

Abstract
Cytotoxic T lymphocytes (CTLs) are primary mediators of viral clearance, but high viral burden can result in deletion of antigen-specific CTLs. We previously reported a potential mechanism for this deletion: tumor necrosis factor (TNF)-α–mediated apoptosis resulting from stimulation with supraoptimal peptide–major histocompatibility complex. Here, we show that although death is mediated by TNF-α and its receptor (TNF-RII), surprisingly neither the antigen dose dependence of TNF-α production nor that of TNF-RII expression can account for the dose dependence of apoptosis. Rather, a previously unrecognized effect of supraoptimal antigen in markedly decreasing levels of the antiapoptotic protein Bcl-2 was discovered and is likely to account for the gain in susceptibility or competence to sustain the death signal through TNF-RII. This decrease requires a signal through the TCR, not just through TNF-RII. Although death mediated by TNF-RII is not as widely studied as that mediated by TNF-RI, we show here that it is also dependent on proteolytic cleavage by caspases and triggered by a brief initial encounter with antigen. These results suggest that determinant density can regulate the immune response by altering the sensitivity of CTLs to the apoptotic effects of TNF-α by decreasing Bcl-2 levels.
Keywords: T lymphocytes, cytotoxic; apoptosis; protooncogene; proteins c-bel-2; tumor necrosis factor
hen confronted with extremely high levels of antigen, the immune system is often unable to function optimally. Historically, such a challenge was shown to result in a state of nonresponsiveness termed high zone tolerance (1). More recently, in a lymphocytic choriomeningitis virus (LCMV) model in which the infection is not cleared and very high titers of virus are produced within cells, it has been reported that CTLs are deleted from the periphery (2). This phenomenon has been termed clonal exhaustion and is thought to be due to the repetitive stimulation of CTLs by specific antigen. During HIV infection, as in the LCMV model, extremely high viral loads can be obtained that correlate with a loss of HIV-specific CTLs (3–5). It has been postulated that clonal exhaustion may also be occurring in this system.
In previous studies, we selectively expanded immune spleen cells to obtain high and low avidity CTLs with different efficacies in clearing viral infections (6), and showed that the stimulation of high avidity CTLs with APCs presenting supraoptimal densities of peptide–MHC results in the apoptotic deletion of CTLs (7). The appearance of apoptotic nuclei is maximal at 40–48 h and is mediated by TNF-α. Similarly, antibodies against CD3 have also been shown to induce a TNF-α–mediated apoptosis of bulk lymph node CD8+ T cells (8). Therefore, it is tempting to speculate that the in vivo death that occurs when the viral load is very high is the result of exposure to supraoptimal peptide–MHC determinant density similar to the death observed in our in vitro model.
Perhaps it is not surprising that CTLs are restricted in the level of stimulation that will result in their activation and proliferation. It is now well documented that CD4+8+ thymocytes can be readily stimulated to mature into single positive cells (CD4+8− or CD4−8+) if presented with the appropriate peptide (9, 10). However, the concentration of the peptide used and its affinity for the MHC molecule are critical, as too little or too much will result in death of the immature lymphocyte. Similarly, the capacity of a mature T cell to tolerate only a defined range of signal strength is likely the result of the same qualities required for appropriate thymic selection.
In this report, we further investigate the mechanism by which apoptotic death occurs as a result of stimulation with high dose peptide. The death is mediated by TNF-RII rather than TNF-RI, the receptor usually associated with TNF-mediated apoptotic death (11–14). Interestingly, although both TNF and its receptor were upregulated by antigen stimulation, neither the threshold for production of TNF-α nor the threshold for receptor expression was found to correlate with the susceptibility to apoptotic death. Thus although both TNF and TNF-RII are necessary for high dose induction of apoptotic death of CTL, under usual conditions they are not sufficient. This suggested that either another molecule contributing to death is upregulated or a protective molecule is downregulated. To pursue this, two protective molecules, Bcl-2 and Bcl-XL, were examined. Bcl-2 and Bcl-XL have previously been shown to be potent inhibitors of apoptotic death induced under a variety of conditions (15), although their role in TNF-α–mediated death is unknown. We found that a previously unrecognized effect of supraoptimal antigen levels was to decrease the level of Bcl-2 protein in CTLs and thus to render them competent to be killed by exposure to the TNF-α they secrete. Death required a signal, not just through TNF-RII but also through the TCR. This finding suggests that a decrease in Bcl-2 may account for the TNF-α–mediated apoptotic death triggered by supraoptimal peptide–MHC stimulation.
Materials and Methods
Mice and Antibodies.
BALB/c mice were purchased from The Jackson Laboratory (Bar Harbor, ME). P815 is a DBA/2-derived mastocytoma. Blocking antibodies for the TNF-RI and TNF-RII were a gift of Dr. Robert Schreiber (Washington University School of Medicine, St. Louis, MO) or were purchased from Genzyme (Cambridge, MA). The biotinylated anti–TNF-R antibodies were purchased from HyCult Biotechnology (Uden, The Netherlands). FITC-avidin secondary antibody, anti–TNF-α capture antibody, biotin conjugated anti–TNF-α detection antibody, anti–Bcl-2, and anti–Bcl-X were obtained from PharMingen (San Diego, CA) or Genzyme. Antivinculin and antiactin were purchased from Sigma Chemical Co. (St. Louis, MO).
Peptides, Protein, and Inhibitors.
Peptides were synthesized on an automated peptide synthesizer (no. 430A; PE Applied Biosystems, Foster City, CA) using t-boc chemistry (16) and purified as previously described (17). The I10 peptide (RGPGRAFTVI) represents the immunodominant CTL epitope in the V3 loop of HIV-1 IIIB gp160 in mice of the H-2d haplotype (17–19). Soluble recombinant Dd (H-2Dd [α3]) was isolated as previously described (20) and was the generous gift of L.F. Boyd and D.H. Margulies (NIAID, Bethesda, MD). Boc-Asp-fluoromethyl ketone (BD-FMK)1 and CBZ-Phe-Ala-fluoromethyl ketone (βZFA-FMK) were obtained from Enzyme System Products, (Livermore, CA).
Generation of CTL Lines.
7.5 × 106 responding BALB/c spleen cells from mice previously immunized with a recombinant vaccinia virus expressing the gp160 protein from HIV-1IIIB were cocultured either with 3.5 × 106 stimulating irradiated (3,000 rads) BALB/c splenocytes previously pulsed with various concentrations (100, 0.1, or 0.0001 μM) of I10 peptide or with irradiated nonpulsed splenocytes in the presence of 1 μM free peptide in 24-well plates containing 2 ml/well of a 1:1 mixture of RPMI 1640 and Eagle-Hanks Amino Acid (EHAA) medium supplemented with L-glutamine, sodium pyruvate, nonessential amino acids, penicillin, streptomycin, 5 × 10−5 M β-mercaptoethanol, 10% FCS, and 10% T-stim (Collaborative Biomedical Products, Bedford, MA). CTL lines were established from primary cultures and were maintained by weekly restimulation of 3–5 × 105 cells/well in the presence of 5 × 106 irradiated (3,000 rads) BALB/c spleen cells pulsed with the appropriate concentration of I10 peptide.
Proliferation Assays.
CTLs were plated at 5 × 104/well in a 96-well round-bottomed microtiter plate. Irradiated (3,000 rads) BALB/c splenocytes previously pulsed with I10 peptide and washed three times were added at 3 × 105/well. Supernatant from the final wash of stimulators pulsed with 100 μM I10 peptide was reserved and added to wells with 0.001-μM-pulsed stimulators at a final dilution of 1:1 to ensure that effects seen with 100-μM-pulsed stimulators were not due to residual free I10 peptide that might have bound to the CTLs directly. In some cases, anti–TNF-RI or anti–TNF-RII blocking antibodies (21) were added at 5 μg/well. Proliferation was measured by addition of 1 μCi [3H]thymidine per well at 24 and 48 h and plates were harvested at 48 and 72 h, respectively. Results obtained at the second harvest were qualitatively similar. Results were expressed as the geometric mean of triplicate cultures.
Assay for TNF-α Production.
CTLs were stimulated with pulsed spleen APCs, as for proliferation assays, or with substrate-bound soluble Dd. To measure soluble TNF-α, culture supernatant was harvested at 24 h and assayed for TNF-α by an ELISA using capture and biotinylated detecting antibodies from PharMingen as described in their Cytokine ELISA Protocol. Surface-bound TNF-α was detected at various times after stimulation using anti–TNF-α or isotypic control FITC- or PE-conjugated antibodies.
Apoptotic Death Assays.
To detect apoptotic nuclei, CTLs were enriched by centrifuging lines over ficoll to remove irradiated APCs. After washing, 5 × 104 CTLs were added along with 3 × 105 peptide-pulsed stimulators (depleted of Thy 1.2+ cells) in 0.2 ml of medium that contained 10% T-stim as a source of IL-2 per well of a 96-well plate in triplicate. After 40–45 h, cells were harvested and incubated with biotin-conjugated Thy 1.2 mAb and FcBlock followed by PE-avidin (to identify CTLs), washed, and incubated for 30 min with 5 μg/ml Hoechst 33342 (Molecular Probes, Inc., Eugene, OR) at 37°C. Cells were then centrifuged and the pellet was resuspended in 20 μl PBS. Apoptotic nuclei were identified by fluorescent microscopy (22). Apoptosis was also estimated by the reduction in CTL proliferation at 24 or 48 h and the decrease in forward and side scatter detected by flow cytometry.
Flow Cytometry.
For flow cytometric analysis, 2 × 105 cells were washed and resuspended in PBS containing 0.2% BSA and 0.1% sodium azide. Cells were incubated on ice with the appropriate antibody for 30 min and then washed. Where necessary, a secondary reagent was then added for an additional 30 minutes and the cells were again washed. Samples were analyzed on a FACScan® (Becton Dickinson, Mountain View, CA). Background staining was assessed by use of a similarly conjugated isotypic control antibody or by staining with the secondary antibody alone or by the secondary antibody after an unconjugated isotypic control primary antibody.
Western Blot Analysis.
Immulon IV plates were coated with soluble Dd protein (0.4–0.5 μg/well) or anti–TNF-RII antibody (0.5 μg/well) in PBS for 2–3 h at 37°C or overnight at 4°C. Wells were washed, blocked with RPMI containing 10% FCS for 1 h, and washed again. I10 peptide in RPMI plus 10% FCS at the appropriate concentration was added to wells coated with soluble Dd and allowed to bind overnight at 37°C. Wells were then washed extensively. CTLs harvested 3–5 d after routine stimulation were ficolled to remove APCs and added at 1.5 × 105/well. 5 × 106 CTLs were used per antigen concentration tested. CTLs were cultured for 24 h, and then harvested and lysed with 0.5% Triton X-100 containing leupeptin, aprotinin, PMSF, and iodoacetamide. After 45 min on ice, samples were centrifuged and the supernatant was removed and pellet discarded. Sodium deoxycholate (10% final) and SDS (0.2% final) were added to the supernatant. 6 × 105 cell equivalents per sample were run on either a 13 or a 4–12% gradient polyacrylamide gel in either Tris-glycine or MES [2-(N-morpholino) ethane sulfonic acid] buffer systems (Novex, San Diego, CA). Separated proteins were transferred onto nitrocellulose membrane and the blots were blocked overnight with either powdered milk/bovine albumin or SuperBlock-TBS (Pierce Chemical Co., Rockford, IL). After incubation with rabbit antiactin or antivinculin, and rabbit anti–Bcl-2 or anti–Bcl-XL for 1 h, the blots were washed, and then FITC- or alkaline phosphatase–conjugated secondary goat anti–rabbit was added for 1 h. After a further wash, FITC-conjugated secondary antibody was directly quantitated with a Storm fluoroimaging system using ImageQuant (Molecular Dynamics, Sunnyvale, CA). Alternatively, alkaline phosphatase–conjugated secondary was indirectly visualized photographically by reaction with a chemiluminescent substrate.
Results
A Single Brief Exposure to High Dose Antigen Is Adequate to Irreversibly Trigger CTL Death.
In a previous study from this laboratory, we described the TNF-α–mediated apoptotic death of CD8+ CTLs in response to high (supraoptimal) peptide–MHC determinant density (7). The induction of the death signal in resting and recently activated CTLs was found to be identical, and death required ∼40 h in both cases. These observations of the death process in CD8+ CTLs differ from those for the activation-induced cell death (AICD) of CD4+ T cells (23–26). Studies of AICD in CD4+ T cells suggest that antigen can induce death only in cycling cells. Resting cells would thus require two signals, the first to induce entry into the cell cycle and the second to actually initiate apoptosis. To distinguish between these two mechanisms in the case of CD8+ T cells, we investigated the required length of time for antigen presentation to CTLs in order to trigger apoptosis by two approaches. First, to ensure a discrete, limited exposure to MHC–peptide, resting CTLs (11 d after stimulation) were activated by plate-bound, recombinant soluble Dd molecules pulsed with high dose peptide antigen (Fig. 1 A). CTLs were removed from the antigen at various time points and replated in wells with normal medium in the absence of specific peptide–MHC (I10/Dd). At 44 h the CTLs were analyzed by Hoechst staining for the presence of apoptotic nuclei. As little as 2 h of exposure to antigen was adequate to initiate the cell death pathway in a significant portion of the population, and maximal death was induced by only 4 h exposure to supraoptimal antigen (Fig. 1 A). These data suggest that the initial exposure to high dose (supraoptimal) antigen is sufficient for the induction of the death signal. Second, to explore the role of continued antigen exposure, resting CTLs were exposed to APCs pulsed with either high or low doses of peptide for 6 h, and were then moved to different conditions. APCs were removed from the cultures by passage over an R&D T cell enrichment affinity column (coated with Ig-anti-Ig to remove surface Ig and Fc receptor-bearing cells; R&D Systems, Inc., Minneapolis, MN), and the CTLs were replated with fresh APCs presenting either a high or low determinant density. FACS® analysis showed that the recovered CTLs were essentially free of APCs; >98% were I-Ad negative. Also, a control supernatant from the last wash of the APCs was tested for its ability to induce apoptosis to show that no free peptide was carried over to be presented by the CTLs themselves (data not shown). CTLs that were exposed to high-dose antigen for 6 h and then replated with APCs bearing high or low density antigen or no antigen underwent apoptotic death to the same extent as cells that were continuously exposed to APCs pulsed with high dose antigen (Fig. 1 B). These data demonstrate that an initial encounter with high (supraoptimal) antigen induces a signaling event quite different from that induced by an encounter with low (optimal) antigen. Furthermore, they distinguish the apoptotic mechanisms of CD8+ T cells stimulated with supraoptimal antigen from the AICD seen in CD4+ T cells.
Figure 1.
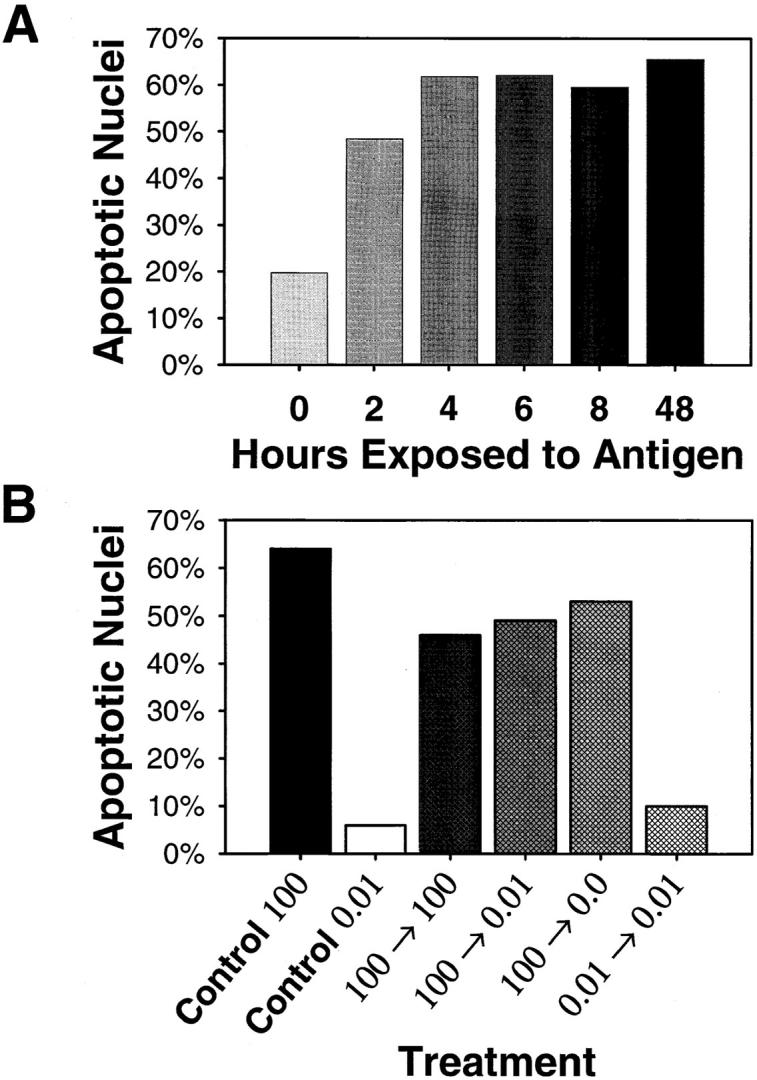
The trigger for apoptotic death is a result of the initial encounter with supraoptimal peptide–MHC determinant density. (A) Wells of an Immulon IV plate were coated with 0.5 μg of recombinant Dd, blocked, and pulsed with 50 μM I10 peptide. After extensive washing, 105 resting CTLs were added to each well. CTLs were transferred to wells without Dd/ I10 at the times indicated and the culture period continued. At 45 h after initiation of the culture, CTLs were harvested and assessed for apoptotic nuclear morphology by Hoechst 33342 staining. The CTLs were irreversibly triggered to undergo apoptotic death after as little as 2 h of antigen exposure. (B) Resting high avidity CTLs were cultured for 6 h in the presence of stimulators pulsed with a high (100 μM) or low (0.01 μM) concentration of I10 peptide. CTLs were then recovered by passage over an R & D T cell purification column, replated with fresh stimulators previously pulsed with various concentrations of I10 peptide, and cultured for an additional 37 h. As controls for the induction of apoptotic death, CTLs were continuously cultured in the presence of stimulators pulsed with high concentrations of peptide (positive control) or low concentrations of peptide (negative control) for 45 h. CTLs exposed to supraoptimal peptide–MHC determinant density for only 6 h underwent apoptotic death to the same extent as CTLs cultured in the continuous presence of APCs that presented high density peptide–MHC, clearly demonstrating that the initial encounter with high peptide–MHC determinant density can signal death in CTLs.
Production of TNF-α by High Avidity CTLs Does Not Correlate with Susceptibility to Apoptotic Death.
What is the nature of the signaling event that distinguishes high and low dose antigen? The most ready explanation for the TNF-α–mediated death after stimulation by APCs pulsed with high doses of antigen was that supraoptimal stimulation was required to induce production of TNF-α by the CTLs. Fig. 2 shows that these CTLs did indeed produce TNF-α when stimulated with high doses of peptide antigen that induced inhibition of proliferation; however, maximal TNF-α production also occurred at lower antigen concentrations where maximal proliferation was observed. We have previously shown that inhibition of the proliferative response by high dose antigen is correlated with the induction of apoptotic death (7). Thus, the threshold for production of TNF-α did not solely account for the death observed with high dose antigen. Furthermore, addition of exogenous TNF-α in the presence of stimulators pulsed with an optimal (low) concentration of antigen did not induce death (data not shown). Because TNF-α can exist in a membrane-bound form as well as a soluble form, and since membrane-bound TNF-α has been suggested as the active form interacting with TNF-RII (27), we also asked whether levels of surface TNF-α could be correlated with the high dose antigen induction of apoptosis. However, we found that, as with soluble TNF-α, surface TNF-α also reaches a plateau at low antigen density, two logs lower than does apoptosis, and does not increase further at high antigen density (Fig. 3). As the concentration of antigen required for TNF-α production did not correlate with the concentration that resulted in apoptotic death, the induction of TNF-receptor by high dose antigen was investigated.
Figure 2.

The threshold of stimulation resulting in TNF-α production does not correlate with the threshold required for apoptotic death. CTLs were assayed concurrently for production of TNF-α and for proliferation in response to stimulation with splenocytes pulsed with various concentrations of I10 peptide. Production of TNF-α was quantitated by ELISA of culture supernatants. CTLs were stimulated by APCs pulsed with the indicated concentration of peptide antigen. After 24 h of culture, supernatant was collected and assayed for TNF-α. Proliferation was assessed as the incorporation of [3H]thymidine between 24 and 48 h of culture. As peptide concentration increased, TNF-α production rose concurrently with the rate of proliferation until both reached maximums at similar concentrations of peptide. With further increases in peptide concentration, proliferation decreased, characteristic of the high antigen induction of apoptosis, whereas TNF-α production remained elevated. Because TNF-α production was maximal at antigen concentrations too low to observe apoptosis or inhibition of proliferation, the requirement for high dose antigen to trigger apoptotic death is not the result of the stimulation threshold for TNF-α production.
Figure 3.

Surface-bound TNF-α is dependent upon antigen density and is not correlated with apoptosis. 5 d after stimulation, high avidity CTLs were stimulated with Thy 1.2–depleted spleen cells pulsed with various concentrations of I10 peptide. After 24 h in culture, cells were harvested and washed, and surface-bound TNF-α was detected by FITC-conjugated antibody using flow cytometry, gating only Thy 1.2–positive cells to exclude antigen presenting cells. The mean fluorescence was determined for CTLs in both the viable and apoptotic gates based on forward versus side scatter. Apoptosis was measured in the same samples as the number of CTLs in the apoptotic gate taken as a percentage of the total CTLs in both viable and apoptotic gates. Surface-bound TNF-α increased at optimal levels of peptide, two logs lower than peptide concentrations sufficient to cause apoptosis under these conditions. Similar results were obtained in three additional experiments.
The Signal Required for Death Is Mediated by Binding of TNF-α to the TNF-RII Receptor.
Our previous studies indicated that anti–TNF-α antibody could block the apoptotic death of high avidity CTLs induced by stimulation with a high concentration of antigen (7). It seemed likely that this was mediated by TNF-RI, since the majority of TNF-α–mediated apoptotic death has been shown to involve signaling via the TNF-RI receptor (11–14). However, in several instances, including AICD of bulk CD8+ lymph node cells stimulated with anti-CD3, TNF-RII has been demonstrated to be the mediator of the death signal (8). To determine which receptor was involved in the TNF-α–mediated inhibition of proliferation and the correlated apoptotic death in our system, we used blocking antibodies to TNF-RI and TNF-RII. Addition of antibodies during proliferation assays demonstrated that binding of TNF-α to TNF-RII was required for inhibition, but that there appeared to be no role for TNF-RI (Fig. 4).
Figure 4.

The apoptotic death resulting from stimulation with supraoptimal peptide–MHC determinant density is mediated by binding of TNF-α to TNF-RII. To determine whether TNF-α transduces the death signal via TNF-RI or TNF-RII, CTLs were stimulated with high (100 μM) or low (0.001 μM) concentrations of peptide in the presence of blocking antibodies to either TNF-RI or TNF-RII (each at 5 μg/well). Proliferation after stimulation with APCs pulsed with high concentrations of peptide antigen was restored only in the presence of antibodies to TNF-RII. The inability of the anti-TNF-RI antibody to prevent death was not due to an inability to block signaling through this receptor as this antibody could block TNF-α–mediated death in a TNF-α–sensitive L929 cell line (data not shown).
TNF-α Receptor Expression.
Since the amount of antigen required for either soluble or surface-bound TNF-α production could not explain the regulation of apoptotic death, it was possible that the differential susceptibility to TNF-α after stimulation with different determinant densities was manifested in the expression of TNF-RII. Accordingly, CTLs exposed to various concentrations of peptide were analyzed by flow cytometry for the surface expression of TNF-RII. As shown in Fig. 5, the results were similar to those seen for TNF-α. At the high peptide concentration that induced a decrease of proliferation, TNF-RII levels were indeed high, but they were also high at lower (optimal) concentrations of peptide. Furthermore, levels of TNF-RII increased with a dose–response curve that mirrored the one for proliferation. Therefore, as was observed for the production of TNF-α, expression of TNF-RII becomes maximal at too low an antigen density to account for the susceptibility to TNF-α–mediated death at high doses of antigen. These results suggested that if neither the expression of TNF-α nor that of its receptor is sufficient to account for the high antigen density-induced apoptosis then the engagement of TCR by high antigen density must potentiate the susceptibility of CTLs to a subsequent death by signals through the TNF-RII receptor. This potentiation could be the result of an increase in an actively apoptotic molecule or, conversely, the decrease in a protective molecule (such as Bcl-2 or Bcl-XL).
Figure 5.

TNF-α receptor regulation does not control the susceptibility to TNF-α–mediated apoptotic death. CTLs were analyzed for TNF-RII expression and for the rate of proliferation after stimulation with splenocytes pulsed with graded concentrations of I10 peptide. Concentrations of peptide that induced maximal expression of TNF-RII did not correlate with peptide concentrations required to induce death: both TNF-RII and proliferation increased in parallel to reach maximums at similar peptide concentrations. Thus, TNF-RII density was maximal at peptide concentrations insufficient to induce decreased proliferation (apoptosis) and receptor upregulation can not account for the switch to the TNF-α–sensitive phenotype.
Regulation of Bcl-2 and Bcl-XL by Antigen Dose.
There is significant evidence that the antiapoptotic proteins Bcl-2 and Bcl-XL are important in the regulation of cell death in T lymphocytes (15). Therefore, the expression of these two molecules in CTLs undergoing apoptotic death was analyzed after stimulation with supraoptimal peptide–MHC. To preclude any contribution from stimulator cells to the analysis, immobilized recombinant H-2Dd protein pulsed with various peptide concentrations was used to stimulate the CTLs. High avidity CTLs were incubated for 24 h in the presence of immobilized H-2Dd molecules pulsed with 50 μM I10 peptide, 0.005 μM I10 peptide, or no peptide. After incubation, CTLs were harvested and the levels of Bcl-2 and Bcl-XL extracted with Triton X-100 were analyzed by Western blot analysis. After stimulation with high dose antigen, the level of Bcl-2 was significantly reduced compared with cultures stimulated with optimal antigen doses (Fig. 6). This reduction ranged from 65 to 99% depending on the individual assay and method of detection. In contrast, when Bcl-XL was analyzed by Western blots under parallel conditions, there was little or no difference in concentration under any conditions (data not shown). As expected, low avidity CTLs, which do not undergo apoptotic death after stimulation with high dose antigen, showed no decrease in Bcl-2 or Bcl-XL with any antigen concentration. Correspondingly, under optimal (low) dose antigen conditions where high avidity CTLs also show no increase in apoptosis, levels of Bcl-2 were likewise not decreased. Therefore, it is likely that a decrease in Bcl-2 levels is the third process induced by high dose antigen stimulation of the TCR that allows the cells to undergo TNF-mediated apoptosis.
Figure 6.

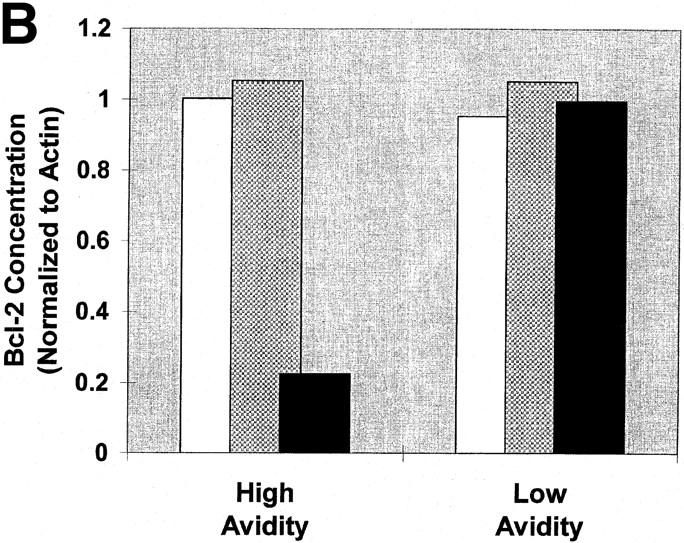
The induction of apoptotic death is associated with a decrease in Bcl-2 levels. High and low avidity CTLs were cultured in the presence of immobilized Dd (0.5 μg/well) pulsed with either 50 μM or 0.005 μM I10 peptide, or unpulsed. After 24 h, CTLs were harvested and solubilized with 0.5% Triton X-100 lysis buffer containing protease inhibitors. Lysates were analyzed by Western blot analysis for expression of Bcl-2. Actin was used as a control to ensure equivalent loading among the samples. High avidity CTLs stimulated with high peptide–MHC determinant density have significantly decreased expression of Bcl-2 compared with CTLs stimulated with low peptide–MHC determinant density. Low avidity CTLs stimulated with these same conditions showed no change in expression of Bcl-2. (A) Visualization of FITC-conjugated antibodies after transfer of protein bands to nitrocellulose. In this experiment, Bcl-2 is not demonstrable in the high avidity CTL line after culture with high I10 peptide despite a relatively higher amount of cell extract loaded on that lane (as assessed by the densities of the actin bands). (B) In a replicate experiment, binding of FITC-conjugated antibodies was quantitated and the binding to Bcl-2 normalized by comparison with binding to actin. Quantitation in several experiments showed that Bcl-2 in CTLs cultured with high peptide decreased to between 0 and 35% of the Bcl-2 present in controls. White bars, no peptide; gray bars, 0.005 μM; black bars, 50 μM.
The Decrease in Bcl-2 Is Dependent upon TNF-α Signaling, but this Signal Is Enabled Only by TCR Stimulation with High Dose Antigen.
Because the high dose antigen that causes a decrease in Bcl-2 levels also induces production of TNF-α and expression of TNF-RII, we asked whether TNF-α/ TNF-RII signaling was necessary for this decrease. Our initial expectation was that TNF-RII engagement by itself would not be sufficient to cause the observed decrease in Bcl-2 levels. If it were, then the low (optimal) dose of antigen, which stimulates both TNF-α and TNF-RII to maximal levels, would also be expected to decrease Bcl-2 levels and it does not. Nevertheless, to address this question, we carried out two types of experiments (Fig. 7). First, we asked whether the decrease in Bcl-2 levels seen with high (supraoptimal) peptide stimulation in high avidity lines was affected by blocking TNF-α signaling with anti–TNF-α. As predicted by the ability of anti–TNF-α to prevent the apoptosis of high avidity CTLs under these conditions, anti–TNF-α also prevented the decrease in Bcl-2 levels induced by high dose antigen (Fig. 7; compare the third and fourth bars), demonstrating that TNF-α signaling was a necessary requirement for the observed decrease in Bcl-2 levels. Second, we asked whether signaling by cross-linking TNF-RII with antibody was sufficient to decrease Bcl-2. To test this question, we first had to expose the high avidity CTLs to low (optimal dose) antigen to upregulate TNF-RII. 6 h after stimulation, we transferred the cells to wells that were either untreated or coated with anti–TNF-RII for a further 22–24 h. No decrease in Bcl-2 was observed (Fig. 7; compare fifth and sixth bars). Thus, even though TNF-α/TNF-RII signaling is necessary to induce apoptosis, it is not by itself sufficient to cause either apoptosis or a decrease in Bcl-2 levels in the absence of a potentiating signal engendered by the prior engagement of TCR by high (supraoptimal) MHC–peptide.
Figure 7.
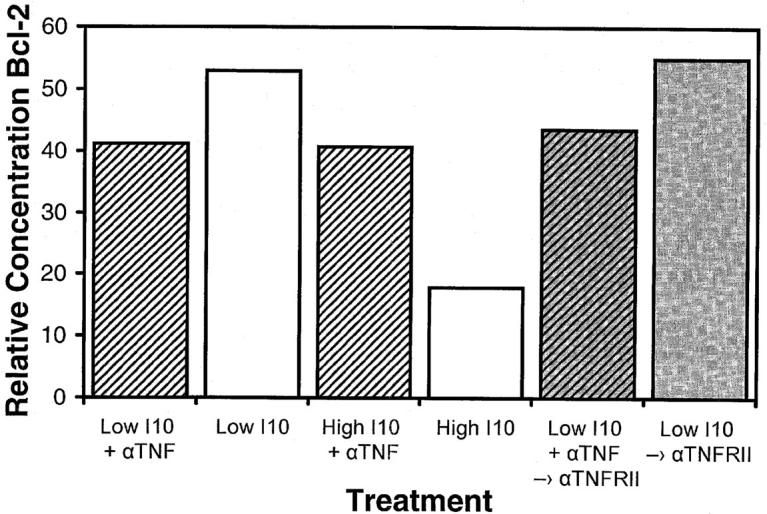
Signaling through TNF-RII is not sufficient to decrease Bcl-2 levels, but TNF-α signaling contributes to this effect. High avidity CTLs were stimulated by transfer to soluble Dd coated wells pulsed with optimal (0.0005 μM) or supraoptimal (50 μM) I10 peptide in either the presence or absence of anti–TNF-α. After 6 h, cells were transferred to either untreated wells or wells coated with anti–TNF-RII and incubated for a further 22–24 h under their initial media conditions. CTLs were then harvested and solubilized with 0.5% Triton X-100 lysis buffer containing protease inhibitors. Lysates were analyzed by Western blot analysis for determination of Bcl-2 concentrations. Actin was used as a control to ensure equivalent loading among the samples. Anti–TNF-α blocked the decrease in Bcl-2 that occurred after high avidity CTLs were stimulated with high peptide–MHC determinant density. Substrate-bound anti–TNF-RII caused no decrease in the levels of Bcl-2, either in the presence or in the absence of anti– TNF-α, indicating that these conditions were not equivalent to stimulation through the TCR by supraoptimal levels of peptide–MHC.
The Apoptotic Death of High Avidity CTLs Induced by Supraoptimal Stimulation Is Caspase Dependent.
Although the role of caspases in Fas-mediated apoptotic death is well documented (28–30), less is known about the involvement of these proteases in the TNF-RII death pathway. To determine the role of caspases in the apoptotic death resulting from stimulation with supraoptimal peptide–MHC, we used Boc-Asp-fluoromethyl ketone (BD-FMK), an irreversible inhibitor of caspases. Recognition of the inhibitors by caspases depends on the presence of an aspartic acid at the NH2 terminus. ZFA-FMK, which lacks the target aspartic acid, was used as a control. These cell permeant peptide-fluoromethyl ketone caspase inhibitors have been previously shown to block apoptotic death induced in T lymphocytes by a number of stimuli (31). The presence of BD-FMK prevented the induction of apoptotic nuclear morphology in a dose titratable fashion (Fig. 8), whereas the βZFA-FMK control inhibitor had no effect. Thus the pathway used by the TNF-RII receptor when it signals death in CD8+ T lymphocytes is caspase dependent and shares a common pathway with other death inducing receptors, e.g., Fas and TNF-RI (28–30).
Figure 8.

The apoptotic death of CTLs stimulated with high peptide– MHC determinant density can be blocked by caspase inhibitors. High avidity CTLs were stimulated with splenocytes either pulsed with 100 or 0.01 μM I10 peptide or unpulsed. BD-FMK or the control inhibitor (βZFA-FMK) was added at the indicated concentrations. Apoptotic nuclear morphology was assessed by staining with Hoechst 33342. The presence of BD-FMK during stimulation of CTLs with supraoptimal concentrations of peptide greatly inhibited apoptotic death. No inhibition of apoptotic nuclear morphology was observed with the control inhibitor, βZFA-FMK, which lacks the target aspartic acid.
Discussion
The results presented herein demonstrate that high avidity CD8+ CTLs can undergo apoptotic deletion as a result of supraoptimal peptide–MHC stimulation via secretion of TNF-α and its subsequent binding to TNF-RII. The preponderance of literature reporting the induction of cell death as a result of exposure to TNF-α suggests that the apoptotic signal is mediated by the TNF-RI receptor, which contains a death domain similar to that in Fas (for review see reference 32). However, TCR-induced apoptosis of activated CD8+ T cells appears different from the apoptosis seen in other cell types in that it uses TNF-RII (8 and this paper). Thus, TNF-RII may play a specialized role in this subset of T cells. Furthermore, our results are the first to demonstrate the caspase dependence of the TNF-RII signaling pathway.
The death observed as a result of supraoptimal engagement of the TCR on CTLs appears to result from a brief initial encounter with antigen (Fig. 1). This suggests that the signal generated is qualitatively different from that observed when the same cell engages optimal antigen levels. One major outcome of this different signal is that CTLs are rendered susceptible to the apoptotic action of TNF-α. During stimulation with optimal antigen concentrations, CTLs produce both surface and secreted TNF-α and upregulate TNF-RII (Figs. 2, 3, and 5). This would suggest that TNF-α should be able to signal through TNF-RII at optimal antigen levels. However, under these circumstances either the TNF-α signal is ignored by the CTLs or, alternatively, it may contribute to the activation of the CTLs. Although TNF-RII lacks the death domain found in TNF-RI and Fas, several “adapter” molecules have now been identified that link TNF-RII with the Fas and TNF-RI signaling pathways. TRAF (TNF receptor–associated factor) proteins associate with TNF-RII in the region that is required for signal transduction (33). TRAF2 can interact with TRADD, which has been shown to trigger apoptotic death when over-expressed (13). Similarly, TRAF1 has also been described as mediating apoptosis after cross-linking of CD3 (34). Two additional proteins, IAP (inhibitor of apoptosis)-1 and IAP-2, associate with TNF-RII through their interaction with TRAF1 and TRAF2 and are thought to be involved in signaling (33). Certainly the pathway leading to death via TNF-RII signaling is complex and the regulation of any one of a number of molecules in the pathway may contribute to the differential signal resulting from high versus low dose stimulation.
The lack of correlation between apoptosis and the production of TNF-α (secreted or surface) or of TNF-RII expression, despite the necessity of TNF-α/TNF-RII engagement, is an intriguing issue. The fact that a TNF-α/ TNF-RII signal is necessary but not sufficient suggests that alteration in levels of a third molecule must be postulated to account for the observed apoptosis. The surprising induction by high dose antigen of a decrease in Bcl-2 levels may explain, in part, the increased susceptibility to death after high dose antigen stimulation. Consistent with this interpretation, in preliminary experiments we found that cells exposed to high dose antigen, but prevented from undergoing TNF-α–mediated apoptotic death by addition of anti–TNF-α, were more susceptible to other agents that induced apoptosis, such as ionizing radiation (data not shown). These observations are consistent with the accumulation of a putative potentiating agent produced by stimulation of the TCR with supraoptimal antigen. Whatever this agent may be, it must act upstream of Bcl-2 regulation, allowing Bcl-2 levels to be decreased in conjunction with TNF-α/TNF-RII engagement.
Several studies have demonstrated the importance of Bcl-2 for the survival of lymphocytes (15, 35–37). Early studies reported the ability of Bcl-2 to protect pre-B cell lines from growth factor–induced apoptotic death (38). Furthermore, in mice deficient in Bcl-2 T cells disappear from the periphery within the first 4 wk of life, suggesting a requirement for Bcl-2 in lymphocyte survival (35). In resting T cells, Bcl-2 is expressed at low levels and is upregulated in response to stimulation with mitogen or IL-2 (36). Using antigen doses capable of inducing apoptotic deletion of CD8+ cytotoxic T cells, we find that there is a specific reduction in the level of Bcl-2, but little or no observable decrease in Bcl-XL. This result is in agreement with a study using the human T leukemia lines HL-60 and U937 (37) wherein TNF-α–mediated death was correlated with downregulation of the bcl-2 gene. We find that a decrease in Bcl-2 levels depends on a supraoptimal antigen-induced signal through the TCR, because the presence of TNF-α and its receptor TNF-RII are not sufficient to induce this decrease, and direct cross-linking of TNF-RII with surface-bound antibodies, even after stimulation with an optimal dose of antigen, does not induce a decrease of Bcl-2 levels (Fig. 7). Furthermore, the Bcl-2 decrease is prevented if antibody to TNF-α is present during stimulation with supraoptimal antigen, demonstrating that TNF-α signaling is necessary, even though it is not sufficient. Thus Bcl-2 may be a major participant in the control of CTL survival during the course of the immune response and may contribute to the loss of CTLs observed when very high viral burdens are attained, as is the case during infection with HIV.
Despite its direct correlation with apoptosis, the precise mechanism controlling Bcl-2 levels is not clear. Clearly, all three events, increase in surface and/or secreted TNF-α, increase in surface TNF-RII, and decrease in Bcl-2, are necessary in the course of high dose antigen induction of apoptosis. However, both surface and secreted TNF-α and TNF-RII increase to maximal levels at low antigen doses that do not cause apoptosis. Of these three events, only the decrease in Bcl-2 levels occurs solely at the higher antigen doses that induce apoptosis. Thus, a high dose antigen signal through the TCR is necessary for induction of the Bcl-2 decrease, but a TNF-α signal is also necessary for the Bcl-2 decrease, as indicated by the ability to block with antibody to TNF-α. Therefore, the situation is symmetrical in these two requirements, as both are necessary and one cannot tell which is potentiating for the other. We conclude that an interaction of signals from the TCR and TNF-α leads to the downregulation of Bcl-2 and thus to apoptosis. Future studies will be needed to dissect the signal transduction pathways from both these receptors to determine the intracellular pathways involved. However, whatever the initiating signals, our study also demonstrates that the final pathway for this TNF-α/TNF-RII–mediated death involves caspases, as is already known for death through fas or TNF-RI. Understanding this pathway will be important for developing ways to expand the repertoire of high avidity CTLs without deleting them by inducing apoptosis, and to preserve high avidity CTLs in the presence of high viral burdens, since high avidity CTLs appear to be the key CTLs necessary for control of viral infection (6, 39).
Acknowledgments
We thank Dr. Robert Schreiber for a kind gift of blocking antibodies for the TNF-RI and TNF-RII, Dr. David Margulies and Lisa Boyd for a generous gift of soluble recombinant H-2Dd, Dr. Lou Staudt for assistance with the Storm fluoroimaging system, and Drs. Colin Duckett and Michael Lenardo for critical reading of the manuscript.
Abbreviations used in this paper
- AICD
activation-induced cell death
- BD-FMK
Boc-Asp-fluoromethyl ketone
- βZFA-FMK
CBZ-Phe-Ala-fluoromethyl ketone
- TRAF
TNF receptor–associated factor
Footnotes
Martha A. Alexander-Miller's current address is Department of Microbiology and Immunology, Wake Forest University School of Medicine, Winston-Salem, NC 27157-1064.
References
- 1.Mitchison NA. Induction of immunological paralysis in two zones of dosage. Proc R Soc Ser B. 1964;1964:275–292. doi: 10.1098/rspb.1964.0093. [DOI] [PubMed] [Google Scholar]
- 2.Moskophidis D, Lechner F, Pircher H, Zinkernagel RM. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature. 1993;362:758–761. doi: 10.1038/362758a0. [DOI] [PubMed] [Google Scholar]
- 3.Hoffenbach A, Langlade-Demoyen P, Dadaglio G, Vilmer E, Michel F, Mayaud C, Autran B, Plata F. Unusually high frequencies of HIV-specific cytotoxic T lymphocytes in humans. J Immunol. 1989;142:452–462. [PubMed] [Google Scholar]
- 4.Carmichael A, Jin X, Sissons P, Borysiewicz L. Quantitative analysis of the human immunodeficiency virus type 1 (HIV-1)–specific cytotoxic T lymphocyte (CTL) response at different stages of HIV-1 infection: differential CTL responses to HIV-1 and Epstein-Barr virus in late disease. J Exp Med. 1993;177:249–256. doi: 10.1084/jem.177.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pantaleo G, De Maria A, Koenig S, Butini L, Moss B, Baseler M, Lane HC, Fauci AS. CD8+T lymphocytes of patients with AIDS maintain normal broad cytolytic function despite the loss of human immunodeficiency virus-specific cytotoxicity. Proc Natl Acad Sci USA. 1990;87:4818–4822. doi: 10.1073/pnas.87.12.4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alexander-Miller MA, Leggatt GR, Berzofsky JA. Selective expansion of high or low avidity cytotoxic T lymphocytes and efficacy for adoptive immunotherapy. Proc Natl Acad Sci USA. 1996;93:4102–4107. doi: 10.1073/pnas.93.9.4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alexander-Miller MA, Leggatt GR, Sarin A, Berzofsky JA. Role of antigen, CD8, and CTL avidity in high dose antigen induction of apoptosis of effector CTL. J Exp Med. 1996;184:485–492. doi: 10.1084/jem.184.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng L, Fisher G, Miller RE, Peschon J, Lynch DH, Lenardo MJ. Induction of apoptosis in mature T cells by tumour necrosis factor. Nature. 1995;377:348–351. doi: 10.1038/377348a0. [DOI] [PubMed] [Google Scholar]
- 9.Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 10.Jameson SC, Hogquist KA, Bevan MJ. Specificity and flexibility in thymic selection. Nature. 1994;369:750–752. doi: 10.1038/369750a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith CA, Farrah T, Goodwin RG. The TNF receptor superfamily of cellular and viral proteins: activation, costimulation, and death. Cell. 1994;76:959–962. doi: 10.1016/0092-8674(94)90372-7. [DOI] [PubMed] [Google Scholar]
- 12.Wang C-Y, Mayo MW, Baldwin AS., Jr TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-κB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- 13.Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD signals cell death and NF-κB activation. Cell. 1995;81:495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 14.Tartaglia LA, Rothe M, Hu Y-F, Goeddel DV. Tumor necrosis factor's cytotoxic activity is signaled by the p55 TNF receptor. Cell. 1993;73:213–216. doi: 10.1016/0092-8674(93)90222-c. [DOI] [PubMed] [Google Scholar]
- 15.Strasser A. Life and death during lymphocyte development and function: evidence for two distinct killing mechanisms. Curr Opin Immunol. 1995;7:228–234. doi: 10.1016/0952-7915(95)80007-7. [DOI] [PubMed] [Google Scholar]
- 16.Stewart, J.M., and J.D. Young. 1984. Solid Phase Peptide Synthesis. Pierce Chemical Company, Rockford, Illinois.
- 17.Takeshita T, Takahashi H, Kozlowski S, Ahlers JD, Pendleton CD, Moore RL, Nakagawa Y, Yokomuro K, Fox BS, Margulies DH, Berzofsky JA. Molecular analysis of the same HIV peptide functionally binding to both a class I and a class II MHC molecule. J Immunol. 1995;154:1973–1986. [PubMed] [Google Scholar]
- 18.Takahashi H, Cohen J, Hosmalin A, Cease KB, Houghten R, Cornette J, DeLisi C, Moss B, Germain RN, Berzofsky JA. An immunodominant epitope of the HIV gp160 envelope glycoprotein recognized by class I MHC molecule-restricted murine cytotoxic T lymphocytes. Proc Natl Acad Sci USA. 1988;85:3105–3109. doi: 10.1073/pnas.85.9.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kozlowski S, Corr M, Takeshita T, Boyd LF, Pendleton CD, Germain RN, Berzofsky JA, Margulies DH. Serum angiotensin-1 converting enzyme activity processes an HIV 1 gp160 peptide for presentation by MHC class I molecules. J Exp Med. 1992;175:1417–1422. doi: 10.1084/jem.175.6.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kozlowski S, Takeshita T, Boehncke W-H, Takahashi H, Boyd LF, Germain RN, Berzofsky JA, Margulies DH. Excess β2-microglobulin promotes functional peptide association with purified soluble class I MHC molecules. Nature. 1991;349:74–77. doi: 10.1038/349074a0. [DOI] [PubMed] [Google Scholar]
- 21.Sheehan KCF, Pinckard JK, Arthur CD, Dehner LP, Goeddel DV, Schreiber RD. Monoclonal antibodies specific for murine p55 and p75 tumor necrosis factor receptors: identification of a novel in vivo role for p75. J Exp Med. 1995;181:607–617. doi: 10.1084/jem.181.2.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sarin A, Adams DH, Henkart PA. Protease inhibitors selectively block T cell receptor–triggered programmed cell death in a murine T cell hybridoma and activated peripheral T cells. J Exp Med. 1993;178:1693–1700. doi: 10.1084/jem.178.5.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lenardo MJ. Interleukin-2 programs mouse αβ T lymphocytes for apoptosis. Nature. 1991;353:858–861. doi: 10.1038/353858a0. [DOI] [PubMed] [Google Scholar]
- 24.Critchfield JM, Racke MK, Zúñiga-Pflücker JC, Cannella B, Raine CS, Goverman J, Lenardo MJ. T cell deletion in high antigen dose therapy of autoimmune encephalomyelitis. Science. 1994;263:1139–1143. doi: 10.1126/science.7509084. [DOI] [PubMed] [Google Scholar]
- 25.Critchfield JM, Zúñiga-Pflücker JC, Lenardo MJ. Parameters controlling the programmed death of mature mouse T lymphocytes in high-dose suppression. Cell Immunol. 1995;160:71–78. doi: 10.1016/0008-8749(95)80011-7. [DOI] [PubMed] [Google Scholar]
- 26.Boehme SA, Zheng L, Lenardo MJ. Analysis of the CD4 coreceptor and activation-induced costimulatory molecules in antigen-mediated mature T lymphocyte death. J Immunol. 1995;155:1703–1712. [PubMed] [Google Scholar]
- 27.Grell M, Douni E, Wajant H, Löhden M, Clauss M, Maxeiner B, Georgopoulos S, Lesslauer W, Kollias G, Pfizenmaier K, Scheurich P. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell. 1995;83:793–802. doi: 10.1016/0092-8674(95)90192-2. [DOI] [PubMed] [Google Scholar]
- 28.Chinnaiyan AM, Dixit VM. The cell-death machine. Curr Biol. 1996;6:555–562. doi: 10.1016/s0960-9822(02)00541-9. [DOI] [PubMed] [Google Scholar]
- 29.Nagata S. Apoptosis: telling cells their time is up. Curr Biol. 1996;6:1241–1243. doi: 10.1016/s0960-9822(02)70706-9. [DOI] [PubMed] [Google Scholar]
- 30.Yuan J. Transducing signals of life and death. Curr Opin Cell Biol. 1997;9:247–251. doi: 10.1016/s0955-0674(97)80069-5. [DOI] [PubMed] [Google Scholar]
- 31.Sarin A, Wu M-L, Henkart PA. Different interleukin-1b converting enzyme (ICE) family protease requirements for the apoptotic death of T lymphocytes triggered by diverse stimuli. J Exp Med. 1996;184:2445–2450. doi: 10.1084/jem.184.6.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ware CF, VanArsdale S, VanArsdale TL. Apoptosis mediated by the TNF-related cytokine and receptor families. J Cell Biochem. 1996;60:47–55. doi: 10.1002/(SICI)1097-4644(19960101)60:1%3C47::AID-JCB8%3E3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 33.Rothe M, Pan M-G, Henzel WJ, Ayres TM, Goeddel DV. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell. 1995;83:1243–1252. doi: 10.1016/0092-8674(95)90149-3. [DOI] [PubMed] [Google Scholar]
- 34.Speiser DE, Lee SY, Wong B, Arron J, Santana A, Kong YY, Ohashi PS, Choi Y. A regulatory role for TRAF1 in antigen-induced apoptosis of T cells. J Exp Med. 1997;185:1777–1783. doi: 10.1084/jem.185.10.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakayama K-i, Nakayama K, Negishi I, Kuida K, Shinkai Y, Louie MC, Fields LE, Lucas PJ, Stewart V, Alt FW, Loh DY. Disappearance of the lymphoid system in Bcl-2 homozygous mutant chimeric mice. Science. 1993;261:1584–1588. doi: 10.1126/science.8372353. [DOI] [PubMed] [Google Scholar]
- 36.Reed JC, Tsujimoto Y, Alpers JD, Croce CM, Nowell PC. Regulation of bcl-2 proto-oncogene expression during normal human lymphocyte proliferation. Science. 1987;236:1295–1299. doi: 10.1126/science.3495884. [DOI] [PubMed] [Google Scholar]
- 37.Chen M, Quintans J, Fuks Z, Thompson C, Kufe DW, Weichseibaum RR. Suppression of Bcl-2messenger RNA production may mediate apoptosis after ionizing radiation, tumor necrosis factor α, and ceramide. Cancer Res. 1995;55:991–994. [PubMed] [Google Scholar]
- 38.Nunez G, London L, Hockenbery D, Alexander M, McKearn JP, Korsmeyer SJ. Deregulated Bcl-2 gene expression selectively prolongs survival of growth factor- deprived hemopoietic cell lines. J Immunol. 1990;144:3602–3610. [PubMed] [Google Scholar]
- 39.Gallimore A, Dumrese T, Hengartner H, Zinkernagel RM, Rammensee HG. Protective immunity does not correlate with the hierarchy of virus-specific cytotoxic T cell responses to naturally processed peptides. J Exp Med. 1998;187:1647–1657. doi: 10.1084/jem.187.10.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]