. Freedman, and H. Ni. 2005. Vitronectin stabilizes thrombi and vessel occlusion but plays a dual role in platelet aggregation. J. Thromb. Haemost. 3:875–883. [DOI] [PubMed] [Google Scholar]