

Abstract
Activation of autoreactive T cells can lead to autoimmune diseases such as insulin-dependent diabetes mellitus (IDDM). The initiation and maintenance of IDDM by dendritic cells (DC), the most potent professional antigen-presenting cells, were investigated in transgenic mice expressing the lymphocytic choriomeningitis virus glycoprotein (LCMV-GP) under the control of the rat insulin promoter (RIP-GP mice). We show that after adoptive transfer of DC constitutively expressing the immunodominant cytotoxic T lymphocyte (CTL) epitope of the LCMV-GP, RIP-GP mice developed autoimmune diabetes. Kinetic and functional studies of DC-activated CTL revealed that development of IDDM was dependent on dose and timing of antigenic stimulation. Strikingly, repeated CTL activation by DC led to severe destructive mononuclear infiltration of the pancreatic islets but also to de novo formation of islet-associated organized lymphoid structures in the pancreatic parenchyma. In addition, repetitive DC immunization induced IDDM with lymphoid neogenesis also in perforin-deficient RIP-GP mice, illustrating that CD8+ T cell–dependent inflammatory mechanisms independent of perforin could induce IDDM. Thus, DC presenting self-antigens not only are potent inducers of autoreactive T cells, but also help to maintain a peripheral immune response locally; therefore, the induction of autoimmunity against previously ignored autoantigens represents a potential hazard, particularly in DC-based antitumor therapies.
Keywords: dendritic cells, autoimmunity, lymphoid organogenesis, diabetes, cytotoxic T cells
Many autoimmune diseases are initiated and maintained by sensitized and activated autoreactive T cells, which destroy target cells harboring corresponding tissue-specific antigens. In insulin-dependent diabetes mellitus (IDDM),1 T cell reactivity against a number of islet β cell proteins, e.g., insulin and glutamic acid decarboxylase, has been shown (1, 2). Genetic (3, 4) and environmental factors, e.g., viruses (5) and superantigens (6), may contribute to the activation of self-reactive lymphocytes. However, the precise mechanisms involved in the initial sensitization of T cells are only incompletely understood. It has been established that many self-antigens exclusively expressed by cells not reaching blood and lymphoid tissues are possibly ignored by the immune system (7). On the other hand, it is striking that target organs of several autoantibody-dependent autoimmune diseases, such as Hashimoto's thyroiditis, exhibit lymphoid structures including lymphoid follicles and germinal centers.
Antigen presentation by professional APCs, particularly dendritic cells (DC), is of key importance for the initiation of primary immune responses (8). This special capacity derives from their ability to transport antigen from the periphery to draining lymphoid organs and accessory molecules that support induction of T cell responses (9). The potency of DC in eliciting primary immune responses in vivo is demonstrated by studies in which immune responses against alloantigens (10) and HY antigens (11) were induced with as few as 104–105 DC.
Multiple lines of evidence indicate that DC may also participate in the onset of autoimmune diseases. DC are the first cells to infiltrate the thyroid gland during development of thyroid autoimmune disease in BioBreeding rats (12) and are found to establish direct contact with follicular epithelium during autoimmune thyroiditis in Graves' disease (13). Furthermore, Knight et al. (14) showed that DC presenting thyroglobulin enhanced the development of autoimmune thyroiditis in genetically susceptible CBA mice.
Due to their high immunostimulatory capacity, it has been proposed to use DC pulsed with tumor peptides or crude tumor protein homogenates as an efficient immunotherapeutic means of inducing immune responses against tumors (15, 16). However, such protocols may include the possible consequence of autoimmune reactions when anti-self reactions are induced, as suggested by induction of autoreactive T cells and vitiligo in patients with metastatic melanoma receiving immunotherapy (17).
To investigate the involvement of DC in the initiation and maintenance of an autoimmune disease, we used transgenic mice expressing lymphocytic choriomeningitis virus glycoprotein (LCMV-GP) exclusively in pancreatic β cells (RIP-GP mice). RIP-GP mice do not spontaneously develop autoimmune diabetes, defined as hyperglycemia, insulitis, and destruction of pancreatic islets, because this self-antigen is immunologically ignored. However, these mice develop an acute form of nonlethal autoimmune diabetes when infected with LCMV (18, 19). Thus, in the RIP-GP, mice lymphocytes are not deleted in the thymus and T cells react when appropriately activated by antigen in the lymphoid tissues. In this model situation, where acute infection triggers disease, primarily CD8+ CTL acting via perforin and the immunostimulatory cytokine IFN-γ are responsible for the development of IDDM (18, 20–23). We report here on the development of diabetes in RIP-GP and perforin-deficient RIP-GP mice after immunization of DC from transgenic mice constitutively expressing the immunodominant epitope GP33 of LCMV (H8-mice). Repetitive injection of the antigen-expressing DC initiated an autoimmune response leading to destruction of β islet cells. Interestingly, and comparable to Hashimoto's thyroiditis in man, evolution of this model autoimmune disease in mice correlated with the local development of organized lymphoid tissues.
Materials and Methods
Mice.
C57BL/6 mice were obtained from the Institut für Labortierkunde (University of Zürich). Mice expressing LCMV-GP under the control of the rat insulin promoter (RIP-GP) and perforin-deficient mice crossed with RIP-GP mice (PKO × RIP-GP) have been described previously (18, 22). Mice transgenic for a Vα2/Vβ8 TCR specific for H2-Db and the major LCMV-GP epitope, GP33-41 (GP33), were used as donors of transgenic T cells (24). Transgenic mice expressing the LCMV GP33 epitope in all tissues were generated using a construct containing the H-2Kb regulatory elements and the coding sequence for amino acids 1–60 of the LCMV glycoprotein (H8-mice [25]). All animals were kept under specific pathogen–free conditions. Experiments were carried out with age- (8–16 wk) and sex-matched animals.
Viruses, Cell Lines, and Peptides.
LCMV-WE was originally obtained from Dr. F. Lehmann-Grube (University of Hamburg, Germany) and was propagated on L929 cells. EL-4 (H-2b), a thymoma cell line, was used as target cell. LCMV GP33-41 (KAVYNFATM; GP33) was produced by Neosystem Laboratoire (Strasbourg, France).
Preparation of DC.
Generation of DC from C57BL/6 and H8 bone marrow cultures has been described previously (26). In brief, bone marrow was flushed from femurs and tibias and subsequently depleted of red cells with ammonium chloride. Bone marrow cells were depleted of lymphocytes, B cells, and I-Ab positive cells using a cocktail of mAbs (YTS191.1, YTS169.4.2, RA3-3A1/6.1, B21-2) and goat anti–rat IgG–coated Dynabeads (Dynal A.S., Oslo, Norway). Cells were plated at 0.5 × 106/ml in RPMI 1640 supplemented with 5% FCS, penicillin/streptomycin, 10 ng/ml recombinant murine (rm)GM-CSF (supplied by Novartis Pharma Schweiz AG, Bern, Switzerland) and IL-4–containing supernatant from cell line X63-IL4 (provided by Dr. M. Kopf, Basel Institute of Immunology, Basel, Switzerland) to a final concentration of 100 ng/ml. At days 2 and 4 of culture, 50% of the supernatant was removed, and fresh medium and fresh cytokines were added. At day 6, nonadherent cells were collected and further purified over metrizamide (14.5% in RPMI 1640/5% FCS; Sigma Chemical Co., St. Louis, MO) to remove cell debris and high density cells. The resulting DC populations showed a purity of 80–90% as determined by cytofluorometry using mAb against MHC class I and II antigens, costimulatory molecules CD80 and CD86, and the DC marker NLDC-145 (not shown). Cells were washed three times with BSS (balanced salt solution), resuspended at 2 × 106/ml in BSS, and serial 10-fold dilutions were made. DC were intravenously injected in a volume of 0.5 ml.
Cytotoxicity Assay.
For detection of primary ex vivo cytotoxicity, effector cell suspensions were prepared from spleens of immunized mice at the indicated time point after priming. EL-4 cells were labeled with GP33 (10−6 M) and 250 μCi 51Cr for 1.5 h at 37°C. 104 target cells per well were incubated in 96-well round-bottomed plates with serial threefold dilutions of spleen effector cells, starting with an E/T ratio of 90:1, for 15 h. EL-4 cells without peptide served as controls. The supernatant of the cytotoxicity cultures was counted in a gamma counter (Cobra II; Canberra Packard, Berkshire, UK). Percentage of specific lysis was calculated as (experimental release − spontaneous release)/ (total release − spontaneous release) × 100. Spontaneous release was always <29%.
CTL Precursor Assay.
Quantification of GP33-specific precursor CTL (CTLp) per spleen was performed by limiting dilution analysis as described (27). Responder spleen cells were titrated and cultured with 104 LCMV-infected, irradiated peritoneal macrophages and 105 irradiated feeder spleen cells in IMDM/10% FCS and 10% Con A supernatant using 16 wells per dilution step. After 6 d of culture, cytotoxicity was tested on GP33-labeled or unlabeled EL-4 cells in a standard 51Cr-release assay and CTLp frequencies were calculated.
Cytofluorometry.
To detect expansion of transgenic TCR- expressing T cells (24), peripheral blood cells, spleen cells, and lymph node cells from hepatic lymph nodes were stained for CD8, Vα2, and Vβ8.1 using FITC-conjugated rat anti–mouse CD8, PE-conjugated rat anti–mouse Vα2a, and biotinylated rat anti–mouse Vβ8.1 (all from PharMingen). Erythrocytes were lysed with FACS® lysis solution (Becton Dickinson, San Jose, CA), and the cell suspensions were analyzed on a FACScan® flow cytometer (Becton Dickinson) using logarithmic scales. Viable lymphocytes were gated using a combination of forward light scatter and 90° side scatter.
Immunohistology.
Freshly removed organs were immersed in HBSS and snap-frozen in liquid nitrogen. Tissue sections of 5-μm thickness were cut in a cryostat and fixed in acetone for 10 min. Sections were incubated with anti–mouse mAb against CD4+ cells (YTS191.1 [28]), CD8+ cells (YTS169.4.2 [28]), B cells (RA3-3A1/6.1; American Type Culture Collection, Rockville, MD), follicular DC (4C11 [29]); CD11c+ DC (30), peripheral node addressin (PNAd) (MECA-79; PharMingen), and polyclonal guinea pig antibodies against insulin (Dakopatts A/S, Glostrup, Denmark). Alkaline phosphatase–labeled, species-specific goat antibodies (Tago Inc., Burlingame, CA) were used as secondary reagents. The substrate for the red color reaction was AS-BI phosphate/New Fuchsin. Sections were counterstained with hemalum. 15–20 islets from 2–3 histological sections of each mouse were evaluated.
Measurement of Blood Glucose.
The glucose concentration in blood obtained from a tail vein was measured using Haemo-Glucotest strips (Boehringer Mannheim, Mannheim, Germany). Mice were considered diabetic with values >14 mM at two consecutive measurements.
Results
Induction of Diabetes in RIP-GP Mice by DC Immunization.
To study whether DC can initiate and promote IDDM in the RIP-GP mouse, different doses of DC from H8-mice (H8-DC) were administered intravenously using various protocols, and blood glucose levels were monitored. With a single injection of a high dose of 106 H8-DC, mice remained normoglycemic or displayed only transient hyperglycemia with intermediate blood glucose levels of 15–20 mM and eventually returned to normoglycemia within a few days (Fig. 1 A). In some mice (2/8), blood glucose quickly rose to values >44 mM (Fig. 1 A), followed by death after 4–6 d. After a single injection of an intermediate dose of 105 H8-DC, diabetes did not develop (Fig. 1 B). In contrast, repetitive injection of intermediate doses of H8-DC, i.e., three doses of 105 DC in 6-d intervals (Fig. 1 C) or four doses of 104 DC in 2-d intervals (Fig. 1 D), caused development of diabetes. 50% of the mice repetitively immunized developed diabetes between days 10 and 14, whereas 40% of the mice developed hyperglycemia later, by days 18–21. One out of eight mice repetitively immunized with 104 DC showed a transient hyperglycemia, and one out of eight mice from this group remained normoglycemic. 4 out of 16 mice repetitively immunized with H8-DC rapidly developed diabetes and died before day 21. This was surprising because the same mice infected with LCMV developed diabetes (with blood glucose values >44 mM), but survived usually for 40 d or more (not shown). Thus, under the conditions used, a severe and rapidly lethal IDDM was induced by DC, in a dose- and timing-dependent fashion.
Figure 1.

Blood glucose levels in RIP-GP mice after single or repetitive intravenous immunization with H8-DC. Mice were immunized with either a single dose of (A) 106 DC (n = 8) on day 0 or (B) 105 DC (n = 4) on day 0, or repetitive doses of (C) 105 DC (n = 8) on days 0, 6, and 12 or (D) 104 DC (n = 8) on days 0, 2, 4, and 6. Arrows, Day of DC injection. Values of four representative mice per group are shown.
CTL Activity and Expansion after Single versus Repetitive DC Immunization.
To determine how dose and timing of antigen delivery by DC influences CTL responses, CTL activity and expansion at various time points after single high dose (106), single low dose (104), and repetitive intermediate doses (105) of DC was monitored. Injection of a single high dose of DC induced CTL activity by day 4, as determined by direct ex vivo cytotoxicity in an overnight 51Cr-release assay; maximum CTL activity was reached by day 8 and waned between days 12 and 16. With low doses of DC, CTL activity was lower and the period during which CTL activity could be detected was shorter (Table 1). Importantly, repetitive stimulation with DC on days 0 and 8 prolonged CTL activity, leading to a weak but detectable CTL activity also on day 16 (Table 1).
Table 1.
Primary Activity and Frequency of GP33-specific CTL in Spleen after Immunization with H8-DC
| Single immunization | Repetitive immunization | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Group | day 4 | day 8 | day 12 | day 16 | day 16 | |||||||
| 106 H8-DC | CTL activity | 35 ± 5 | 81 ± 7 | 25 ± 3 | 2 ± 1 | n.d. | ||||||
| CTLp frequency | 1/5,000 | 1/18,000 | 1/51,000 | 1/63,000 | n.d. | |||||||
| 105 H8-DC | CTL activity | n.d. | 74 ± 6 | n.d. | 2 ± 1 | 34 ± 5 | ||||||
| CTLp frequency | n.d. | 1/22,000 | n.d. | 1/73,000 | 1/83,000 | |||||||
| 104 H8-DC | CTL activity | 7 ± 2 | 59 ± 8 | 4 ± 1 | 1 ± 0 | n.d. | ||||||
| CTLp frequency | 1/15,000 | 1/45,000 | 1/60,000 | 1/119,000 | n.d. | |||||||
C57BL/6 mice were immunized intravenously at day 0 (single immunization) or at days 0 and 8 (repetitive immunization) with different numbers of H8-DC. At the indicated time point, effector spleen cells were prepared (three mice per group), and an overnight 51Cr-release assay was performed as detailed in Materials and Methods to determine DC-induced CTL activity and a limiting dilution assay was done to assess the CTLp frequency. CTL activity is shown as mean specific target cell lysis ± SD at an E/T ratio of 90:1. Data from one of three experiments are shown. n.d., Not done.
To follow the expansion of GP33-specific CTLp in the spleen during the activation by DC, CTLp frequencies were determined by limiting dilution assay. The maximal expansion of GP33-specific CTLp was detected on day 4, followed by a continuous decrease until day 16 (Table 1). A 100-fold increase of DC used for immunization resulted only in a modest elevation of CTLp in the spleen, e.g., 1:15,000 and 1:5,000 at day 4 for 104 and 106 H8-DC, respectively (Table 1). Repetitive inoculation of 105 DC did not further enhance CTLp frequency (Table 1).
To test whether the limited expansion of CTL after high dose or repetitive DC immunization was due to a lack of GP33-specific precursor cells or whether CTL leave the spleen immediately after activation by DC, we followed the expansion of H8-DC–activated GP33-specific TCR transgenic CTL (24). After adoptive transfer of 5 × 105 spleen cells from TCR transgenic mice into C57BL/6 mice, 2–3% of the CD8+ T cells expressed the transgenic Vα2/Vβ8 TCR. Single DC immunization led to a maximal expansion of these cells by day 4 to ∼8% in spleen and blood and 5% in the hepatic lymph node (Fig. 2). Similarly, repetitive DC administration in 2-d intervals induced maximal expansion on day 4, with 22% in spleen and blood and 14% in hepatic lymph nodes (Fig. 2). In both single and repetitive immunization protocols, TCR transgenic T cell expansion was transient and TCR transgenic T cells persisted at slightly elevated levels between 3.5 and 5% (Fig. 2). After intravenous injection, H8-DC have been shown to migrate mainly to the spleen and, to a smaller proportion, to hepatic lymph nodes (26). Thus, it is reasonable to assume that GP33-specific T cells were first activated and expanded by H8-DC in spleen and hepatic lymph nodes and subsequently entered the blood to emigrate into peripheral nonlymphoid tissues.
Figure 2.

Expansion of GP33-specific TCR transgenic T cells by H8-DC. 5 × 105 spleen cells from TCR transgenic mice were adoptively transferred into C57BL/6 mice on day −1. Mice were immunized intravenously once with (A) 105 H8-DC on day 0 or repetitively with (B) 105 H8-DC on days 0, 2, 4, and 6. The expansion of the TCR transgenic T cells in blood, spleen, and hepatic lymph nodes (hep. Ln) was followed by three-color FACS® analysis and is depicted as percentage of Vα2+ Vβ8.1+ (tg-TCR +) cells in the CD8+ compartment (mean of three animals per time point). Arrows, Day of injection of H8-DC. Data from one of two experiments are shown.
Cellular Composition and Organization of DC-induced Autoimmune Lesions.
Using immunohistochemical analysis, we evaluated the cellular composition and organization of pancreatic infiltrates in the course of DC-induced autoimmune diabetes. To this end, RIP-GP mice were immunized intravenously at days 0, 6, and 12 with 105 H8-DC, and pancreata and submandibular salivary glands were analyzed on days 7, 14, and 21 for the presence of CD8+ and CD4+ T cells, B220+ B cells, F4/80+ macrophages, CD11c+ DC, and insulin-producing islet cells. Mice analyzed by immunohistochemistry on day 7 were normoglycemic, whereas mice analyzed on day 14 had been hyperglycemic for 1–2 d (blood glucose 17–25 mM), and mice analyzed on day 21 had been hyperglycemic for at least 7 d (blood glucose >44 mM). At the early time point, CD8+ T cells infiltrating the pancreatic islets clearly outnumbered CD4+ T cells, and B cells were barely detectable (Fig. 3, A1–5). Interestingly, APCs characteristic of lymphoid organs could be detected in these early infiltrates. Particularly DC staining positive with N418, an antibody that stains the gp150/ 90 αx integrin CD11c (30), were found in the infiltrates (Fig. 3 A5). F4/80+ macrophages were less abundant in these early infiltrates (not shown). The infiltrates were always associated with insulin-positive islets or periductal in their close proximity (Fig. 3, A1–5). At day 14, more cells had infiltrated the insulin-producing islets, particularly CD4+ T cells, B cells, and N418+ dendritic cells (Fig. 3, B1–5). Correlating with the increased infiltration by immune cells, staining for insulin decreased (Fig. 3 B1), indicating the progressive destruction of islet cells. By day 21, infiltrating cells had nearly completely replaced islet cells (Fig. 3, C1–5). Control stainings of submandibular salivary glands in DC-immunized RIP-GP mice (Fig. 3 D) and liver and pancreas sections of DC-immunized C57BL/6 mice (not shown) showed only a few scattered CD8+ and CD4+ T cells. A striking finding was the formation of lymphoid structures in close proximity to remaining insulin-producing islets (Fig. 4). These newly formed lymphoid tissues were found mainly periductally in the pancreatic parenchyma (Fig. 4 A, arrow), whereas pancreatic lymph nodes were clearly located outside of and loosely attached to the pancreas (Fig. 4 A, arrowhead). CD4+ T cell infiltrates were found in insulin-positive islets as well as in the newly formed lymphoid structures (Fig. 4 B). In contrast, B cell infiltrates were detected mainly in the periductal lymphoid structures (Fig. 4 C), where they showed distinct compartmentalization particularly with CD8+ T cells (Fig. 4, E and F), comparable to that of peripheral lymphoid organs. Cells staining with 4C11, an antibody specific for follicular DC, were restricted to the newly formed lymphoid tissues and colocalized with B cells (Fig. 4, G and H). Their presence in the DC-induced pancreatic infiltrates is particularly interesting, since follicular DC are involved in B cell Ig-switch, affinity maturation, and memory. PNAd, an adhesion molecule found on endothelial cells in peripheral lymph nodes and involved in recirculation of lymphocytes into peripheral lymphoid tissues, was expressed in blood vessels in the periductal organized lymphoid structures (Fig. 4 I). Pancreatic endothelial cells outside these lymphoid structures did not express PNAd.
Figure 3.
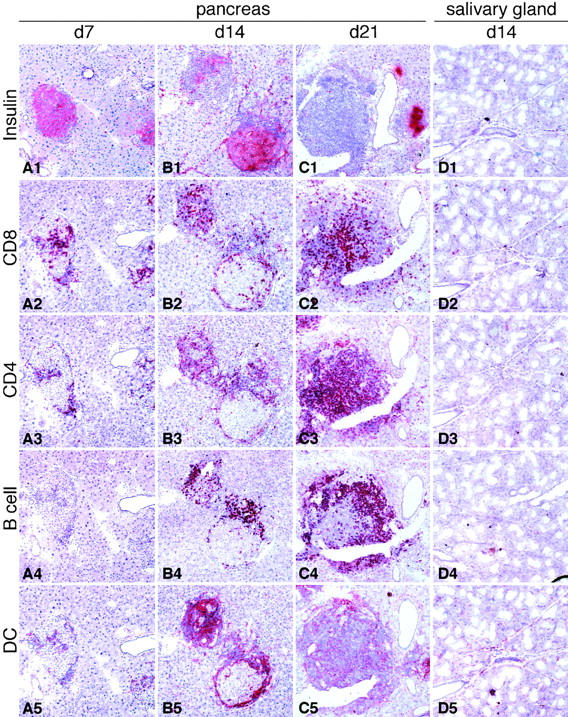
Immunohistological analysis of pancreatic islets and submandibular salivary glands in RIP-GP mice after H8-DC immunization. 105 H8-DC were adoptively transferred on days 0, 6, and 12, and pancreata were analyzed on day 7 (A), day 14 (B), and day 21 (C) for the presence of insulin-producing islet cells, CD8+ and CD4+ T cells, B220+ B cells, and CD11c+ DC. Pancreas sections from untreated RIP-GP mice were also stained but were essentially negative except for insulin. (D) Control staining of submandibular salivary gland of a diabetic RIP-GP mouse on day 14 with the respective markers. Original magnification: ×100.
Figure 4.
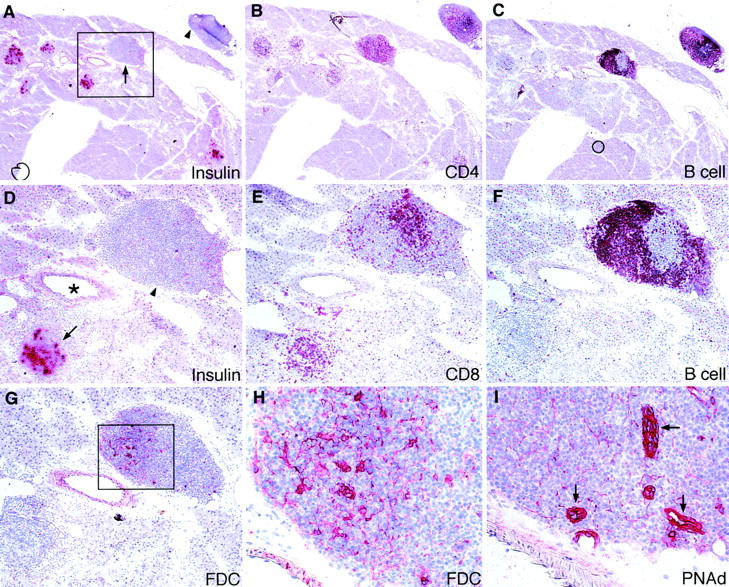
Organization of newly formed lymphoid tissues in RIP-GP mice after repetitive priming with H8-DC. Pancreata were analyzed by immunohistochemistry on day 21 by staining for the indicated markers. (A) Newly formed organized lymphoid tissue was found periductally in the pancreatic parenchyma (arrow); a pancreatic lymph node located outside the pancreatic parenchyma is also shown (arrowhead). Distribution of infiltrating (B) CD4+ T cells and (C) B cells in islets and newly formed lymphoid tissues. (D) Magnification of the boxed area in A showing an insulin-positive islet (arrow) and periductal, de novo–formed lymphoid tissue (arrowhead) in close vicinity to an artery (*). Distinct spatial distribution of (E) CD8+ T cells, (F) B cells, and (G) follicular DC (FDC) in the newly formed lymphoid tissue. (H) Magnification of the boxed area in G showing the 4C11+ follicular DC network. (I) Staining for PNAd. The region corresponding to the boxed area in G was photographed showing PNAd+ blood vessels in the newly formed lymphoid tissue. Original magnifications: A–C, ×24; D–G, ×63; H and I, ×250.
It has been shown previously that MHC-restricted destruction of β cells by perforin-dependent CTL is an important initiating event for autoimmune diabetes in RIP-GP mice (22). To evaluate whether contact-dependent T cell cytotoxicity is involved in DC-induced autoimmune diabetes, we studied perforin-deficient RIP-GP mice (PKO × RIP-GP; reference 22). 45% of the PKO × GP mice that were repetitively immunized with H8-DC developed diabetes between days 12 and 18 (Table 2). Immunohistological analysis revealed that all PKO × RIP-GP mice developed insulitis with >50% of the islets affected. Furthermore, PKO × RIP-GP mice rapidly progressed to diabetes and, like perforin-competent RIP-GP mice, showed lymphoid neogenesis in periductal areas in proximity to remaining insulin-producing islet cells (summarized in Table 2). In contrast to this and confirming earlier results (22), LCMV infection of RIP-GP mice with a disrupted perforin gene did not lead to the development of autoimmune diabetes (Table 2). These mice showed a pronounced peri- and intrainsular infiltrate; however, some islet cells remained intact, and lymphoid neogenesis did not develop (Table 2). Taken together, these results show that DC immunization with the appropriate self-antigen leads to destructive autoimmune diabetes accompanied by local formation of organized lymphoid tissues.
Table 2.
Development of Insulitis, Diabetes, and Lymphoid Neogenesis in RIP-GP and PKO × RIP-GP Mice after DC Immunization
| Immunization protocol* | Recipient mice | Incidence of diabetes‡ | Assessment of insulitis§ | Lymphoid neogenesis‖ | ||||
|---|---|---|---|---|---|---|---|---|
| Repetitive H8-DC | RIP-GP | 14/16 | +++ | Yes | ||||
| Repetitive H8-DC | PKO × RIP-GP | 7/16 | ++ or +++ | Yes | ||||
| LCMV-WE | PKO × RIP-GP | 0/4 | +++ | No | ||||
| Single H8-DC | RIP-GP | 3/8 | ++ to +++ | Yes | ||||
| Repetitive B6-DC | RIP-GP | 0/3 | − | No | ||||
| Repetitive H8-DC | C57BL/6 | 0/3 | − | No |
Mice were immunized intravenously either repetitively with 105 DC on days 0, 6, and 12 or with 104 DC on days 0, 2, 4, and 6; single intravenous injection was with 106 DC on day 0; 200 PFU LCMV (WE strain) were given on day 0.
Mice were scored diabetic with blood glucose >14 mM on two consecutive measurements.
Insulitis was classified on day 21 by immunohistochemistry as strong (+++) with severe destructive infiltration in >90% of the islets, moderate (++) with destructive infiltration in 50–90% of the islets, and weak (+) with <50% destructive infiltration and mainly periinsular and perivascular infiltration; −, no insulitis. Two to five mice were evaluated per group. 15–20 islets were evaluated from 2–3 histological sections per mouse.
Criteria for lymphoid neogenesis by day 21: massive infiltration with T cells, B cells, CD11c+ cells, and organized lymphoid structure. Two to five mice were evaluated per group.
Discussion
A major question in the pathogenesis of autoimmune disorders is to define the triggering events leading to activation of previously indifferent but potentially autoreactive T cells. Expression of the LCMV-GP in pancreatic β cells using the rat insulin promoter (RIP-GP mice) allowed us to study the role of virus infections as possible triggers for breaking tolerance against peripheral self-antigens leading to destruction of pancreatic islets (18, 19). Development of virus-induced autoimmune diabetes in RIP-GP mice is largely dependent on the number of activated self-reactive CTL; e.g., only double-transgenic mice expressing the LCMV-GP in the pancreas and a GP33-specific TCR on many of their CTL (RIP-GP × TCR), but not single transgenic mice (RIP-GP), become acutely diabetic after infection with the less immunogenic LCMV-GP recombinant vaccinia virus, whereas both develop diabetes upon LCMV infection (20). Therefore, it is surprising that immunization of RIP-GP mice with DC constitutively expressing the endogenous peptide led to development of a slow form of clinically manifest and rapidly lethal diabetes, despite the fact that they induced 10–100-fold lower CTL activity and CTLp frequencies compared with LCMV infection (26). Although this lower response corresponded to about that induced by LCMV-GP recombinant vaccinia virus, the data here suggest that the CTL stimulation by repetitive DC immunization was an important prerequisite for the eventual progression to IDDM. Thus, with the number of autoreactive cytotoxic T cells (18, 20), the duration of antigenic stimulation by professional APCs, i.e., the integral of CTL activity over time, determines the disease outcome in this model of autoimmune diabetes. Although perforin usually plays a key role also in the induction of acute diabetes in the RIP-GP model (22), repetitive immunization with GP33-presenting DC overcame the need for perforin in causing diabetes in 45% of the mice.
Cell types normally found in peripheral lymphoid tissues were present in DC-induced islet infiltrates and in periductal lymphoid structures, including T cells, B cells, bone marrow–derived APCs, i.e., DC, and macrophages with a distinct compartmentalization into T and B cell areas. Follicular DC were found exclusively in these newly formed lymphoid structures, suggesting their functional involvement in local B cell responses. Only mice developing early infiltrations and de novo formation of lymphoid follicles developed diabetes, suggesting that autoimmunity is linked to lymphoid neogenesis in the target organ. Similarly, de novo formation of lymphoid tissue can be observed in mice expressing lymphotoxin-α under the control of the RIP (31); however, despite massive pancreatic infiltrates, these mice do not develop diabetes (32). Therefore, it seems likely that the appropriate and repeated activation of CTL, e.g., via DC, together with development of lymphoid structures in the target organs, plays a major role in the development of destructive autoimmunity.
Human IDDM is often associated with chronic infiltration of mononuclear cells in the diseased tissue, leading to tissue destruction (33, 34) and occasional development of lymphoid structures (35). Additional evidence for the possible involvement of DC in the induction of IDDM comes from the nonobese diabetic (NOD) mouse model. In prediabetic NOD mice, DC form an organized network in and around pancreatic islets, facilitating efficient stimulation of infiltrating lymphocytes (36). Taken together, the presented and the discussed findings suggest that DC are not only crucially involved in the initiation of an anti-self immune response but are also essential for the maintenance of autoimmune lesions in peripheral organs. Several antibody-dependent organ-specific autoimmune diseases (including Hashimoto's thyroiditis and IDDM) and the data shown here support the notion that peripheral self-antigen, usually ignored by the immune system, can trigger an immunopathological, autoimmune response if the antigen enters draining organized lymphoid tissues in sufficient quantity and for a sufficient time. This autoimmune response is transient if antigen ceases to reach organized lymphoid tissues. It is maintained if sufficient antigen reaches draining lymphoid organs, or, alternatively, if lymphoid tissue is formed within the target organ itself.
These results may impinge on our rationales to use repetitive immunization with DC against tumors. Due to their high immunostimulatory capacity and their efficient migration into organized lymphoid tissues, autologous DC are well suited for immunization against tumors (15, 16). In mice, antitumor immunity can be induced using DC exogenously pulsed with tumor-derived peptides (37), preincubated with crude tumor cell preparations (38, 39), transfected with tumor cell-derived mRNA (39, 40), infected with recombinant viruses (41, 42), or by fusion of DC with tumor cells (43). By presenting tumor antigens together with other self-antigens by DC or by using tumor antigens that are not exclusively expressed on tumor cells, there is a potential of inducing undesirable anti-self responses, as exemplified in this study. Therefore, it may be important to use antigens solely expressed on tumor cells for DC-based anti-tumor therapy to avoid possible autoimmune complications.
Acknowledgments
We thank Maries van den Broek, Peter Aichele, and Paul Klenerman for helpful discussions and critical reading of the manuscript, Lenka Vlk and Anne Henzelin for expert technical assistance, and Norbert Wey for excellent microphotography.
This work was supported by the Swiss National Science Foundation, the Deutsche Forschungsgemeinschaft (to B. Ludewig), and the Kanton Zürich.
Abbreviations used in this paper
- CTLp
CTL precursor(s)
- DC
dendritic cell(s)
- GP
glycoprotein
- IDDM
insulin-dependent diabetes mellitus
- PNAd
peripheral node addressin
- RIP
rat insulin promoter
- LCMV
lymphocytic choriomeningitis virus
References
- 1.Daniel D, Gill RG, Schloot N, Wegmann D. Epitope specificity, cytokine production profile and diabetogenic activity of insulin-specific T cell clones isolated from NOD mice. Eur J Immunol. 1995;25:1056–1062. doi: 10.1002/eji.1830250430. [DOI] [PubMed] [Google Scholar]
- 2.Tisch R, Yang XD, Singer SM, Liblau RS, Fugger L, McDevitt HO. Immune response to glutamic acid decarboxylase correlates with insulitis in non-obese diabetic mice. Nature. 1993;366:72–75. doi: 10.1038/366072a0. [DOI] [PubMed] [Google Scholar]
- 3.Acha-Orbea H, McDevitt HO. The first external domain of the nonobese diabetic mouse class II I-A beta chain is unique. Proc Natl Acad Sci USA. 1987;84:2435–2439. doi: 10.1073/pnas.84.8.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Todd JA, Bell JI, McDevitt HO. HLA-DQ beta gene contributes to susceptibility and resistance to insulin-dependent diabetes mellitus. Nature. 1987;329:599–604. doi: 10.1038/329599a0. [DOI] [PubMed] [Google Scholar]
- 5.Dahlquist G, Frisk G, Ivarsson SA, Svanberg L, Forsgren M, Diderholm H. Indications that maternal coxsackie B virus infection during pregnancy is a risk factor for childhood-onset IDDM. Diabetologia. 1995;38:1371–1373. doi: 10.1007/BF00401772. [DOI] [PubMed] [Google Scholar]
- 6.Conrad B, Weidmann E, Trucco G, Rudert WA, Behboo R, Ricordi C, Rodriquez-Rilo H, Finegold D, Trucco M. Evidence for superantigen involvement in insulin-dependent diabetes mellitus aetiology. Nature. 1994;371:351–355. doi: 10.1038/371351a0. [DOI] [PubMed] [Google Scholar]
- 7.Zinkernagel RM. Immunology taught by viruses. Science. 1996;271:173–178. doi: 10.1126/science.271.5246.173. [DOI] [PubMed] [Google Scholar]
- 8.Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol. 1991;9:271–296. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- 9.Austyn JM. New insights into the mobilization and phagocytic activity of dendritic cells. J Exp Med. 1996;183:1287–1292. doi: 10.1084/jem.183.4.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knight SC, Mertin J, Stackpoole A, Clark J. Induction of immune responses in vivo with small numbers of veiled (dendritic) cells. Proc Natl Acad Sci USA. 1983;80:6032–6035. doi: 10.1073/pnas.80.19.6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boog CJ, Kast WM, Timmers HT, Boes J, de Waal LP, Melief CJ. Abolition of specific immune response defect by immunization with dendritic cells. Nature. 1985;318:59–62. doi: 10.1038/318059a0. [DOI] [PubMed] [Google Scholar]
- 12.Voorby HA, Kabel PJ, de Haan M, Jeucken PH, van der Gaag RD, de Baets MH, Drexhage HA. Dendritic cells and class II MHC expression on thyrocytes during the autoimmune thyroid disease of the BB rat. Clin Immunol Immunopathol. 1990;55:9–22. doi: 10.1016/0090-1229(90)90065-x. [DOI] [PubMed] [Google Scholar]
- 13.Molne J, Jansson S, Ericson LE, Nilsson M. Adherence of RFD-1 positive dendritic cells to the basal surface of thyroid follicular cells in Graves' disease. Autoimmunity. 1994;17:59–71. doi: 10.3109/08916939409014659. [DOI] [PubMed] [Google Scholar]
- 14.Knight SC, Farrant J, Chan J, Bryant A, Bedford PA, Bateman C. Induction of autoimmunity with dendritic cells: studies on thyroiditis in mice. Clin Immunol Immunopathol. 1988;48:277–289. doi: 10.1016/0090-1229(88)90021-9. [DOI] [PubMed] [Google Scholar]
- 15.Schuler G, Steinman RM. Dendritic cells as adjuvants for immune-mediated resistance to tumors. J Exp Med. 1997;186:1183–1187. doi: 10.1084/jem.186.8.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Young JW, Inaba K. Dendritic cells as adjuvants for class I major histocompatibility complex–restricted antitumor immunity. J Exp Med. 1996;183:7–11. doi: 10.1084/jem.183.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawakami Y, Rosenberg SA. Immunobiology of human melanoma antigens MART-1 and gp100 and their use for immuno-gene therapy. Int Rev Immunol. 1997;14:173–192. doi: 10.3109/08830189709116851. [DOI] [PubMed] [Google Scholar]
- 18.Ohashi PS, Oehen S, Buerki K, Pircher H, Ohashi CT, Odermatt B, Malissen B, Zinkernagel RM, Hengartner H. Ablation of “tolerance” and induction of diabetes by virus infection in viral antigen transgenic mice. Cell. 1991;65:305–317. doi: 10.1016/0092-8674(91)90164-t. [DOI] [PubMed] [Google Scholar]
- 19.Oldstone MB, Nerenberg M, Southern P, Price J, Lewicki H. Virus infection triggers insulin-dependent diabetes mellitus in a transgenic model: role of anti-self (virus) immune response. Cell. 1991;65:319–331. doi: 10.1016/0092-8674(91)90165-u. [DOI] [PubMed] [Google Scholar]
- 20.Ohashi PS, Oehen S, Aichele P, Pircher H, Odermatt B, Herrera P, Higuchi Y, Buerki K, Hengartner H, Zinkernagel RM. Induction of diabetes is influenced by the infectious virus and local expression of MHC class I and tumor necrosis factor-α. J Immunol. 1993;150:5185–5194. [PubMed] [Google Scholar]
- 21.von Herrath MG, Dockter J, Oldstone MB. How virus induces a rapid or slow onset insulin-dependent diabetes mellitus in a transgenic model. Immunity. 1994;1:231–242. doi: 10.1016/1074-7613(94)90101-5. [DOI] [PubMed] [Google Scholar]
- 22.Kagi D, Odermatt B, Ohashi PS, Zinkernagel RM, Hengartner H. Development of insulitis without diabetes in transgenic mice lacking perforin-dependent cytotoxicity. J Exp Med. 1996;183:2143–2152. doi: 10.1084/jem.183.5.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.von Herrath MG, Oldstone MB. Interferon-γ is essential for destruction of β cells and development of insulin-dependent diabetes mellitus. J Exp Med. 1997;185:531–539. doi: 10.1084/jem.185.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pircher H, Burki K, Lang R, Hengartner H, Zinkernagel RM. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature. 1989;342:559–561. doi: 10.1038/342559a0. [DOI] [PubMed] [Google Scholar]
- 25.Ehl S, Hombach J, Aichele P, Rulicke T, Odermatt B, Hengartner H, Zinkernagel R, Pircher H. Viral and bacterial infections interfere with peripheral tolerance induction and activate CD8+T cells to cause immunopathology. J Exp Med. 1998;187:763–774. doi: 10.1084/jem.187.5.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ludewig B, Ehl S, Karrer U, Odermatt B, Hengartner H, Zinkernagel RM. Dendritic cells efficiently induce protective antiviral immunity. J Virol. 1998;72:3812–3818. doi: 10.1128/jvi.72.5.3812-3818.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moskophidis D, Assmann-Wischer U, Simon MM, Lehmann-Grube F. The immune response of the mouse to lymphocytic choriomeningitis virus. V. High numbers of cytolytic T lymphocytes are generated in the spleen during acute infection. Eur J Immunol. 1987;17:937–942. doi: 10.1002/eji.1830170707. [DOI] [PubMed] [Google Scholar]
- 28.Cobbold SP, Jayasuriya A, Nash A, Prospero TD, Waldmann H. Therapy with monoclonal antibodies by elimination of T-cell subsets in vivo. Nature. 1984;312:548–551. doi: 10.1038/312548a0. [DOI] [PubMed] [Google Scholar]
- 29.Gray D, Kosco M, Stockinger B. Novel pathways of antigen presentation for the maintenance of memory. Int Immunol. 1991;3:141–148. doi: 10.1093/intimm/3.2.141. [DOI] [PubMed] [Google Scholar]
- 30.Metlay JP, Witmer-Pack MD, Agger R, Crowley MT, Lawless D, Steinman RM. The distinct leukocyte integrins of mouse spleen dendritic cells as identified with new hamster monoclonal antibodies. J Exp Med. 1990;171:1753–1771. doi: 10.1084/jem.171.5.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kratz A, Campos-Neto A, Hanson MS, Ruddle NH. Chronic inflammation caused by lymphotoxin is lymphoid neogenesis. J Exp Med. 1996;183:1461–1472. doi: 10.1084/jem.183.4.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Picarella DE, Kratz A, Li CB, Ruddle NH, Flavell RA. Insulitis in transgenic mice expressing tumor necrosis factor beta (lymphotoxin) in the pancreas. Proc Natl Acad Sci USA. 1992;89:10036–10040. doi: 10.1073/pnas.89.21.10036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bottazzo GF, Dean BM, McNally JM, MacKay EH, Swift PG, Gamble DR. In situ characterization of autoimmune phenomena and expression of HLA molecules in the pancreas in diabetic insulitis. N Engl J Med. 1985;313:353–360. doi: 10.1056/NEJM198508083130604. [DOI] [PubMed] [Google Scholar]
- 34.Sibley RK, Sutherland DE, Goetz F, Michael AF. Recurrent diabetes mellitus in the pancreas iso- and allograft. A light and electron microscopic and immunohistochemical analysis of four cases. Lab Invest. 1985;53:132–144. [PubMed] [Google Scholar]
- 35.Jansen A, Voorbij HA, Jeucken PH, Bruining GJ, Hooijkaas H, Drexhage HA. An immunohistochemical study on organized lymphoid cell infiltrates in fetal and neonatal pancreases. A comparison with similar infiltrates found in the pancreas of a diabetic infant. Autoimmunity. 1993;15:31–38. doi: 10.3109/08916939309004836. [DOI] [PubMed] [Google Scholar]
- 36.Lo D, Reilly CR, Scott B, Liblau R, McDevitt HO, Burkly LC. Antigen-presenting cells in adoptively transferred and spontaneous autoimmune diabetes. Eur J Immunol. 1993;23:1693–1698. doi: 10.1002/eji.1830230744. [DOI] [PubMed] [Google Scholar]
- 37.Zitvogel L, Mayordomo JI, Tjandrawan T, DeLeo AB, Clarke MR, Lotze MT, Storkus WJ. Therapy of murine tumors with tumor peptide–pulsed dendritic cells: dependence on T cells, B7 costimulation, and T helper cell 1–associated cytokines. J Exp Med. 1996;183:87–97. doi: 10.1084/jem.183.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Flamand V, Sornasse T, Thielemans K, Demanet C, Bakkus M, Bazin H, Tielemans F, Leo O, Urbain J, Moser M. Murine dendritic cells pulsed in vitro with tumor antigen induce tumor resistance in vivo. Eur J Immunol. 1994;24:605–610. doi: 10.1002/eji.1830240317. [DOI] [PubMed] [Google Scholar]
- 39.Ashley DM, Faiola B, Nair S, Hale LP, Bigner DD, Gilboa E. Bone marrow–generated dendritic cells pulsed with tumor extracts or tumor RNA induce antitumor immunity against central nervous system tumors. J Exp Med. 1997;186:1177–1182. doi: 10.1084/jem.186.7.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boczkowski D, Nair SK, Snyder D, Gilboa E. Dendritic cells pulsed with RNA are potent antigen-presenting cells in vitro and in vivo. J Exp Med. 1996;184:465–472. doi: 10.1084/jem.184.2.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Song W, Kong HL, Carpenter H, Torii H, Granstein R, Rafii S, Moore MA, Crystal RG. Dendritic cells genetically modified with an adenovirus vector encoding the cDNA for a model antigen induce protective and therapeutic antitumor immunity. J Exp Med. 1997;186:1247–1256. doi: 10.1084/jem.186.8.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Specht JM, Wang G, Do MT, Lam JS, Royal RE, Reeves ME, Rosenberg SA, Hwu P. Dendritic cells retrovirally transduced with a model antigen gene are therapeutically effective against established pulmonary metastases. J Exp Med. 1997;186:1213–1221. doi: 10.1084/jem.186.8.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gong J, Chen D, Kashiwaba M, Kufe D. Induction of antitumor activity by immunization with fusions of dendritic and carcinoma cells. Nat Med. 1997;3:558–561. doi: 10.1038/nm0597-558. [DOI] [PubMed] [Google Scholar]