

Abstract
Immunoglobulin (Ig) genes expressed in mature B lymphocytes can undergo somatic hypermutation upon cell interaction with antigen and T cells. The mutation mechanism had previously been shown to depend upon transcription initiation, suggesting that a mutator factor was loaded on an RNA polymerase initiating at the promoter and causing mutations during elongation (Peters, A., and U. Storb. 1996. Immunity. 4:57–65). To further elucidate this process we have created an artificial substrate consisting of alternating EcoRV and PvuII restriction enzyme sites (EPS) located within the variable (V) region of an Ig transgene. This substrate can easily be assayed for the presence of mutations in DNA from transgenic lymphocytes by amplifying the EPS insert and determining by restriction enzyme digestion whether any of the restriction sites have been altered. Surprisingly, the EPS insert was mutated many times more frequently than the flanking Ig sequences. In addition there were striking differences in mutability of the different nucleotides within the restriction sites. The data favor a model of somatic hypermutation where the fine specificity of the mutations is determined by nucleotide sequence preferences of a mutator factor, and where the general site of mutagenesis is determined by the pausing of the RNA polymerase due to secondary structures within the nascent RNA.
Keywords: Ig genes, somatic hypermutation, RNA secondary structure, hot spots, RNA polymerase pausing
The diversity of immunoglobulin variable regions is created from the germline variable (V),1 diversity (D), and joining (J) gene sequences by V(D)J recombination and somatic hypermutation. The latter is initiated when a mature B lymphocyte interacts with specific antigen and T lymphocytes in a germinal center response (1, 2). The B cell undergoes Ig gene mutation during several rounds of rapid proliferation and is subsequently selected for functional antibody production by interaction with antigen bound to follicular dendritic cells (1, 2).
The mechanism of somatic hypermutation is not understood. The mutations are mainly point mutations with very rare deletions and even rarer insertions (3). Mutations are found from near the start of the gene and occur over ∼1.5–2 kb. The 3′ region of the gene, including the constant (C) region, is generally not mutated (4). Transcription of the Ig gene is required (5, 6). We have determined recently that duplication of the Ig promoter upstream of the C region leads to transcription from this internal site and somatic mutations in the C region at a similar frequency as in the V region (7). The region upstream of the duplicate promoter is not mutated. Based on these data, we proposed a model of somatic hypermutation where a mutator factor (MuF) becomes associated with the RNA polymerase at an Ig promoter that is interacting with an Ig enhancer (7, 8).
Whereas the general model of coupling somatic mutation to the transcription of an Ig gene is most likely correct, the mechanism of how the mutations arise during transcription elongation is not known. We had postulated that the MuF causes pausing of the elongating RNA polymerase and that the paused complex is treated like a lesion in the DNA, eliciting a transcription coupled repair mechanism (7, 8). However, we now know that many proteins required for two major mechanisms of transcription coupled repair, nucleotide excision repair and mismatch repair, are not required for somatic hypermutation (9–15; Kim, N., and U. Storb, unpublished data). Although there are possibly other mechanisms of transcription-coupled repair that play a role in somatic hypermutation, this aspect of the model may have to be modified.
To further investigate the molecular mechanism responsible for somatic hypermutation of Ig genes, we created an artificial mutation substrate which has an unusual repeat structure containing seven EcoRV restriction sites alternating with six PvuII sites inserted into the middle of the V region of an Igκ transgene (16). As reported here, the analysis of the restriction enzyme site repeats has revealed that they are hypermutable relative to the flanking Ig sequences and has provided new clues to the mechanism of somatic hypermutation.
Materials and Methods
Production of PEPS4 Transgenic Mice, Immunization, and Isolation of PNAhi, B220+ B Cells.
These methods were performed as previously described (16).
Cloning and EPS Analysis of the PEPS4 Transgene.
The transgenes were cloned from the DNA of B220+ PNAhi FACS®-sorted spleen cells of mice immunized with sheep red blood cells by amplification with PFU DNA polymerase (Stratagene, La Jolla, CA) using JRH2 (5′-GACCACGCTACCTGCAG) as the 3′ primer (see Fig. 1 B). The 5′ primer was Vmu+1 (5′-CCTGGGGGTGCTTATGTTCTAGATCTCTGG). The PCR products were digested with BglII (New England Biolabs Inc., Beverly, MA). The 5′ BglII sites (underlined) were introduced by a single base change from the transgenic sequence. The BglII fragment was cloned into the BamHI site of pUC18. Clones were screened for the presence of mutations within the EPS fragment by amplification with Taq DNA polymerase using EKVK1 (5′-ATTTGATGTCCACCCGTG) and EKVK2 (5′-TCAACTGATAATGAGCCCTC) or Vk8B (5′-GTTTCAGCTCCAGCTTG) and Vk9B (5′-CTCCTCAGCTCCTGATC) for 30 cycles of 94°C 15 s, 60°C 20 s, 72°C 30 s. The PCR product was digested with either EcoRV or PvuII. The samples were run on an 18% acrylamide/5% glycerol gel that was subsequently stained with ethidium bromide. Amplifications were repeated to confirm that the mutations detected were not due to Taq DNA polymerase errors.
Figure 1.

Maps of the EPS transgene and primers. (A) The EPS transgene and sequence of the EPS insert (not to scale). (B) The EPS transgene from the leader to the 5′ end of the J-C intron. Primers used for cloning and sequencing are shown below the transgene. (C) Maps of the EcoRV and PvuII sites within the EPS. Numbers indicate distances between the cut sites.
Sequencing of the EPS and Flanking Transgenic DNA.
To confirm the mutations within the restriction enzyme sites and to identify flanking mutations, the cloned transgenes were sequenced using Sequenase Version 1.0 and the dGTP Nucleotide Kit. For manual sequencing combinations of the following primers (see Fig. 1 B) were used: 1233, 1224, 1212 (New England Biolabs Inc.), EKVK1, EKVK2, EPSAS (5′-ATACACACCCACATCCTCAGCC), Vk7 (5 ′-GAGTGAAGGCTGAGGATGTG), JRH1 (5′-GATGTAGATTCAGGTGC), seq4 (5′-CAGGAGCTGAGGAGATTGTC), and Vk10A (5′-CTGCAGGAGATGGAAAC). A subset of the sequences were obtained by automated sequencing with dye terminators using a ABI Prism model 377 sequencer. The sequencing primers were Vκ234 (5′-TGATGGCCCAGATGATTCCTA) and V κ950(5′-GAGCCCTCTCCATTTTCTCAAGAT). Both strands were sequenced and analyzed with the Macintosh version 3.0 of Sequencher (GeneCodes, Ann Arbor, MI).
Mutability Quotient.
This represents the observed/expected ratio of mutations in dinucleotides (see Fig. 4), or trinucleotides (see Figs. 3 and 4) as tabulated in Tables II and III (columns A/J + Literature), respectively, in Smith et al. (17). The data in Smith et al. (17) were obtained from Jκ and JH 3′ flanking sequences that could not be selected because they do not code for protein sequences.
Figure 4.

Relationship of mutations to hydrogen bonds and mutability of di- and trinucleotides within the EcoRV and PvuII sites. The columns show (black) the number of mutations from Fig. 3 in the EcoRV sites (EB to EG) and PvuII sites (PA to PF); (gray) the hydrogen bonds between the unmutated restriction site NTs G/C, C/G, A/T, and T/A base pairs. Below the graph are shown the mutability quotients (MQ; see Materials and Methods) of somatic mutations in di- and trinucleotides from a large database of normal Ig gene mutations (17). The numbers for dinucleotides are placed between two NTs in the EPS dinucleotide; the numbers for trinucleotides are placed below the central NT in the EPS trinucleotide. Bold numbers: the MQ is greatly higher than 1 (see legend Fig. 3). * Numbers: the MQ is greatly lower than 1. The first and last NT written in parentheses is the most frequent flanking NT. In the calculation of the MQ for given di- or trinucleotides of the first or last NTs, the MQs were averaged for all flanking NTs. The white boxes above the first G in the EcoRV site and the last G in the PvuII site indicate a NT that is overlapping both sites between PA and EB (see Fig. 1).
Figure 3.

Mutations in the EPS and flanks. The original transgene sequence from NT 290–900 is shown in uppercase letters; below it in lower case letters are the observed mutations from all 46 sequences. The restriction sites within the EPS are indicated (E, EcoRV; P, PvuII; the seven Eco and six Pvu sites are labeled A–G and A–F, respectively). 3×, mutations in the EA site were found by restriction analysis in three clones that were lost before DNA sequencing could be performed; since in these clones the PA site was not mutated, the mutations can only be in position 1–5 of EA; the 6 mutations in EA are excluded from Fig. 4, but included in Fig. 6. The symbols below each sequence indicate the mutability quotient (MQ; see Materials and Methods) of the trinucleotide starting at the indicated NT (after reference 17). MQ symbols above the line: high column = very high MQ (≥1.44), low column = high MQ (1.36–1.43); MQ symbols below the line: high column = very low MQ (≤0.60), low column = low MQ (0.68–0.61); no symbol: MQ 0.69–1.35 (i.e., the observed mutability does not differ greatly from the expected mutability).
Results
The EPS Transgene.
Several transgenic lines were produced with the EPS transgene (16). One line, PEPS4, containing three to four copies of the transgene, is the subject of this report. The nucleotide sequence of the 108 base EPS and a diagram of its placement within the Ig κ transgene are shown in Fig. 1 A. Amplification with flanking primers (Fig. 1 B) and complete digestion with either EcoRV or PvuII results in two larger fragments and a ladder of small DNA fragments (Figs. 1 C and 2). Fig. 2 shows an example of the PCR products for 10 transgene clones obtained from PNAhi B lymphocytes (germinal center B cells) of a PEPS4 mouse. Digestion with EcoRV or PvuII results in two larger bands representing the 5′ and 3′ EcoRV and PvuII fragments, respectively (the smaller [5′] and larger [3′] bands on top of the gel in Fig. 2, respectively). In addition, the EcoRV digestion results in fragments of 10, 12, 14, 16, 18, and 20 base pairs in length, and the PvuII digestion gives fragments of 11, 13, 15, 17, and 19 base pairs in length. The smaller fragments (underlined) are not visible on the gel, presumably due to melting of the DNA strands. Loss of one of the restriction sites due to a point mutation will result in the loss of two smaller bands and the appearance of a larger band. For example, clone G8 at the right end of the gel, has both an EcoRV and a PvuII mutation. Changes in the first Eco or Pvu sites at the 5′ end of the cluster lead to a size increase of the smaller fragment at the top of the gel. 76 bases of the EPS lie within restriction sites and thus, mutation of these bases are detectable by digestion. The restriction sites are labeled by letter (Fig. 1 C), thus the EcoRV site mutated in clone G8 is site EF and the PvuII site is site PE (compare Fig. 2 with Fig. 3).
Figure 2.

PAGE of EPS PCR products from 10 clones digested with PvuII or EcoRV. The EPS in B1 and A1 is not mutated. P, PvuII; E, EcoRV.
Somatic Hypermutation in the EPS Transgene.
In a DNA preparation obtained from the PNAhi cells of a hyperimmunized PEPS4 mouse, on average 1 in 19 clones showed a change in an EPS restriction enzyme site (see below). Clones mutated in the EPS were sequenced, including ∼500 NTs of flanking sequence. Fig. 3 summarizes the mutations in the EPS and the flanks. Sequencing confirmed all the mutations that had been observed within the EcoRV or PvuII sites by gel electrophoresis of PCR amplified DNA. In addition, five mutations were found between the restriction sites in the EPS. 27 of 46 clones also contained one or more mutations in the flanking regions. Strikingly, nucleotides in the EPS region on average were mutated seven times more frequently (17 ± 8.6 mutations/ 1,000 NTs) than those in the flanks (2.3 ± 3.5 mutations/ 1,000 NTs). The seven mutations found between the restriction sites in the EPS (Fig. 3) were included in the EPS mutations since these sites were mutated at a frequency more similar to that of the average nucleotide within the restriction sites than those within the flanks. The frequency of mutations in the flanks is similar 5′ and 3′ of the EPS. The hypermutability of the EPS is also apparent in individual transgene copies; 19 of 46 clones have 1–3 mutations in the EPS, but none in the flanks. The highest EPS/flank mutation ratio was >31.3. Only one clone had the same mutation frequency in the flank and EPS (6 mutations/574 NTs in the flank and 1 mutation/96 NTs in the EPS). In all other clones the EPS is mutated at least 2.5× more than the flank. 13 of 46 clones have more than one mutation in the flank. 10 of these have more than one mutation in the EPS.
One mutation in the flanks, position 534, was found to be mutated from G to A in about one of three sequenced clones. This mutation was considered to be a germline mutation. The frequency of its occurrence in one-third of the clones supports the transgene copy number of about four determined by Southern blotting of genomic DNA from the PEPS4 transgenic line (16). The transgene copies with G at position 534 were named copy A, the ones with A were named copy B. The mutation frequency in A and B copies is about the same (not shown).
None of the EPS mutations are likely to be a germline mutation, because in >200 DNA clones obtained from two batches of nonimmune B cells analyzed by restriction enzyme analysis, none were found to be mutated (not shown). These large numbers of unmutated transgene sequences also show that the EPS is not generally hypermutable, but that EPS mutations occur only in the context of somatic hypermutation accompanying immunization.
The data presented here are from one PEPS4 mouse. Similar results were obtained in a small sample of sequences from another PEPS4 mouse, and from a different mouse, PEPS 3 (not shown). As with all transgenes, position effects can be important: some other transgenic lines carrying the same insert in a κ transgene show low or no detectable mutations in the EPS or the flanking sequences (16).
Are the Transgenes without EPS Mutations Mutated in the Flanking V/J Region?
To understand the impact of the artificial EPS sequence on the process of somatic mutation it was important to know if the transgene copies that lacked mutations within the EPS contained mutations in the flanking regions. Sequencing was carried out on 10 clones from PNAhi cells that had shown no EPS mutations by PCR/restriction enzyme analysis. A region of ∼700 bp was sequenced that encompassed the 3′ 250 bp of the L-V intron, the V region (300 bp), the EPS (108 bp), the J region (37 bp), and the first 30 bases of the J-C intron. None of the clones contained any mutations, either in the EPS or in the flanks (not shown).
To assess the probability of the existence of clones that might be highly mutated in the flanks but not in the EPS, the sequencing information from mutated and unmutated clones was submitted to a statistical analysis. Because it was prohibitive to sequence the V/J regions for all clones unmutated in the EPS, and because most of the clones sequenced were not a random subsample, but had EPS mutations, we were unable to use standard statistical methods to test the null hypothesis that the mutation rate was the same in the EPS as in the flank. Therefore, the following simulation was performed. First, a negative binomial model was fit to the observed distribution of the number of mutations within the 96 nucleotides of the EPS using the data from the first 466 clones (24 with EPS mutations, and the rest without). This model may be viewed as a two-step process by which a different mutation rate for each clone is first chosen from an underlying γ distribution (a family of probability distributions taking only positive values), and then mutations on each clone occur randomly according to a Poisson process with the chosen rate. A comparison of the observed distribution of mutations within the 96 NTs of the EPS to that predicted by the negative binomial model suggests that the model provides a reasonable fit to the data.
We then used the fitted model to randomly generate data for 466 hypothetical clones. The number of mutations in the V/J regions were generated according to a Poisson process with the same rate as in the corresponding EPS region. Thus, the model reflects the null hypothesis that the mutation rate for each clone is the same in the EPS as in the flank. As in the actual data, a subsample was then chosen by taking all clones for which there was at least one EPS mutation and a random subsample of 10 of the remaining clones. The difference in the observed mutation rates between the EPS and the flank was then computed using only this subsample. This process was repeated 10,000 times.
In none of the 10,000 repetitions did we observe a difference between the EPS and flank as large as that observed in our data. Thus, we may conclude that the statistical analysis provides strong evidence that the mutation rates in the two regions are different (details available upon request).
Thus, the statistical analysis leads to the conclusion that the EPS is mutated at a higher rate than the flanks and that most of the DNA clones that did not show mutations in the EPS also would not contain mutations in the flanks. This suggests that within this transgene, the EPS is a hypermutable sequence (see below). It appears likely that the transgene copies that did not show any mutations were from cells in which the mutation mechanism was not activated.
Differential Mutability of Different Nucleotides within the EPS Sequence.
There is a clear bias in the nucleotides within each restriction site within the EPS that are targeted for mutation (Fig. 4). The G and C in positions 3 and 4 of the PvuII site and the T and A in positions 3 and 4 of the EcoRV site are the most highly mutable nucleotides. The T and G in positions 5 and 6 of the PvuII site are the least mutated. All sequenced PvuII and EcoRV sites show these differential mutabilities, regardless of their position within the repeats (Fig. 3).
Discussion
The EPS sequence is hypermutable compared with the flanking V and J regions. This is apparent for the individual transgenes analyzed copies as well as for the total sample. Because of its ease of analysis, this artificial mutation substrate will be a useful test substrate in combination with Ig transgenes to define more completely the cis-acting control elements and transacting factors required for somatic mutation. Furthermore, modifications of the EPS will permit a more refined analysis of the DNA sequence and overall structure that is conducive to hypermutability.
The Position of Mutations within the Restriction Sites Reflects Preferences of a MuF.
The six nucleotides within the EcoRV or PvuII sites, respectively, are not mutated at an equivalent frequency. There is a preference for G3 and C4 in the Pvu site and T3 and A4 in the Eco site. In addition, the 3′ terminal nucleotides in the Pvu site are rarely, if at all, mutated. The mutability does not correlate with the strength of hydrogen bonding of the individual nucleotides (Fig. 4): three hydrogen bonds can form in a G/C (C/G) base pair, and only two between A/T (T/A). Furthermore, if the energy of pairing between like restriction sites were the cause for the preferential nucleotide mutability, the mutations would be expected to be symmetrical across each restriction site. Clearly this is not the case (Fig. 4).
Another possibility is that the restriction sites themselves, being palindromic in nature, may be the direct targets for somatic mutation. We believe this to be unlikely because then the nonpalindromic sequences between the restriction sites should not be more highly mutable than the flanking Ig sequences, but they are. Also, the mutations should be symmetrical across each restriction site, but they are not. Similarly, Goyenchea and Milstein (18) found that shortening a palindrome to a length where it cannot form a stem/ loop anymore does not alter the hypermutability of a hotspot.
However, there is a good correlation between the mutability of individual EPS nucleotides and the sequence of known hotspots of somatic hypermutation (17, 19, 20). Smith et al. (17) compiled data of somatic mutations in the 3′ flanks of functionally rearranged Jκ genes. These sequences are outside of the Ig coding region and therefore are not selected based on protein function. Fig. 4 shows a comparison of the EPS mutations with the mutability of dinucleotides and trinucleotides from natural immunoglobulin genes surveyed by Wysocki et al. (17). The two most highly mutated nucleotides in both the Pvu and Eco sites are partners in dinucleotide and trinucleotide combinations that are preferentially mutable in Ig genes (17). Interestingly, the three positions in the trinucleotides AGC and GCT that occur in the center of the PvuII site were found to be mutated in the Wysocki survey in the following order: A < G < C; G < C >> T. These are exactly the relative mutabilities found in the PvuII sites of the EPS. On the other hand, the less mutated residues are within dinucleotides and trinucleotides that are disfavored for somatic hypermutation.
We propose from these comparisons that the frequently and rarely mutated sites within the EPS reflect preferences of a postulated MuF (see below).
The Hypermutability of the EPS Over the Flanking Sequences Appears to Be Related to RNA Secondary Structure.
The most highly mutable trinucleotides in the EcoRV and PvuII sequences are among the most highly mutable trinucleotides in conventional Ig genes (17). To determine whether their concentration was significantly higher within the EPS than in the flanks, we have tallied the mutability of all trinucleotides over 600 NTs of flanks and EPS (Fig. 3 and Table 1). The EPS containing NTs 608–707 have a slightly higher number of trinucleotides with a very high mutability quotient than the flanking sequences 408–507 and 808– 907 (Table 1). They also have a smaller number of trinucleotides with a very low mutability quotient than these two stretches flanking sequences, but they are mutated 6 and 11 times more highly. Although currently it is not possible to quantitatively assess the relative importance of the various hotspots and coldspots for mutability, given these small differences in mutability quotients it seems unlikely that they alone are responsible for the hypermutability of the EPS. Furthermore, there are several regions of very hot trinucleotides in the flanks that are not mutated, but every hotspot is mutated in the EPS (Fig. 3). These findings suggest that besides the primary sequence something inherent in the overall structure of the EPS may induce hypermutability.
Table 1.
Mutability of EPS Transgene
| NT sequence | Mutability Quotient | Mutations | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Very low (≤0.60) | Total low (≤0.68) | Total high (≥1.36) | Very high (≥1.44) | |||||||
| 308–407 | 30 | 37 | 32 | 20 | 22 | |||||
| 408–507 | 31 | 40 | 28 | 25 | 12 | |||||
| 508–607 | 37 | 50 | 14 | 11 | 7 | |||||
| 608–707 | 26 | 26 | 30 | 30 | 78 | |||||
| 708–807 | 29 | 32 | 22 | 17 | 8 | |||||
| 808–907 | 33 | 37 | 25 | 23 | 7 | |||||
The first column shows the 100 NTs analyzed in each row (see Fig. 3). Columns 2–5 show the number of trinucleotides in each sequence that have the mutability quotient (see Materials and Methods) shown at the top of the column (after Table III in reference 17). In bold are the data for the 100 NTs that include the EPS.
It appeared possible that the repeated restriction sites that can form high stability RNA stems (and DNA stems, see below) may play a role. We have used the Mfold program (21) as adapted to the Wisconsin Package to scan the predicted structures for the nascent RNA that would be synthesized across the EPS and sequenced flanks. A reiterative application of Mfold (Martin, T.E., and D. Wang, unpublished) can conveniently scan the predicted folding energies for various RNA sequence windows at chosen NT steps across the sequences, possibly approximating the way the transcription machinery would encounter nascent RNA structures. Determination of the folding energies around the predicted energy minima (largest values of −ΔG) for windows of 20, 30, 40, 50, 60, 70, 80, 90, and 100 NTs suggested that 50 NTs gave favorable signal-to-noise values (not shown). The data shown in Fig. 6 are derived from scans using a 50 NT window and 5 NT steps (an example structure is shown in Fig. 5).
Figure 6.
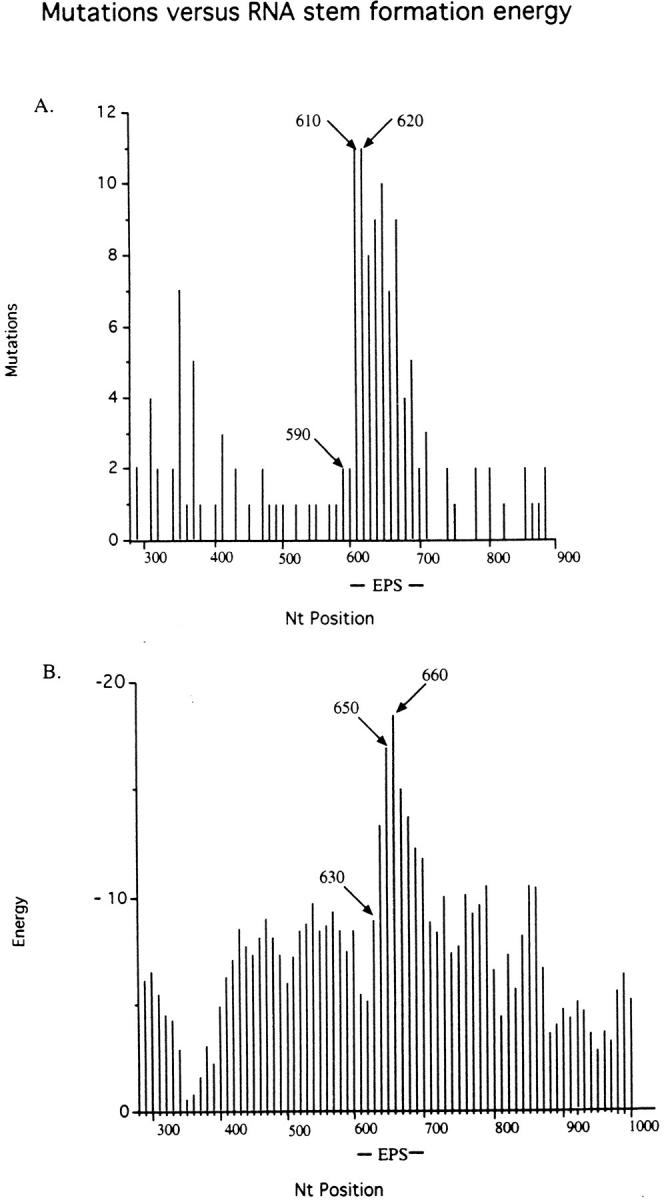
Mutations and RNA stem formation energies. (A) Mutations. NTs 290–900 from the start of the leader peptide sequence are shown (data are shown in Fig. 3). Numbers of point mutations in intervals of 10 nucleotides were summed. NTs 590 and 610/620 indicate the start and peak of the EPS related hypermutability, respectively. The EPS comprises nucleotides 608–703. (B) Energy. An example of a predicted RNA secondary structure and its folding energy is shown in Fig. 5. The RNA folding energies (expressed as negative numbers) were determined for the transgene sequence from position 290–900 for a window of 50 nucleotides at 5 nucleotide intervals. For the plot, energies of 10 nt intervals were derived by averaging the energies from three readings of the most 3′ positions within ±5 nucleotides of the plotted position. The start and peak of the EPS-related energy is indicated as 630 and 650/660, respectively.
Figure 5.

One example of the determination of the predicted energy of RNA folding in a 50 NT window from a reiterative scanning of the Mfold program. The nascent RNA is reportedly paired with the transcribed DNA for 2–12 NTs (26), or 9–10 NTs (27). Upstream of this DNA/RNA hybrid a potential RNA double stem of high predicted stability may arrest the elongating RNA polymerase located at the 3′ end of the nascent RNA (see Fig. 7).
Clearly, there is a strong increase in the predicted stem formation (as large negative energies) within the EPS compared with the flanks. Interestingly, the rise and peak in the predicted stabilities of RNA secondary structures is shifted 3′ by ∼40 nucleotides relative to the rise and peak of the mutations. DNA stem-loop structures have been proposed to be targets for somatic mutation (20, 22). From our data we conclude, instead, that stem-loops act as structures that cause RNA polymerase pausing (see below).
A Model for Somatic Hypermutation.
Previous work has shown that somatic hypermutation is linked to transcription initiation (7). When the Ig promoter upstream of the V region was duplicated upstream of the C region, mutations were also observed over the C region, but not between V and C. The frequency of mutations was similar in the V and C regions, as were the numbers of transcripts originating from the upstream and downstream promoters. Based on these findings we have proposed that a MuF associates with the RNA polymerase and during transcription elongation causes mutations (7). How the mutations are introduced is not known; however, the data presented here have given new clues for the mutation mechanism. Two major predictions for somatic hypermutation can be proposed from the results reported in this paper. First, the exact nucleotide changes depend on the nucleotide preferences of the MuF that is specific for the somatic hypermutation of Ig genes. Second, the overall region where point mutations occur may be determined by the secondary structure of the nascent RNA (or perhaps DNA). These findings have led to the model shown in Fig. 7.
Figure 7.

Model of somatic hypermutation of Ig genes. MuF, mutator factor; E, Ig enhancer; IEBP, Ig enhancer binding proteins; pol, RNA polymerase II; exo, DNA exonuclease; x, point mutation; Fen1, flap endonuclease 1.
The postulated MuF associates with RNA polymerase II (pol) that is initiating transcription at an Ig gene promoter (Fig. 7 a). MuF binding to pol is only possible when certain (unknown) enhancer binding proteins have interacted with the basal transcription machinery (23). During transcript elongation MuF travels with pol (Fig. 7 b). If pol encounters a block to transcription (shown is a hairpin in the nascent transcript), it pauses (Fig. 7 c). The conformation of the polymerase changes, resulting in the transfer of MuF to the DNA (Fig. 7 d).
How the MuF then induces mutations is rather speculative, but a testable possibility is that MuF binds to double stranded DNA upstream of pol (Fig. 7 e). MuF causes a nick in the nontranscribed strand, it remains associated with the newly created 5′ end and also with one or several nucleotides on the transcribed strand (Fig. 7 f). Because of the bound MuF, the single stranded ends cannot be ligated. Exonuclease trims back the single stranded 3′ end (Fig. 7 g). A DNA polymerase fills in the gap creating mutation(s) (Fig. 7, x) opposite the MuF associated base(s) (Fig. 7 h). The DNA polymerase continues past the MuF bound residues, creating a 5′ flap of the nontranscribed strand (Fig. 7 i). The flap is cut by endonuclease Fen1 (DNase IV; references 24, 25). The free DNA ends are ligated. MuF has been removed with the flap (Fig. 7 j).
Most of the proposed steps are hypothetical, but all are compatible with previous observations and the results of the present study. The Ig promoter can be replaced with certain other promoters, but the Ig enhancer appears to be required for high mutability of Ig genes (5, 6). The postulated MuF would only be produced during the short window in the life of the B cell when somatic hypermutation is ongoing. It can only bind to pol initiating at a promoter that is activated by transcription factors bound to the Ig enhancer, or bound to motifs shared with the Ig enhancer (23), not randomly with pol or DNA.
During transcript elongation, pol may stop transcription without releasing the transcript (26). Such pausing is presumably due to secondary structure of the template or hairpins formed in the nascent RNA (26). The EPS or its nascent transcript is an array of multiple repeats that can form very stable stem-loops (Figs. 5 and 6). Similar, but less frequent repeats are present in natural Ig genes. Transcription through a region that is transcribed into RNA that can form hairpins is prone to becoming stalled. The pause inducing structure is most likely an RNA hairpin, at least in the EPS, rather than a DNA hairpin. The DNA transcription bubble comprises only ∼15 single stranded NTs (27). In order for two identical restriction sites in the EPS DNA to hybridize, 16 to 26 nucleotides are required to be single stranded in the transcription bubble. In natural Ig genes pausing may be due to either DNA or RNA secondary structure (26). During pausing the conformation of pol appears to change, presumably under the influence of elongation factors (28, 29). We propose that only when transferred by a pol with such an altered conformation can MuF bind to DNA. Since the profile of mutagenesis is shifted by ∼40 nucleotides 5′ with respect to the pattern of predicted RNA secondary structure stabilities (Fig. 6, A and B) the MuF appears to be deposited on the DNA ∼40 nucleotides upstream of the paused pol.
We propose that the mutations may arise due to errors made during repolymerization of the excised single stranded region when the polymerase is copying a base to which the MuF is bound on the template strand. There appear to be clear target preferences, e.g., for certain NTs within di- or trinucleotides (Fig. 4). These preferences could represent sites at which the MuF prefers to nick the DNA, sites at which the MuF prefers to bind on the opposite strand, thus modifying the template for the DNA polymerase, or sites which when combined with the MuF make the DNA polymerase most error prone. These questions need to be addressed in future studies.
At the end of the reaction the MuF would be removed with the short DNA segment. We suggest that the MuF would not be able to reload on the RNA polymerase in the elongation mode, as it can only bind to pol that is initiating transcription. This mode of MuF/pol interaction is suggested by the loading of mRNA cleavage/polyadenylation factors to initiation competent pol (30). The requirement for loading MuF to an initiating pol, coupled with a high chance for pausing within the first 1 kb or so of transcribed DNA, explains the extent of somatic hypermutations over only 1–2 kb of the 5′ region of the Ig gene with sparing of the 3′ region (reviewed in reference 3).
In the PEPS4 transgene (and others described in reference 16 the EPS sequence is inserted in the middle of the V region. Presumably, insertion at another position may increase or decrease the differential mutability between EPS and flanks, depending on the secondary structures formed in the nascent RNA. These questions can be addressed by further studies with the artificial substrate EPS or modifications thereof.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grant GM38649. E. Klotz was the recipient of NIH training grants GM07183 and AI07090. The DNA sequencing, biostatistics, and transgenic facilities are supported by the University of Chicago Cancer Research Center. Supported by NIH Cancer Center support grant P30(A 14599).
Abbreviations used in this paper
- C
constant
- D
diversity
- EPS
alternating EcoRV and PvuII restriction enzyme sites
- J
joining
- MuF
mutator factor
- pol
RNA, polymerase II
- V
variable
Footnotes
We are greatly indebted to Phil Schumm for the statistical analysis. We are grateful to Peter Engler, Nancy Michael, Larry Loeb, David Roth, and Kevin Struhl for critical reading of the manuscript.
Dr. Klotz's current address is NCI, NIH, Bethesda, MD 20892. Dr. Hackett's current address is Abbott Laboratories, North Chicago, IL 60064.
References
- 1.Kelsoe G. The germinal center reaction. Immunol Today. 1995;16:324–326. doi: 10.1016/0167-5699(95)80146-4. [DOI] [PubMed] [Google Scholar]
- 2.MacLennan I. Germinal centers. Annu Rev Immunol. 1994;112:117–139. doi: 10.1146/annurev.iy.12.040194.001001. [DOI] [PubMed] [Google Scholar]
- 3.Storb U. The molecular basis of somatic hypermutation of immunoglobulin genes. Curr Opin Immunol. 1996;8:206–214. doi: 10.1016/s0952-7915(96)80059-8. [DOI] [PubMed] [Google Scholar]
- 4.Hackett J, Rogerson B, O'Brien R, Storb U. Analysis of somatic mutations in κ transgenes. J Exp Med. 1990;172:131–137. doi: 10.1084/jem.172.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Betz A, Milstein C, Gonzalez-Fernandes R, Pannell R, Larson T, Neuberger M. Elements regulating somatic hypermutation of an immunoglobulin κ gene: critical role for the intron enhancer/matrix attachment region. Cell. 1994;77:239–248. doi: 10.1016/0092-8674(94)90316-6. [DOI] [PubMed] [Google Scholar]
- 6.Tumas-Brundage K, Manser T. The transcriptional promoter regulates hypermutation of the antibody heavy chain locus. J Exp Med. 1997;185:239–250. doi: 10.1084/jem.185.2.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peters A, Storb U. Somatic hypermutation of immunoglobulin genes is linked to transcription initiation. Immunity. 1996;4:57–65. doi: 10.1016/s1074-7613(00)80298-8. [DOI] [PubMed] [Google Scholar]
- 8.Storb U, Peters A, Klotz E, Kim N, Shen HM, Kage K, Rogerson B. Somatic hypermutation of immunoglobulin genes is linked to transcription. Curr Topics Microbiol Immunol. 1998;229:11–19. doi: 10.1007/978-3-642-71984-4_2. [DOI] [PubMed] [Google Scholar]
- 9.Wagner S, Elvin J, Norris P, McGregor J, Neuberger M. Somatic hypermutation of Ig genes in patients with xeroderma pigmentosum (XP-D) Int Immunol. 1996;8:701–705. doi: 10.1093/intimm/8.5.701. [DOI] [PubMed] [Google Scholar]
- 10.Shen HM, Cheo DL, Friedberg E, Storb U. The inactivation of the XP-C gene does not affect somatic hypermutation or class switch recombination of immunoglobulin genes. Mol Immunol. 1997;34:527–533. doi: 10.1016/s0161-5890(97)00064-3. [DOI] [PubMed] [Google Scholar]
- 11.Kim N, Kage K, Matsuda F, Lefranc M-P, Storb U. B lymphocytes of xeroderma pigmentosum or Cockayne syndrome patients with inherited defects in nucleotide excision repair are fully capable of somatic hypermutation of immunoglobulin genes. J Exp Med. 1997;186:413–419. doi: 10.1084/jem.186.3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacobs H, Fukita Y, van der Horst G, de Boer J, Weeda G, Essers J, de Wind N, Engelward B, Samson L, Verbeek S, et al. Hypermutation of immunoglobulin genes in memory B cells of DNA repair-deficient mice. J Exp Med. 1998;187:1735–1743. doi: 10.1084/jem.187.11.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Phung Q, Winter D, Cranston A, Tarone R, Bohr W, Fishel R, Gearhart P. Increased hypermutation at G and C nucleotides in immunoglobulin variable genes from mice deficient for the MSH2 mismatch repair protein. J Exp Med. 1998;187:1745–1751. doi: 10.1084/jem.187.11.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Winter D, Phung Q, Umar A, Baker S, Tarone R, Tanaka R, Liskay R, Kunkel T, Bohr V, Gearhart P. Altered spectra of hypermutation in antibodies from mice deficient for the DNA mismatch repair protein PMS2. Proc Natl Acad Sci USA. 1998;95:6953–6958. doi: 10.1073/pnas.95.12.6953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim N, Storb U. The role of DNA repair in somatic hypermutation of immunoglobulin genes. J Exp Med. 1998;187:1729–1733. doi: 10.1084/jem.187.11.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klotz E, Hackett JJ, Storb U. Somatic hypermutation of an artificial test substrate within an Ig kappa transgene. J Immunol. 1998;161:782–790. [PubMed] [Google Scholar]
- 17.Smith D, Creadon G, Jena P, Portanova J, Kotzin B, Wysocki L. Di- and trinucleotide target preferences of somatic mutagenesis in normal and autoreactive B cells. J Immunol. 1996;156:2642–2652. [PubMed] [Google Scholar]
- 18.Goyenchea B, Milstein C. Modifying the sequence of an Immunoglobulin V-gene alters the resulting pattern of hypermutation. Proc Natl Acad Sci USA. 1996;93:13979–13984. doi: 10.1073/pnas.93.24.13979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doerner T, Brezinschek H-P, Brezinschek R, Foster S, Domiati-Saad R, Lipsky P. Analysis of the frequency and pattern of somatic mutations within nonproductively rearranged human variable heavy chain genes. J Immunol. 1997;158:2779–2789. [PubMed] [Google Scholar]
- 20.Rogozin I, Kolchanov N. Somatic hypermutagenesis in immunoglobulin genes. II. Influence of neighboring base sequences on mutagenesis. Biochim Biophys Acta. 1992;1171:11–18. doi: 10.1016/0167-4781(92)90134-l. [DOI] [PubMed] [Google Scholar]
- 21.Zucker M. On finding all suboptimal folding of an RNA molecule. Science. 1989;244:48–52. doi: 10.1126/science.2468181. [DOI] [PubMed] [Google Scholar]
- 22.Golding G, Gearhart P, Glockman B. Patterns of somatic mutation in immunoglobulin variable genes. Genetics. 1987;115:169–176. doi: 10.1093/genetics/115.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen H, Peters A, Baron B, Zhu X, Storb U. Mutation of the BCL6 gene in normal B cells by the process of somatic hypermutation of immunoglobulin genes. Science. 1998;280:1750–1752. doi: 10.1126/science.280.5370.1750. [DOI] [PubMed] [Google Scholar]
- 24.Harrington J, Lieber M. The characterization of a mammalian DNA structure-specific endonuclease. EMBO (Eur Mol Biol Organ) J. 1994;13:1235–1246. doi: 10.1002/j.1460-2075.1994.tb06373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klungland A, Lindahl T. Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1) EMBO (Eur Mol Biol Organ) J. 1997;16:3341–3348. doi: 10.1093/emboj/16.11.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uptain S, Kane C, Chamberlin M. Basic mechanisms of transcript elongation and its regulation. Annu Rev Biochem. 1997;66:117–172. doi: 10.1146/annurev.biochem.66.1.117. [DOI] [PubMed] [Google Scholar]
- 27.Nudler E, Mustaev A, Lukhtanov E, Goldfarb A. The RNA-DNA hybrid maintains the register of transcription by preventing backtracking of RNA polymerase. Cell. 1997;89:33–41. doi: 10.1016/s0092-8674(00)80180-4. [DOI] [PubMed] [Google Scholar]
- 28.Kane, C.M. 1994. Transcript elongation and gene regulation in eukaryotes. In Transcription: Mechanisms and Regulation. R.C. Conaway and J.W. Conaway, editor. Raven Press, Ltd., New York, NY. 279–296.
- 29.Reines, D. 1994. Nascent RNA cleavage by transcription elongation complexes. In Transcription: Mechanisms and Regulation. R.C. Conaway and J.W. Conaway, editor. Raven Press, Ltd., New York, NY. 263–278.
- 30.McCracken S, Fong N, Yankulov K, Ballantyne S, Pan G, Greenblatt J, Patterson S, Wickens M, Bentley D. The COOH-terminal domain of RNA polymerase II couples mRNA processing to transcription. Nature. 1997;385:357–361. doi: 10.1038/385357a0. [DOI] [PubMed] [Google Scholar]