

Abstract
The products of recombination activating gene (RAG)1 and RAG2 initiate the lymphoid-specific phase of the V(D)J recombination by creating a DNA double-strand break (dsb), leaving hairpin-sealed coding ends. The next step uses the general DNA repair machinery of the cells to resolve this dsb. Several genes involved in both V(D)J recombination and DNA repair have been identified through the analysis of in vitro mutants (Chinese hamster ovary cells) and in vivo situations of murine and equine severe combined immunodeficiency (scid). These studies lead to the description of the Ku–DNA-dependent protein kinase complex and the XRCC4 factor. A human SCID condition is characterized by an absence of B and T lymphocytes. One subset of these patients also demonstrates an increased sensitivity to the ionizing radiation of their fibroblasts and bone marrow precursor cells. This phenotype is accompanied by a profound defect in V(D)J recombination with a lack of coding joint formation, whereas signal joints are normal. Functional and genetic analyses distinguish these patients from the other recombination/repair mutants, and thus define a new group of mutants whose affected gene(s) is involved in sensitivity to ionizing radiation and V(D)J recombination.
Keywords: human, immunodeficiency, V(D)J recombination, DNA repair, severe combined immunodeficiency
Immunoglobulin and TCR genes are composed of variable (V),1 diversity (D) and joining (J) segments scattered along the chromosome that require somatic rearrangement before their expression (1). The V(D)J recombination process is common to both B and T cells and can be divided into three steps. The initiation of the V(D)J recombination involves the recognition of recombination-specific sequences (RSS) flanking the V, D, and J segments by the lymphoid specific recombination activating gene (RAG)1 and RAG2 proteins. RAG1 and RAG2 then introduce a DNA double-strand break (dsb) at the site of the RSS, leaving covalently sealed (hairpin) coding ends (2). The critical role for the RAG proteins in V(D)J recombination has been demonstrated via targeting experiments in mice (3, 4), and more recently through the description of human Rag mutants suffering from SCID (reference 5 and our unpublished observations). An intermediate phase in the V(D)J recombination process is characterized by the recruitment of the DNA-dependent protein kinase (DNA-PK) complex at the site of the DNA dsb (6). DNA-PK is composed of a 450-kD catalytic subunit (DNA-PKcs) and a DNA targeting subunit called Ku. Ku itself is a heterodimer of the 70- and 80-kD proteins (Ku70 and Ku80, respectively), and displays DNA end–binding activity in vitro (7). The whole DNA-PK complex is essential for the V(D)J recombination/DNA repair processes as illustrated by the analysis of (a) the murine and equine scid models (8–10), (b) several Chinese hamster ovary cell (CHO) mutants (11–14), (c) Ku80 knockout mice (15, 16), and (d) Ku70 knockout experiments (17–19). More specifically, nonsense and frame-shift mutations in the DNA-PKcs encoding gene are responsible for the murine and equine scid conditions, respectively (20, 21), whereas point mutations in the Ku80 gene have been found in the x-ray cross-complementing (XRCC)5 complementation group of CHO mutants (22, 23). Alteration of either one of the DNA-PK complex partners results in a defect in both V(D)J recombination and DNA repair, identified by an increased sensitivity to any DNA dsb–causing agent. The product of the XRCC4 gene (24) is another important effector, as a defect in this gene is responsible for the impairment of V(D)J recombination and DNA repair in the XR1-CHO mutant (11, 12). The final step in V(D)J recombination results in the rejoining of the coding ends by a molecular mechanism that is largely unknown thus far, although several recent studies have suggested the possible involvement of DNA ligase I and/or DNA ligase IV in this process (25–29).
A human SCID condition of autosomal recessive inheritance is characterized by an absence of circulating T and B lymphocytes (T−B−), whereas NK cells are present (30). Absent or aberrant pattern of DQ52-Jh rearrangement of the immunoglobulin heavy chain locus were demonstrated in some of the patients with this condition (31), whereas bone marrow–derived, EBV-transformed B cell lines obtained in some cases displayed germline status of Ig genes (32). We demonstrated that marrow cells and fibroblasts from one subset of T−B− SCID patients exhibited increased radiosensitivity (33). In normally radiosensitive T−B− SCID, mutations in the RAG1 or RAG2 genes were recently described (reference 5 and our unpublished results). Abnormally radiosensitive human SCID resembles the murine scid condition, as patients are devoid of mature lymphocytes and display ubiquitous increased sensitivity to ionizing radiation (33). However, normal kinetics of DNA repair after a 50-Gray γ-radiation, as judged by pulse-field gel electrophoresis, together with normal DNA-PK activity in vitro, have cast doubt on the hypothesis that this condition truly corresponds to a V(D)J recombination/DNA repair defect group of mutants (34). To address this question more directly, we analyzed the V(D)J recombination activity in RAG1/2-transfected fibroblasts from T−B− SCID patients using extrachromosomal recombination substrates. Moreover, we took advantage of a consanguineous family with three affected children to perform genetic studies using polymorphic markers flanking genes involved in V(D)J recombination/DNA repair processes.
Materials and Methods
Patients and Cells.
Patients P1, P2, P5, P6, P10, P11, P12, and P15 were characterized by an absence of peripheral CD3+ T cells and CD19+ B cells (34). For all T−B− SCID patients except P11 and P12 radiosensitivity status was determined on marrow cells (33, 34). P11 and P12 are the brother and sister of P5 and were born to related parents (see Fig. 1 A). P10 is a T−B− SCID patient caused by mutation in the RAG2 gene (our unpublished observation) with no increased cell radiosensitivity. Primary skin fibroblast cell lines were obtained as previously described (34) and maintained in complete culture medium consisting of RPMI 1640 (Glutamax; GIBCO BRL, Gaithersburg, MD) supplemented with 15% FCS. SV40-transformed fibroblast lines were obtained by transfection of the SV40 large T antigen encoding plasmid pLAS-wt (35) and by overgrowth of transformed cells over nontransformed fibroblasts without other means of selection.
Figure 1.
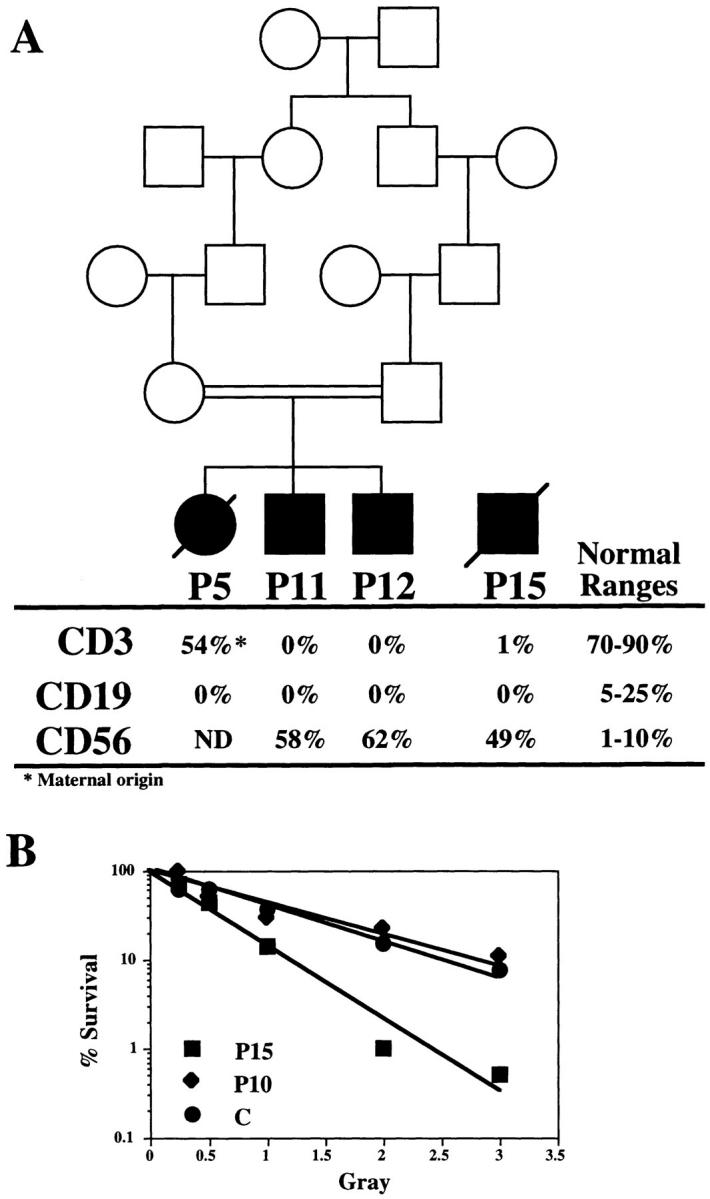
(A) Pedigree and biological status of P5, P11, P12, and P15. Peripheral blood counts of mature T, B, and NK cells were determined by FACS® immunostaining with anti-CD3, anti-CD19, and anti-CD56 mAbs, respectively. (B) Sensitivity to ionizing radiation in CFU-GM from P10 and P15. Plating efficiency of marrow cell cultures was determined after increasing doses (0.5–3 Gray) of γ-radiation. Results are expressed as percentage of survival relative to unirradiated cells. C represents the survival curve obtained with cells from an age-matched control. P10 shows normal radiosensitivity, whereas the survival curve from P15 cells is representative of the group of patients showing increased cell radiosensitivity.
DNA End–binding and DNA-PK Activity Assays.
DNA-PK activity was determined in fibroblast cell extracts as previously described (34). In brief, 5 μg of cellular extract were incubated with a p53 peptide (Promega, Charbonnieres, France) with or without 200 ng of linear DNA as activator. Incorporation of γ[32P] into the peptide was monitored. The MO59J and MO59K human cell lines (36) were used as negative and positive controls, respectively. For the DNA end–binding assay, 1 μg whole cell extracts prepared in DNA-PK lysis buffer were incubated with 0.5 ng of a 32P-labeled double-strand oligonucleotide probe (5′-CGCGTGGTACGTAGGTCACTCTC-3′) in 1XGR buffer (20 mM Tris, pH 7.5, 2 mM MgCl2, 0.1 mM EDTA, 0.25 mM dithiothreitol, and 5% glycerol) with 800 ng of circular plasmid DNA. After 15 min incubation at 22°C the reactions were analyzed by 4% PAGE in 1× TGE buffer (50 mM Tris, pH 8.5, 380 mM glycine, and 2 mM EDTA). For supershift experiments, whole cell extracts were preincubated (30 min at 4°C) with 500 ng anti-Ku70/80 (clone 162, Ab-3 NeoMarkers) mAb or anti-Rag1 (clone G189-1417; PharMingen, San Francisco, CA) as control before the addition of the oligonucleotide probe.
Transient Transfection Assay for V(D)J Recombination.
V(D)J recombination assay was performed on SV40-transformed fibroblasts using the V(D)J extrachromosomal substrates pHRecCJ and pHRecSJ (see Fig. 3 A). pHRecCJ was constructed by replacing the polyoma early region (large T antigen and origin of replication) from pBlueRec (37) with analogous sequences from SV40 to allow for replication in human cells (38). The recombination of the pHRecCJ plasmid leads to the formation of a coding joint upon deletion of the two RSS sites. For signal joint formation analysis, the two RSS sites were inverted in the pHRecSJ plasmid. Recombination of the pHRecSJ plasmid leads to the fusion of the two RSS (signal-joint) creating a diagnostic ApalI restriction site. 5 × 106 exponentially growing SV40-transformed fibroblasts were electroporated (Easyject, Eurogentec, Angers, France; setting: 250 V, 1,500 μF, ∞ Ω) in 400 μl of complete culture medium with 2.5 μg of either pHRecCJ or pHRecSJ along with 6 μg of RAG1, and 4.8 μg of RAG2-encoding genes in eukaryotic expression vector (gift of M. Oettinger, Department of Molecular Biology, Massachusetts General Hospital, Boston, MA; reference 39). 48 h after transfection, extrachromosomal DNA was prepared by rapid alkaline lysis (40), digested with DpnI to eliminate nonreplicated plasmids, and used for transfection of bacteria or PCR analysis using T3 and T7 primers. The percentage of recombination was determined by transforming DH10B bacteria in the presence of Xgal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) and IPTG (isopropyl-β-thiogalactopyranoside) and counting the blue colonies. Integrity of the recombined substrates following V(D)J recombination was analyzed by DNA sequencing using T3 primer.
Figure 3.

(A) Structure of the V(D)J recombination extrachromosomal substrates. pHRecCJ and pHRecSJ constructs were adapted from pBlueRec (37). They contain the SV40 large T coding sequence and the SV40-Ori for autonomous replication in human cells, and a LacZ gene interrupted by a 310-bp DNA stuffer flanked by RSS on both sides. pHRecCJ is used for studying the coding joint formation. In the pHRecSJ plasmid, the two RSS were inverted to allow for the analysis of signal joint formation. (B) Analysis of signal joints after pHRecSJ recombination. The recovered pHRecSJ reporter plasmids were PCR-amplified with T3 and T7 primers, digested (+) or not (−) by ApalI, and run on a 2% agarose gel. The 500-bp band represents the PCR band obtained from unrecombined pHRecSJ. The 190-bp band represents the fusion of the RSS in the recombined plasmid in the absence of treatment with ApalI. This band is further digested to a 100-bp fragment with ApalI.
XRCC4 cDNA Sequence Analysis.
Total RNA was extracted from fibroblasts and reverse transcribed using random hexamers and Moloney murine leukemia virus reverse transcriptase without RNase H according to manufacturer's recommendations (GIBCO BRL). XRCC4 cDNA was PCR-amplified using XRF1 5′ GGGCTGCCTCTTTAAATAAC and XRR1 5′ ATGATGTTTTCTTTGTCCTGTTTT primers and TaqAdvantage (Clontech Labs., Inc., Palo Alto, CA) enzyme. Cycling parameters were 30 s of denaturation at 94°C, 30 s of annealing at 50°C, and 2.5 min of extension at 72°C for 30 cycles. PCR products were cloned into pGemT (Promega) and sequenced using the DYEnamic cycle sequencing kit (Amersham Pharmacia Biotech, Piscataway, NJ) and the Dye terminator cycle sequencing kit (Perkin-Elmer Corp., Norwalk, CT) on an ABI377 sequencer (Perkin-Elmer).
Genotype Analysis.
DNA from P5, P11, P12, and their parents were genotyped using 12 polymorphic microsatellite PCR markers referenced at Généthon (Evry, France; reference 41): D2S137 and D2S301 for Ku80; D22S276 and D22S537 for Ku70; D8S519 and D8S1745 for the DNA-PKcs; D5S2029 and D5S428 for XRCC4; D13S173 and D13S285 for DNA ligase IV; and D19S412 and D19S866 for DNA ligase I. The forward primers of each pair were end-labeled with γ-[32P]ATP using polynucleotide kinase. The amplifications were performed in a final reaction volume of 17.5 μl containing 100 ng template DNA, 0.2 mM deoxynucleotides, 3 pmol unlabeled reverse primer, 1.5 pmol unlabeled forward primer, 1.5 pmol end-labeled forward primer, and 0.07 U Taq polymerase (ATGC Biotechnologie, Noisy-le-Grand, France). PCR conditions consisted of 30 cycles, each consisting of 45 s at 94°C, 1 min at the annealing temperature specific for each pair of primers, and 45 s at 72°C. The PCR products were analyzed by electrophoresis on 5% denaturing polyacrylamide gels.
Results
Increased Radiosensitivity in Human T−B− SCID with Normal DNA-PK Activity.
We previously reported on the occurrence of human immune deficiency characterized by a lack of both B and T lymphocytes, accompanied by an increase in radiosensitivity of bone marrow precursor cells and fibroblasts (33, 34). We here extended our series of patients with three new cases (P11, P12, and P15). P11 and P12 are two brothers of the previously described patient, P5, and were born to related parents (Fig. 1 A). These three patients were devoid of both mature T and B lymphocytes (Fig. 1 A). Bone marrow cells from P5 were previously shown to be abnormally radiosensitive and cells from P15 showed the same extent of increased sensitivity to γ-rays (Fig. 1 B). The radiosensitivity status of marrow cells from P11 and P12 was not assessed as they are brothers of P5, but according to the following results (V(D)J recombination assay) one can assume that they would display increased radiosensitivity as well. One striking finding in our previous report was the normal DNA-PK activity in cellular extracts from T−B− SCID with increased cell radiosensitivity (34). DNA-PK activity was also normal in cellular extracts from the four patients described here (Fig. 2 B), in contrast to the absence of activity in the DNA-PK negative MO59J cell line. Given the normal DNA-PK activity in these patients, it was not surprising to find a functional Ku70/Ku80 complex in these cellular extracts as tested by the DNA end–binding assay (Fig. 2 A). The real nature of the Ku–DNA complex was further proven by supershift experiments using anti-Ku70/80 mAb, whereas the anti-Rag1 control antibody did not result in a shift of this complex. Indeed, any defect in this complex would have severely compromised the activation of the DNA-PKcs and ultimately resulted in the absence of kinase activity as is the case in various CHO mutants (for review see reference 42). Altogether, the findings concerning the three newly studied patients have confirmed and extended the results of our previous studies.
Figure 2.

(A) DNA end–binding assay using cellular extracts from T−B− SCID patients P5, P11, P12, and P15. Whole cell extracts were preincubated for 30 min at 4°C or not with specific mAb for Ku70/80 (lanes 3, 6, 9, and 12) or Rag1 (lanes 4, 7, 10, and 13) as control. (B) DNA-PK activity in cellular extracts from P5, P11, P12, and P15. MO59J and MO59K cell lines (36) lacking and containing active DNA-PKcs, respectively, were used as controls. DNA-PK activity was determined in the absence (−) or presence (+) of linear DNA as specific activator.
Defective V(D)J Recombination in T−B−SCID Patients with Increased Cell Radiosensitivity.
The “DNA-repair” phase of the V(D)J recombination can be analyzed by transient transfection of RAG1, RAG2, and an extrachromosomal substrate into fibroblasts. The two recombination substrates used here to analyze both coding and signal joint formation are depicted in Fig. 3 A. These plasmids were derived from pBlueRec (37), in which the polyoma large T antigen and replication origin sequences were replaced by those of SV40 to allow for replication in human cells (38). In pHRecCJ, the LacZ gene is interrupted by a DNA stuffer flanked by two RSS that will be deleted upon coding joint formation. In pHRecSJ, the two RSS sequences were inverted so that the fusion of the two RSS (signal joint) remains on the plasmid after recombination. A functional LacZ gene is reconstituted upon recombination of the two plasmids that lead to the formation of blue colonies when introduced into bacteria and plated on Xgal- and IPTG-containing medium. The ratio of blue colonies to white colonies is therefore indicative of the capacity of the cells to fully accomplish V(D)J recombination. In the case of the coding joint formation, only one-third of the recombined substrates will lead to blue colonies because of random nibbling of the coding ends, leading to shifts in the LacZ gene open reading frame (37). In contrast, the signal joint is perfect without deletion of nucleotides, so that the number of blue colonies represent the total number of recombined substrates. SV40-transformed fibroblast lines from T−B− radiosensitive SCID patients and two control cell lines (P10 and Hela) were transiently transfected with RAG1 and RAG2 expression plasmids together with either pHRecCJ or pHRecSJ. 48 h later the reporter constructs were recovered, digested with DpnI to eliminate plasmids that did not replicate in the fibroblasts and reintroduced into bacteria, and the ratio of blue to white colonies was determined (Table 1). The recombination frequency ranged from 3.86 × 10−3 to 11.06 × 10−3 in P10 and Hela cells, respectively, using the coding joint–specific plasmid pHRecCJ. Sequence analysis of these recombination products revealed that they were bona fide coding joints with limited deletions at the junction (data not shown). P10 is a T−B− SCID patient with mutations in the RAG2 gene (our unpublished observation). P10 belongs to a different group of patients whose DNA repair machinery is not affected and whose marrow cells do not display increased radiosensitivity (Fig. 1 B). Analysis of signal joint formation in Hela cells using the pHRecSJ plasmid leads to a recombination frequency of 4.28 × 10−3. The V(D)J recombination was assessed in fibroblast lines from seven T−B− radiosensitive SCID patients and the coding joint formation was found to be profoundly affected in all cases with recombination frequencies <0.08 × 10−3 (Table 1). Interestingly, the frequency of signal joint formation, determined with the pHRecSJ construct, was not significantly different in these patients compared with the Hela control (mean: 2.55 × 10−3). The perfect fusion of the heptamer sequences at the signal joint causes the formation of an ApalI site. This ApalI diagnostic site was used to check for the integrity of the signal joints formed in T−B− SCID cells (Fig. 3 B). The pHRecSJ plasmid was PCR-amplified using T3 and T7 primers after transfection into fibroblasts. The signal joints resulted in a 190-bp PCR fragment that could always be digested into a 100-bp ApalI fragment, indicating the integrity of these joints in SCID cells. Altogether, these results clearly establish that the DNA repair machinery necessary for the completion of the V(D)J recombination is profoundly impaired in T−B− SCID with increased cell radiosensitivity.
Table 1.
V(D)J Recombination Assay in Human T−B−SCID Fibroblasts
| pHRecCJ coding joints | pHRecSJ signal joints | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Blue | Total | R (× 10−3)* | Blue | Total | R (× 10−3)‡ | |||||||||
| Hela | Exp. 1 | 38 | 10,300 | 11.06 | 48 | 11,200 | 4.28 | |||||||
| P10 | Exp. 1 | 209 | 96,000 | 6.53 | – | – | – | |||||||
| Exp. 2 | 38 | 29,500 | 3.86 | – | – | – | ||||||||
| P1 | Exp. 1 | 2 | 109,000 | 0.05 | – | – | – | |||||||
| P2 | Exp. 1 | 0 | 54,000 | <0.02 | – | – | – | |||||||
| P5 | Exp. 1 | 8 | 1,250,000 | 0.02 | 24 | 18,600 | 1.29 | |||||||
| Exp. 2 | 4 | 390,000 | 0.03 | 15 | 2,410 | 6.20 | ||||||||
| P6 | Exp. 1 | 3 | 217,000 | 0.04 | 11 | 6,600 | 1.66 | |||||||
| Exp. 2 | 2 | 560,000 | 0.01 | 243 | 420,000 | 0.58 | ||||||||
| P11 | Exp. 1 | 3 | 242,000 | 0.04 | 8 | 6,500 | 1.23 | |||||||
| Exp. 2 | 3 | 730,000 | 0.01 | 49 | 41,000 | 1.19 | ||||||||
| P12 | Exp. 1 | 7 | 370,000 | 0.05 | 14 | 5,000 | 2.80 | |||||||
| Exp. 2 | 3 | 790,000 | 0.01 | 196 | 370,000 | 0.53 | ||||||||
| P15 | Exp. 1 | 2 | 72,000 | 0.08 | 23 | 8,100 | 2.84 | |||||||
| Exp. 2 | 6 | 500,000 | 0.03 | 36 | 5,000 | 7.20 | ||||||||
R (coding joints) = 3 × (Blue col.)/(Total col.) × 1,000.
R (signal joints) = (Blue col.)/(Total col.) × 1,000.
The Ku80–DNA-PK Complex Is Not Affected in T−B− SCID Patients with Increased Cell Radiosensitivity.
In light of the molecular defects responsible for the various CHO mutants and the murine and equine scid conditions (8–12, 14, 43), the functional assay for DNA-PK and Ku activity in human T−B− SCID (Fig. 2) implies that the Ku70 and Ku80 proteins are present and capable of binding DNA and recruiting the DNA-PKcs in our series of patients. This also indicates that the DNA-PKcs is expressed with a functional kinase domain. However, this in vitro assay does not rule out the possibility of mutations in other, as yet unknown, domains of these three proteins involved in V(D)J recombination and DNA repair. This limitation is of particular importance when considering the size (450 kD) of the DNA-PKcs protein. P5, P11, and P12 are three siblings from a consanguineous family. In a consanguineous family it can be assumed that a mutated allele for a given gene will be inherited as a homozygous trait in affected children and will be heterozygous in both parents and in healthy siblings. The same holds true for nearby flanking markers. Therefore, we looked for homozygosity of highly polymorphic markers flanking the Ku–DNA-PK complex encoding genes in P5 family members. Results of this analysis (Fig. 4, A–C) showed independent segregation patterns between these markers and the disease locus in the different affected children. These results therefore irrevocably exclude the Ku70, Ku80, and DNA-PKcs genes in the T−B− SCID condition, at least in this family.
Figure 4.

Genotype analysis of P5 family members with highly polymorphic microsatellite markers. Lane 1, father; lane 2, mother; lane 3, P5; lane 4, P11; lane 5: P12. For each gene upstream and downstream markers were used. None of the markers that are heterozygous in the parents are found homozygous in the three affected children.
XRCC4, Ligase I, and Ligase IV Are Not Involved in T−B− SCID.
Among the various CHO mutants that were shown to be defective in V(D)J recombination and DNA repair (for review see reference 44), the XR1 cell line is characterized by defects in both coding and signal joint formation, but does not show any defect in DNA end–binding and DNA-PK activities (8, 45, 46). A human cDNA, called XRCC4, that complements the XR1 defect has been cloned (24). Although cells from human radiosensitive T−B−SCID show normal signal joint formation (Table 1 and Fig. 3 B), the possible involvement of the XRCC4 gene in the pathology was assessed by cDNA sequence analysis in four unrelated patients (P1, P2, P6, and P11) and by genotype analysis of flanking polymorphic markers in P5, P11, and P12 (Fig. 4 D) and in P15 (data not shown). Normal cDNA sequences (data not shown) and the absence of cosegregation between flanking markers and disease inheritance excluded an XRCC4 gene defect as being responsible for this condition in all patients tested. Recently, the XRCC4 protein has been found to interact with DNA ligase IV in mammalian cells and stimulate its activity in vitro (26, 27). Moreover, the yeast counterpart of DNA ligase IV plays a critical role in DNA dsb repair by nonhomologous end joining as demonstrated by the increase of radiosensitivity of yeast strains carrying DNA ligase IV mutations in a Rad52 defective background (25, 28, 47). Altogether, these reports make ligase IV an attractive candidate for the religation process during V(D)J recombination. Genotype analysis of DNA ligase IV gene flanking markers in P5, P11, and P12 excluded this gene as being implicated in the T−B− SCID condition (Fig. 4 E). Moreover, cDNA sequence analysis of ligase IV in P15 did not reveal any mutation in the coding region (data not shown). This result was perhaps not surprising provided that the signal joint formation, which can be considered as a nonhomologous end joining event, was not affected in these patients (Table 1 and Fig. 3 B). Lastly, another DNA ligase (ligase I) has been shown to enhance cell-free V(D)J recombination in vitro (29) and represents another potential player of the V(D)J recombination reaction. Again, genotype analysis excluded this gene as being defective in P5, P11, and P12 (Fig. 4 F). Moreover, an impaired DNA ligase I activity does not result in a T−B− SCID phenotype (48–50) nor does it lead to faulty V(D)J recombination activity in vitro (51, 52).
Discussion
Several in vivo conditions are characterized by an absence of B and T lymphocyte development associated with a defect in DNA dsb repair. In the murine and equine SCID conditions a lack of DNA-PK activity caused by mutations in the DNA-PKcs coding gene are responsible for the lack of V(D)J recombination (21, 53, 54). Specific gene targeting of the Ku80 gene also results in a T−B− phenotype in mice (15, 16), in accord with the V(D)J defect first noted in CHO cells harboring Ku80 mutations (for review see reference 42). Finally, mice with a targeted deletion of the Ku70 gene (18, 19) harbor a somewhat surprising phenotype: an absence of B cells but an almost normal number of mature T lymphocytes. This last result contrasts with the profound defect of V(D)J recombination noted in vitro in embryonic stem cells homozygous for a Ku70 gene deletion (17) and suggests the existence of T cell lineage specific factors that complemented the defect in vivo. The human T−B− SCID condition we describe is yet another setting in which early arrest in T and B lymphocyte development is most probably caused by a primary defect in the DNA repair/recombination machinery. However, the factor(s) involved in this pathology are clearly different from the Ku70-80–DNA-PK complex and from XRCC4, another factor found to be critical for both V(D)J recombination and DNA repair in vitro in CHO-XR1 cell line but for which no in vivo model are yet available (24). Human RAG1+RAG2+ T−B− SCID patients therefore define a new complementation group of mutants whose affected gene(s) are involved in sensitivity to ionizing radiation and V(D)J recombination. The finding of a normal activity of signal joint formation in fibroblasts from SCID patients in vitro indicates that the ultimate ligation step of the broken ends is probably not affected in these patients. The exclusion of DNA ligase I and DNA ligase IV as candidate genes in three patients certainly strengthens this assumption. The defect in V(D)J recombination in these patients appears to be specific of the coding joint formation and may therefore involve enzymatic activities acting around the resolution of the hairpin-sealed coding ends. A more general survey of which kind of broken ends cells from these patients are able or not to repair should help clarifying the role of the defective molecule(s).
Acknowledgments
We thank F. Alt for communication of the XRCC4 sequence before publication; M. Oettinger for the phRAG1 and pmRAG2 expression plasmids; and L. Daya-Grosjean (Institut de Recherches sur le Cancer, Villejuif, France) for the pLAS-wt construct. We thank C. Muller for advice on Ku assay.
This work was supported by institutional grants from Institut National de la Santé et de la Recherche Médicale and Ministère de l'Education Nationale, de l'Enseignement Supérieur, de la Recherche et de l'Insertion Professionnelle, and grants from Association de Recherche sur le Cancer (ARC), Groupe de Recherche et d'étude sur le génome (GREG), and Association contre les myopathies (AFM). N. Nicolas is supported by a doctoral fellowship from Ministère de la Recherche et de la Technologie (France). D. Moshous is supported by Deutsche Forschungsgemeinschaft (Germany).
Abbreviations used in this paper
- CHO
Chinese hamster ovary
- cs
catalytic subunit
- D
diversity
- dsb
double-strand break
- J
joining
- PK
protein kinase
- RAG
recombination activating gene
- RSS
recombination-specific sequences
- T−B−
absence of circulating T and B lymphocytes
- V
variable
- XRCC
x-ray cross-complementing
References
- 1.Alt FW, Oltz EM, Young F, Gorman J, Taccioli G, Chen J. VDJ recombination. Immunol Today. 1992;13:306–314. doi: 10.1016/0167-5699(92)90043-7. [DOI] [PubMed] [Google Scholar]
- 2.McBlane JF, van Gent DC, Ramsden DA, Romeo C, Cuomo CA, Gellert M, Oettinger MA. Cleavage at a V(D)J recombination signal requires only RAG1 and RAG2 proteins and occurs in two steps. Cell. 1995;83:387–395. doi: 10.1016/0092-8674(95)90116-7. [DOI] [PubMed] [Google Scholar]
- 3.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaionnou VE. RAG-1 deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 4.Shinkai Y, Rathbun G, Lam KP, Oltz EM, Stewart V, Mendelsohn M, Charron J, Datta M, Young F, Stall AM, Alt FW. RAG-2 deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell. 1992;68:855–867. doi: 10.1016/0092-8674(92)90029-c. [DOI] [PubMed] [Google Scholar]
- 5.Schwarz K, Gauss GH, Ludwig L, Pannicke U, Li Z, Linder D, Friedrich W, Seger RA, Hansen-Hagge TE, Desiderio S, et al. RAG mutations in human B cell–negative SCID. Science. 1996;274:97–99. doi: 10.1126/science.274.5284.97. [DOI] [PubMed] [Google Scholar]
- 6.Jackson SP, Jeggo PA. DNA double-strand break repair and V(D)J recombination; involvement of DNA-PK. Trends Biol Sci. 1995;20:412–415. doi: 10.1016/s0968-0004(00)89090-8. [DOI] [PubMed] [Google Scholar]
- 7.Mimori T, Hardin JA, Steitz JA. Characterization of the DNA-binding protein antigen Ku recognized by autoantibodies from patients with rheumatic disorders. J Biol Chem. 1986;261:2274–2278. [PubMed] [Google Scholar]
- 8.Blunt T, Finnie NJ, Taccioli GE, Smith GCM, Demengeot J, Gottlieb TM, Mizuta R, Varghese AJ, Alt FW, Jeggo PA, Jackson SP. Defective DNA-dependent protein kinase activity is linked to V(D)J recombination and DNA repair defects associated with the murine scid mutation. Cell. 1995;80:813–823. doi: 10.1016/0092-8674(95)90360-7. [DOI] [PubMed] [Google Scholar]
- 9.Kirchgessner CU, Patil CK, Evans JW, Cuomo CA, Fried LM, Carter T, Oettinger MA, Brown MA. DNA-dependent kinase (p350) as a candidate gene for the murine scid defect. Science. 1995;267:1178–1183. doi: 10.1126/science.7855601. [DOI] [PubMed] [Google Scholar]
- 10.Willer R, Leber R, Moore BB, van Dyk L, Perryman LE, Meek K. Equine severe combined immunodeficiency: a defect in V(D)J recombination and DNA-dependent protein kinase activity. Proc Natl Acad Sci USA. 1995;92:11485–11489. doi: 10.1073/pnas.92.25.11485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taccioli GE, Rathbun G, Oltz E, Stamato T, Jeggo PA, Alt FW. Impairment of V(D)J recombination in double-strand break repair mutants. Science. 1993;260:207–210. doi: 10.1126/science.8469973. [DOI] [PubMed] [Google Scholar]
- 12.Pergola F, Zdzienicka MZ, Lieber MR. V(D)J recombination in mammalian cell mutants defective in DNA double-strand break repair. Mol Cell Biol. 1993;13:3464–3471. doi: 10.1128/mcb.13.6.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taccioli GE, Cheng HL, Varghese AJ, Whitmore G, Alt FW. A DNA repair defect in Chinese hamster ovary cells affects V(D)J recombination similarly to the murine scid mutation. J Biol Chem. 1994;269:7439–7442. [PubMed] [Google Scholar]
- 14.Lee SE, Pulaski CR, He DM, Benjamin DM, Voss M, Um J, Hendrickson EA. Isolation of mammalian cell mutants that are X-ray sensitive, impaired in DNA double-strand break repair and defective for V(D)J recombination. Mutat Res. 1995;336:279–291. doi: 10.1016/0921-8777(95)00002-2. [DOI] [PubMed] [Google Scholar]
- 15.Zhu C, Bogue MA, Lim DS, Hasty P, Roth DB. Ku86-deficient mice exhibit severe combined immunodeficiency and defective processing of V(D)J recombination intermediates. Cell. 1996;86:379–389. doi: 10.1016/s0092-8674(00)80111-7. [DOI] [PubMed] [Google Scholar]
- 16.Nussenzweig A, Chen C, da Costa V, Soares, Sanchez M, Sokol K, Nussenzweig MC, Li GC. Requirement for Ku80 in growth and immunoglobulin V(D)J recombination. Nature. 1996;382:551–555. doi: 10.1038/382551a0. [DOI] [PubMed] [Google Scholar]
- 17.Gu Y, Jin S, Gao Y, Weaver DT, Alt FW. Ku70-deficient embryonic stem cells have increased ionizing radiosensitivity, defective DNA end-binding activity, and inability to support V(D)J recombination. Proc Natl Acad Sci USA. 1997;94:8076–8081. doi: 10.1073/pnas.94.15.8076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ouyang H, Nussenzweig A, Kurimasa A, da Costa V, Soares, Li X, Cordon-Cardo C, Li W-h, Cheong N, Nussenzweig M, Iliakis G, et al. Ku70 is required for DNA repair but not for T cell antigen receptor gene recombination in vivo. J Exp Med. 1997;186:921–929. doi: 10.1084/jem.186.6.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu Y, Seidl KJ, Rathbun GA, Zhu C, Manis JP, van der Stoep N, Davidson L, Cheng HL, Sekiguchi JM, et al. Growth retardation and leaky SCID phenotype of Ku70-deficient mice. Immunity. 1997;7:653–665. doi: 10.1016/s1074-7613(00)80386-6. [DOI] [PubMed] [Google Scholar]
- 20.Blunt T, Gell D, Fox M, Taccioli GE, Lehmann AR, Jackson SP, Jeggo PA. Identification of a nonsense mutation in the carboxyl-terminal region of DNA-dependent protein kinase catalytic subunit in the scid mouse. Proc Natl Acad Sci USA. 1996;93:10285–10290. doi: 10.1073/pnas.93.19.10285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shin EK, Perryman LE, Meek K. A kinase-negative mutation of DNA-PK(CS) in equine SCID results in defective coding and signal joint formation. J Immunol. 1997;158:3565–3569. [PubMed] [Google Scholar]
- 22.Mizuta R, Taccioli GE, Alt FW. The V(D)J recombination defect in the xrs-6 cell line results from a point mutation in the Ku80 gene. Int Immunol. 1996;8:1467–1471. doi: 10.1093/intimm/8.9.1467. [DOI] [PubMed] [Google Scholar]
- 23.Errami A, Smider V, Rathmell WK, He DM, Hendrickson EA, Zdzienicka MZ, Chu G. Ku86 defines the genetic defect and restores X-ray resistance and V(D)J recombination to complementation group 5 hamster cell mutants. Mol Cell Biol. 1996;16:1519–1526. doi: 10.1128/mcb.16.4.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Z, Otevrel T, Gao Y, Cheng HL, Seed B, Stamato TD, Taccioli GE, Alt FW. The XRCC4 gene encodes a novel protein involved in DNA double-strand break repair and V(D)J recombination. Cell. 1995;83:1079–1089. doi: 10.1016/0092-8674(95)90135-3. [DOI] [PubMed] [Google Scholar]
- 25.Schar P, Herrmann G, Daly G, Lindahl T. A newly identified DNA ligase of Saccharomyces cerevisiaeinvolved in RAD52–independent repair of DNA double-strand breaks. Genes Dev. 1997;11:1912–1924. doi: 10.1101/gad.11.15.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Critchlow SE, Bowater RP, Jackson SP. Mammalian DNA double-strand break repair protein XRCC4 interacts with DNA ligase IV. Curr Biol. 1997;7:588–598. doi: 10.1016/s0960-9822(06)00258-2. [DOI] [PubMed] [Google Scholar]
- 27.Grawunder U, Wilm M, Wu X, Kulesza P, Wilson TE, Mann M, Lieber MR. Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells. Nature. 1997;388:492–495. doi: 10.1038/41358. [DOI] [PubMed] [Google Scholar]
- 28.Wilson TE, Grawunder U, Lieber MR. Yeast DNA ligase IV mediates non-homologous DNA end joining. Nature. 1997;388:495–498. doi: 10.1038/41365. [DOI] [PubMed] [Google Scholar]
- 29.Ramsden DA, Paull TT, Gellert M. Cell-free V(D)J recombination. Nature. 1997;388:488–491. doi: 10.1038/41351. [DOI] [PubMed] [Google Scholar]
- 30.Stephan JL, Vlekova V, Le Deist F, Blanche S, Donadieu J, De Saint-Basile G, Durandy A, Griscelli C, Fischer A. Severe combined immunodeficiency: a retrospective single-center study of clinical presentation and outcome in 117 patients. J Pediatr. 1993;123:564–572. doi: 10.1016/s0022-3476(05)80951-5. [DOI] [PubMed] [Google Scholar]
- 31.Schwarz K, Hansen-Hagge TE, Knobloch C, Friedrich W, Kleihauer E, Bartram CR. Severe combined immunodeficiency (SCID) in man: B cell negative (B−) SCID patients exhibit an irregular recombination pattern at the Jh locus. J Exp Med. 1991;174:1039–1048. doi: 10.1084/jem.174.5.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abe T, Tsuge I, Kamachi Y, Torii Y, Utsumi K, Akahori Y, Ichihara Y, Kurosawa Y, Matsuoka H. Evidence for defects in V(D)J rearrangements in patients with severe combined immunodeficiency. J Immunol. 1994;152:470–471. [PubMed] [Google Scholar]
- 33.Cavazzana-Calvo M, Le Deist F, De Saint G, Basile, Papadopoulo D, de Villartay JP, Fischer A. Increased radiosensitivity of granulocyte macrophage colony-forming units and skin fibroblasts in human autosomal recessive severe combined immunodeficiency. J Clin Invest. 1993;91:1214–1218. doi: 10.1172/JCI116282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nicolas N, Finnie NJ, Cavazzana-Calvo M, Papadopoulo D, Le Deist F, Fischer A, Jackson SP, de Villartay JP. Lack of detectable defect in DNA double-strand break repair and DNA-dependent protein kinase activity in radiosensitive human severe combined immunodeficiency fibroblasts. Eur J Immunol. 1996;26:1118–1122. doi: 10.1002/eji.1830260524. [DOI] [PubMed] [Google Scholar]
- 35.Daya-Grosjean L. An immortalized xeroderma pigmentosum, group C, cell line which replicates SV40 shuttle vectors. Mutat Res. 1987;183:185–196. doi: 10.1016/0167-8817(87)90061-7. [DOI] [PubMed] [Google Scholar]
- 36.Lees-Miller SP, Godbout R, Chan DW, Weinfeld M, Day RS, III, Barron GM, Allalunis-Turner J. Absence of p350 subunit of DNA-activated protein kinase from a radiosensitive human cell line. Science. 1995;267:1183–1185. doi: 10.1126/science.7855602. [DOI] [PubMed] [Google Scholar]
- 37.Kallenbach S, Goodhardt M, Rougeon F. A rapid test for V(D)J recombinase activity. Nucleic Acids Res. 1990;18:6730. doi: 10.1093/nar/18.22.6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith, J., J.C. Andrau, S. Kallenbach, A. Laquerbe, N. Doyen, and D. Papadopoulo. 1998. Abnormal rearrangements associated with V(D)J recombination in Fanconi anemia. Cancer Res. In press. [DOI] [PubMed]
- 39.Oettinger, M.A., Schatz, D.G., Gorka, C., and Baltimore, D. 1990. RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science. 248:1517–1523. [DOI] [PubMed]
- 40.Birboim HC, Doly J. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 1979;7:1513–1523. doi: 10.1093/nar/7.6.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dib C, Fauré S, Fizames C, Samson D, Drouot N, Viganl A, Millasseau P, Marc S, Hasan J, Seboun E, et al. A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature. 1996;380:152–154. doi: 10.1038/380152a0. [DOI] [PubMed] [Google Scholar]
- 42.Jeggo PA. DNA-PK: at the cross-roads of biochemistry and genetics. Mutat Res. 1997;384:1–14. doi: 10.1016/s0921-8777(97)00009-8. [DOI] [PubMed] [Google Scholar]
- 43.Taccioli GE, Cheng HL, Varghese AJ, Whitmore GF, Alt FW. A DNA repair defect in Chinese ovary cell affects V(D)J recombination similarly to the murine Scid mutation. J Biol Chem. 1994;269:7439–7442. [PubMed] [Google Scholar]
- 44.Taccioli GE, Alt FW. Potential targets for autosomal SCID mutations. Curr Opin Immunol. 1995;7:436–440. doi: 10.1016/0952-7915(95)80085-9. [DOI] [PubMed] [Google Scholar]
- 45.Getts RC, Stamato TD. Absence of Ku-like DNA end binding activity in the xrs double-strand DNA repair-deficient mutant. J Biol Chem. 1994;269:15981–15984. [PubMed] [Google Scholar]
- 46.Rathmell WK, Chu G. A DNA end-binding factor involved in double-strand break repair and V(D)J recombination. Mol Cell Biol. 1994;14:4741–4748. doi: 10.1128/mcb.14.7.4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Teo SH, Jackson SP. Identification of Saccharomyces cerevisiaeDNA ligase IV: involvement in DNA double-strand break repair. EMBO (Eur Mol Biol Organ) J. 1997;16:4788–4795. doi: 10.1093/emboj/16.15.4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lehmann AR, Willis AE, Broughton BC, James MR, Steingrimsdottir H, Harcourt SA, Arlett CF, Lindhal T. Relation between the human fibroblast strain 46BR and cell lines representative of Bloom's syndrome. Cancer Res. 1988;48:6343–6347. [PubMed] [Google Scholar]
- 49.Willis AE, Lindahl T. DNA ligase I deficiency in Bloom's syndrome. Nature. 1987;325:355–357. doi: 10.1038/325355a0. [DOI] [PubMed] [Google Scholar]
- 50.Chan JY, Becker FF, Germain J, Ray JH. Altered DNA ligase I activity in Bloom's syndrome cells. Nature. 1987;325:357–359. doi: 10.1038/325357a0. [DOI] [PubMed] [Google Scholar]
- 51.Hsieh CL, Arlett CF, Lieber MR. V(D)J recombination in ataxia telangectasia, Bloom's syndrome, and a DNA ligase I–associated immunodeficiency disorder. J Biol Chem. 1993;27:20105–20109. [PubMed] [Google Scholar]
- 52.Petrini JH, Donovan JW, Dimare C, Weaver DT. Normal V(D)J coding junction formation in DNA ligase I deficiency syndromes. J Immunol. 1994;152:176–183. [PubMed] [Google Scholar]
- 53.Araki R, Fujimori A, Hamatani K, Mita K, Saito T, Mori M, Fukumura R, Morimyo M, Muto M, Itoh M, et al. Nonsense mutation at Tyr-4046 in the DNA-dependent protein kinase catalytic subunit of severe combined immune deficiency mice. Proc Natl Acad Sci USA. 1997;94:2438–2443. doi: 10.1073/pnas.94.6.2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blunt T, Gell D, Fox M, Taccioli GE, Lehmann AR, Jackson SP, Jeggo PA. Identification of a nonsense mutation in the carboxyl-terminal region of DNA-dependent protein kinase catalytic subunit in the scid mouse. Proc Natl Acad Sci USA. 1996;93:10285–10290. doi: 10.1073/pnas.93.19.10285. [DOI] [PMC free article] [PubMed] [Google Scholar]