

Abstract
Trypanosoma cruzi is the causative agent of Chagas' disease. The major protease, cruzain, is a target for the development of new chemotherapy. We report the first successful treatment of an animal model of Chagas' disease with inhibitors designed to inactivate cruzain. Treatment with fluoromethyl ketone–derivatized pseudopeptides rescued mice from lethal infection. The optimal pseudopeptide scaffold was phenylalanine-homophenylalanine. To achieve cure of infection, this pseudopeptide scaffold was incorporated in a less toxic vinyl sulfone derivative. N-methyl piperazine-Phe-homoPhe-vinyl sulfone phenyl also rescued mice from a lethal infection. Six of the treated mice survived over nine months, three without further treatment. Three mice that had entered the chronic stage of infection were retreated with a 20-d regimen. At the conclusion of the experiments, five of the six mice had repeated negative hemacultures, indicative of parasitological cure. Studies of the effect of inhibitors on the intracellular amastigote form suggest that the life cycle is interrupted because of inhibitor arrest of normal autoproteolytic cruzain processing at the level of the Golgi complex. Parasites recovered from the hearts of treated mice showed the same abnormalities as those treated in vitro. No abnormalities were noted in the Golgi complex of host cells. This study provides proof of concept that cysteine protease inhibitors can be given at therapeutic doses to animals to selectively arrest a parasitic infection.
Keywords: Chagas' disease, Trypanosoma cruzi, cysteine protease, drug design, protease inhibitors
Chagas' disease, the result of infection with the protozoan parasite Trypanosoma cruzi, is the leading cause of heart disease in Latin America (1). Over 1.6 × 107 people are infected, up to 80% in endemic areas, and over 9 × 107 are at risk (2). By definition, the acute phase of Chagas' disease lasts up to 60 d and parasites are easily detected by direct examination of peripheral blood (3). Acute Chagas' disease results in myocarditis in ∼60% of patients with an estimated 9% mortality occurring in endemic areas (4). Chagasic encephalitis, not uncommon in children, is also associated with immunosuppression and AIDS (5–7). Gastrointestinal megasyndromes are common especially in Brazilian patients. Most chagasic patients die from heart failure associated with cardiomyopathy during the chronic phase of the disease (3, 8).
Therapy for Chagas' disease is unsatisfactory. Because of significant toxicity, chemotherapy with nifurtimox or benznidazole must be carried out under close medical supervision. In addition to dermatotoxicity and digestive disorders, benznidazole induces chromosomal damage in chagasic children (9). Long-term use of nifurtimox and benznidazole in humans has not been documented, but its association with malignant lymphomas in experimental animals precludes prolonged treatment of patients including immunocompromised patients (10). Both compounds may shorten the acute phase and decrease mortality, but they achieve parasitologic cures in only ∼60% of acute patients and are not used during the chronic phase of the disease (10). Finally, a large gradient of susceptibility to nifurtimox and benznidazole treatments ranging from 0–100% correlates with geographic region and may delineate distribution of drug-resistant T. cruzi (8, 11, 12).
One approach to novel chemotherapy for Chagas' disease has focused on the development of specific inhibitors of cruzain (a.k.a. cruzipain, gp 57/51), the major cysteine protease of T. cruzi (13–17). Diazomethane or fluoromethyl ketone (FMK)1 cysteine protease inhibitors (CPI) that effectively blocked cruzain activity prevented growth and differentiation of T. cruzi in cell culture models of infection (18–20). A new generation of CPI has been synthesized with chemical modifications aimed at enhancing specificity and in vivo stability and minimizing toxicity. We now report that CPI treatment rescued mice from the acute phase of a lethal experimental T. cruzi infection and cleared parasitemia in chronically infected mice without toxicity to the mammalian host. The inhibitors induced major ultrastructural alterations leading to death of the intracellular amastigote stage that were similar to those previously observed in the extracellular insect stage epimastigotes after exposure to the same protease inhibitors (21).
Materials and Methods
Growth Inhibition of T. cruzi Amastigotes by CPI.
J774 macrophages were cultured in RPMI-1640 medium with 5% heat- inactivated FCS (RPMI medium). For growth inhibition assays, J774 macrophages were irradiated (3,000 rad) to arrest cell growth and cultured on coverglasses within six-well plates for 24 h at 37°C. After infection with T. cruzi trypomastigotes of the Y strain for 3 h, monolayers were washed with RPMI medium and treated with inhibitors at 20 μM in RPMI medium. Inhibitor stocks were made at 20 mM in DMSO and all assays included DMSO (0.01–0.02%, vol/vol) controls. FMK inhibitors (Mu-F-hF-VSφ, Mu-F-K-VSφ, Mu-F-V-VSφ, Mu-F-S(OBzl)-VSφ, Mu-L-hF-VSφ, Mu-Yii-hF-VSφ, Boc-tic-hF-VS, Mu-tic-hF-VS, Mu-Y-hF-VS, Mu-F-hF-FMK, Mu-F-hF-VAmBzl, N-Pip-F-hF-VSφ, Z-F-A-FMK, Mu-bsu-hF-FMK, and Mu-F-hF-FMK) were provided by Prototek (Dublin, CA) and vinyl sulfones were provided by Axys Pharmaceuticals (South San Francisco, CA). CPI were evaluated in T. cruzi–infected macrophage cultures for 21– 30 d. Trypomastigote output, indicative of the completion of the intracellular cycle, was then assayed in treated and untreated cultures to determine growth inhibition of intracellular T. cruzi amastigotes (20).
After this initial inhibitor screen, T. cruzi–infected macrophages were treated with 20 μM Mu-F-hF-VSφ and Mu-F-V-VSφ for up to 76 h. Monolayers were washed, fixed with 4% paraformaldehyde, and then Giemsa stained at determined intervals. To evaluate treatment, the percentage of infected macrophages and the total number of intracellular amastigotes in 100 infected macrophages were quantified. A decrease in the number of intracellular generations indicated inhibition of intracellular growth of T. cruzi amastigotes and was calculated from the total number of intracellular amastigotes per 100 infected macrophages (n = 3).
Effect of CPI on the Survival of T. cruzi–infected Mice.
3-wk-old female C3H mice weighing initially between 17–19 g were used in all experiments. In the first experiment (see Fig. 1), mice (five animals per lot) were infected with 105 trypomastigotes of the Y strain and treated with a 1-mg i.p. injection of FMK inhibitors (Mu-F-hF-FMK, Mu-bsu-hF-FMK, and Z-F-A-FMK) twice per day. Controls included intraperitoneal injection with equal volume of DMSO. Treatment was initiated 24 h after infection and continued until death of the animals or the end of the experiment, as appropriate. Parasitemias were determined every 48 h for each animal on alternating days from 5 μl of blood extracted from the tail and diluted 1/4 (vol/vol) in RPMI medium. The numbers of parasites per milliliter, calculated in a Neubauer chamber, were expressed as a mean of two or three animals per day. The experiment was terminated 18 d after infection.
Figure 1.

Levels of parasitemia and survival of mice treated with peptidomimetic fluoromethyl ketones. CH3 mice were infected with T. cruzi and treated twice daily with 1 mg i.p. of Z-F-Ala-FMK (•); Mu-bsu-hF-FMK (○); Mu-F-hF-FMK (▵); and controls with and without DMSO i.p. (□, ▪). Parasitemias were determined every 48 h in each animal on alternating days. Results are mean of two to three animals per day.
Approximate dosing regimes and different inhibitor chemistries were then analyzed (see Table 3, Experiment 1). Mice (five animals per lot) were infected (intraperitoneally) with 4 × 106 tissue culture–derived trypomastigotes. 24 h after infection, mice were treated with two daily doses of 1 mg i.p. Mu-F-hF-FMK and Mu-F-hF-VSφ for up to 16 d. In a third experiment (see Table 3, Experiment 2), animals were infected with 106 trypomastigotes and treated with the inhibitors with the same regimen for 12 d.
Table 3.
Treatment of T. cruzi–infected C3H Mice with Cysteine Protease Inhibitors
| Experiment | Inhibitor (CPI) | Number of mice | Inoculum (trypomastigotes) | Number of mice rescued* from lethal infection | Maximal Survival | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| d | ||||||||||
| 1‡ | Control | 5 mice per lot | 4 × 106 | — | 5 | |||||
| Mu-F-hF-FMK | 0/5 | 10 | ||||||||
| Mu-F-hF-VSφ | 5/5 | >16§ | ||||||||
| 2‡ | Control | 6 mice per lot | 1 × 106 | — | 10 | |||||
| Mu-F-hF-FMK | 5/6 | 18 | ||||||||
| Mu-F-hF-VSφ | 6/6 | >180‖ | ||||||||
| 3¶ | Control | 6 mice | 1 × 105 | 22 | ||||||
| N-Pip-F-hF-VSφ | 10 mice | 10/10 | >240** |
Effect of cysteine protease inhibitors on the survival of T. cruzi–infected mice. In these series of independent experiments, T. cruzi–infected, C3H mice were treated with Mu-F-hF-FMK, Mu-F-hF-VSφ, and N-Pip-F-hF-VSφ at the doses and regimens indicated. Controls were inoculated intraperitoneally with an equal volume of DMSO solution or not injected.
“Rescued” defined as surviving at least 7 d after all controls died.
Animals treated with 1 mg CPI twice a day (100 mg/kg/d).
Experiment stopped at 16 d.
2/6 animals survived >180 d after infection when experiment was stopped. Blood smears were negative and hemo-cultures were positive.
Animals treated with 0.7 mg CPI three times a day.
6/10 animals survived >240 d after infection; 3/6 mice were both blood smear and hemo-culture negative.
Finally (see Table 3, Experiment 3), mice were infected with 105 T. cruzi trypomastigotes and treated with three daily intraperitoneal doses of N-Pip-F-hF-VSφ (2.1 mg/d) for 24 d. Blood (hemo) cultures of untreated controls (n = 6) and N-Pip-F-hF-VSφ–treated animals (n = 10) were performed 16, 22, and 46 d after infection using arrested macrophages as host cells. In brief, 6-μl aliquots of blood were resuspended in RPMI medium with 5% FCS and antibiotics. Irradiated J774 macrophages were infected with blood dilutions, and incubated for up to 30 d at 37°C in a 5% CO2 atmosphere. Blood cultures were considered positive if T. cruzi infected macrophages and/or free trypomastigotes were observed. Hemocultures were considered negative if no infected host cells and no free trypomastigotes were observed for up to 30 d, after which macrophages died.
Six animals from Table 3, Experiment 3, survived the acute phase of the infection. Three out of six mice had consistently negative blood cultures while the remaining three out of six were positive. After being allowed to establish chronic infection for 3 mo, the latter three mice were retreated with three daily doses (2.1 mg/d i.p.) of N-Pip-F-hF-VSφ for 21 d (see Table 4, Experiment 4). Hemocultures from the six animals were performed repeatedly as described above but with a larger volume of blood sampled (6–50 μl).
Table 4.
Treatment of Chronically Infected C3H Mice with N-Pip-F-hF-VSφ
| Experiment | Number of chronically infected mice | Treatment | Hemoculture* (Number of mice) | Survival after injection | ||||
|---|---|---|---|---|---|---|---|---|
| d | ||||||||
| 4 | 3‡ | N-Pip-F-hF-VSφ | Positive before treatment (3/3) | 300 | ||||
| Negative after treatment (2/3) | ||||||||
| 3§ | No | Negative (3/3) | 300 |
Retreatment of three mice from Table 3 Experiment 3 that had entered chronic phase of disease but remained hemoculture positive. A 21-d oral regimen of 0.7 mg TID N-Pip-F-hF-VSφ was used.
Macrophage hemocultures were described in Materials and Methods.
Chronically infected mice from Table 3 Experiment 3 retreated with i.p. regimen.
Mice treated only as in Table 3 Experiment 3 but remaining hemoculture negative.
T. cruzi–infected, CPI-treated mice, noninfected CPI-treated mice, and untreated T. cruzi–infected animals were necropsied to evaluate toxicity of the inhibitor regimen. Mice were weighed, examined grossly, and all major organs also examined by routine hematoxylin-eosin histopathology. The toxicity of Mu-F-hF-VSφ and N-Pip-F-hF-VSφ was further evaluated in uninfected C3H mice treated for 45 d with 4 mg/d i.p., and 12 mg/d per oral (p.o.), respectively.
Fluorescence Microscopy to Determine the Effect of CPI in the Parasite.
Irradiated macrophages were infected with T. cruzi trypomastigotes for 2 h at 37°C. Monolayers were washed 24 h after infection and reincubated with or without the addition of the CPI Mu-F-hF-VSφ (10 μM). After 48 h, culture medium containing Bodipy FL ceramide (Molecular Probes, OR) was substituted for 15 min at 37°C. Monolayers were subsequently processed according to manufacturer's instructions and observed by fluorescence microscopy (21).
Ultrastructure and Immunocytochemistry of T. cruzi Intracellular Amastigotes.
T. cruzi–infected, irradiated J774 macrophages were treated or not with Mu-F-hF-VSφ (10 μM) for 48 h. Monolayers were trypsinized, washed, fixed, and processed for electronmicroscopy. Cells were fixed with 1.5% glutaraldehyde in 0.66 M sodium cacodylate buffer, pH 7.4, at room temperature for 2 h, embedded in EPONATE 12 (Ted Pella, Inc., Redding, CA), sectioned, stained, and then observed with a Zeiss 10C electronmicroscope (Carl Zeiss Inc., Thornwood, NY; reference 22). The techniques (23) and reagents used for immunocytochemistry have been described previously for T. cruzi epimastigotes (21). Heart muscle from a mouse treated 6 d after infection with Mu-F-hF-VSφ (2 mg/d i.p. for 3 d) was fixed and processed as described above.
Confirmation of Inhibitor Targets by Labeled Inhibitor.
Irradiated J774 macrophages were infected with Y strain trypomastigotes and incubated at 37°C for 48 h to allow intracellular development of amastigotes. T. cruzi–infected macrophages, uninfected macrophage controls, and T. cruzi epimastigotes were radiolabeled with 20 μM [14C]Mu-F-hF-VSφ for 2 h at 37°C or 26°C, as appropriate. For competition experiments, duplicate cultures were treated with 20 μM Mu-F-hF-VSφ for 3 h before addition of the radiolabeled inhibitor. Infected macrophages were scraped and centrifuged at 1,700 g for 12 min at 4°C. Pellets were resuspended and lysed in a tissue grinder to release intracellular amastigotes, centrifuged at 260 g for 5 min, and the amastigote-containing supernatant centrifuged at 2,000 g for 12 min. Amastigote pellets were resuspended, transferred to Eppendorf tubes, and washed three times with PBS before sonication. Radiolabeled, noninfected macrophages and T. cruzi epimastigotes were sonicated as above. Purified recombinant cruzain was also radiolabeled as a control. Samples were boiled in sample buffer (24), electrophoresed in 10% acrylamide gels, and autoradiographed (21). Western blots were developed with anti-cruzain antibody (21).
Results
Effect of CPI on the Intracellular Development of T. cruzi.
The efficacy of a number of CPI on the mammalian stages of the life cycle of T. cruzi was evaluated as the percentage of growth inhibition of intracellular amastigotes in the presence of the inhibitors for up to 21 d (Table 1). The first intracellular cycle of untreated controls was completed within 6 d after infection. Mu-F-hF-VSφ was the most effective compound and inhibited growth 100%. Mu-F-V-VSφ and BOC-tic-hF-VS, which are less effective inhibitors of the protease itself, produced 30% and 60% growth inhibition, respectively.
Table 1.
Effect of Cysteine Protease Inhibitors on the Intracellular Cycle of T. cruzi In Vitro
| Inhibitor (20 μM) | Percentage of growth inhibition | |
|---|---|---|
| None (control) | 0 | |
| Mu-F-hF-VSφ | 100 | |
| Mu-F-K-VSφ | 0 | |
| Mu-F-V-VSφ | 33.4 | |
| Mu-F-S(OBzl)-VSφ | 16.6 | |
| Mu-L-hF-VSφ | 24.9 | |
| Mu-Yii-hF-VSφ | 16.6 | |
| BOC-tic-hF-VS | 58.3 | |
| Mu-Y-hF-VS | 24.9 | |
| Mu-F-hF-FMK | >83 | |
| Mu-F-hF-VAmBzl | 41.5 |
Growth inhibition of intracellular amastigotes by cysteine protease inhibitors. Irradiated J774 macrophages were infected with trypomastigotes of the Y strain of T. cruzi. Cells were treated daily with 20 μM of peptidomimetic inhibitors of cruzain. Cultures were observed daily by contrast phase microscopy for ≤30 d. The first intracellular cycle of controls was completed within 6 d.
Based on this initial screening of inhibitors, Mu-F-hF-VSφ with Mu-F-V-VSφ for comparison were further tested (Table 2). 20 μM concentrations of Mu-F-hF-VSφ cured T. cruzi–infected macrophages. Although intracellular amastigotes were still visible in Giemsa-stained cultures 24– 78 h after infection, their morphology was extremely abnormal 50 h after treatment. Cultures were amastigote free after 12 d. No intracellular amastigotes or release of trypomastigotes were observed in cultures treated for 12 d and maintained without CPI for up to 30 d in numerous independent experiments, indicating that Mu-F-hF-VSφ not only blocked the intracellular development of T. cruzi, but eventually eliminated all parasites. In contrast, amastigotes divided at normal rate in the presence of the chemically related, but less effective inhibitor of cruzain, Mu-F-V-VSφ.
Table 2.
Effect of Two Vinyl Sulfone Inhibitors on the Intracellular Cycle of T. cruzi In Vitro
| Treatment | Duration | Percentage of cells infected | Amastigotes per 100 infected cells | Number of intracellular generations | ||||
|---|---|---|---|---|---|---|---|---|
| h | ||||||||
| Control | 18 | 38% | 76 ± 4 | 0 | ||||
| 50 | 41% | 1,004 ± 6 | 3.3 | |||||
| 72 | 45% | 5,220 ± 6 | 5.6 | |||||
| Mu-F-hF-VSφ (20 μM) | 18 | 41% | 80 ± 5 | 0 | ||||
| 50 | 25% | 42 ± 5 | 0 | |||||
| 76 | 22% | 35 ± 5 | 0 | |||||
| Mu-F-V-VSφ (20 μM) | 18 | 36% | 72 ± 6 | 0 | ||||
| 50 | 35% | 1,106 ± 7 | 3.6 |
Effect of the cysteine protease inhibitors on the intracellular growth of T. cruzi amastigotes. Irradiated J774 macrophages were infected with T. cruzi before treatment with Mu-F-hF-VSφ and Mu-F-V-VSφ. The percentage of infected cells, the total number of intracellular amastigotes in 100 infected cells, and the number of intracellular generations of T. cruzi amastigotes are indicated. Because host cells were irradiated to prevent their replication, a decrease in percentage of cells infected and number of amastigotes/cell is indicative of parasite death and clearance. Mu-F-V-VSφ treatment had no effect on amastigote growth. Cells treated with Mu-F-hF-VSφ were amastigote free after 12 d.
Resolution of Acute Experimental Chagas' Disease.
Levels of parasitemia in T. cruzi–infected C3H mice were first analyzed after treatment with Z-F-A-FMK, Mu-bsu-hF-FMK, and Mu-F-hF-FMK (Fig. 1). These FMK inhibitors were chosen based on results of tissue culture screens (20, Table 1) and included a natural amino acid dipeptide (Z-F-A-FMK) and pseudopeptides designed to increase in vivo half-life (Mu-bsu-hF-FMK and Mu-F-hF-FMK). Control animals died at day 12 after infection while all animals treated with Mu-bsu-hF-FMK and Mu-F-hF-FMK survived throughout the experiment (18 d). The levels of parasitemia were lowest in mice treated with Mu-F-hF-FMK and ranged from 10–500 trypomastigotes per milliliter of blood. This corresponded to a reduction of 3 log units from untreated controls. Mice treated with the related but less effective (in protease substrate assays) dipeptide inhibitor Mu-bsu-hF-FMK had persistent high parasitemia in the range of 4 × 104–5 trypomastigotes per milliliter of blood. Not only was Z-F-A-FMK ineffective, but mice had higher parasitemia and died earlier than controls.
From tissue culture screens (Tables 1 and 2), vinyl sulfone derivatized pseudopeptides with high efficacy but less toxicity than FMK inhibitors were selected for further evaluation in a mouse model of acute Chagas' disease. The effect of CPI-treatment on the survival of T. cruzi infected C3H mice is shown in Table 3. Untreated controls infected with a very high dose (4 × 106) of tissue culture– derived trypomastigotes all died by 4–5 d after infection. All five mice treated with Mu-F-hF-VSφ survived for 14– 16 d at which time the experiment was terminated and animals killed (Table 3, Experiment 1). When the infectious dose was reduced to 1 × 106 T. cruzi trypomastigotes and mice treated with one 24-d regimen of Mu-F-hF-VSφ, two out of six mice survived up to 180 d after infection at which time the experiment was terminated (Table 3, Experiment 2).
From these pilot studies, the most promising lead compound was then evaluated in a new dosing regimen based on a pharmacokinetics analysis of Mu-F-hF-VSφ (25). Mice were treated three times daily with 2.1 mg/d i.p. of N-Pip-F-hF-VSφ (Table 3, Experiment 3). As indicated in Table 3, all 10 mice survived at least 7 d longer than untreated mice and 6 out of 10 mice survived over 270 d after infection. Three of the six long-term surviving mice had consistently negative blood-parasite cultures over the entire treatment period while the remaining three out of six were still positive, although well below controls. After being allowed to establish chronic infection for 3 mo, this latter group of 3 mice were retreated with 2.1 mg/d i.p. of N-Pip-F-hF-VSφ for 21 d (Table 4, Experiment 4). Blood cultures (10–50 μl) were negative for the retreated animals. In summary, 5 of 10 treated mice from this study have now survived for 9 mo without symptomatology and with negative parasitemia and hemocultures (one mouse died of accidental trauma after 100 d). Similar results were obtained with 3-wk-old outbred Swiss mice weighing initially 19– 21 g (data not shown).
In the studies described above, CPI treatment did not induce gross or microscopic abnormalities in either infected or uninfected mice. To further assess toxicity, doses of 4 mg/d i.p. of Mu-F-hF-VSφ and 12 mg p.o. of N-Pip-F-hF-VSφ for 4 d were also evaluated. No gross or microscopic abnormalities were observed in necropsied animals.
Confirmation that CPI Enters Amastigotes and Targets Cruzain.
CPI have been shown to produce a characteristic Golgi abnormality in the extracellular epimastigote stage of T. cruzi (21).
To confirm that the same effect was produced in the treated amastigotes, amastigotes within host macrophages that had been treated with Mu-F-hF-VSφ (10 μM) for 48 h were examined after labeling with Bodipy FL ceramide that accumulates in the Golgi compartment. Cells were observed by contrast phase (Fig. 2, A and B) and fluorescence microscopy (Fig. 2, C and D). No Bodipy FL labeling was apparent in untreated intracellular amastigotes (Fig. 2 C), whereas the larger Golgi complex (G) of untreated host cells was visible. In contrast, the Golgi apparatus (g) of Mu-F-hF-VSφ–treated amastigotes had abnormally large, fluorescent vesicles consistent with ultrastructural alterations in the Golgi complex previously observed in treated epimastigotes (Fig. 2 D). To confirm these results, the normal morphology of control intracellular amastigotes (Fig. 3 A) was compared with the ultrastructure of CPI-treated tissue culture amastigotes (Fig. 3 B). Golgi complex and cytoplasmic vesicle alterations found in amastigotes (Fig. 3 B) resembled those described for the epimastigote stage, and consisted of significant dilation of cisternae (21). Similar ultrastructural alterations were evident in amastigotes isolated from heart muscle of T. cruzi–infected mice treated with Mu-F-hF-VSφ (Fig. 3 C). The amounts of cruzain expressed on the surface of Mu-F-hF-VSφ–treated and untreated amastigotes were quantified by immunoelectronmicroscopy (Fig. 4). A marked decrease in cruzain expressed on the cell surface of CPI-treated cells (Fig. 4 B) was evident compared with untreated amastigotes (Fig. 4 A).
Figure 2.

Treatment of T. cruzi–infected macrophages with a cysteine protease inhibitor and a fluorescent probe specific for the Golgi complex. Phase contrast (A) and fluorescence (C) microphotograph of an untreated T. cruzi–infected macrophage. Several intracellular amastigotes are visible within the cytoplasm of the host cell. The Golgi complex (G) of the host cell is labeled. Bodipy FL does not induce visible fluorescence in the Golgi complex of untreated amastigotes. Phase contrast (B) and fluorescence microphotograph (D) of an infected macrophage treated with 20 μM Mu-F-hF-VSφ for 48 h. Large, fluorescent Golgi vesicles (g) are evident in CPI-treated amastigotes indicative of vesicle dilation abnormality (21). a, amastigote; G, macrophage– Golgi complex; g, amastigote–Golgi complex; N, macrophage nucleus.
Figure 3.
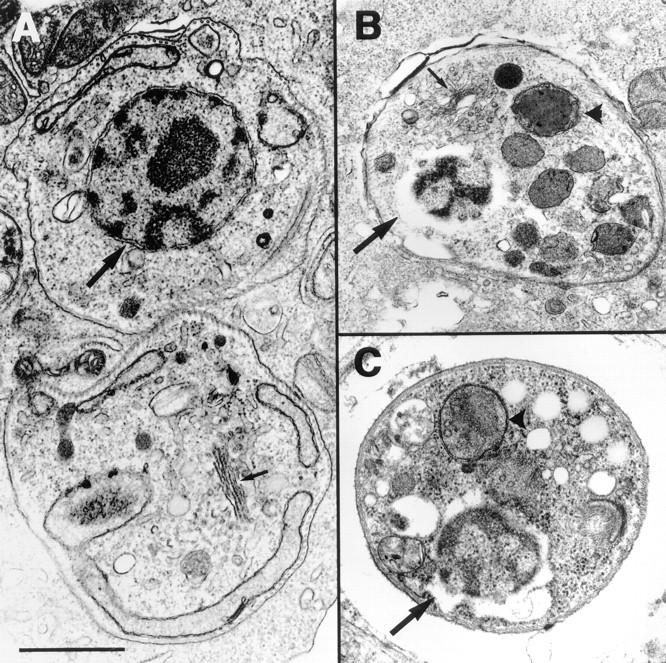
Electronmicroscopy of T. cruzi intracellular amastigotes. T. cruzi amastigotes observed within culture macrophages (A and B) or heart muscle of Mu-F-hF-VSφ–treated mice (C). The normal ultrastructure of untreated intracellular amastigotes within irradiated macrophages (A) contrasts with the altered morphology of intracellular amastigotes treated with 20 μM of Mu-F-hF-VSφ for 48 h (B). More dilated Golgi vesicles and perinuclear membrane similar to that reported in treated epimastigotes (21). Similar ultrastructural alterations were observed in amastigotes isolated from heart muscle (C) of experimentally infected mice treated with 2 mg/d i.p. Mu-F-hF-VSφ for 4 d before necropsy and isolation of heart muscle. A, Untreated amastigotes; B, CPI-treated cell culture amastigote; C, amastigote infecting the heart muscle of a CPI-treated animal. Nuclear membrane (large arrows); Golgi complex (small arrow); vesicle (▴). No abnormalities were noted in Golgi complex or other organelles of host cells. Bar, 1 μm.
Figure 4.
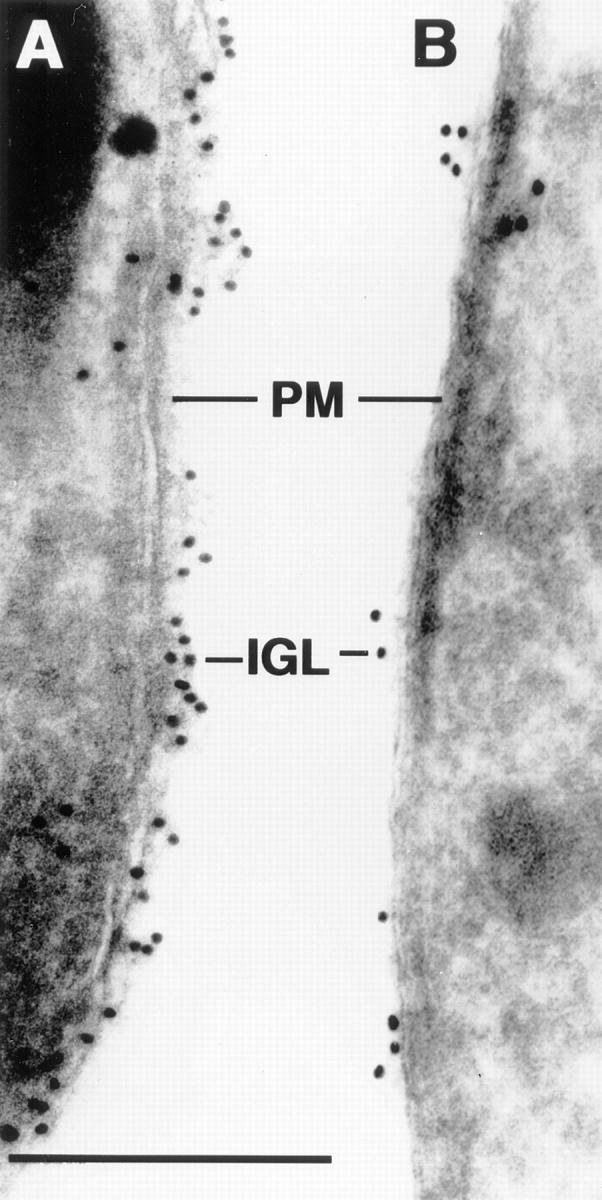
Immunoelectronmicroscopy of cell surface membranes of T. cruzi amastigotes. Cell surface membranes of untreated (A) and Mu-F-hF-VSφ–treated (B) amastigotes were immunocytochemically labeled with a specific anti-cruzain antibody. Note markedly diminished gold label on surface of treated parasite consistent with retention of unprocessed cruzain in Golgi (21). PM, parasite cell surface membrane; IGL, immunogold label. Bar, 0.2 μm.
T. cruzi intracellular amastigotes were incubated with or without cold inhibitor before incubation labeling with [14C]Mu-F-hF-VSφ. T. cruzi epimastigotes and recombinant cruzain were also radiolabeled and autoradiographed as standards (Fig. 5 A). 14C-inhibitor labeling of amastigote cruzain (lane 1) was abolished by preincubation with unlabeled Mu-F-hF-VSφ (lane 2). In amastigotes isolated from infested host cells two protease species were labeled. One comigrated with the epimastigote cruzain species (∼50 kD) known to contain both the catalytic and COOH-terminal domains (13) while the second comigrated with recombinant cruzain containing only the catalytic domain and with a macrophage protease (∼30 kD).
Figure 5.
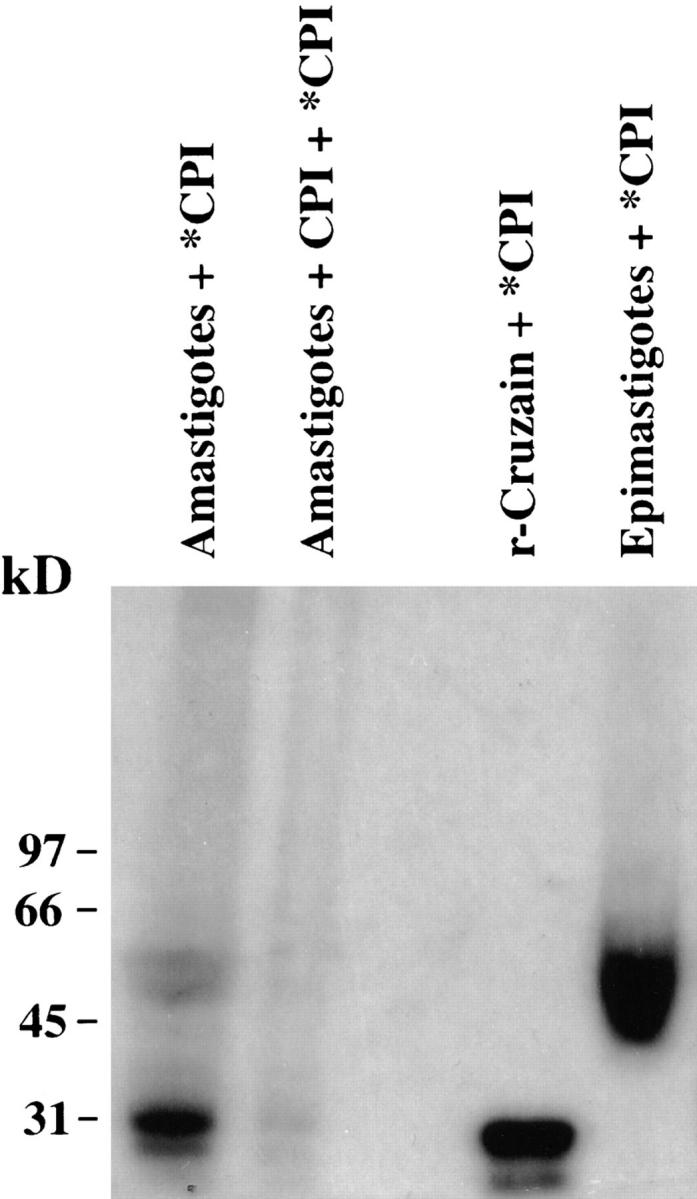
Autoradiogram of extract from radiolabeled [14C]Mu-F-hF-VSφ intracellular amastigotes. Binding of the radiolabeled inhibitor to amastigote cruzain was abolished by preincubation with unlabeled Mu-F-hF-VSφ followed by [14C]CPI. Epimastigote cruzain and recombinant cruzain controls are shown as standards. Note two species labeled in amastigotes that comigrate with either epimastigote cruzain (50 kD) which has both catalytic and COOH-terminal domains (13) or with recombinant cruzain (30 kD) that only has catalytic domain. Lane 1, T. cruzi intracellular amastigotes labeled with [14C]CPI; lane 2, T. cruzi intracellular amastigotes preincubated with unlabeled inhibitor; lane 3, sample buffer; lane 4, recombinant cruzain labeled with [14C]CPI; lane 5, epimastigotes radiolabeled with [14C]Mu-F-hF-VSφ. Note 57/51-kD doublet characteristic of native cruzain which retains COOH-terminal domain (13). Recombinant cruzain lacks the COOH-terminal domain (14).
Discussion
High toxicity and low efficacy make current chemotherapy for Chagas' disease highly unsatisfactory. Moreover, commercial nifurtimox production has been discontinued and benznidazole is at present the only treatment available. Until recently, it was still controversial as to whether drug treatment would have any effect on the more common chronic stage of Chagas' disease because it was unclear whether parasites were still present. However, there is now a clear association between parasitic burden and degree of myocardial damage (26). A recent follow up study in Argentina of benznidazole-treated versus untreated chronic chagasic patients showed less cardiomyopathy in the first group (27). Electrocardiogram patterns of benznidazole-treated patients confirmed an important reduction in disease progression (27). Andrade et al. (28) reported a correlation between heart disease and persistent parasitemia in mice. These studies emphasize the importance of the development of more effective and less toxic chemotherapy for both the acute and the chronic phase of Chagas' disease.
We have targeted cruzain (a.k.a. cruzipain, gp57/51), the major cysteine protease of T. cruzi (reviewed in 13, 29, 30), for the development of new chemotherapy. Previously tested dipeptide-based cruzain inhibitors that mimic substrate (20) were further modified to increase affinity for the catalytic site of cruzain, increase half-life in vivo, provide oral bioavailability, and reduce toxicity. In a previous report, we showed that these CPI induced death in the extracellular epimastigote stage of T. cruzi as a consequence of blocking the autocatalytic processing of cruzain precursor protein. This resulted in Golgi complex and endoplasmic reticulum abnormalities secondary to accumulation of unprocessed cruzain precursor molecules in the vesicle compartments (21). We now present evidence that these CPI also inhibit T. cruzi amastigote growth within macrophages by the same mechanism. Among the inhibitors tested, Mu-F-hF-VSφ most effectively blocked the intracellular cycle of the parasite (Table 2) and resulted in amastigote death in vitro. None of the inhibitors produced abnormalities in the host cells at the concentrations necessary to interrupt the parasite life cycle.
Subsequent testing of a number of CPI in an experimental mouse model of acute Chagas' disease showed that the inhibitors also disrupted the life cycle of T. cruzi in vivo. Initially, T. cruzi–infected animals were treated with the fluoromethyl ketone–derivatized peptidomimetics Mu-F-hF-FMK, Mu-bsu-hF-FMK, and Z-F-A-FMK (Fig. 1). Treatment with Mu-F-hF-FMK reduced parasitemia by 3 log units which resulted in the survival of infected mice throughout the experiment, terminated 18 d after infection. In contrast, animals treated with Mu-bsu-hF-FMK, a less effective inhibitor of the protease itself, and Z-F-A-FMK, a peptide with natural amino acids, had ∼100- and 10,000-fold higher parasitemias, respectively. Survival in the Z-F-A-FMK group (8 d) was significantly lower than in controls (12 d). The increased mortality and parasitemia probably result from a toxic metabolite of the natural amino acid inhibitor. Z-F-A-FMK is cleaved between the phenylalanine and alanine in vivo by an as yet unknown mammalian protease releasing alanine-FMK which, in turn, enters and inhibits the Krebs cycle (31). This fluoride-dependent toxicity results in hypothermia in mice, and T. cruzi has a higher growth rate at 35°C than at 37°C (32, 33). The peptidomimetics containing at least one nonnatural amino acid analogue (e.g., hF) do not undergo this cleavage and metabolism (25). To completely avoid the potential toxicity of FMK derivatives, vinylsulfone analogues were tested and found to extend survival to 120–180 d after infection with 106 trypomastigotes. We further evaluated the more aqueous soluble derivative N-Pip-F-hF-VSφ that is absorbed after oral dosing. 5 out of 10 treated mice have now survived for over 9 mo, 4 of them with repeatedly negative hemocultures indicative of parasitological cure. No toxicity was observed at doses two- to sixfold higher than the therapeutic doses administered p.o. or intraperitoneally for 20 d or when mice were treated for 45 d with the therapeutic dose of 2 mg/d inhibitor.
To elucidate the mechanism of action of CPI versus intracellular amastigotes, the pathogenic stage of T. cruzi, we infected host cells whose cell cycles were arrested. Intracellular amastigotes treated with Mu-F-hF-VSφ showed major morphological alterations as early as 24–48 h after treatment, and macrophage cultures were amastigote free within 12 d. Mu-F-hF-VSφ produced abnormalities in the protein trafficking pathway (nuclear membrane, ER, Golgi complex) and induced the appearance of double-membrane vacuoles with similar morphology to autophagosome vacuoles (Fig. 3). Cruzain normally localizes to the cell surface and lysosome membrane of T. cruzi amastigotes (34, 35). CPI treatment significantly reduced the amounts of cruzain appearing both on the cell membrane (Fig. 4) and in the lysosome consistent with an arrest of cruzain transport. An increase of fluorescence with Bodipi FL of the Golgi complex in treated amastigotes was similar to that described for epimastigotes and correlated with enlargement of the Golgi complex secondary to retention of unprocessed cruzain (Fig. 2; reference 21). Radiolabeled inhibitor was used to confirm entry into amastigotes and specific labeling of the target protease cruzain (Fig. 5). The absence of any host cell or animal toxicity at therapeutic doses suggests that the parasites are more susceptible to inhibitor perhaps because of the redundancy of cysteine proteases in mammalian cells versus the parasite.
We have identified peptidomimetic cysteine protease inhibitors that consistently rescued mice from acute lethal infections of T. cruzi, reduced parasitemia by up to 3 log units, and cured mice in the chronic stage of disease treated by a 21-d regimen. These results provide an important “proof of concept” for the development of cysteine protease inhibitors as chemotherapy for a number of disease entities including cancer cell invasion, inflammation, osteoporosis, and microbial infections where cysteine proteases are thought to play a key role in pathogenesis (36). Although further improvements on vinyl sulfone inhibitor leads should be forthcoming, they clearly demonstrate that animals can tolerate CPI at concentrations and dosing schedules that eliminate an intracellular parasite. The pharmacokinetics of these inhibitors were adequate to sustain therapeutic levels and the N-methyl piperazine derivative demonstrated oral bioavailability. The efficacy and lack of toxicity of CPI in treating both acute and chronic T. cruzi infections supports a call for further development of these leads.
Acknowledgments
The authors thank Drs. M. Zimmerman and R. Smith (Prototek, Dublin, CA), and J. Palmer (Axys Pharmaceuticals, San Francisco, CA) for cysteine protease inhibitors; and G. Gan (Anatomic Pathology Service, VAMC San Francisco) for processing of histological samples.
Abbreviations used in this paper
- Boc
butyloxycarbonyl
- Bsn
benzyl succinic acid
- CPI
cysteine protease inhibitors
- F
phenylalanine
- FMK
fluoromethyl ketone
- hF
homophenylalanine
- Mu
morpholine urea
- Obzl
O-benzyl
- Pip
piperazine
- tic
tetrahydroisoquinoline 3-carboxylic acid
- VAmBzl
valine acetamidomethylbenzyl
- VS
vinyl sulfone
- VSφ
vinyl sulfone phenyl
- Yii
diiodo tyrosine
- Z
benzyloxycarbonyl
- p.o.
per oral
Footnotes
This research was supported by grants from the National Institutes of Health (AI35707-1) and the American Heart Association (no. 93015380). J.H. McKerrow is a Burroughs Wellcome Molecular Parasitology Scholar.
References
- 1.Libow LF, Beltranni VP, Silvers DN, Grossman ME. Post-cardiac transplant reactivation of Chagas' disease diagnosed by skin biopsy. Cutis. 1991;48:37–40. [PubMed] [Google Scholar]
- 2.Godal, T., and J. Nagera. 1990. Tropical diseases. In WHO Division of Control in Tropical Diseases, 12-13. World Health Organization; Geneva, Switzerland.
- 3.The National Foundation of Brazil. Etiological treatment for Chagas' disease. Parasitol Today. 1996;13:127–128. [Google Scholar]
- 4.Parada H, Carrasco HA, Anez N, Inglessis I. Cardiac involvement is a constant finding in acute Chagas' disease: a clinical, parasitological and histopathological study. Int J Cardiology. 1997;60:49–54. doi: 10.1016/s0167-5273(97)02952-5. [DOI] [PubMed] [Google Scholar]
- 5.Chagas, C. 1981. Carlos Chagas: Coletanea de trabalhos cientificos. Editora Universidade de Brasilia. 6:247–258.
- 6.Di Lorenzo GA, Pagano MA, Taratuto AL, Garau ML, Meli FJ, Pomsztein MD. Chagasic granulomatous encephalitis in immunosuppressed patients. Computed tomography and magnetic resonance imaging findings. J Neuroimaging. 1996;6:94–97. doi: 10.1111/jon19966294. [DOI] [PubMed] [Google Scholar]
- 7.Pimentel PC, Handfas BW, Carmignani M. Trypanosoma cruzimeningoencephalitis in AIDS mimicking cerebral metastasis: case report. Arq Neuro-Psiquiatr. 1996;54:102–106. doi: 10.1590/s0004-282x1996000100017. [DOI] [PubMed] [Google Scholar]
- 8.Filardi LS, Brener Z. Susceptibility and natural resistance of Trypanosoma cruzistrains to drugs used clinically in Chagas' disease. Trans Royal Soc Trop Med Hyg. 1987;81:755–759. doi: 10.1016/0035-9203(87)90020-4. [DOI] [PubMed] [Google Scholar]
- 9.Gorla NB, Ledesma OS, Barbieri GP, Larripa IB. Assessment of cytogenetic damage in chagasic children treated with benznidazole. Mutat Res. 1988;206:217–220. doi: 10.1016/0165-1218(88)90163-2. [DOI] [PubMed] [Google Scholar]
- 10.Kirchhoff LV. American Trypanosomiasis (Chagas' disease)—a tropical disease now in the United States. New Engl J Med. 1993;329:639–644. doi: 10.1056/NEJM199308263290909. [DOI] [PubMed] [Google Scholar]
- 11.Cerisola JA, Alvarez M, De Rissio AM. Immunodiagnostico da doenca de Chagas. Evolucao serologica de pacientes com doenca de Chagas. Rev Inst Med Trop Sao Paulo. 1970;18:357–364. [PubMed] [Google Scholar]
- 12.Andrade SG, Magalhaes JB, Pontes AL. Evaluation of chemotherapy with benznidazole and nifurtimox in mice infected with Trypanosoma cruzistrains of different types. Bull W H O. 1985;63:721–726. [PMC free article] [PubMed] [Google Scholar]
- 13.Cazzulo JJ, Stoka V, Turk V. Cruzipain, the major cysteine proteinase from the protozoan parasite Trypanosoma cruzi. . Biol Chem. 1997;378:1–10. doi: 10.1515/bchm.1997.378.1.1. [DOI] [PubMed] [Google Scholar]
- 14.Eakin AE, McGrath ME, McKerrow JH, Fletterick RJ, Craik CS. Production of crystallizable cruzain, the major cysteine protease from Trypanosoma cruzi. . J Biol Chem. 1993;9:6115–6118. [PubMed] [Google Scholar]
- 15.Ring CS, Sun E, McKerrow JH, Lee GK, Rosenthal PJ, Kuntz ID, Cohen FE. Structure-based inhibitor design by using protein models for the development of antiparasitic agents. Proc Natl Acad Sci USA. 1993;90:3583–3587. doi: 10.1073/pnas.90.8.3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eakin AE, McKerrow JH, Craik CS. A cysteine protease is a target for the enzyme structure-based design of antiparasitic drugs. Drug Inf J. 1995;92:1501S–1517S. [Google Scholar]
- 17.McGrath ME, Eakin AE, Engel JC, McKerrow JH, Craik CS, Fletterick RJ. The crystal structure of cruzain: a therapeutic target for Chagas' disease. J Mol Biol. 1995;247:251–259. doi: 10.1006/jmbi.1994.0137. [DOI] [PubMed] [Google Scholar]
- 18.Ashall F, Angliker H, Shaw E. Lysis of trypanosomes by peptidyl fluoromethyl ketones. Biochem Biophys Res Commun. 1990;170:923–929. doi: 10.1016/0006-291x(90)92179-4. [DOI] [PubMed] [Google Scholar]
- 19.Meirelles MN, Juliano L, Carmona E, Silva SG, Costa EM, Murta ACM, Scharfstein J. Inhibitors of the major cysteinyl proteinase (GP57/51) impair host cell invasion and arrest the intracellular development of Trypanosoma cruziin vitro. Mol Biochem Parasitol. 1992;52:175–184. doi: 10.1016/0166-6851(92)90050-t. [DOI] [PubMed] [Google Scholar]
- 20.Harth G, Andrews N, Mills AA, Engel JC, Smith R, McKerrow JH. Peptide-fluoromethyl ketones arrest intracellular replication and intercellular transmission of Trypanosoma cruzi. . Mol Biochem Parasitol. 1993;58:17–24. doi: 10.1016/0166-6851(93)90086-d. [DOI] [PubMed] [Google Scholar]
- 21.Engel JC, Doyle PS, Palmer J, Hsieh I, Bainton DF, McKerrow JH. Cysteine protease inhibitors alter Golgi complex ultrastructure and function in Trypanosoma cruzi. . J Cell Sci. 1998;111:597–606. doi: 10.1242/jcs.111.5.597. [DOI] [PubMed] [Google Scholar]
- 22.Stenberg PE, Schuman MA, Levine SP, Bainton DF. Redistribution of alpha-granules and their contents in thrombin-stimulated platelets. J Biol Chem. 1984;98:748–760. doi: 10.1083/jcb.98.2.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kjeldsen L, Bainton DF, Sengelov H, Borregaard N. Structural and functional heterogeneity among peroxidase-negative granules in human neutrophils: identification of a distinct gelatinase-containing granule subset by combined immunocytochemistry and subcellular fractionation. Blood. 1993;82:3183–3191. [PubMed] [Google Scholar]
- 24.Laemli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;222:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 25.Zhang, D. 1998. The roles of cytochrome P450 3A and P-glycoprotein in the absorption, metabolism and elimination of a novel cysteine protease inhibitor. Ph.D. thesis. University of California, San Francisco.
- 26.Tarleton RL, Zhang L, Downs MO. Autoimmune rejection of neonatal heart transplants in experimental Chaga's disease is a parasite-specific response to infected host-tissue. Proc Natl Acad Sci USA. 1997;94:3932–3937. doi: 10.1073/pnas.94.8.3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Viotti R, Vigliano C, Armenti H, Segura EL. Treatment of chronic Chagas' disease with benznidazole: clinical and serological evolution of patients with long-term follow up. Am Heart J. 1994;127:151–162. doi: 10.1016/0002-8703(94)90521-5. [DOI] [PubMed] [Google Scholar]
- 28.Andrade SG, Stocker-Guerret S, Pimentel AS, Grimaud JA. Reversibility of cardiac fibrosis in mice chronically infected with Trypanosoma cruzi, under specific chemotherapy. Mem Inst Oswaldo Cruz. 1991;86:187–200. doi: 10.1590/s0074-02761991000200008. [DOI] [PubMed] [Google Scholar]
- 29.Robertson CD, Coombs GH, North MJ, Mottram JC. Parasite cysteine proteinases. Perspect Drug Discov Des. 1996;6:1–20. [Google Scholar]
- 30.McKerrow JH, McGrath ME, Engel JC. The cysteine protease of Trypanosoma cruzias a model for antiparasite drug design. Parasitol Today. 1995;11:279–282. doi: 10.1016/0169-4758(95)80039-5. [DOI] [PubMed] [Google Scholar]
- 31.Eichhold TH, Hookfin EB, Taiwo YO, De B, Wehmeyer KR. Isolation and quantification of fluoroacetate in rat tissues following dosing of Z-Phe-Ala-CH2-F, a peptidyl fluoromethyl ketone protease inhibitor. J Pharm Biochem Anal. 1997;16:459–467. doi: 10.1016/s0731-7085(97)00102-7. [DOI] [PubMed] [Google Scholar]
- 32.Marinkelle CJ, Rodriguez E. The influence of environmental temperature on the pathogenicity of Trypanosoma cruziin mice. Exp Parasitol. 1968;23:260–263. doi: 10.1016/0014-4894(68)90067-2. [DOI] [PubMed] [Google Scholar]
- 33.Bertelli MS, Golgher RR, Brener Z. Intraspecific variation in Trypanosoma cruzi: effect of temperature on the intracellular differentiation in tissue culture. J Parasitol. 1977;63:434–437. [PubMed] [Google Scholar]
- 34.Fresno M, Hernandez-Munain C, de Diego J, Rivas L, Scharfstein J, Bonay P. Trypanosoma cruzi:identification of a membrane cysteine proteinase linked through a GPI anchor. Braz J Med Biol Res. 1994;27:431–437. [PubMed] [Google Scholar]
- 35.Nascimento AE, de Souza W. High resolution localization of cruzipain and Ssp4 in Trypanosoma cruziby replica staining label fracture. Biol Cell. 1996;86:53–58. [PubMed] [Google Scholar]
- 36.McKerrow JH, James MNG. Cysteine proteases: evolution, function, and inhibitor design. Perspect Drug Discov Des. 1996;6:1–125. [Google Scholar]