

Abstract
The multidrug resistance protein 1 (MRP1) gene encodes a transporter protein that helps to protect cells against xenobiotics. Elevated levels of MRP1 in tumor cells can result in active extrusion of a wide range of (anticancer) drugs with different cellular targets, a phenomenon called multidrug resistance (MDR). To explore the protective function of the mouse mrp1 protein during drug treatment, we investigated the toxicity caused by the anticancer drug etoposide-phosphate (ETOPOPHOS) in mice lacking the mrp1 gene (mrp1 −/− mice). We show here that the lack of mrp1 protein results in increased etoposide-induced damage to the mucosa of the oropharyngeal cavity and to the seminiferous tubules of the testis. The high concentrations of mrp1 that we find in the basal layers of the oropharyngeal mucosa and in the basal membrane of the Sertoli cells in the testis apparently protect wild-type mice against this tissue damage. We also find drug-induced polyuria in mrp1 −/− mice, which correlates with the presence of mrp1 protein in the urinary collecting tubules, the major site of kidney water reabsorption. Our results indicate that specific inhibitors of MRP1 used to reverse MDR, in combination with carcinostatic drugs transported by MRP1, might lead to drug-induced mucositis, (temporary) infertility, and diabetes insipidus.
Keywords: etoposide, multidrug resistance protein 1, mucositis, polyuria, blood–testis barrier
Multidrug resistance (MDR)1 of tumor cells is an obstacle to successful anticancer treatment. In human cancer cells, MDR can be caused by the enhanced drug efflux mediated by two members of the ATP-binding cassette (ABC) superfamily of transporter proteins, the MDR gene 1 (MDR1) P-glycoprotein (Pgp; reference 1) and MDR protein 1 (MRP1 [2–6]). Pgp can transport hydrophobic amphipathic compounds like anthracyclines, Vinca alkaloids, epipodophyllotoxins, and taxanes in unmodified form. MRP1 can transport negatively charged conjugated hydrophilic compounds with a large hydrophobic moiety as well as hydrophobic amphipathic compounds. Both transporter genes were identified because of their overexpression in MDR tumor cell lines, and it is likely that both proteins contribute to the (intrinsic or acquired) resistance against drugs used in cancer therapy.
MRP1 can act as a glucuronide- or glutathione S-conjugate (GS-X) pump or multispecific organic anion transporter (MOAT [7–13]). Five other members of the MRP family have been found thus far (14): canalicular (c)MOAT or MRP2 (15, 16), MRP3, MRP4, MRP5, and MRP6. It is not yet known whether MRP2–6 play a role in MDR.
To elucidate the physiological functions of the MDR-conferring proteins, mutant mice have been generated lacking Pgp (17, 18) or mrp1 protein (19, 20). These knockout mice provide a useful pharmacological tool to study the effects of complete blockade of these transporters. In addition, these animals are proving helpful in testing the specificity and effectiveness of MDR reversal agents.
Analyses of mice lacking mdr1a Pgp established an important role for this Pgp in the blood–brain barrier, as it protects the brain against entry of xenobiotics. This transporter is also important in the intestine, where it actively excretes xenobiotics from the bloodstream into the intestinal lumen, and limits the entry of these Pgp substrates from the intestinal lumen (17, 18, 21). Mice homozygous for an mrp1 null allele, mrp1 −/−, have an impaired response to an arachidonic acid–induced inflammatory stimulus. This is most likely due to a decreased secretion of leukotriene C4 (LTC4) from mast cells, macrophages, and eosinophilic and basophilic granulocytes (19). We showed that mrp1 actively transports GS-X substrates across the mouse erythrocyte membrane, and mediates drug (e.g., etoposide, vincristine) resistance to bone marrow–derived mast cells in vitro. Furthermore, we and others showed that the mrp1 −/− mice are hypersensitive to the anticancer drug etoposide, resulting in loss of body weight and mortality (19, 20). However, the reason for this hypersensitivity remained unknown.
We show here that mrp1 is present in the basal epithelial layer of the tongue and mucosal layer of the cheek, in the basal plasma membrane of the Sertoli cells, and in the kidney urinary collecting duct cells. We also demonstrate that the etoposide treatment of mrp1 −/− mice leads to damage of the mucosal layer of the tongue and cheek, and to inhibition of spermatogenesis. Our data indicate that an acquired diabetes insipidus and oropharyngeal toxicity are important elements in the etoposide hypersensitivity of the mrp1 −/− mice, and might become clinically relevant if etoposide is combined with an MRP1 blocker.
Materials and Methods
Animals.
The animals used were mrp1-deficient (mrp1 −/−) mice, generated by gene targeting in embryonic stem cells as described by Wijnholds et al. (19). Control and mutant mice were on the genetic background (129/Ola)/FVB (50%/50%). All animals were housed in constant temperature rooms with a 12-h light/12-h dark cycle, and were fed a pelleted chow diet (Hope Farms B.V., Woerden, The Netherlands) and were given acidified water ad libitum. Mouse handling and experimental procedures were conducted in accordance with institutional guidelines for animal care and use.
Drug Sensitivity.
Etoposide-phosphate (100 mg effective etoposide, ETOPOPHOS®; Bristol Myers-Squibb Pharmaceuticals, Princeton, NJ) was dissolved in sterile 0.9% NaCl to obtain a stock solution of 20 mg/ml. Concentrations were adjusted to inject intravenously 5–7 μl/g body wt (40–140 mg/kg) in a tail vein of male mice (11–13 wk old) lightly anesthetized with diethyl ether. Mice were weighed and observed daily for a period of 2 wk. Lethal toxicity occurred between days 2 and 13 after injection. In experiments where urine and feces were separately collected, mice were kept in type M/1 stainless steel metabolic cages (Ruco Metaalindustrie Nederland B.V., Valkenswaard, The Netherlands), and specimens were collected during time intervals of 0–2, 2–4, and 4–6 d.
Histological Analysis.
Tissues were obtained by dissection of mice (n = 5 per genotype) after lethal anesthesia with diethyl ether, fixed in ethanol/acetic acid/formaldehyde in 0.9% NaCl (40:5:10:45, vol/vol/vol/vol), embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin.
Immunohistochemical Analysis.
Cryostat sections were cut 4 μm thick, air-dried overnight, and fixed in acetone for 10 min at room temperature. The slides were incubated with normal rabbit serum for 10 min and incubated with the mAb MRPr1 (5 μg/ml) for 1 h at room temperature (22). All reagents were diluted in PBS containing 1% (wt/vol) BSA (PBS/BSA). Sections were stained by the biotin-streptavidin immunoperoxidase method, using biotinylated rabbit anti–rat IgG (1:100; Dakopatts A/S, Glostrup, Denmark) and avidin/biotin complex (1:200; Dakopatts A/S). Bound peroxidase was developed with 0.02% H2O2 and 0.5 g/liter 3,3′-diaminobenzidine tetrahydrochloride (Sigma Chemical Co., St. Louis, MO). All slides were counterstained with hematoxylin and mounted. Negative controls included both the omission of the first step and substitution of the primary antibody by isotype-matched nonspecific control antibodies.
Clinical Chemistry and Hematology.
Hemoglobin, hematocrit, and erythrocyte, leucocyte, and thrombocyte counts were determined in heparinized blood on a Coulter counter (Coulter Corp., Miami, FL). Total protein, sodium, creatinine, urea, and glucose concentrations in plasma, and urinary glucose, creatinine, sodium, and potassium concentrations were determined on an analyzer according to the instructions of the manufacturer (model 911; Hitachi Instruments, Inc., San Jose, CA).
Measurement of Renal Function.
Glomerular filtration rate (GFR) was calculated as the clearance of creatinine according to the formula, GFR = (UCr × Uvol)/PCr, where UCr and PCr are the urine and plasma creatinine concentrations, respectively, and Uvol is urine volume. Fractional excretion of sodium (FeNa) was calculated according to the formula, FeNa = (UNa × PCr) × 100/ (PNa × UCr), where UNa and PNa are the urine and plasma sodium concentrations, respectively. The renal failure index (RFI) was calculated according to the formula, RFI = UNa/(UCr/PCr). Also calculated were blood urea nitrogen (BUN)/PCr and UCr/PCr.
Statistical Evaluation.
The results are presented as means ± SE. The difference between groups was evaluated with the Student's t test.
Results
Sensitivity to ETOPOPHOS.
It has been shown that mice lacking a functional mrp1 gene are hypersensitive to etoposide, resulting in loss of body weight and lethality (19, 20). The cause of the loss of body weight and lethality was unclear, since we did not find differences in etoposide tissue distribution, the area under the plasma concentration– time curve for the drug, or in hematological parameters, or in biliary, fecal, or urinary excretion of radiolabeled etoposide (19). The mrp1 −/− mice are also hypersensitive to the pro-drug ETOPOPHOS (Table 1; references 19 and 20). After 7 d, the wild-type mice retained 91 ± 2% of body weight (n = 15; 30.0 ± 0.6 g at day 0), whereas the mutant mice that survived retained 74 ± 2% (P < 0.01) of body weight (n = 14; 29.8 ± 0.4 g at day 0), after a single intravenous dose of 100 mg/kg of ETOPOPHOS. The symptoms observed in the mutant mice after drug treatment included a partial closure of the eyes and a soft testis, but no clear gross abnormalities were observed in the appearance or size of the liver, kidney, intestinal tract, spleen, lung, heart, brain, or other tissues. Mice that survived for 7 d (14 out of 15 mice) were more active than wild-type mice, showed a mucus substance around the mouth, and showed alopecia specifically around the mouth, chin, and forelegs, possibly due to the observed abnormally active polishing. Not before 4 d (19, 20) but after 7 d of drug treatment, the number of white blood cells dropped to a significantly lower level in drug-treated wild-type as well as mrp1 −/− mice compared with drug-naive controls, whereas the number of thrombocytes increased (see Table 2). However, in microscopic analysis of tissues, we did not find symptoms of gross infection or occult bleeding. The mucus excretion from the mouth indicated a possible defect in the oropharyngeal cavity. To determine the cause of the loss of body weight after drug treatment, we examined parameters for dehydration, food uptake, and fecal and urinary excretion.
Table 1.
ETOPOPHOS Toxicity in Male Wild-type and mrp1-deficient Mice (11–13 wk old)
| ETOPOPHOS dose | Survival of mice | |||
|---|---|---|---|---|
| Wild-type | mrp1 −/− | |||
| (mg/kg) | ||||
| 40 | — | 3/3 | ||
| 60 | 3/3 | 6/6 | ||
| 80 | 3/3 | 11/11 | ||
| 100 | 3/3 | 1/3 | ||
| 110 | — | 0/3 | ||
| 115 | — | 0/3 | ||
| 120 | 8/8 | 0/3 | ||
| 140 | 0/3 | 0/3 | ||
The number of surviving animals per group at each intravenous ETOPOPHOS dose is listed. mrp1 −/− mice show increased sensitivity to ETOPOPHOS; P < 0.01.
Table 2.
Plasma and Urinary Variables in ETOPOPHOS-treated and Naive Mice
| Drug-naive | Drug-treated | |||||||
|---|---|---|---|---|---|---|---|---|
| Wild-type | mrp1 −/− | Wild-type | mrp1 −/− | |||||
| Hematocrit | 0.45 ± 0.01 | 0.43 ± 0.01 | 0.40 ± 0.02 | 0.42 ± 0.03 | ||||
| Leukocytes, 109/liter | 9.3 ± 0.5** | 9.5 ± 0.7‡‡ | 5.8 ± 0.6 | 2.5 ± 0.4§§ | ||||
| Thrombocytes, 109/liter | 48 ± 5* | 165 ± 76‡ | 369 ± 142 | 696 ± 148 | ||||
| Plasma values | ||||||||
| Protein, g/liter | 57 ± 1 | 56 ± 1‡ | 55 ± 2 | 71 ± 9§ | ||||
| Creatinine, μmol/liter | 33 ± 2 | 37 ± 1‡ | 43 ± 4 | 49 ± 4 | ||||
| Sodium, mmol/liter | 138 ± 2* | 139 ± 1‡ | 147 ± 4 | 150 ± 4 | ||||
| Urea, mmol/liter | 8.9 ± 0.4 | 9.7 ± 0.5‡ | 11.0 ± 1.2 | 15.7 ± 2.0 | ||||
| Glucose, mmol/liter | 4.7 ± 0.4 | 4.9 ± 0.5 | 4.0 ± 1.2 | 4.2 ± 0.4 | ||||
| Urine values | ||||||||
| Vol (d1–2), ml/48 h | 2.4 ± 0.3** | 2.7 ± 0.5‡‡ ‡ | 4.4 ± 0.5 | 7.2 ± 0.6§ | ||||
| Vol (d3–4), ml/48 h | 3.2 ± 0.5 | 3.7 ± 0.7‡ | 1.8 ± 0.4 | 6.6 ± 0.9§§ | ||||
| Vol (d5–6), ml/48 h | 3.1 ± 0.5 | 3.8 ± 0.6‡‡ | 2.3 ± 0.5 | 7.5 ± 0.4§§ § | ||||
| Creatinine, mmol/liter | 7.8 ± 1.0 | 6.5 ± 0.8‡‡ | 8.6 ± 1.4 | 2.3 ± 0.4§§ | ||||
| μmol/48 h | 21.0 ± 1.8 | 21.8 ± 2.0 | 16.2 ± 2.4 | 16.8 ± 2.0 | ||||
| Glucose, mmol/liter | 7.0 ± 1.2 | 6.0 ± 1.7‡ | 10.2 ± 3.1 | 1.0 ± 0.5§ | ||||
| μmol/48 h | 17.9 ± 1.7 | 18.3 ± 3.5‡ | 17.1 ± 2.8 | 6.6 ± 3.0§ | ||||
| Sodium, mmol/liter | 241 ± 26 | 176 ± 20‡‡ | 167 ± 40 | 56 ± 9§ | ||||
| μmol/48 h | 663 ± 62** | 602 ± 68 | 267 ± 60 | 387 ± 48 | ||||
| Potassium, mmol/liter | 358 ± 31 | 287 ± 32‡‡ | 294 ± 52 | 96 ± 16§§ | ||||
| μmol/48 h | 1018 ± 111* | 987 ± 106 | 547 ± 75 | 685 ± 72 | ||||
| FeNa, % | 0.74 ± 0.03* | 0.73 ± 0.03 | 0.46 ± 0.09§ | 0.81 ± 0.07 | ||||
| RFI, mmol/liter | 1.0 ± 0.0* | 1.0 ± 0.0 | 0.7 ± 0.1§ | 1.2 ± 0.1 | ||||
| GFR, ml/48 h | 655 ± 65* | 598 ± 65 | 399 ± 62 | 365 ± 90 | ||||
| UCr/PCr | 242 ± 31 | 175 ± 19‡‡ | 228 ± 50 | 52 ± 16§ | ||||
| BUN/PCr | 273 ± 9 | 262 ± 12 | 258 ± 8 | 318 ± 30 | ||||
Plasma values 7 d after ETOPOPHOS administration (100 mg/kg). Urinary values are for the day 5–6 sample. Statistical significance is indicated as follows:
drug-treated versus drug-naive wild-type;
drug-treated versus drug-naive mrp1 −/−;
drug-treated mrp1 −/− versus drug-treated wild-type mice. A single symbol (e.g.,
*) denotes P < 0.05; a double symbol (e.g.,
) P < 0.01; a triple symbol (e.g.,
) P < 0.001. In all comparisons, n = 6. BUN, Blood urea nitrogen; d, day.
Drug-induced Hypotonic Polyuria in ETOPOPHOS-treated mrp1− /− Mice.
The food uptake and fecal excretion during 7 d were not significantly different between drug-treated mutant and wild-type mice (food uptake: 22 ± 3 and 19 ± 3 g; fecal excretion: 11 ± 1 and 8 ± 1 g, mutant versus wild-type mice, respectively; n = 6). However, drug-treated mice tended to become dehydrated, as indicated by the data presented in Table 2. Plasma sodium, creatinine, and urea were modestly increased in drug-treated mice, and this increase was significant at P < 0.05 in the mrp1 −/− mice. Plasma protein was increased as well in the knockout mice. The hematocrit and mean cell volumes did not differ significantly between drug-treated and -naive animals.
Without drug treatment, the total 48-h urine volumes were similar in wild-type and mutant mice. After drug treatment, the mutant mice excreted a markedly larger urine volume than the wild-type mice, indicative of a drug-induced acquired polyuria. Values for urinary sodium, potassium, creatinine, and glucose in 48-h urine collected on days 5 and 6 were normal and similar in drug-naive wild-type and mutant mice. Compared with the drug-treated wild-type and drug-naive mice, the concentrations of sodium, potassium, and creatinine in the urine (mmol/ liter) of the drug-treated mutant mice were significantly lower, but the total excretion of these electrolytes (μmol/ 48 h) did not differ significantly, indicative of dilute urine. The drug-treated mutant mice excreted significantly less glucose in the urine, indicating that glucose reabsorption by the mutant kidney was functional. We also tested whether drug-naive old female mice (14 mo of age) had developed a spontaneous polyuria, but no significant differences were detected between wild-type and mutant mice (3.1 ± 0.8 and 3.9 ± 0.6 ml/48 h, respectively). The FeNa, GFR, and RFI (Table 2) indicate a modest drug-induced renal failure in wild-type but not in mutant mice. However, the drug-treated mutant mice acquired hypotonic polyuria.
Oropharyngeal Morphology of ETOPOPHOS-treated mrp1− /− Mice.
To investigate the abnormal mucus production in the mouth, we performed a histological examination on sections through the head. Staining with hematoxylin and eosin revealed that the epithelial layer of the mutant tongue is heavily damaged, with a disappearance of all tongue papillae (Fig. 1, A–D). In addition, the mucosal layer of the cheek is distorted (Fig. 1, E and F). Some mice show skin infections around the mouth, a beginning parodontitis, and infections on the surface of the tongue without signs of deep penetration of bacteria into the tissue. Wild-type mice treated with the drug do not show a changed morphology or disappearance of the papillae or a distortion of the mucosal layer of the cheek, and do not show oropharyngeal bacterial infections. No microscopic abnormalities were observed in the salivary or other excretory glands, the muscular layer of the tongue, the palatum, the eye, the nose area, the brain, or other parts of the head. Also, no abnormalities were observed in the mucosal layer of the esophagus, or in the small or large intestines. However, the stomach was empty in most of the mutant animals, and a regeneration of crypts was occurring specifically around the pylorus (Fig. 1, G and H). These findings show that the mutant mice treated with ETOPOPHOS suffer from oropharyngeal toxicity, and that mrp1 protects drug-sensitive epithelial layers in the oropharyngeal cavity of wild-type mice.
Figure 1.
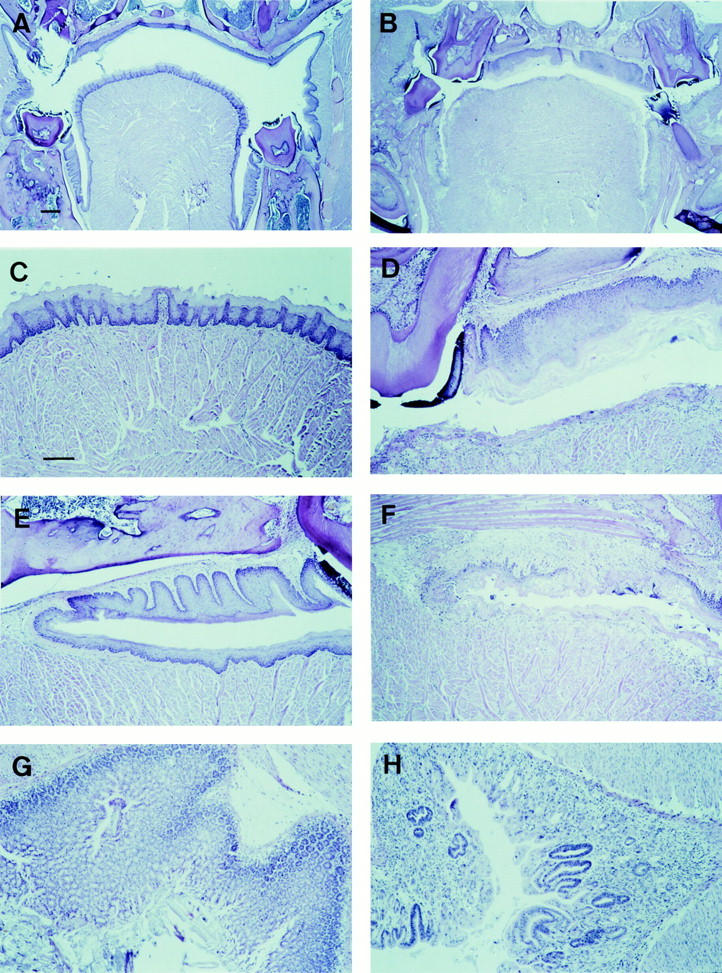
Hematoxylin and eosin–stained paraffin sections of tissues from ETOPOPHOS-treated wild-type and mrp1-deficient mice. Wild-type (A, C, E, and G) and mrp1-deficient (B, D, F, and H) mice 7 d after ETOPOPHOS administration. (A and B) Sections of the oropharyngeal cavity through the tongue, molars, and palatum showing a degeneration of the mucosal layers of the tongue and cheek (B). (C and D) Higher magnification showing at the bottom the tongue and mucosal layer. (E and F) Higher magnification of A and B showing at the bottom the tongue and mucosal layer, and above it the mucosal layer of the cheek. (G and H) Sections through the stomach pyloric region. The dark staining reveals a regeneration of crypts (H). Same magnification in A and B (bar, 200 μm), and in C–H (bar, 100 μm).
Testicular Morphology of ETOPOPHOS-treated mrp1− /− Mice.
We reported previously that mice lacking mrp1 are viable and fertile for at least 1 yr under normal animal facility conditions, with no obvious difference in litter size, indicating that these mice do not show abnormal breeding characteristics. However, the testis of drug-treated mutant mice looked abnormal, and we therefore examined the weight of the testis. Testis from drug-naive wild-type and mutant mice did not differ significantly in weight (187 ± 15 and 204 ± 7 mg in seven mice, respectively). Drug treatment resulted in a lower testis weight for wild-type as well as mutant mice (143 ± 9 and 106 ± 3 mg in six mice, P < 0.05 and P < 0.001, respectively). Weight loss differed significantly between drug-treated mutant and wild-type mice (P < 0.01).
Spermatogenesis is a tightly organized differentiation process in the seminiferous tubules that occurs according to a cycle of subsequent stages, with specific cell associations at different cross sections of the testicular tubules (the spermatogenic cycle). Histological sections stained with hematoxylin and eosin indicated that the testis of drug-treated mutant mice showed largely distorted spermatogenesis with no signs of meiotic divisions (Fig. 2, A and B), and an increased number of prematurely released round germ cells, including many spermatids, with very few spermatozoa in the epididymis (Fig. 2, C and D). The testis of drug-treated wild-type mice showed a partially distorted spermatogenesis, but meiotic divisions still occurred and the epididymis contained many spermatozoa and several prematurely released round germ cells. These findings indicate that mrp1 could play a direct protective role in the blood–testis barrier.
Figure 2.
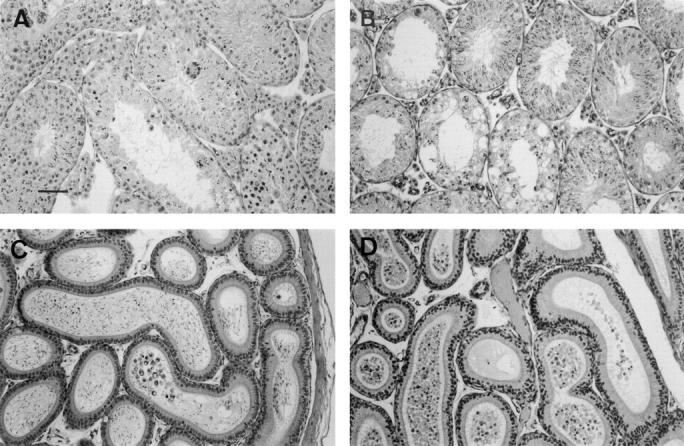
Hematoxylin and eosin–stained paraffin sections of testis and epididymis from ETOPOPHOS-treated wild-type and mrp1-deficient mice. Wild-type (A and C) and mrp1-deficient (B and D) mice 7 d after ETOPOPHOS administration. (A and B) Sections of the testis showing vacuoles and disrupted spermatogenesis (B). (C and D) Sections of the epididymis showing reduced numbers of intact spermatids in the lumen (D). Bar, 100 μm.
Further histological examination indicated that the thymus was degenerated upon drug treatment in both genotypes, and that the mutant bone marrow was rich in cells and showed in a few cases megakaryocytosis. The liver, kidney, lung, bronchi, heart, pancreas, esophagus, intestinal tract, urinary bladder, adrenal gland, thyroid, bone and cartilage, salivary glands, secondary sex organs, mammary glands, spleen, and lymph nodes showed no significant differences between drug-naive and drug-treated animals.
mrp1 in the Tongue, Testis, and Kidney.
In humans, MRP1 protein is detected in sweat and sebaceous glands of the skin, in the ducts of the salivary glands and pancreas, in epithelia of the esophagus, colon, stomach, lung, and tonsil, in muscular layers of the large intestine, heart, smooth and skeletal muscle, in the cortex of the adrenal glands, in islets of Langerhans of the pancreas, and in Leydig and Sertoli cells of the testis, and weak immunoreactivity has been reported for the tubules of the kidney (22). No protein has been detected in the capillary endothelial cells. In situ RNA hybridization has shown that mrp1 RNA is present in some of the seminiferous tubules of the testis (23).
To correlate the observed drug-induced hypotonic polyuria and the damage in the oropharyngeal cavity and the testis with mrp1 protein location, we stained sections with the mAb MRPr1 directed against an NH2-terminal part of the human MRP1 (22). As negative controls, we used mice lacking mrp1 (19), and isotype control antibodies.
Mouse mrp1 is present in high amounts in the mucosal layer of the tongue, whereas very low levels were detected in the muscular layer (Fig. 3, A and B). High levels were also found in the mucosal layers of the cheek (Fig. 3, C and D), esophagus (Fig. 3, E and F), and lung (Fig. 3, G and H). Interestingly, mrp1 mainly resides in the basolateral membranes of the mouse lung epithelium, rather than in subapical vesicles as in human bronchial epithelium (22).
Figure 3.
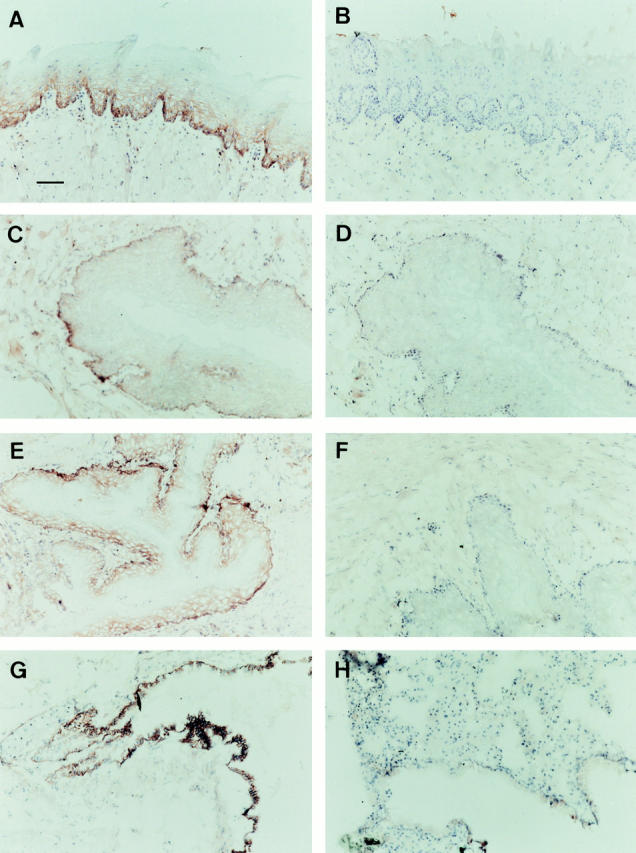
mrp1 staining of frozen sections of drug-naive wild-type and mrp1-deficient tissues. Sections were counterstained with hematoxylin. Wild-type (A, C, E, and G) and mrp1-deficient (B, D, F, and H) mice. (A and B) Sections of the tongue showing mrp1 staining in the mucosal layer (A). (C and D) Sections of the mucosal layer of the cheek. (E and F) Sections of the esophagus. (G and H) Sections of the lung. Bar, 50 μm.
Immunostaining revealed high levels of mrp1 protein in the epithelial layers of the limb of Henle and the urinary collecting ducts in the marrow and cortex of the wild-type kidney, whereas the glomeruli and the proximal convoluted tubules showed no immunoreactivity (Fig. 4, A–D).
Figure 4.
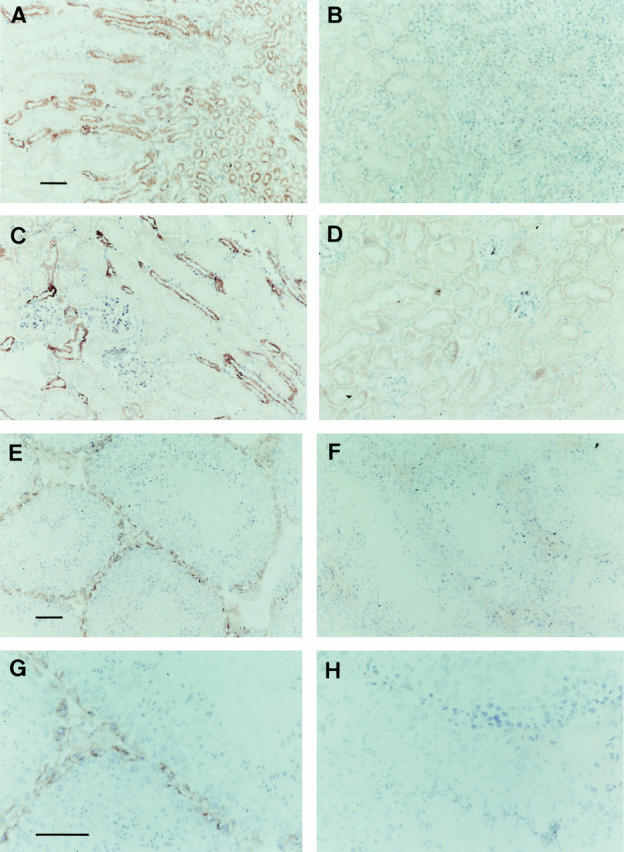
mrp1 staining of frozen sections of drug-naive wild-type and mrp1-deficient tissues. Sections were counterstained with hematoxylin. Wild-type (A, C, E, and G) and mrp1-deficient (B, D, F, and H) mice. (A–D) Sections of the kidney showing mrp1 staining in the limb of Henle and urinary collecting ducts, and the absence of staining in the glomeruli and proximal convoluted tubules (C and D). (E–H) Sections of the testis showing mrp1 staining in the Leydig and Sertoli cells. (G and H) Higher magnification of E and F. Same magnification in A–D (bar, 100 μm), in E and F (bar, 50 μm), and in G and H (bar, 50 μm).
In the testis, the Leydig cells in the interstitial tissue outside of the seminiferous tubules, and the basal plasma membrane but not the apical or the lateral membranes of all the Sertoli cells in the seminiferous tubules contain mrp1 (Fig. 4, E–H). The tight junctions in between neighboring Sertoli cells, unlike other polarized cells, are located near the basal plasma membrane, on the adluminal side of the spermatogonia (24), explaining the absence of mrp1 in the adluminal part of lateral membranes. No staining is observed in the endothelial layer of the blood vessels in the testis, a position where Pgp is known to reside (25, 26). The location of the drug pump mrp1 in the basal plasma membrane of the Sertoli cells supports a protective function in spermatogenesis, since Sertoli cells form a Sertoli–Sertoli cell barrier, the so-called blood–testis barrier, against xenobiotics (24, 27).
Discussion
Our data show that the cellular drug exporter mrp1 protects mice against oropharyngeal and testicular damage by the anticancer drug etoposide. This protective function will most likely extend to several xenobiotics, carcinogens, and (anti)cancer drugs, and possibly also to chemical substances reducing fertility. Like the mdr1a Pgp (17, 18), mrp1 appears to be an important element in the defense against potentially harmful compounds in the diet and toxins released during bacterial or fungal infections. The aggravating effect of mrp1 deficiency becomes pronounced after drug treatment or an inflammatory stimulus (19), indicating that mrp1 is involved in ancient (cellular drug extrusion) as well as more sophisticated (cellular LTC4 extrusion) mechanisms of cellular protection. As the function and tissue distribution of murine mrp1 and human MRP1 are similar, we expect our mouse results to be relevant for drug-induced damage in humans.
Is mrp1 of Importance for the Protection of Kidney Function?
The drug-induced hypotonic polyuria in mrp1-deficient mice could be either renal or due to excessive drinking caused by the oral mucositis. Since the drug-treated mutant mice show symptoms of dehydration with relatively high plasma levels of protein, sodium, creatinine, and urea, it is unlikely that primary polydipsia can explain the polyuria, although it may contribute. Therefore, we focused on the kidney. The data indicate either a reduced responsiveness of the urinary collecting ducts to vasopressin, or a reduced effectiveness or plasma level of vasopressin. A water-deprivation test did not help to distinguish between a nephrogenic or central diabetes insipidus, since the majority of drug-treated mutant mice did not survive (four out of seven mice; data not shown) a 20-h water deprivation 48 h after drug administration. Since mrp1 is not expressed in the magnocellular neurons of the anterior hypothalamus which secrete vasopressin (22), there is no reason why the osmoregulatory and vasopressin-producing cells would be more sensitive to drug in the mrp1 mutant than in wild-type mice. Therefore it is unlikely that the polyuria is caused by central diabetes insipidus.
The urinary collecting ducts are the site of water reabsorption regulated by vasopressin, whereas passive water transport occurs in the proximal convoluted tubules and in the descending limb of Henle. The ascending limb of Henle and the distal convoluted tubes are largely impermeable to water. The permeability of the urinary collecting ducts to water in the absence of vasopressin is low but can be enhanced in the presence of vasopressin (28). mrp1 is present in the basolateral membranes of these collecting ducts. Even though we did not observe histological abnormalities in these ducts, we propose that etoposide treatment of mrp1 mutant mice causes cell damage or persistent subtle changes in the presence or function of vasopressin receptors in the basolateral membrane of the collecting ducts, leading to reduced renal tubular responsiveness to vasopressin. This interpretation implies that the polyuria is caused, at least in part, by drug-induced acquired nephrogenic diabetes insipidus.
mrp1 Plays a Major Role in the Protection against Chemotherapy-induced Mucositis.
Oropharyngeal mucosal injury due to cytotoxic agents is caused by damage to stem cells in the basal epithelium, where these cells are subsequently depleted, paving the way for infections and inflammation (29). Mucositis is a common, painful condition which occurs in the majority of patients who receive high-dose chemotherapy for the treatment of cancer. In fact, mucositis is the dose-limiting toxicity for many combinations of anticancer drugs clinically in use (29, 30). The factors determining oropharyngeal toxicity have remained largely unknown (29). Our data show that the drug transporter mrp1 is one of these factors. The absence of this protein results in a severe disruption of the mucosal layer of the tongue and cheek during a high-dose etoposide treatment.
What Might Be the Cause of Death of mrp1 Mutant Mice during the Etoposide Mortality Test?
The mrp1 −/− mice are killed at lower etoposide doses than wild-type mice (19, 20), but the cause of this hypersensitivity has remained unclear, as we found no evidence for increased drug exposure or bone marrow depression in our initial studies (19). We now find a significant decrease in leukocytes 7 d after etoposide administration (Table 2). Combined with the oropharyngeal damage and the decreased inflammatory response due to defective LTC4 secretion (19), this could compromise the ability of mrp1 −/− mice to cope with opportunistic infections. The hemoconcentration observed might also jeopardize survival. However, it is possible that there are additional drug targets protected by mrp1 that contribute to the hypersensitivity of the mrp1 −/− mice and that remain to be found.
We attribute the etoposide-induced toxicities in the mrp1 −/− mice to the absence of a functional mrp1 drug transporter in the affected cells. An alternate explanation might be the presence of an unnoticed subtle defect leading indirectly to the toxicities described. This seems unlikely, since the etoposide hypersensitivity was observed in two independently derived mrp1 −/− mouse lines (19, 20). It is also unlikely to be a secondary effect of altered drug pharmacokinetics, as there were no differences between wild-type and mrp1 −/− mice (19). Moreover, the toxicities described in this study affect cells which normally contain mrp1. However, the definitive experiment might come from transgenic mrp1 −/− mice in which the etoposide sensitivity is rescued by overexpressing a wild-type mrp1 gene under the control of a Sertoli cell–specific, oropharyngeal or renal collecting tubular epithelium–specific promoter.
Our data show that some tissues are mainly protected against etoposide by the drug transporter mrp1. Other tissues, e.g., epithelia of esophagus and intestines (21) and endothelia of blood brain capillaries (17, 18), might be mainly protected against etoposide by one or several Pgps or other members of the drug-transporter family. The identification and subsequent ablation of each of these drug transporters in mice might reveal tissues uniquely or redundantly protected by drug transporters. Indeed, mice which lack the three drug transporters mdr1a, mdr1b, and mrp1 are viable and fertile but show higher sensitivity to etoposide than mice lacking only one of the transporters (J.Wijnholds, A.H. Schinkel, and P. Borst, unpublished data).
Implications for Treatment of Patients with Carcinostatic Drugs Transported by MRP1.
Mucosal protectants which lower the rate of basal cell proliferation lower the susceptibility of the epithelium to drugs (29) and might be clinically useful in reducing the oral toxic side effects, but this needs to be proven. Our finding that mrp1 plays an important role in the protection against drug-induced oral injury could potentially be clinically exploited. If it were possible to transiently elevate levels of mrp1 in the basal epithelial layers of the tongue and cheek by drugs, this might reduce the susceptibility of the epithelium to anticancer drugs.
Significance of mrp1 in the Protective Role of the Blood–Testis Barrier.
We show here that mrp1 is present in the basal plasma membrane of all Sertoli cells. The etoposide-induced impairment of spermatogenesis and premature release of spermatogenic cells in mrp1-deficient testis indicates that mrp1 plays an important role in the protection of the Sertoli–germ cell communication network (24, 27, 31). The seminiferous tubules of the testis contain two cell types: germ cells, which constitute the male contribution to the reproductive cycle, and Sertoli cells, which support the growth and differentiation of germ cells. Sertoli cells abut onto the basal lamina of the tubules, and are joined near their bases by tight or occluding junctions, to create a permeability barrier between the extratubular and intratubular compartments. The blood–testis barrier is formed by the tight junctions as well as by the body of the Sertoli cells (24). A proper cell number ratio between maturing germ cells and Sertoli cells is essential for proper spermatogenesis, and disruption of this complex network might result in permanently reduced function.
High levels of mrp1, and MRP1 (22), are also detected in the Leydig (or interstitial) cells which are grouped in the supportive connective tissue between the seminiferous tubules next to blood vessels and nerves. No histological differences were detected in the Leydig cells after drug treatment. The seminiferous tubules are partially under hormonal control by the Leydig cells. Therefore, we cannot exclude that damage of these cells contributes to the drug-induced reduction of spermatogenesis.
In humans, persistent fertility problems can occur after chemotherapy or consumption of food toxins or after medications (32). We hypothesize that MRP1 deficiency or malfunction might play a role in reduced viable sperm numbers. Lowered levels of MRP1 in the Sertoli–Sertoli cell barrier would lead to decreased efflux of infertility-inducing MRP1 substrates (such as etoposide) from the Sertoli cells. Interestingly, MDR1 Pgp is present in the capillary endothelium (25, 26) and might play a role in the blood– testis barrier as well. In conclusion, our results using the mrp1 mutant mice imply that the clinical use of inhibitors of MRP1 (drug reversal agents) in combination with chemotherapy increases the risk of reduction in fertility.
What Is the Physiological Role of mrp1 Localized in the Basal or Basolateral Plasma Membranes of Polarized Cells?
Our demonstration that mrp1 resides in the basolateral membranes of the lung epithelium and in the basal plasma membrane of the Sertoli cells extends previous work showing the basolateral localization of human MRP1 in transfected kidney cells (33) and of murine mrp1 in liver (34). This contrasts with the apical location of other drug pumps, such as the MDR1 Pgp or the GS-X pump cMOAT (or MRP2 [16, 25]). The presence of mrp1 in the basal but not the apical plasma membrane of Sertoli cells makes sense, as it helps to protect not only the Sertoli cells themselves, but also the other cells in the seminiferous tubules from xenotoxins. In fact, we expect mrp1 to be an essential component of the Sertoli–Sertoli cell barrier, in a similar way as the MDR1-type Pgp acts as a guardian of the blood–brain barrier (17, 18). In addition, mrp1 might help to clear metabolic waste products (e.g., hormone metabolites) from the seminiferous tubule. Our results also indicate that the drug-clearing role of mrp1 may be more important than previously thought. Drug-clearing by cmoat, mdr1a, and mdr1b has always been rather obvious, as these transporters are located in apical membranes and directly secrete into luminal spaces. The basolateral location of mrp1 seems less suitable for this purpose and more appropriate for the secretion of useful molecules, or for the retrieval of valuable metabolites, e.g., from urine. However, our results with oral mucosa and testis show that removal of toxins from cells into the interstitial fluid may be effective in protecting vital epithelial structures against damage. For instance, in the lung, mrp1 in the epithelium could assist in the clearance of toxins coming from the luminal or interstitial fluid (back) into the interstitial fluid. In other polarized epithelia flanking lumens, mrp1 may have similar protective functions.
Acknowledgments
We thank Drs. J.A. Grootegoed (Erasmus University, Rotterdam, The Netherlands), S. Rodenhuis (The Netherlands Cancer Institute), J.J. Weening (Academic Medical Center, Amsterdam, The Netherlands), and our colleagues Drs. R. Evers, Z. Holló, and M. Kool for critical advice on the manuscript, and A.J. Schrauwers for excellent biotechnical assistance.
This work was supported in part by grants from the Dutch Cancer Society.
Abbreviations used in this paper
- cMOAT and cmoat
human and mouse canalicular multispecific organic anion transporter, respectively
- ETOPOPHOS
etoposide-phosphate
- FeNa
fractional excretion of sodium
- GFR
glomerular filtration rate
- GS-X
glutathione S-conjugate
- LTC4
leukotriene C4
- MDR
multidrug resistance
- MDR1 and mdr1
human and mouse multidrug resistance gene 1, respectively
- MRP1 and mrp1
human and mouse multidrug resistance protein 1, respectively
- Pgp
P-glycoprotein
- RFI
renal failure index
- UCr and PCr
urine and plasma creatinine
References
- 1.Gottesman MM, Hrycyna CA, Schoenlein PV, Germann UA, Pastan I. Genetic analysis of the multidrug transporter. Annu Rev Genet. 1995;29:607–649. doi: 10.1146/annurev.ge.29.120195.003135. [DOI] [PubMed] [Google Scholar]
- 2.Zaman, G.J.R., and P. Borst. 1996. MRP, mode of action and role in MDR. In Multidrug Resistance in Cancer Cells. S. Gupta and T. Tsuruo, editors. John Wiley & Sons Ltd., Chichester, UK. 95–107.
- 3.Loe, D.W., R.G. Deeley, and S.P.C. Cole. 1996. Biology of the multidrug resistance-associated protein, MRP. Eur. J. Cancer. 32A:945–957. [DOI] [PubMed]
- 4.Cole SPC, Bhardwaj G, Gerlach JH, Mackie JE, Grant CE, Almquist KC, Stewart AJ, Kurz EU, Duncan AMV, Deeley RG. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science. 1992;258:1650–1654. doi: 10.1126/science.1360704. [DOI] [PubMed] [Google Scholar]
- 5.Zaman GJR, Flens MJ, van Leusden MR, de Haas M, Mülder HS, Lankelma J, Pinedo HM, Scheper RJ, Baas F, Broxterman HJ, Borst P. The human multidrug resistance-associated protein MRP is a plasma membrane drug-efflux pump. Proc Natl Acad Sci USA. 1994;91:8822–8826. doi: 10.1073/pnas.91.19.8822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cole SPC, Sparks KE, Fraser K, Loe DW, Grant CE, Wilson GM, Deeley RG. Pharmacological characterization of multidrug resistant MRP-transfected human tumor cells. Cancer Res. 1994;54:5902–5910. [PubMed] [Google Scholar]
- 7.Leier I, Jedlitschky G, Buchholz U, Cole SPC, Deeley RG, Keppler D. The MRP gene encodes an ATP-dependent export pump for leukotriene C4and structurally related conjugates. J Biol Chem. 1994;269:27807–27810. [PubMed] [Google Scholar]
- 8.Jedlitschky G, Leier I, Buchholz U, Barnouin K, Kurz G, Keppler D. Transport of glutathione, glucuronate, and sulfate conjugates by the MRPgene-encoded conjugate export pump. Cancer Res. 1996;56:988–994. [PubMed] [Google Scholar]
- 9.Ishikawa T, Li Z-S, Lu Y-P, Rea PA. The GS-X pump in plant, yeast, and animal cells: structure, function, and gene expression. Biosci Rep. 1997;17:189–207. doi: 10.1023/a:1027385513483. [DOI] [PubMed] [Google Scholar]
- 10.Müller M, Meijer C, Zaman GJR, Borst P, Scheper RJ, Mulder NH, de Vries E, Jansen PLM. Overexpression of the gene encoding the multidrug resistance-associated protein results in increased ATP-dependent glutathione S-conjugate transport. Proc Natl Acad Sci USA. 1994;91:13033–13037. doi: 10.1073/pnas.91.26.13033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pulaski L, Jedlitschky G, Leier I, Buchholz U, Keppler D. Identification of the multidrug-resistance protein (MRP) as the glutathione-S-conjugate export pump of erythrocytes. Eur J Biochem. 1996;241:644–648. doi: 10.1111/j.1432-1033.1996.00644.x. [DOI] [PubMed] [Google Scholar]
- 12.Zaman GJR, Cnubben NHP, Bladeren PJ, Evers R, Borst P. Transport of the glutathione conjugate of ethacrynic acid by the human multidrug resistance protein MRP. FEBS Lett. 1996;391:126–130. doi: 10.1016/0014-5793(96)00718-1. [DOI] [PubMed] [Google Scholar]
- 13.Zaman GJR, Lankelma J, van Tellingen O, Beijnen J, Dekker H, Paulusma C, Baas F, Borst P. Role of glutathione in the export of compounds from cells by the multidrug-resistance-associated protein. Proc Natl Acad Sci USA. 1995;92:7690–7694. doi: 10.1073/pnas.92.17.7690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kool M, de Haas M, Scheffer GL, Scheper RJ, van Eijk MJT, Juijn JA, Baas F, Borst P. Analysis of expression of cMOAT (MRP2), MRP3, MRP4, and MRP5, homologues of the multidrug resistance-associated protein gene (MRP1), in human cancer cell lines. Cancer Res. 1997;57:3537–3547. [PubMed] [Google Scholar]
- 15.Paulusma CC, Bosma PJ, Zaman GJR, Bakker CTM, Otter M, Scheffer GL, Scheper RJ, Borst P, Oude RPJ, Elferink Congenital jaundice in rats with a mutation in a multidrug resistance-associated protein gene. Science. 1996;271:1126–1128. doi: 10.1126/science.271.5252.1126. [DOI] [PubMed] [Google Scholar]
- 16.Evers R, Kool M, van Deemter L, Jansen H, Calafat J, Oomen LCJM, Paulusma CC, Oude RPJ, Elferink, Baas F, Schinkel AH, Borst P. Drug export activity of the human canalicular multispecific organic anion transporter in polarized kidney MDCK cells expressing cMOAT (MRP2)cDNA. J Clin Invest. 1998;101:1310–1319. doi: 10.1172/JCI119886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schinkel AH, Mayer U, Wagenaar E, Mol CAAM, van Deemter L, Smit JJM, van der Valk M, Voordouw AC, Spits H, van Tellingen O, et al. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc Natl Acad Sci USA. 1997;94:4028–4033. doi: 10.1073/pnas.94.8.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schinkel AH, Smit JJ, van Tellingen O, Beijnen JH, Wagenaar E, van Deemter L, Mol CAAM, van der Valk MA, Robanus-Maandag EC, te Riele HPJ, et al. Disruption of the mouse mdr1aP-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell. 1994;77:491–502. doi: 10.1016/0092-8674(94)90212-7. [DOI] [PubMed] [Google Scholar]
- 19.Wijnholds J, Evers R, van Leusden MR, Mol CAAM, Zaman GJR, Mayer U, Beijnen JH, van der Valk M, Krimpenfort P, Borst P. Increased sensitivity to anticancer drugs and decreased inflammatory response in mice lacking the multidrug resistance-associated protein. Nat Med. 1997;3:1275–1279. doi: 10.1038/nm1197-1275. [DOI] [PubMed] [Google Scholar]
- 20.Lorico A, Rappa G, Finch RA, Yang D, Flavell RA, Sartorelli AC. Disruption of the murine MRP(multidrug resistance protein) gene leads to increased sensitivity to etoposide (VP-16) and increased levels of glutathione. Cancer Res. 1997;57:5238–5242. [PubMed] [Google Scholar]
- 21.Sparreboom A, van Asperen J, Mayer U, Schinkel AH, Smit JW, Meijer DKF, Borst P, Nooijen WJ, Beijnen JH, van Tellingen O. Limited oral bioavailability and active epithelial excretion of paclitaxel (taxol) caused by P-glycoprotein in the intestine. Proc Natl Acad Sci USA. 1997;94:2031–2035. doi: 10.1073/pnas.94.5.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flens MJ, Zaman GJR, van der Valk P, Izquierdo MA, Schroeijers AB, Scheffer GL, van der Groep P, de Haas M, Meijer CJLM, Scheper RJ. Distribution of the multidrug resistance-associated protein (MRP) in normal and malignant human tissues. Am J Pathol. 1996;148:1237–1247. [PMC free article] [PubMed] [Google Scholar]
- 23.Stride BD, Valdimarsson G, Gerlach JH, Wilson GM, Cole SPC, Deeley RG. Structure and expression of the messenger RNA encoding the murine multidrug resistance protein, an ATP-binding cassette transporter. Mol Pharmacol. 1996;49:962–971. [PubMed] [Google Scholar]
- 24.Russell LD, Peterson RN. Sertoli cell junctions: morphological and functional correlates. Int Rev Cytol. 1985;94:177–211. doi: 10.1016/s0074-7696(08)60397-6. [DOI] [PubMed] [Google Scholar]
- 25.Cordon-Cardo C, O'Brien JP, Boccia J, Casals D, Bertino JR, Melamed MR. Expression of the multidrug resistance gene product (P-glycoprotein) in human normal and tumor tissues. J Histochem Cytochem. 1990;38:1277–1287. doi: 10.1177/38.9.1974900. [DOI] [PubMed] [Google Scholar]
- 26.Thiebaut F, Tsuruo T, Hamada H, Gottesman MM, Pastan I, Willingham MC. Cellular localization of the multidrug resistance gene product in normal human tissues. Proc Natl Acad Sci USA. 1987;84:7735–7738. doi: 10.1073/pnas.84.21.7735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tindall DJ, Rowley DR, Murthy L, Lipshultz LI, Chang CH. Structure and biochemistry of the Sertoli cell. Int Rev Cytol. 1985;94:127–149. doi: 10.1016/s0074-7696(08)60395-2. [DOI] [PubMed] [Google Scholar]
- 28.Brenner, B.M. 1996. The Kidney. 5th ed. B.M. Brenner and F.C. Rector, editors. W.B. Saunders Co., Philadelphia. 2653 pp.
- 29.Marks JE. Mucosal protectants and their application for head and neck chemoirradiation. Curr Opin Oncol. 1997;9:267–273. doi: 10.1097/00001622-199709030-00009. [DOI] [PubMed] [Google Scholar]
- 30.Postmus PE, Mulder NH, Sleijfer DT, Meinesz AF, Vriesendorp R, de Vries EGE. High-dose etoposide for refractory malignancies: a phase 1 study. Cancer Treat Rep. 1984;68:1471–1474. [PubMed] [Google Scholar]
- 31.Steinberger A, Klinefelter G. Sensitivity of Sertoli and Leydig cells to xenobiotics in in vitro models. Reprod Toxicol. 1993;7:23–37. doi: 10.1016/0890-6238(93)90066-g. [DOI] [PubMed] [Google Scholar]
- 32.Lipshultz, L.I., and S.S. Howards. 1991. Infertility in the Male. 3rd ed. Mosby, St. Louis. 576 pp.
- 33.Evers R, Zaman GJR, van Deemter L, Jansen H, Calafat J, Oomen LCJM, Oude RPJ, Elferink, Borst P, Schinkel AS. Basolateral localization and export activity of the human multidrug resistance–associated protein in polarized pig kidney cells. J Clin Invest. 1996;97:1211–1218. doi: 10.1172/JCI118535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mayer R, Kartenbeck J, Büchler M, Jedlitschky J, Leier I, Keppler D. Expression of the MRP gene-encoded conjugate export pump in liver and its selective absence from the canalicular membrane in transport-deficient mutant hepatocytes. J Cell Biol. 1995;131:137–150. doi: 10.1083/jcb.131.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]