

Abstract
During episodes of inflammation, polymorphonuclear leukocyte (PMN) transendothelial migration has the potential to disturb vascular barrier function and give rise to intravascular fluid extravasation and edema. However, little is known regarding innate mechanisms that dampen fluid loss during PMN-endothelial interactions. Using an in vitro endothelial paracellular permeability model, we observed a PMN-mediated decrease in endothelial paracellular permeability. A similar decrease was elicited by cell-free supernatants from activated PMN (FMLP 10−6 M), suggesting the presence of a PMN-derived soluble mediator(s). Biophysical and biochemical analysis of PMN supernatants revealed a role for PMN-derived 5′-adenosine monophosphate (AMP) and its metabolite, adenosine, in modulation of endothelial paracellular permeability. Supernatants from activated PMN contained micromolar concentrations of bioactive 5′-AMP and adenosine. Furthermore, exposure of endothelial monolayers to authentic 5′-AMP and adenosine increased endothelial barrier function more than twofold in both human umbilical vein endothelial cells and human microvascular endothelial cells. 5′-AMP bioactivity required endothelial CD73-mediated conversion of 5′-AMP to adenosine via its 5′-ectonucleotidase activity. Decreased endothelial paracellular permeability occurred through adenosine A2B receptor activation and was accompanied by a parallel increase in intracellular cAMP. We conclude that activated PMN release soluble mediators, such as 5′-AMP and adenosine, that promote endothelial barrier function. During inflammation, this pathway may limit potentially deleterious increases in endothelial paracellular permeability and could serve as a basic mechanism of endothelial resealing during PMN transendothelial migration.
Keywords: adenosine, adenosine monophosphate, ectonucleotidase, endothelium, neutrophil
Neutrophil-mediated tissue injury is a prominent component in organ damage during inflammation (1). Endothelial cells, which line the inner lumen of blood vessels and largely determine overall vascular permeability, are primary targets for PMN during uncontrolled inflammatory responses (2). Endothelial injury may result in increased endothelial paracellular permeability and decreased endothelial barrier function. The resultant local tissue edema and intravascular loss of fluid and solutes may further exacerbate tissue injury via interference with cellular function and limitation of regional blood flow (3). At present, mechanisms that may limit fulminant vascular leak during PMN-endothelial interactions remain largely unknown.
Although PMN possess an extensive cytotoxic arsenal (1), their interactions with the endothelium do not inevitably result in increased endothelial paracellular permeability (4). In fact, in the absence of PMN-endothelial adhesion, no significant change in endothelial paracellular permeability is observed (5, 6), and in vitro models, PMN-mediated changes in endothelial paracellular permeability do not occur in the absence of prior endothelial activation (5). Endothelial paracellular permeability may remain unchanged even with transendothelial PMN migration (7). And in vivo, increased endothelial paracellular permeability is not apparent despite extensive noninflammatory PMN-endothelial adhesion and subsequent transendothelial PMN migration (8).
In this study of PMN–endothelial interactions, we observed an increase in endothelial barrier function upon exposure to activated PMN. Further examination revealed that an increase in barrier function was elicited by a small, stable PMN-derived soluble mediator(s). Biochemical analysis revealed that 5′-adenosine monophosphate (AMP)1 and adenosine fulfill criteria as PMN-derived bioactive compounds which promote endothelial barrier function. From this work, we propose a pathway by which PMN regulate endothelial paracellular permeability during such cell–cell crosstalk.
Materials and Methods
Endothelial Cell Isolation and Culture.
Human umbilical vein endothelial cells (HUVEC) were harvested and cultured by a modification of methods previously described (9, 10). In brief, HUVEC were harvested with 0.1% collagenase and incubated at 37°C in 95% air/5% CO2. Culture medium was supplemented with heat-inactivated bovine calf serum, penicillin, streptomycin, Hepes, heparin, l-glutamine, and endothelial mitogen factor. For preparation of experimental HUVEC monolayers, confluent endothelial cells (passages 1, 2, or 3) were seeded at ∼2 × 105 cells/ cm2 onto either permeable polycarbonate inserts or 6-well plates precoated with 0.1% gelatin. Endothelial cell purity was assessed by phase microscopic “cobblestone” appearance and uptake of fluorescent acetylated low density lipoprotein. Human microvascular endothelial cells (HMVEC), a microvascular endothelial primary culture isolated from adult dermis, were obtained from Cascade Biologics (Portland, OR) and cultured as recommended. Where indicated, T84 intestinal epithelial cells were cultured and passaged as previously described (11).
Endothelial Macromolecule Paracellular Permeability Assay.
Using a modification of methods previously described (9), HUVEC or HMVEC on polycarbonate permeable inserts (0.4-μm pore, 6.5-mm diam; Costar Corp., Cambridge, MA) were studied 7–10 d after seeding (2–5 d after confluency). Inserts were placed in HBSS-containing wells (1 ml), and HBSS (alone or with PMN, PMN supernatant, 5′-AMP, or adenosine) was added to inserts (150 μl). In experiments using adenosine receptor antagonists, 5′-ectonucleotidase inhibitors, or NBTI, these compounds were added 20 min before addition of 5′-AMP or adenosine. At the start of the assay (t = 0), FITC-labeled dextran 70 kD (concentration 3.5 μM) was added to fluid within the insert. In experiments examining HMVEC paracellular permeability, the initial concentration of FITC-labeled dextran 70 kD was increased to 7 μM due to increased baseline barrier function of HMVEC (∼20-fold) compared with HUVEC. The size of FITC-dextran, 70 kD, approximates that of human albumin, both of which have been used in similar endothelial paracellular permeability models (12, 13). Monolayers were stirred via a rotating platform (60 rotations/min, Clinical Rotater; Fisher Scientific Co., Pittsburgh, PA) and serosal fluid from each well was sampled (50 μl) over 60 min (t = 5, 10, 15, 20, 30, 60 min); sample volume was replaced with HBSS. Fluorescence intensity of each sample was measured (excitation, 485 nm; emission, 530 nm; Cytofluor 2300; Millipore Corp., Waters Chromatography, Bedford, MA) and FITC-dextran concentrations were determined from standard curves generated by serial dilution of FITC-dextran. Paracellular flux was calculated by linear regression of sample fluorescence (Excel 5.0, Microsoft WA, Power Macintosh 7200; reference 14). Consistent with observations of other investigators, control experiments demonstrated decreased paracellular permeability with forskolin and 8-bromo-cAMP (15) and increased paracellular permeability with thrombin and hydrogen peroxide (16; data not shown).
Isolation of Human Neutrophils.
PMN were freshly isolated from whole blood obtained by venipuncture from human volunteers and anticoagulated with acid citrate/dextrose (11). Plasma and mononuclear cells were removed by aspiration from the buffy coat after centrifugation (400 g × 20 min) at 25°C. Erythrocytes were removed using a 2% gelatin sedimentation technique. Residual erythrocytes were removed by lysis in cold NH4Cl buffer. Remaining cells were >90% PMN as assessed by microscopic evaluation. PMN were studied within 2 h of their isolation.
PMN-HUVEC Adhesion Assay.
PMN, activated with FMLP (10-6 M), was added to HUVEC grown on permeable inserts. After incubation for 60 min at 37°C, each monolayer was gently washed with 1 ml of Dulbecco's PBS to remove nonadherent cells. The contents of each monolayer were then solubilized in 0.5% Triton X-100. Adherent PMN were quantified by myeloperoxidase assay (11).
Preparation of Activated PMN Supernatants.
Freshly isolated PMN (108 cells/ml in HBSS with 10-6 M FMLP) were placed in a glass culture tube and agitated (Adams Nutator; Clay Adams Inc., Nutley, NJ) for one minute. PMN suspensions were then immediately spun (1,000 g × 20 s, 4°C), filtered (0.45 μm; Phenomenex, Torrance, CA), and frozen (−80°C) until studied. In experiments measuring supernatant concentrations of 5′-AMP and adenosine, 100-μl samples were taken from PMN suspensions (t = 0, 1, 2.5, 5, 10, 15, and 20 min), immediately spun (1,000 g × 20 s, 4°C), filtered (0.45 μm), and frozen (−80°C) until analysis via HPLC.
Measurement of 5-Ectonucleotidase Activity.
Based on a modification of the method of Bonitati et al. (17), HBSS with or without 1E9 (mAb anti-CD73; 10 mcg/ml), C5/D5 (mAb anti-CD47; 10 mcg/ml), or APCP (3 μM) was added to HUVEC monolayers on 6-well plates. After 10 min, E-AMP (10 μM) was added. After 10 min, fluid was removed, acidified to pH 3.5 with HCl, spun (1,000 g × 20 s, 4°C), filtered (0.45 μm), and frozen (−80°C) until analysis via HPLC. Endothelial 5′-ectonucleotidase activity was assessed by measuring the conversion of E-AMP to E-ADO.
Characterization of PMN-derived Mediators.
To estimate the size range of PMN-derived mediators, activated PMN supernatants were passed through progressively smaller nominal molecular mass cut-off filters (Amicon, Danvers, MA) under N2 pressure (18). To investigate the stability of PMN-derived mediators to extremes in temperature, activated PMN supernatants were placed in 1.5-ml eppendorf tubes and either boiled (15 min) or repeatedly (3×) frozen (−80°C) and thawed.
High Performance Liquid Chromatography.
A Hewlett-Packard HPLC (model 1050) with an HP 1100 diode array detector was used with a reverse phase HPLC column (Luna 5μ C18(2) 150 × 4.60 mm; Phenomenex, CA). 5′-AMP was measured with a NaPhos 0.1 M, pH 6.0, mobile phase (1 ml/min). Adenosine was measured with a H2O:CH3CN (both with 0.01% acetic acid) 96:4 mobile phase (1 ml/min). E-AMP and E-ADO were measured with a 0–50% methanol/H2O gradient (10 min) mobile phase (2 ml/min). Absorbance was measured at 260 nm. UV absorption spectra were obtained at chromatographic peaks. Adenosine and adenine nucleotides were identified by their chromatographic behavior (retention time, UV absorption spectra, and coelution with standards; 19).
CD73 Immunoprecipitation.
Confluent cells were labeled with biotin, lysed, and cell debris removed by centrifugation (9). Lysates were precleared with 50 μl preequilibrated protein G–Sepharose (Amersham Pharmacia Biotechnology, Piscataway, NJ). Immunoprecipitation of CD73 was performed with mAb 1E9 (1:1,000) followed by addition of 50 μl preequilibrated protein G–Sepharose and overnight incubation. Washed immunoprecipitates were boiled in nonreducing sample buffer (2.5% SDS, 0.38 M Tris, pH 6.8, 20% glycerol, and 0.1% bromophenol blue), separated by nonreducing SDS-PAGE, transferred to nitrocellulose, and blocked overnight in blocking buffer. Biotinylated proteins were labeled with streptavidin-peroxidase and visualized by enhanced chemiluminescence (ECL; Amersham Pharmacia Biotechnology).
Western Blotting of A2B Receptor.
Confluent cells were lysed and cell debris removed by centrifugation (9). Samples of lysates were separated by nonreducing SDS-PAGE, transferred to nitrocellulose, and blocked overnight in blocking buffer (250 nM NaCl, 0.02% Tween-20, 5% normal goat serum, and 3% BSA). Primary Ab to A2BR (1:1,000 polyclonal; reference 20) was added for 3 h followed by wash and addition of peroxidase-conjugated secondary Ab (1:3,000 goat anti–chicken antibody; Kirkegaard & Perry Laboratories, Inc., Gaithersburg, MD). Labeled bands were detected by ECL.
Intracellular cAMP.
HBSS with or without 5′-AMP or adenosine was added to HUVEC monolayers on 6-well plates in the presence of Ro 20-1724 (0.5 mM), a non-xanthine phosphodiesterase inhibitor (21). After 15 min, cells were cooled to 4°C, washed, and cAMP extracted with lysis buffer (EtOH 66%, H2O 33%, Ro 20-1724 0.1 mM). Lysates were cleared by centrifugation (10,000 g × 20 min, 4°C), vacuum dried to remove EtOH, rehydrated in water, and stored (−80°C) until analyzed. Cyclic AMP was quantitated by a displacement ELISA (Amersham Pharmacia Biotechnology).
Materials.
Adenosine was obtained from Calbiochem-Novabiochem Corp. (La Jolla, CA). Adenosine amine congener (ADAC), 5′-(N-cyclopropyl)-carboxamidoadenosine (CPCA), 8-(3-chlorostyryl) caffeine (CSC), and alloxazine were obtained from Research Biochemical International (Natick, MA). Ethenoadenosine-5′-monophosphate (E-AMP), ethenoadenosine (E-Ado), α,β-methylene ADP (APCP), 5′-(N-ethylcarboxamido)adenosine (NECA), nitrobenzylthioinosine (NBTI), and 5′-adenosine monophosphate (5′-AMP) were obtained from Sigma Chemical Co. (St. Louis, MO). FITC-dextran was obtained from Molecular Probes (Eugene, OR). Antibodies 1E9 (mAb anti-CD73) and anti-A2BR were generous gifts from Dr. Linda Thompson (Oklahoma Medical Research Institute, Oklahoma City, OK). Antibody C5/D5 (mAb anti-CD47) was raised by the senior author as previously described (22). Transwell™ inserts were obtained from Corning Costar Corp. (Cambridge, MA).
Results
Activated PMN and Supernatants Derived from Activated PMN Decrease Endothelial Paracellular Permeability.
To investigate the influence of activated PMN on endothelial paracellular permeability, PMN with FMLP (10−6 M) and FITC-dextran 70 kD were added to HUVEC monolayers grown on permeable inserts. The maximal PMN dose (107/monolayer) resulted in a decrease in FITC-dextran transendothelial flux (Fig. 1). This decrease in FITC-dextran flux occurred despite an increase in PMN–HUVEC adhesion, as addition of 105, 106, and 107 PMN/monolayer resulted in 5.8 ± 2.0 × 104, 6.6 ± 1.8 × 105, 2.5 ± 0.7 × 106 adherent PMN/monolayer, respectively. To determine whether a decrease in paracellular flux was mediated by a PMN-derived soluble mediator, cell-free supernatants of activated PMN were added to HUVEC monolayers. Similar to intact PMN, supernatants of activated PMN decreased transendothelial flux of FITC-dextran in a PMN concentration-dependent manner (Fig. 2, ANOVA, P < 0.05). Subsequent experiments revealed no apparent endothelial uptake of FITC-dextran, no influence of FMLP (10−6–10−10 M) on transendothelial FITC-dextran flux, and no quenching of FITC-dextran fluorescence by activated PMN supernatants (data not shown). Therefore, we concluded that a PMN-derived soluble mediator(s) decreases endothelial paracellular permeability.
Figure 1.
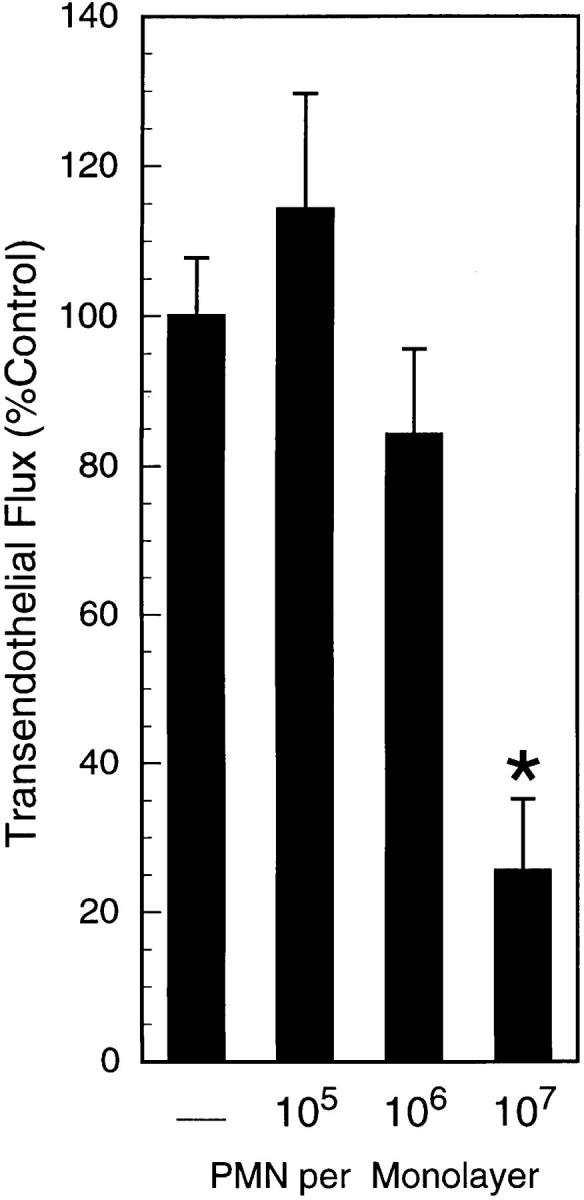
Activated PMN decrease endothelial paracellular permeability. PMN with FMLP (10−6 M) and FITC-dextran 70 kD were added to monolayers. Transendothelial flux was calculated by linear regression (6 samples over 60 min) and normalized as percent of control (HBSS). Data are derived from six monolayers in each condition; results are expressed as mean of percent control flux ± SEM. An asterisk indicates P < 0.05 by ANOVA.
Figure 2.
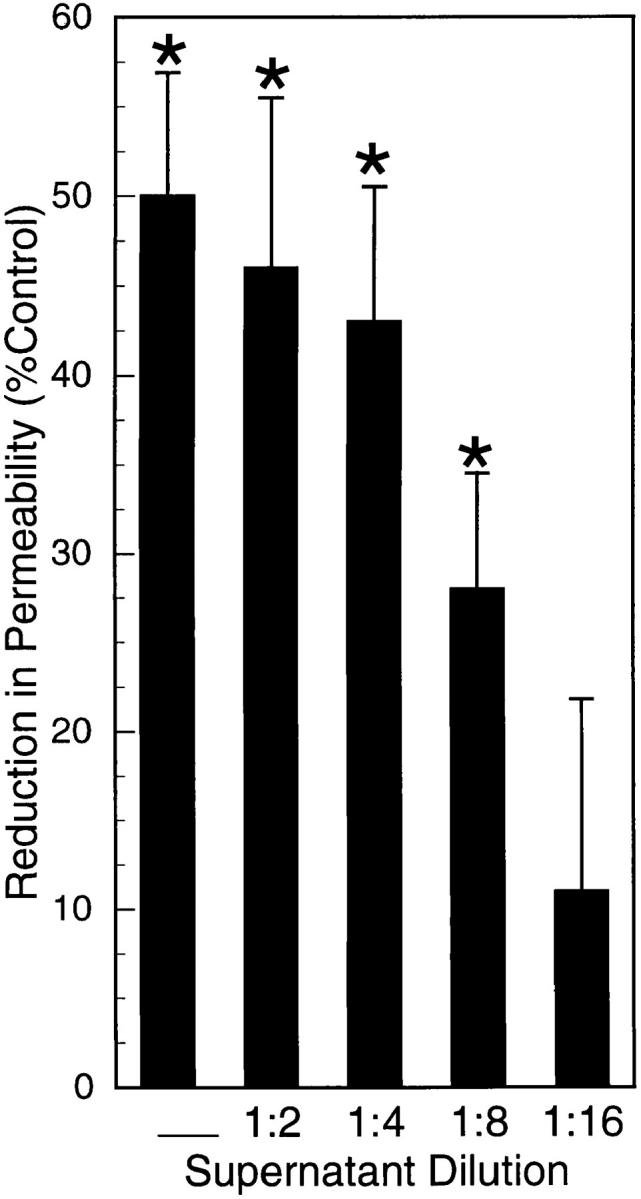
Supernatants from activated PMN decrease endothelial paracellular permeability. Confluent HUVEC on permeable supports were exposed (apical surface only) to cell-free supernatants from activated (FMLP 10−6 M) PMN. Supernatants from activated PMN were added to monolayers. Supernatants, undiluted or diluted (as indicated) decreased transendothelial flux of FITC-dextran (*P < 0.05 vs. control [HBSS]; ANOVA). Supernatants (undiluted) decreased flux by 50 ± 7%. Data are from 10 monolayers in each condition. Results are expressed as mean reduction in permeability ± SEM.
Activated PMN Supernatants Contain 5′-AMP and Adenosine.
After demonstrating that a PMN-derived soluble mediator(s) promotes endothelial barrier function, we examined its basic physical and chemical properties. First, to gain insight into the molecular mass of this mediator(s), activated PMN supernatants were passed through progressively smaller molecular mass cut-off filters (range 1–10 kD). These experiments revealed that supernatants passaged through filters with a molecular mass cut-off as low as 1 kD, when added to endothelial monolayers, decreased endothelial permeability (51 ± 12% decrease in paracellular permeability compared with control [HBSS]). We next addressed the stability of this mediator(s). Supernatants that had undergone boiling or repeated cycles of freeze/thaw, when added to endothelial monolayers, also decreased endothelial paracellular permeability (38 ± 9% and 57 ± 8% decrease in permeability, respectively, compared with control). These supernatant-mediated reductions in FITC-dextran flux were similar to that elicited by untreated, activated PMN supernatants (45 ± 8%, P = NS, ANOVA). Such findings suggested that the PMN-derived soluble mediator(s) was a small (<1 kD), stable molecule. These biophysical features are similar to those of 5′-AMP and adenosine, previously identified PMN-derived factors (18). Therefore, using HPLC analysis, we examined 5′-AMP and adenosine in PMN supernatants under our defined activation conditions (Fig. 3). Indeed, this analysis revealed 5′-AMP and adenosine in activated PMN supernatants by retention times and UV spectra similar to authentic standards (Fig. 3, insets). Supernatant concentrations of 5′-AMP and adenosine peaked within minutes of PMN activation (Fig. 4). After 1 min of PMN activation (FMLP 10−6 M), 5′-AMP concentrations increased from undetectable levels (<20 nM) to 7.3 ± 1.3 μM. The concentration of 5′-AMP declined thereafter, reaching 1.7 ± 0.1 μM by 20 min. In comparison, supernatant concentrations of adenosine reached a maximum of 4.9 ± 0.5 μM at 2.5 min of PMN activation and declined to 1.4 ± 0.3 μM at 20 min. These results demonstrate that PMN activation results in relatively high extracellular concentrations (i.e., micromolar) of both 5′-AMP and adenosine. As with other PMN-derived mediators, local in vivo concentrations of these compounds are likely greater due to restricted diffusion from the PMN-endothelial microenvironment (23, 24).
Figure 3.

Chromatographic identification of 5′-AMP and adenosine in activated PMN supernatants. 5′-AMP and adenosine were identified by their characteristic retention times and UV absorption spectra. Representative chromatograms and UV spectra (insets) of authentic 5′-AMP (A) and adenosine (B) standards are shown by dashed lines. Representative chromatograms and UV spectra of activated PMN supernatants are shown by solid lines.
Figure 4.

5′-AMP and adenosine concentrations in activated PMN supernatants. PMN (108/ml) were activated in HBSS with FMLP (10−6 M) in glass tubes. PMN suspensions were sampled at indicated time points after activation. 5′-AMP and adenosine concentrations were measured via HPLC. Data are from eight donors. Results are expressed as mean concentration ± SEM.
5′-AMP and Adenosine Decrease Endothelial Paracellular Permeability.
After determining that activated PMN supernatants contain micromolar concentrations of 5′-AMP and adenosine, we examined the influence of native compounds on endothelial paracellular permeability. Previous studies have yielded conflicting results with regard to the influence of adenosine on endothelial paracellular permeability (21, 25). Furthermore, we are unaware of studies investigating the influence of 5′-AMP on endothelial paracellular permeability. Addition of either 5′-AMP or adenosine to HUVEC monolayers decreased FITC-dextran flux in a concentration-dependent manner (Fig. 5). Similar results were observed with the addition of 5′-AMP or adenosine to HMVEC monolayers (Fig. 5). In both endothelial cell types, the concentration response curves of 5′-AMP and adenosine were equivalent (P = NS; ANOVA). In subsequent investigations, 5′-AMP and adenosine were studied at their approximate EC50 (50 μM).
Figure 5.

5′-AMP and adenosine decrease endothelial paracellular permeability. 5′-AMP or adenosine were added to HUVEC monolayers (left) or HMVEC monolayers (right). Both 5′-AMP and adenosine decreased transendothelial FITC-dextran flux in a dose-dependent manner (*P < 0.05; ANOVA). 5′-AMP and adenosine were equally potent. Data are from 6–8 monolayers in each condition. Results are expressed as mean ± SEM.
Inhibition of HUVEC 5′-Ectonucleotidase Attenuates the Endothelial Paracellular Permeability Response to 5′-AMP.
Based on the equivalent potencies of 5′-AMP and adenosine, the absence of a known purinoreceptor for 5′-AMP (26), and the 5′-ectonucleotidase enzymatic activity of CD73 in endothelial cells (27), we investigated whether the 5′-AMP– mediated decrease in endothelial paracellular permeability required conversion of 5′-AMP to adenosine at the endothelial cell surface. First, we confirmed the presence of extracellular CD73 expression in this model. As shown in Fig. 6 A, a ∼70-kD cell surface protein consistent with CD73 was immunoprecipitated in HUVEC and HMVEC by the mAb 1E9, similar to that immunoprecipitated from T84 cells, an intestinal epithelia cell line in which CD73 is constitutively expressed (28). Next, 5′-AMP was added to HUVEC monolayers pretreated with increasing concentrations of the 5′-ectonucleotidase inhibitor APCP (29; Fig. 6 B). Pretreatment with APCP diminished (300 nM) or abolished (3 μM) the 5′-AMP–mediated decrease in endothelial paracellular permeability. Furthermore, pretreatment with APCP (3 μM) did not influence the adenosine-mediated decrease in permeability (52 ± 12% without APCP vs. 48 ± 14% with APCP; [adenosine] = 50 μM). Additionally, we examined the influence of the anti-CD73 mAb (1E9; 10 μg/ml) on 5′-AMP bioactivity (Fig. 6 C). Pretreatment of HUVEC monolayers with 1E9 abolished ([5′-AMP] = 10 μM) or attenuated ([5′-AMP] = 100 μM, 1 mM) the 5′-AMP–mediated decrease in permeability. In contrast, 1E9 pretreatment (10 μg/ml) of HUVEC did not influence the adenosine-mediated decrease in permeability (Fig. 6 C). Furthermore, pretreatment of HUVEC with C5/D5 (10 μg/ml), an anti-CD47 endothelial binding control mAb (22), did not influence the 5′-AMP–mediated decrease in endothelial paracellular permeability (62 ± 4% without C5/D5 vs. 58 ± 5% with C5/D5; [5′-AMP] = 1 mM). Prior reports have suggested that 1E9 inhibits 5′-ectonucleotidase activity in a human endothelial cell line (30) but may not inhibit 5′-ectonucleotidase activity in placental tissue (27). Therefore, using HPLC analysis of CD73 substrate, we examined 1E9 inhibition of HUVEC 5′-ectonucleotidase activity by measuring endothelial conversion of the adenine nucleotide E-AMP to E-ADO (17; Fig. 6 D). After pretreatment with 1E9 (10 μg/ml), 5′-ectonucleotidase activity decreased by 58 (4% vs. HBSS. By comparison, pretreatment with anti-CD47 mAb C5/D5 did not influence 5′-ectonucleotidase activity. These results suggest a prominent role for endothelial CD73 5′-ectonucleotidase activity in the 5′-AMP–mediated decrease in endothelial paracellular permeability.
Figure 6.

5′-Ectonucleotidase inhibition reverses the 5′-AMP–mediated decrease in endothelial paracellular permeability. (A) CD73 is expressed on the cell surface of confluent HUVEC and HMVEC as demonstrated by mAb 1E9 immunoprecipitation of biotinylated protein (arrow). CD73 was also immunoprecipitated from confluent T84 cells, an intestinal epithelia cell line previously known to express this ectoenzyme. (B) Addition of 5′-AMP to HUVEC monolayers decreased transendothelial FITC-dextran flux (*P < 0.05 vs. HBSS; ANOVA). APCP, a 5′-ectonucleotidase inhibitor, reversed this decrease in endothelial paracellular permeability (‡ P < 0.05; ANOVA). Data are from six monolayers in each condition. (C) Addition of 5′-AMP or adenosine to HUVEC monolayers decreased transendothelial FITC-dextran flux (*P < 0.05 vs. control [HBSS]; ANOVA). 1E9, an anti-CD73 mAb, attenuated this decrease in permeability for 5′-AMP but not for adenosine (‡ P < 0.05 vs. no 1E9; ANOVA). Data are from 8 monolayers in each condition. (D) HUVEC 5′-ectonucleotidase activity was decreased (*P < 0.05 vs. control) by APCP (3 μM) or 1E9 (10 mcg/ml) pretreatment. Data are from 12 monolayers in each condition. Results are expressed as mean ± SEM.
Endothelial Paracellular Permeability Response to 5′-AMP Is Mediated via Adenosine Receptor Activation.
Based on our observations that the mechanism of the 5′-AMP–mediated decrease in endothelial paracellular permeability may involve extracellular conversion of 5′-AMP to adenosine, the role of adenosine receptor activation was examined. HUVEC monolayers were pretreated (12 h) with NECA, a nonmetabolizable and potent nonselective adenosine receptor agonist; such pretreatment desensitizes adenosine receptor-mediated responses (28). Pretreatment with NECA diminished (300 nM) or abolished (3 μM) the 5′-AMP mediated decrease in endothelial paracellular permeability (Fig. 7). Because endothelial cells are also capable of rapid cellular uptake of extracellular adenosine (31), the potential role of endothelial cell signaling via adenosine uptake was examined. HUVEC monolayers were pretreated with nitrobenzylthioinosine (NBTI, 10 μM) before the addition of 5′-AMP; NBTI is an inhibitor of facilitated membrane transport of purine nucleotides (32). Pretreatment with NBTI did not influence the 5′-AMP–mediated reduction in permeability (53 ± 5% with NBTI vs. 47 ± 4% without NBTI; [5′-AMP] = 50 μM). Therefore, the 5′-AMP– mediated decrease in permeability was not dependent upon endothelial adenosine uptake. We conclude from these data that 5′-AMP decreases endothelial paracellular permeability via its extracellular conversion to adenosine and subsequent endothelial adenosine receptor activation.
Figure 7.
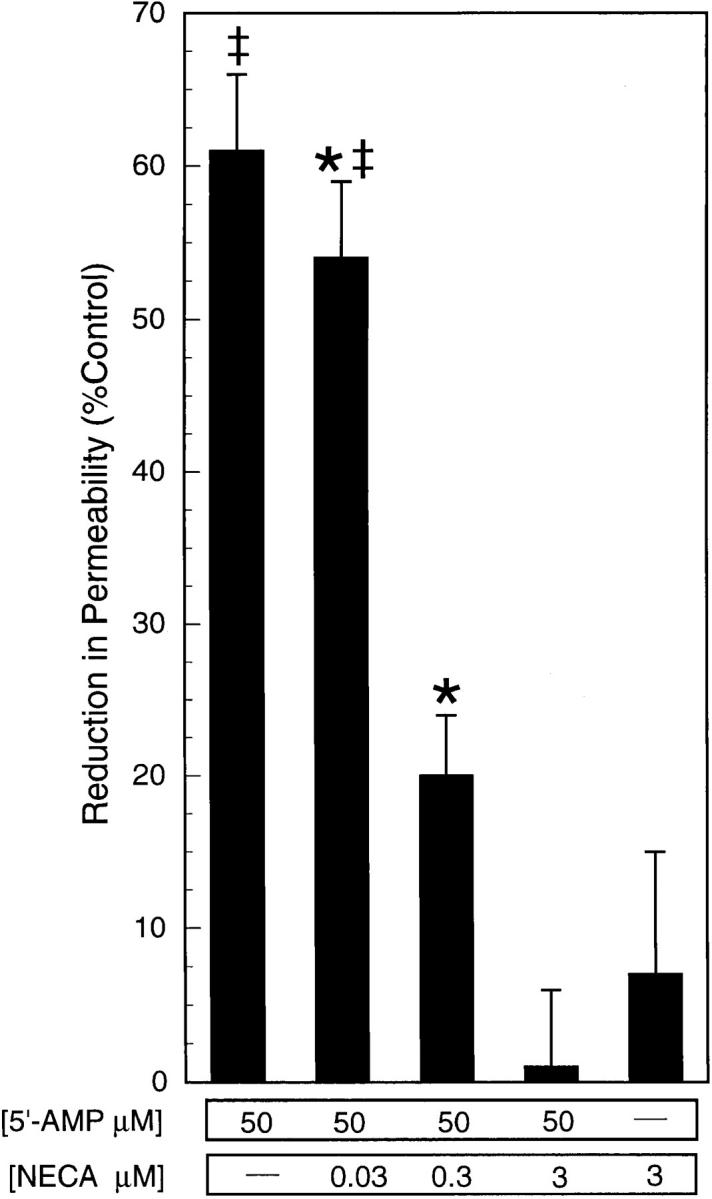
NECA pretreatment reverses the 5′-AMP– mediated decrease in endothelial paracellular permeability. Addition of 5′-AMP (50 μM) to HUVEC monolayers decreased transendothelial FITC-dextran flux (*P < 0.05 vs. HBSS). Pretreatment of HUVEC monolayers with NECA diminished ([NECA] = 300 nM) or abolished ([NECA] = 3 μM) this decrease in flux (‡ P < 0.05 vs. 5′-AMP without NECA). Data are from eight monolayers in each condition. Results are expressed as mean ± SEM.
5′-AMP Decreases Endothelial Paracellular Permeability via Adenosine A2B Receptor Activation.
To further define the purine receptor involved in the adenosine-mediated decrease in endothelial paracellular permeability, increasing concentrations of either the A1-specific agonist ADAC or the A2-specific agonist CPCA were examined in our endothelial paracellular permeability model (Fig. 8). Whereas the A1 agonist ADAC did not influence FITC-dextran transendothelial flux, the A2 agonist CPCA decreased flux in a manner similar to 5′-AMP and adenosine. At present, a specific A2B receptor agonist is not available (20). Therefore, A2A and A2B receptor antagonists were used to further define the A2 receptor subtype. 5′-AMP was added to HUVEC monolayers pretreated with increasing concentrations of either CSC (A2A antagonist) or alloxazine (A2B antagonist; Fig. 9). Whereas pretreatment with CSC had no significant influence (Fig. 9 A), pretreatment with alloxazine significantly diminished the 5′-AMP–mediated decrease in endothelial paracellular permeability (B). We conclude that 5′-AMP decreases permeability via conversion to adenosine with subsequent adenosine A2B receptor activation. Western blot analysis confirmed the presence of the adenosine A2B receptor in both HUVEC and HMVEC (Fig. 9 C), similar to T84 intestinal epithelia cells (20).
Figure 8.
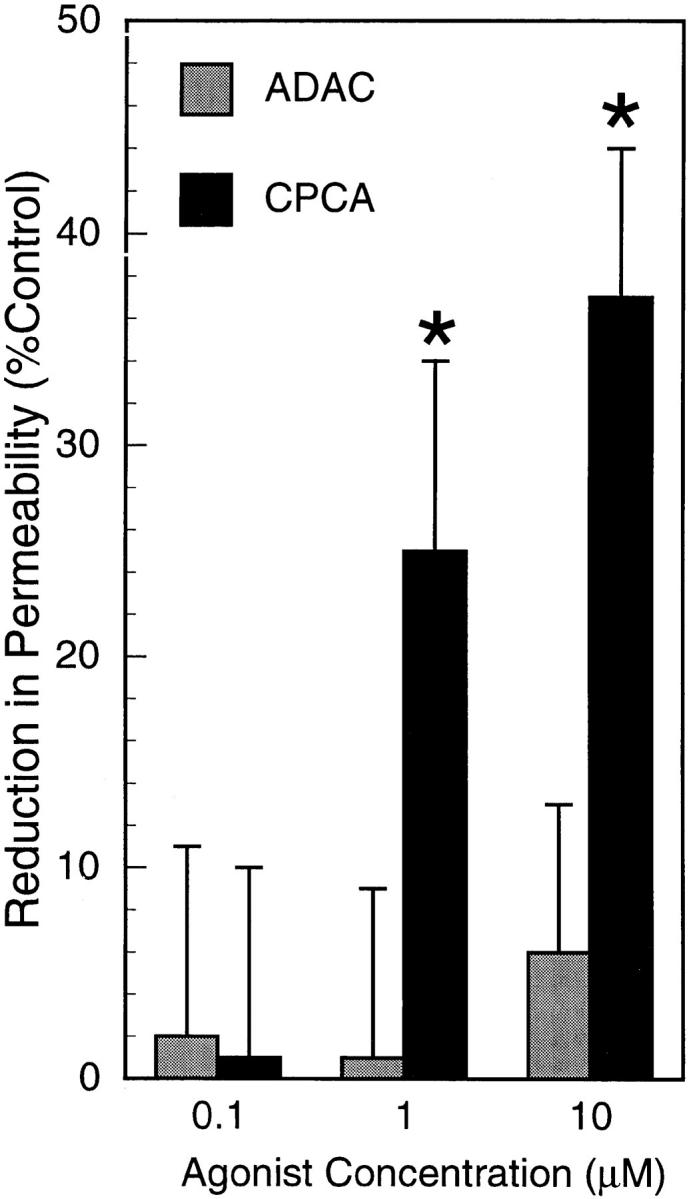
Adenosine A2 receptor activation decreases endothelial paracellular permeability. ADAC and CPCA, adenosine receptor agonists, were added to HUVEC monolayers. Only CPCA (A2 agonist) decreased transendothelial FITC-dextran flux (*P < 0.05 vs. control [HBSS]). Data are from 6–8 monolayers in each condition. Results are expressed as mean ± SEM.
Figure 9.

Adenosine A2B receptor antagonist diminishes the 5′-AMP–mediated decrease in endothelial paracellular permeability. Addition of 5′-AMP (50 μM) to HUVEC monolayers resulted in a decrease in transendothelial FITC-dextran flux (*P < 0.05 vs. control [HBSS]). (A) CSC (A2A receptor antagonist) had no effect on the 5′-AMP–mediated decrease in endothelial paracellular permeability. (B) Alloxazine (A2B receptor antagonist) inhibited the 5′-AMP–mediated decrease in endothelial paracellular permeability (B; ‡ P < 0.05 vs. 5′-AMP only). Data are from eight monolayers in each condition; results are expressed as mean ± SEM. (C) Adenosine A2B receptor is present in confluent HUVEC, HMVEC, and T84 cells as shown by cell lysate Western blot (arrow).
5′-AMP and Adenosine Increase Levels of Endothelial cAMP.
Activation of adenosine A2 receptors results in increased intracellular levels of cAMP via stimulation of adenylate cyclase through receptor interaction with Gαs (26). Therefore, adenylate cyclase activity in response to 5′-AMP (50 μM) or adenosine (50 μM) was investigated by determining the cumulative production of cAMP over 15 min in the presence of the non-xanthine phosphodiesterase inhibitor Ro 20-1724 (0.5 mM; reference 21). Compared with control (HBSS), administration of 5′-AMP and adenosine resulted in similar increases in intracellular cAMP levels (287 ± 23% and 321 ± 21% increase, respectively), suggesting that 5′-AMP and adenosine are equipotent in their stimulation of adenylate cyclase activity.
Discussion
Endothelial cells, the primary determinants of vascular permeability (33), may suffer PMN-mediated injury and dysfunction during inflammation. Such injury and dysfunction is typically manifested by a decrease in endothelial barrier function (2). In this study, we examined the influence of PMN on endothelial paracellular permeability. This study revealed that intact PMN promote endothelial barrier function. Similarly, small (<1 kD), stable PMN-derived mediators also promote endothelial barrier function. These soluble mediators were identified as 5′-AMP and adenosine based on their physical and chemical characteristics and chromatographic behavior. Further studies revealed that 5′-AMP undergoes conversion to adenosine at the endothelial cell surface. Adenosine, whether from CD73-mediated conversion of 5′-AMP or PMN-derived, activates A2B receptors, increases intracellular cAMP, and results in increased endothelial barrier function.
Neutrophil-mediated endothelial cell injury may occur during inflammation but requires PMN-endothelial cell adhesion (5, 6). However, PMN-endothelial adhesion does not necessarily result in endothelial injury. For example, in vitro, PMN-endothelial adhesion or transendothelial PMN migration does not lead to increased permeability without prior endothelial cell activation (5, 7, 34). Furthermore, in vivo, ∼100 billion PMN normally exit the circulation per day (8) via PMN-endothelial cell adhesion and transendothelial PMN migration, without clear adverse consequences. In the studies presented here, PMN interactions with unactivated endothelium resulted in enhanced endothelial barrier function. To eliminate the possible direct physical obstruction to tracer flux by PMN, we examined the influence of cell-free supernatants from activated PMN. Under these conditions, PMN supernatants increased endothelial barrier function by as much as twofold. Thus, activated PMN release soluble mediators that dramatically promote endothelial barrier function. This is the first report of PMN-mediated enhancement of endothelial barrier function and is contrary to the dogma that PMN-endothelial interactions result in endothelial injury. Other investigators using similar models have not observed such an influence of activated PMN or activated PMN supernatants (35, 36), possibly due to their use of lower PMN concentrations. Indeed, in these experiments, low numbers of PMN (105 or 106 per monolayer) or dilute PMN supernatants (>1:8 dilution) failed to influence endothelial paracellular permeability, suggesting that these PMN-derived soluble mediators act locally.
Upon activation, PMN release numerous compounds, either via exocytosis of intracellular granules or directly across the cellular membrane (37). Based on biophysical features of this PMN-derived mediator (i.e., stable, MW <1 kD) and previous work demonstrating actions of PMN-derived 5′-AMP and its metabolite, adenosine, in an epithelial cell model (18), we measured concentrations of these compounds in supernatants from activated PMN. Concentrations of 5′-AMP and adenosine were in a range sufficient to influence endothelial paracellular permeability. However, because the concentrations of these compounds in supernatants do not fully account for the observed changes in endothelial paracellular permeability, their effects are likely augmented by or additive with other, unidentified PMN-derived compounds. As with other PMN-derived mediators, local concentrations of 5′-AMP and adenosine are greater within the PMN-endothelial microenvironment (23, 38). The elevated concentrations of 5′-AMP in activated PMN supernatants are likely due to extracellular metabolism of ATP and ADP released by viable PMN (39, 40). Similarly, the elevated concentrations of adenosine in activated PMN supernatants are likely due to extracellular metabolism of PMN-derived 5′-AMP (40). The mechanisms by which PMN release adenine nucleotides and adenosine into the extracellular environment are not clearly defined but appear to involve direct transport or diffusion across the cell membrane (19). The concentrations of 5′-AMP and adenosine in activated PMN supernatants reported here are consistent with those reported by others (18, 39), and as with previous reports (39), even higher concentrations of these compounds were observed upon PMN activation with PMA (data not shown).
Although we investigated PMN-derived 5′-AMP and adenosine, other cell types may contribute to intravascular concentrations of these compounds and thereby modulate endothelial paracellular permeability. Endothelial cells themselves release adenine nucleotides and adenosine (19) that have the potential to influence permeability through an autocrine pathway. Eosinophils also release 5′-AMP and adenosine in quantities similar to that produced by PMN (41). Platelets release adenine nucleotides and adenosine (42). Platelet-derived ATP and ADP concentrations may reach 20 μM in serum and even higher (i.e., mM) at the endothelial cell surface (43). Through endothelial cell surface conversion of ATP and ADP to 5′-AMP via ecto-ATP diphosphohydrolase (CD39; reference 44) and subsequent conversion to adenosine via 5′-ectonucleotidase/ CD73, such platelet-derived ATP and ADP may indirectly modulate endothelial paracellular permeability (44). Similar enzymatic conversion of ATP to adenosine may modulate endothelial paracellular permeability in conditions where serum ATP concentrations reach 20–100 μM such as organ injury (45) and traumatic shock (46).
Endothelial cell surface metabolism of 5′-AMP to adenosine requires 5′-ectonucleotidase/CD73; other ectonucleotidases are not capable of metabolizing 5′-AMP (44). Through inhibition of endothelial 5′-ectonucleotidase/ CD73 with APCP or mAb 1E9, our data demonstrate that the 5′-AMP–mediated decrease in endothelial paracellular permeability requires endothelial cell surface conversion of 5′-AMP to adenosine. The competitive inhibitor APCP abolished the influence of 5′-AMP, whereas the less potent, noncompetitive inhibitor mAb 1E9 substantially diminished the influence of 5′-AMP. In comparison, neither 5′-ectonucleotidase/CD73 inhibitor had any influence on the adenosine-mediated decrease in endothelial paracellular permeability. Thus, 5′-ectonucleotidase/CD73 influences endothelial cell permeability in addition to its previously characterized role as a cell adhesion molecule (47). This role of 5′-ectonucleotidase in modulating endothelial paracellular permeability via conversion of 5′-AMP to adenosine and subsequent adenosine receptor activation is supported by its previously reported roles in modulating the intestinal chloride secretory effects of PMN-derived 5′-AMP (18) and the anti-inflammatory effects of endothelial-derived 5′-AMP (19). Although this study was restricted to the role of endothelial CD73 in the 5′-AMP–mediated decrease in endothelial paracellular permeability, other cell types may also enhance this 5′-AMP–mediated decrease in endothelial paracellular permeability. For example, PMN have themselves been demonstrated to contribute 5′-ectonucleotidase activity (40). At present, it is not known whether such 5′-ectonucleotidases are regulated or whether such regulation contributes to modulation of vascular permeability in vivo. The CD73 promoter contains a binding site for the nuclear transcription factor NF-κB (48), establishing the possibility that this enzyme may be regulated by proinflammatory mediators (17). Such a mechanism is well established for other endothelial proteins (49).
Adenosine A2B receptor activation, either directly by adenosine or after conversion of 5′-AMP, results in an increase in endothelial barrier function. This increase in endothelial barrier function is the opposite of that expected with endothelial injury but is consistent with adenosine's other, antiinflammatory roles. Although adenosine promotes PMN chemotaxis and PMN-endothelial adherence at low concentrations (i.e., picomolar), adenosine at higher concentrations (i.e., nanomolar) decreases PMN reactive oxygen species and leukotriene generation and inhibits PMN-endothelial adhesion (50, 51). These latter direct effects of adenosine on PMN function may account for the inhibition of PMN-mediated organ injury observed with adenosine administration both in vitro (52) and in vivo (53). Therefore, adenosine may also indirectly promote endothelial barrier function during inflammation via inhibition of PMN cytotoxicity. Other direct antiinflammatory effects of adenosine include inhibition of monocyte and endothelial release of inflammatory cytokines, inhibition of endothelial cell adhesion molecule expression, inhibition of PMN leukotriene biosynthesis, and activation of cellular antioxidant defense systems (51, 54). Adenosine's antiinflammatory effects extend into the postinflammatory phase via its promotion of wound healing (55). Thus, the adenosine-mediated increase in endothelial barrier function demonstrated in this study appears to represent part of an overall antiinflammatory role of this molecule (54).
In our model, adenosine primarily derived from 5′-AMP promoted endothelial barrier function. Adenosine receptor subtypes may be classified as A1, A2A, A2B, or A3. The presence of A2B receptors in HUVEC was demonstrated by RT-PCR by Montesinos (55) and confirmed by Western blot in this study. Adenosine A2 receptor activation (either A2A or A2B) increases intracellular levels of cAMP via stimulation of adenylate cyclase and receptor interaction with Gαs (26), whereas adenosine A1 or A3 receptor activation decreases intracellular levels of cAMP. In our model, adenosine decreased endothelial paracellular permeability and increased intracellular levels of cAMP via adenosine A2B receptor activation. Therefore, our observations are consistent with the known consequences of adenosine A2 receptor activation and prior observations demonstrating a close association between endothelial paracellular permeability antagonists and increased intracellular cAMP (56). Previous studies have yielded conflicting results on the influence of adenosine receptor activation on endothelial paracellular permeability. These differences may be due to differences in endothelial cell origin or proportional expression of adenosine receptor subtypes (21, 25). Our data do not explain the dependence of enhanced endothelial barrier function upon activation of low-affinity A2B receptors rather than activation of high-affinity A2A receptors (26). Possibly, this difference may be due to either increased HUVEC A2B receptor expression or intracellular cAMP compartmentalization.
Although the mechanism by which adenosine A2B receptor activation decreases endothelial paracellular permeability is not clear, it is attractive to propose that it is mediated through increased levels of intracellular cAMP for the following reasons. First, activation of adenosine A2B receptors increase intracellular cAMP (26). And second, there is an association between permeability antagonists and increased intracellular cAMP (56). In endothelial cells from different origins, increased intracellular cAMP (and decreased permeability) is accompanied by a rapid increase in polymerized actin, an increase in cell surface area, a loss of stress fibers, and increased phosphorylation of intermediate filaments, suggesting that cAMP–mediated changes in endothelial paracellular permeability may be due to alterations in cellular cytoskeleton (57). In turn, these cytoskeletal alterations most likely are dependent upon the activity of Rho GTPase proteins (58). Ultimately, these intracellular processes alter endothelial paracellular permeability via their actions on the intercellular junctions (59). Tight junctions and adherens junctions determine paracellular permeability in endothelial cells (4). In endothelial cells where baseline permeability is relatively high such as in HUVEC, organized tight junctions are not expressed and therefore, adherens junctions may be the predominate determinant of permeability (5). The importance of adherens junctions in determining endothelial paracellular permeability is supported by recent observations of a close correlation of permeability with phosphorylation of VE-cadherin, an adherens junction protein (60).
Therefore, based on the data presented here, we propose that PMN-derived 5′-AMP decreases endothelial paracellular permeability via CD73-mediated conversion of 5′-AMP to adenosine (Fig. 10). Adenosine, whether from endothelial CD73-mediated conversion of 5′-AMP or PMN-derived, activates endothelial adenosine A2B receptors. Receptor activation stimulates adenylate cyclase and leads to an increase in intracellular cAMP. This increase in cAMP results in cytoskeletal changes that decrease paracellular permeability via actions at interendothelial junctions. This decrease in endothelial paracellular permeability prevents edema and intravascular loss of fluid and solutes. Such a model suggests an inherent mechanism to dampen increased endothelial paracellular permeability during PMN-endothelial interactions.
Figure 10.

Proposed model of PMN-derived 5′-AMP–mediated decrease in endothelial paracellular permeability. PMN-derived 5′-AMP undergoes conversion to adenosine via endothelial 5′-ectonucleotidase (CD73). Adenosine activates the endothelial A2B receptor (A2BR), and via elevated intracellular cAMP levels, decreases endothelial paracellular permeability through actions at the endothelial intercellular junction.
Acknowledgments
We wish to thank Dr. Linda Thompson for supplying antibodies and Ms. Margaret Morrissey, Ms. Nana Fueki, and Ms. Andrea Dzus for their technical assistance.
This work was supported by National Institutes of Health grants KO8 HL03688-01 (to P.F. Lennon), DK50189 (to S.P. Colgan), DK09699 (to C.T. Taylor), and HL52886 and HL56086 (to G.L. Stahl). P.F. Lennon is a New Investigator of the Foundation for Anesthesia Education and Research (FAER).
Abbreviations used in this paper
- ADAC
adenosine amine congener
- AMP
adenosine monophosphate
- APCP
α,β-methylene ADP
- CPCA
5′-(N-cyclopropyl)-carboxamidoadenosine
- CSC
8-(3-chlorostyryl) caffeine
- E-ADO
ethenoadenosine
- E-AMP
ethenoadenosine-5′-monophosphate
- ECL
enhanced chemiluminescence
- HMVEC
human microvascular endothelial cells
- HUVEC
human umbilical vein endothelial cells
- NBTI
nitrobenzylthioinosine
- NECA
5′-(N-ethylcarboxamido)adenosine
References
- 1.Hansen PR. Role of neutrophils in myocardial ischemia and reperfusion. Circulation. 1995;91:1872–1885. doi: 10.1161/01.cir.91.6.1872. [DOI] [PubMed] [Google Scholar]
- 2.Takano T, Clish CB, Gronert K, Petasis N, Serhan CN. Neutrophil-mediated changes in vascular permeability are inhibited by topical application of aspirin-triggered 15-epi-lipoxin A4 and novel lipoxin B4stable analogues. J Clin Invest. 1998;101:819–826. doi: 10.1172/JCI1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sunnergran KP, Rovetto MJ. Myocyte and endothelial injury with ischemia reperfusion in isolated rat hearts. Am J Physiol. 1987;252:H1211–H1217. doi: 10.1152/ajpheart.1987.252.6.H1211. [DOI] [PubMed] [Google Scholar]
- 4.Dejana E, Corada M, Lampugnani MG. Endothelial cell-to-cell junctions. FASEB J. 1995;9:910–918. [PubMed] [Google Scholar]
- 5.Del Maschio A, Zanetti A, Corada M, Rival Y, Ruco L, Lampugnani MG, Dejana E. Polymorphonuclear leukocyte adhesion triggers the disorganization of endothelial cell-to-cell adherens junctions. J Cell Biol. 1996;135:497–510. doi: 10.1083/jcb.135.2.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hernandez LA, Grisham MB, Twohig B, Arfors KE, Harlan JM, Granger DN. Role of neutrophils in ischemia-reperfusion-induced microvascular injury. Am J Physiol. 1987;253:H699–H703. doi: 10.1152/ajpheart.1987.253.3.H699. [DOI] [PubMed] [Google Scholar]
- 7.Huang AJ, Furie MB, Nicholson SC, Fischbarg J, Liebovitch LS, Silverstein SC. Effects of human neutrophil chemotaxis across human endothelial cell monolayers on the permeability of these monolayers to ions and macromolecules. J Cell Physiol. 1988;135:355–366. doi: 10.1002/jcp.1041350302. [DOI] [PubMed] [Google Scholar]
- 8.Dancey JT, Deubelbeiss KA, Harker LA, Finch CA. Neutrophil kinetics in man. J Clin Invest. 1976;58:705–715. doi: 10.1172/JCI108517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zünd G, Uezono S, Stahl GL, Dzus AL, McGowan FX, Hickey PR, Colgan SP. Hypoxia enhances induction of endothelial ICAM-1: role for metabolic acidosis and proteasomes. Am J Physiol. 1997;273:C1571–C1580. doi: 10.1152/ajpcell.1997.273.5.C1571. [DOI] [PubMed] [Google Scholar]
- 10.Gimbrone MA. Culture of vascular endothelium. Prog Hemostasis Thromb. 1978;3:1–28. [PubMed] [Google Scholar]
- 11.Colgan SP, Dzus AL, Parkos CA. Epithelial exposure to hypoxia modulates neutrophil transepithelial migration. J Exp Med. 1996;184:1003–1015. doi: 10.1084/jem.184.3.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Albelda SM, Sampson PM, Haselton FR, McNiff JM, Mueller SM, Williams SK, Fishman AP, Levine EM. Permeability characteristics of cultured endothelial cell monolayers. J Appl Physiol. 1988;64:308–322. doi: 10.1152/jappl.1988.64.1.308. [DOI] [PubMed] [Google Scholar]
- 13.Mizuno-Yagyu Y, Hashida R, Mineo C, Ikegami S, Ohkuma S, Takano T. Effect of PGI2on transcellular transport of fluorescein dextran through an arterial endothelial monolayer. Biochem Pharmacol. 1987;36:3809–3813. doi: 10.1016/0006-2952(87)90442-4. [DOI] [PubMed] [Google Scholar]
- 14.Sanders SE, Madara JL, McGuirk DK, Gelman DS, Colgan SP. Assessment of inflammatory events in epithelial permeability: a rapid screening method using fluorescein dextrans. Epithelial Cell Biol. 1995;4:25–34. [PubMed] [Google Scholar]
- 15.Casnocha SA, Eskin SG, Hall ER, McIntire LR. Permeability of human endothelial monolayers: effect of vasoactive agonists and cAMP. J Appl Physiol. 1989;67:1997–2005. doi: 10.1152/jappl.1989.67.5.1997. [DOI] [PubMed] [Google Scholar]
- 16.Garcia JGN, Siflinger-Birnboim A, Bizios R, Del Vecchio PJ, Fenton JW, Malik AB. Thrombin-induced increase in albumin permeability across the endothelium. J Cell Physiol. 1986;128:96–104. doi: 10.1002/jcp.1041280115. [DOI] [PubMed] [Google Scholar]
- 17.Bonitati AE, Agarwal KC, Rounds S. A simple assay for ecto-5′-nucleotidase using intact pulmonary artery endothelial cells. Biochem Pharmacol. 1993;46:1467–1473. doi: 10.1016/0006-2952(93)90113-b. [DOI] [PubMed] [Google Scholar]
- 18.Madara JL, Patapoff TW, Gillece-Castro B, Colgan SP, Parkos CA, Delp C, Mrsny RJ. 5′-Adenosine monophosphate is the neutrophil-derived paracrine factor that elicits chloride secretion from T84 intestinal epithelial cell monolayers. J Clin Invest. 1993;91:2320–2325. doi: 10.1172/JCI116462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morabito L, Montesinos MC, Schreibman DM, Balter L, Thompson LF, Resta T, Carlin G, Huie MA, Cronstein BN. Methotrexate and sulfasalazine promote adenosine release by a mechanism that requires ecto-5′-nucleotidase conversion of adenine nucleotides. J Clin Invest. 1998;101:295–300. doi: 10.1172/JCI1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Puffinbarger NK, Hansen KR, Resta R, Laurent AB, Knudsen TB, Madara JL, Thompson LF. Production and characterization of multiple antigenic peptide antibodies to the adenosine A2b receptor. Mol Pharmacol. 1995;47:1126–1132. [PubMed] [Google Scholar]
- 21.Watanabe H, Kuhne W, Schwartz P, Piper HM. A2-adenosine receptor stimulation increases macromolecular permeability of coronary endothelial cells. Am J Physiol. 1992;262:H1174–H1181. doi: 10.1152/ajpheart.1992.262.4.H1174. [DOI] [PubMed] [Google Scholar]
- 22.Parkos CA, Colgan SP, Liang A, Nusrat A, Bacarra AE, Carnes DK, Madara JL. CD47 mediates post-adhesive events required for neutrophil migration across polarized intestinal epithelia. J Cell Biol. 1996;132:437–450. doi: 10.1083/jcb.132.3.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tatsumi T, Fliss H. Hypochlorous acid and chloramine increase endothelial permeability: possible involvement of cellular zinc. Am J Physiol. 1994;267:H1597–H1607. doi: 10.1152/ajpheart.1994.267.4.H1597. [DOI] [PubMed] [Google Scholar]
- 24.Kaslovsky RA, Lai L, Parker K, Malik AB. Mediation of endothelial injury following neutrophil adherence to extracellular matrix. Am J Physiol. 1993;264:L401–L405. doi: 10.1152/ajplung.1993.264.4.L401. [DOI] [PubMed] [Google Scholar]
- 25.Paty PSK, Sherman PF, Shephard JM, Malik AB, Kaplan JE. Role of adenosine in platelet-mediated reduction in pulmonary vascular permeability. Am J Physiol. 1992;262:H771–H777. doi: 10.1152/ajpheart.1992.262.3.H771. [DOI] [PubMed] [Google Scholar]
- 26.Fredholm BB, Abbracchio MP, Burnstock G, Daly JW, Harden TK, Jacobson KA, Leff P, Williams M. Nomenclature and classification of purinoceptors. Pharmacol Rev. 1994;46:143–156. [PMC free article] [PubMed] [Google Scholar]
- 27.Thompson, L.F., A.B. Laurent, M.K. Franklin, W. Gutensohn, and R. Resta. 1995. CD73 workshop panel report. In Leucocyte Typing V. Vol. 1. S.F. Schlossman, L. Boumsell, W. Gilks, J.M. Harlan, T. Kishimoto, C. Morimoto, J. Ritz, S. Shaw, R. Silverstein, T. Springer, T.F. Tedder, and R.F. Todd, editors. Oxford University Press, New York. 564– 567.
- 28.Strohmeier GR, Lencer WI, Patapoff TW, Thompson LF, Carlson SL, Mow SJ, Carnes DK, Mrsny RJ, Madara JL. Surface expression, polarization, and functional significance of CD73 in human intestinal epithelia. J Clin Invest. 1997;99:2588–2601. doi: 10.1172/JCI119447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pearson JD, Carleton JS, Gordon JL. Metabolism of adenine nucleotides by ectoenzymes of vascular endothelial and smooth-muscle cells in culture. Biochem J. 1980;190:421–429. doi: 10.1042/bj1900421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Airas L, Niemela J, Salmi M, Puurunen T, Smith DJ, Jalkanen S. Differential regulation and function of CD73, a glycosyl-phosphatidylinositol-linked 70-kD adhesion molecule, on lymphocytes and endothelial cells. J Cell Biol. 1997;136:421–431. doi: 10.1083/jcb.136.2.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moser GH, Schrader J, Deussen A. Turnover of adenosine in plasma of human and dog blood. Am J Physiol. 1989;256:C799–C806. doi: 10.1152/ajpcell.1989.256.4.C799. [DOI] [PubMed] [Google Scholar]
- 32.Matthews JB, Tally KJ, Smith JA, Zeind AJ, Hrnjez BJ. Activation of Cl secretion during chemical hypoxia by endogenous release of adenosine in intestinal epithelial monolayers. J Clin Invest. 1995;96:117–125. doi: 10.1172/JCI118010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramirez CA, Colton CK, Smith KA, Stemerman MB, Lees RS. Transport of 125I-albumin across normal and deendothelialized rabbit thoracic aorta in vivo. Arteriosclerosis. 1984;4:283–291. doi: 10.1161/01.atv.4.3.283. [DOI] [PubMed] [Google Scholar]
- 34.Allport JR, Ding H, Collins T, Gerritsen ME, Luscinskas FW. Endothelial-dependent mechanisms regulates leukocyte transmigration: a process involving the proteasome and disruption of the vascular endothelial-cadherin complex at endothelial cell-to-cell junctions. J Exp Med. 1997;186:517–527. doi: 10.1084/jem.186.4.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gibbs LS, Lai L, Malik AB. Tumor necrosis factor enhances the neutrophil-dependent increase in endothelial permeability. J Cell Physiol. 1990;145:496–500. doi: 10.1002/jcp.1041450315. [DOI] [PubMed] [Google Scholar]
- 36.Harlan JM, Schwartz BR, Reidy MA, Schwartz SM, Ochs HD, Harker LA. Activated neutrophils disrupt endothelial monolayer integrity by an oxygen radical-independent mechanism. Lab Invest. 1985;52:141–150. [PubMed] [Google Scholar]
- 37.Henson, P.M., J.E. Henson, C. Fittschen, D.L. Bratton, and D.W.H. Riches. 1992. Degranulation and Secretion by Phagocytic Cells. In Inflammation: Basic Principles and Clinical Correlates. J.I. Gallin, I.M. Goldstein, and R. Snyderman, editors. Raven Press, LTD., New York. 511–539.
- 38.Siflinger-Birnboim A, Malik AB. Neutrophil adhesion to endothelial cells impairs the effects of catalase and glutathione in preventing endothelial injury. J Cell Physiol. 1993;155:234–239. doi: 10.1002/jcp.1041550203. [DOI] [PubMed] [Google Scholar]
- 39.van Waeg G, Van den Berghe G. Purine catabolism in polymorphonuclear neutrophils. J Clin Invest. 1991;87:305–312. doi: 10.1172/JCI114987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kitakaze M, Hori M, Morioka T, Takashima S, Minamino T, Sato H, Inoue M, Kamada T. Attenuation of ecto-5′-nucleotidase activity and adenosine release in activated human polymorphonuclear leukocytes. Circ Res. 1993;73:524–533. doi: 10.1161/01.res.73.3.524. [DOI] [PubMed] [Google Scholar]
- 41.Resnick MB, Colgan SP, Patapoff TW, Mrsny RJ, Awtrey CS, Delp-Archer C, Weller PF, Madara JL. Activated eosinophils evoke chloride secretion in model intestinal epithelia primarily via regulated release of 5′-AMP. J Immunol. 1993;151:5716–5723. [PubMed] [Google Scholar]
- 42.McGarrity ST, Stephenson AH, Hyers TM, Webster RO. Inhibition of neutrophil superoxide anion generation by platelet products: role of adenine nucleotides. J Leukoc Biol. 1988;44:411–421. doi: 10.1002/jlb.44.5.411. [DOI] [PubMed] [Google Scholar]
- 43.Gordon EL, Pearson JD, Slakey LL. The hydrolysis of extracellular adenine nucleotides by cultured endothelial cells from pig aorta. J Biol Chem. 1986;261:15496–15504. [PubMed] [Google Scholar]
- 44.Marcus AJ, Broekman MJ, Drosopoulos JHF, Islam N, Alyonycheva TN, Safier LB, Hajjar KA, Posnett DN, Schoenborn MA, et al. The endothelial cell ecto-ADPase responsible for inhibition of platelet function is CD39. J Clin Invest. 1997;99:1351–1360. doi: 10.1172/JCI119294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Born GVR, Kratzer MAA. Source and concentration of extracellular adenosine triphosphate during haemostasis in rats, rabbits and man. J Physiol (Lond) 1984;354:419–429. doi: 10.1113/jphysiol.1984.sp015385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kalckar HM, Lowry OH. The relationship between traumatic shock and the release of adenine compounds. Am J Physiol. 1947;149:240–245. doi: 10.1152/ajplegacy.1947.149.1.240. [DOI] [PubMed] [Google Scholar]
- 47.Airas L, Hellman J, Salmi M, Bono P, Puurunen T, Smith DJ, Jalkanen S. CD73 is involved in lymphocyte binding to the endothelium: characterization of lymphocyte-vascular adhesion protein 2 identifies it as CD73. J Clin Invest. 1995;182:1603–1608. doi: 10.1084/jem.182.5.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hansen KR, Resta R, Webb CF, Thompson LF. Isolation and characterization of the promoter of the human 5′-nucleotidase (CD73)-encoding gene. Gene (Amst) 1995;167:307–312. doi: 10.1016/0378-1119(95)00574-9. [DOI] [PubMed] [Google Scholar]
- 49.Collins T, Read MA, Neish AS, Whitley MZ, Thanos D, Maniatis T. Transcriptional regulation of endothelial cell adhesion molecules: NF-κB and cytokine-inducible enhancers. FASEB J. 1995;6:899–909. [PubMed] [Google Scholar]
- 50.Cronstein BN, Levin RI, Philips M, Hirschhorn R, Abramson SB, Weissmann G. Neutrophil adherence to endothelium is enhanced via adenosine A1 receptors and inhibited via adenosine A2receptors. J Immunol. 1992;148:2159–2162. [PubMed] [Google Scholar]
- 51.Krump E, Picard S, Mancini J, Borgeat P. Suppression of leukotriene B4 biosynthesis by endogenous adenosine in ligand-activated human neutrophils. J Exp Med. 1997;186:1401–1406. doi: 10.1084/jem.186.8.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cronstein BN, Levin RI, Belanoff J, Weissmann G, Hirschorn R. Adenosine: an endogenous inhibitor of neutrophil-mediated injury to endothelial cells. J Clin Invest. 1986;78:760–770. doi: 10.1172/JCI112638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adkins WK, Barnard JW, Moore TM, Allison RC, Prasad VR, Taylor AE. Adenosine prevents PMN-induced lung injury via an A2receptor mechanism. J Appl Physiol. 1993;74:982–988. doi: 10.1152/jappl.1993.74.3.982. [DOI] [PubMed] [Google Scholar]
- 54.Bouma MG, van den Wildenberg FAJM, Buurman WA. The anti-inflammatory potential of adenosine in ischemia-reperfusion injury: established and putative beneficial actions of a retaliatory metabolite. Shock. 1997;8:313–320. doi: 10.1097/00024382-199711000-00001. [DOI] [PubMed] [Google Scholar]
- 55.Montesinos MC, Gadangi P, Longaker M, Sung J, Levine J, Nilsen D, Reibman J, Li M, Jiang C-K, Hirschhorn R, et al. Wound healing is accelerated by agonists of adenosine A2 (Gαs-linked) receptors. J Exp Med. 1997;186:1615–1620. doi: 10.1084/jem.186.9.1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Siflinger-Birnboim A, Bode DC, Malik AB. Adenosine 3′,5′-cyclic monophosphate attenuates neutrophil-mediated increase in endothelial permeability. Am J Physiol. 1993;264:H370–H375. doi: 10.1152/ajpheart.1993.264.2.H370. [DOI] [PubMed] [Google Scholar]
- 57.Stelzner TJ, Weil JV, O'Brien RF. Role of cyclic adenosine monophosphate in the induction of endothelial barrier properties. J Cell Physiol. 1989;139:157–166. doi: 10.1002/jcp.1041390122. [DOI] [PubMed] [Google Scholar]
- 58.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 59.Lum H, Malik AB. Regulation of vascular endothelial barrier function. Am J Physiol. 1994;267:L223–L241. doi: 10.1152/ajplung.1994.267.3.L223. [DOI] [PubMed] [Google Scholar]
- 60.Dejana E. Endothelial adherens junctions: implications in the control of vascular permeability and angiogenesis. J Clin Invest. 1996;98:1949–1953. doi: 10.1172/JCI118997. [DOI] [PMC free article] [PubMed] [Google Scholar]