

Abstract
The cytotoxicity of reactive oxygen intermediates (ROIs) has been implicated in the destruction of pancreatic β cells in insulin-dependent diabetes mellitus (IDDM). Thioredoxin (TRX), a redox (reduction/oxidation)-active protein, has recently been shown to protect cells from oxidative stress and apoptosis. To elucidate the roles of oxidative stress in the development of autoimmune diabetes in vivo, we produced nonobese diabetic transgenic mice that overexpress TRX in their pancreatic β cells. In these transgenic mice, the incidence of diabetes was markedly reduced, whereas the development of insulitis was not prevented. Moreover, induction of diabetes by streptozotocin, an ROI-generating agent, was also attenuated by TRX overexpression in β cells. This is the first direct demonstration that an antioxidative and antiapoptotic protein protects β cells in vivo against both autoimmune and drug-induced diabetes. Our results strongly suggest that oxidative stress plays an essential role in the destruction of β cells by infiltrating inflammatory cells in IDDM.
Keywords: oxidative stress, nonobese diabetic mouse, insulitis, cytokine, apoptosis
Insulin-dependent diabetes mellitus (IDDM)1 is caused by the autoimmune destruction of insulin-producing pancreatic β cells (1). By the clinical onset of this disease, β cells are almost completely gone, requiring affected individuals to inject insulin daily to sustain their lives. Nonobese diabetic (NOD) mice spontaneously develop autoimmune diabetes with remarkable similarity to human IDDM (2; for a review, see reference 3). A common pathological feature is the infiltration of inflammatory cells into the islets, termed insulitis, followed by the selective destruction of β cells. Infiltrating cells are composed of T and B lymphocytes, macrophages, and NK cells, among which T cells and macrophages are believed to play the major role in β cell destruction. However, the molecular mechanisms of β cell destruction by these infiltrating cells are poorly understood. Elucidation of these mechanisms is important not only for understanding the pathogenesis of IDDM and other autoimmune diseases, but also for developing effective means of preventing these diseases. Several reports have suggested that locally produced reactive oxygen intermediates (ROIs) are involved in these effector mechanisms of β cell destruction (4–9). In vitro, ROIs such as nitric oxide (NO), hydroxyl radical (OH·), hydrogen peroxide (H2O2), and superoxide radical (O2 −·) were shown to be induced in β cells by cytokines such as IL-1, IFN-γ, and TNF-α, which are known to be secreted by T cells and macrophages that infiltrate the islets (5–9). ROIs, either administered exogenously or induced in β cells by cytokines, have been shown to cause the destruction of β cells in vitro, and apoptosis has recently been suggested to be the mechanism involved (10–14). Among endocrine cells in pancreatic islets, insulin-producing cells appear particularly vulnerable to oxidative stress, probably due to their low levels of key enzymes that scavenge ROIs (15– 19). Therefore, the targeted expression of antioxidative and antiapoptotic molecules in pancreatic β cells would be expected to protect them from destruction and to prevent the development of IDDM.
Thioredoxin (TRX) is a low molecular mass (∼12 kD) redox (reduction/oxidation)-active protein found in both prokaryotic and eukaryotic cells (for a review, see reference 20). This protein has a conserved active site containing cysteine residues that are reversibly reduced by the NADPH-dependent seleno-flavoprotein, thioredoxin reductase (21). TRX, originally studied as a reducing cofactor for ribonucleotide reductase (22), is one of the essential proteins in DNA synthesis and repair (23–25). Recent studies showed that TRX is induced by various types of stress, such as viral infection, ischemic insult, UV light, x-ray irradiation, and H2O2, that it acts as a scavenger of ROIs, and that it can repair proteins oxidized by ROIs (for a review, see references 20 and 26). In vitro studies have demonstrated a protective effect of TRX against the cytotoxicity of ROIs (26). An augmented expression of TRX is often observed in neoplastic cells (26, 27) and appears to attenuate the cytotoxicity of ROI-generating antineoplastic agents such as cis- diamminedichloroplatinum (II) (CDDP), adriamycin, etoposide, and mitomycin C (28, 29). TRX is also involved in the intracellular signaling that combats oxidative stress and apoptosis (26) through the regulation of a class of transcription factors related to the growth and apoptosis of cells (for a review, see references 30 and 31), such as nuclear factor κB, activator factor 1 (AP-1), and nuclear redox factor 1 (Ref-1) (24, 26, 32–34).
To directly assess the implications of oxidative stress in pancreatic β cell destruction in autoimmune diabetes, we have generated NOD transgenic mice with targeted overexpression of the antioxidative protein, TRX, in pancreatic β cells. The protective effect of TRX against β cell destruction was also tested in experimental diabetes induced by the ROI-generating agent, streptozotocin (STZ). Pancreatic β cells were significantly protected from destruction in both models, indicating the involvement of oxidative stress and apoptosis in the destruction of β cells in autoimmune as well as STZ-induced diabetes.
Materials and Methods
Construction of the TRX Transgene.
The Ins-TRX transgene (Fig. 1) consisted of the human insulin promoter (35), the rabbit β globin gene sequences from the second exon to the polyadenylation signal, and human TRX cDNA inserted at the unique EcoRI site in the β globin sequences.
Figure 1.

The construction of the Ins-TRX transgene. The transgene contains the insulin promoter, an intron from the rabbit β globin gene, human TRX cDNA, and a poly A signal from the rabbit β globin gene. The DNA positions to which the primers for RT-PCR analysis anneal are shown.
Generation of NOD Transgenic Mice.
The Ins-TRX transgene cassette was purified and microinjected into the pronuclei of one-cell embryos of NOD mice (CLEA Japan Inc., Tokyo, Japan) as described previously (36). Founders (F0) of generated NOD transgenic mice were identified by PCR analysis of tail DNA. The primers used for transgenic mouse screening were as follows: forward 5′-GCTGGTTATTGTGCTGTCTC-3′ and reverse 5′-TCATCCACATCTACTTCAAGGA-3′. These primers amplify the transgene sequences between the third exon of the β globin gene and human TRX cDNA. Transgenic mice were bred with NOD mice. Male NOD transgenic mice were also mated with female C57BL/6J (B6) mice (CLEA Japan Inc.) to produce (NOD × B6) F1 mice. Transgenic mice were screened by PCR. Nontransgenic littermates were used as controls.
Reverse Transcription PCR Analysis.
RNA was extracted from major organs of NOD transgenic mice by the guanidine thiocyanate-cesium chloride centrifugation method (37). Complementary DNA was synthesized using oligo dT and reverse transcriptase Superscript II (GIBCO BRL, Gaithersburg, MD). The Ins-TRX gene expression was analyzed by reverse transcription (RT)-PCR with the following primers: forward 5′-GGATCCTGAGAACTTCAGG-3′ and reverse 5′-TCATCCACATCTACTTCAAGGA-3′ (see Fig. 1). To standardize the amount of mRNA in each sample, RT-PCR of hypoxanthine-guanine phosphoribosyl transferase (hprt) mRNA was performed in parallel. The primers for hprt mRNA detection were as follows: forward 5′-CTCGAAGTGTTGGATACAGG-3′ and reverse 5′-TGGCCTATAGGCTCATAGTG-3′. These primers were designed to encompass intron sequences so that the possible PCR products from the contaminating genomic DNA could be distinguished.
Isolation of Pancreatic Islets.
8-wk-old (NOD × B6) F1 transgenic mice, their nontransgenic littermates, and BALB/c mice (CLEA Japan Inc.) were anesthetized with an intraperitoneal injection of pentobarbital. Pancreatic islets were isolated by stationary collagenase (Sigma Chemical Co., St. Louis, MO) digestion of the pancreas, followed by Ficoll 400 (Amersham Pharmacia Biotech, Uppsala, Sweden) density gradient purification (38).
Western Blotting.
Isolated islets and dissected pancreata were lysed in lysis buffer (0.15 M NaCl, 10 mM Tris-HCl, pH 7.4, and 1% NP-40) with a protease inhibitor, PMSF, and were appropriately diluted with cold PBS for immunoblotting. Protein concentration was determined using a protein assay kit (Bio-Rad Laboratories, Hercules, CA). The lysates were incubated at 100°C for 5 min with a third volume of the sample buffer (75 mM Tris-HCl, pH 6.8, 6% SDS, 15% glycerol, 15% 2-ME, and 0.015% bromophenol blue). These samples were subjected to electrophoresis on an SDS/15% polyacrylamide gel and electrophoretically transferred onto a polyvinylidene difluoride membrane (Millipore Corp., Bedford, MA). To assess the levels of human TRX expression, known amounts of recombinant human TRX (>95% pure) prepared from human TRX–producing Escherichia coli (39) were simultaneously loaded on the gel. Mouse anti– human TRX mAb provided by Fujirebio Inc. (Tokyo, Japan) was used as a probe to detect human TRX. Endogenous murine TRX was detected using rabbit anti–murine TRX polyclonal Ab raised against a synthetic polypeptide of the COOH-terminal 10 amino acid residues of murine TRX. This Ab recognized murine TRX but not human TRX (J. Yodoi, unpublished data). The second Ab was rabbit anti–mouse IgG Ab or goat anti–rabbit IgG Ab conjugated with horseradish peroxidase (both from Zymed Laboratories, Inc., South San Francisco, CA). Human and murine TRX were visualized using an ECL detection kit (Amersham Pharmacia Biotech).
Insulin Secretion for Islets.
Isolated islets of 12-wk-old (NOD × B6) F1 transgenic mice (line 90, female) and their nontransgenic littermates were measured for insulin secretion according to the batch incubation method (40). Each batch of 10 islets was incubated at 37°C for 3 h in Krebs-Ringer solution containing 0.3% BSA with 3 different glucose concentrations. Five batches of transgenic and nontransgenic islets were simultaneously tested at each glucose concentration (5, 10, and 25 mM). After removal of the islets, the insulin concentration of each batch was determined using a mouse insulin ELISA kit (Seikagaku Corp., Tokyo, Japan).
Diagnosis of Diabetes.
Female NOD transgenic mice (F2, F3) and their nontransgenic littermates were monitored from 12 wk of age with weekly measurements of urine glucose using TesTape (Eli Lilly and Company, Indianapolis, IN). When mice were glucosuric for two consecutive weeks, they were diagnosed as diabetic.
Evaluation of Insulitis.
Pancreata were removed from 12-wk-old female NOD transgenic mice (line 90, n = 10) and their TRX-negative littermates (n = 8), and were fixed with 20% formalin. After paraffin-embedded fixation, 3-μm slices of each pancreas were prepared for hematoxylin and eosin staining. 25 islets of each pancreas were randomly chosen for microscopic examination of insulitis. According to the classification system of Kurasawa et al. (41), each islet was scored as follows: 0, an islet without infiltration; 1, <25% area of an islet was occupied by immunocytes; 2, ≥25 and <50%; 3, ≥50%.
Production of STZ-induced Diabetic Mice.
STZ (Wako Pure Chemical Industries Ltd., Osaka, Japan) was dissolved in 0.05 M citrate buffer (pH 4.5; reference 42), and immediately injected intraperitoneally into 8-wk-old (NOD × B6) F1 transgenic mice and their TRX-negative littermates at a dose of 250 mg/kg. These mice had been starved for 18 h before STZ treatment, and were then fed ad libitum.
Measurements of Blood Glucose Levels and Pancreatic Insulin Content.
7 d after STZ injection, blood sugar levels of (NOD × B6) F1 transgenic mice and their TRX-negative littermates were measured with Glutest E (KDK Corp., Kyoto, Japan), whose range of measurable blood glucose concentrations is between 40 and 500 mg/dl. 10 d after STZ treatment, pancreata were removed from 6-h-fasted (NOD × B6) F1 transgenic mice and their TRX-negative littermates. Pancreatic insulin was extracted according to the acid ethanol method (43), and the extracted insulin was dissolved in cold PBS. Insulin concentration was determined using a mouse insulin ELISA kit. Insulin content was indicated as the amount of pancreatic insulin (in nanograms) divided by the pancreatic wet weight (in milligrams).
Statistical Analysis.
Data were expressed as mean ± SD or mean ± SEM. Statistical analysis was performed by Mann-Whitney U test, unpaired t test, and Kaplan-Meier methods using StatView version 4.5 for the Macintosh (Abacus Concepts Inc., Berkeley, CA).
Results
Transgenic Mice Overexpressing Human TRX.
The Ins-TRX transgene containing a human TRX cDNA under the human insulin promoter (Fig. 1) was microinjected into fertilized eggs of NOD mice to generate NOD transgenic mice. In the F1 generation, PCR analysis of tail DNA showed that the Ins-TRX transgene was inherited by three NOD transgenic strains, lines 21, 48, and 90. The expression of human TRX mRNA was detected exclusively in the pancreas of transgenic mice using RT-PCR analysis (Fig. 2). For the quantification of human TRX expression, islets were isolated from (NOD × B6) F1 transgenic mice, which were produced by intercrossing female B6 mice with male NOD transgenic mice of each strain. Western blot analysis using a human TRX–specific mAb as probe showed a high level of human TRX expression in the transgenic islets of lines 21 and 90 (Fig. 3, top). For this analysis, 15 μg of islet protein was loaded on each lane. By comparing the densities of these bands with those of known amounts of recombinant human TRX (Fig. 3, top), the ratio of human TRX to total protein in the islets was estimated to be 1.7 ng/μg in both lines 21 and 90. Although human TRX was not detectable in the islets of line 48, prolonged exposure of the membrane revealed a low-level expression of human TRX in these mice (not shown). The expression of endogenous murine TRX was also examined by Western blot analysis using rabbit anti– mouse TRX serum as the probe, which was not reactive with human TRX. The levels of endogenous TRX expression were found to be much lower in the islets than in the pancreas (Fig. 3, bottom).
Figure 2.

RT-PCR analysis of Ins-TRX transgene expression in various organs. RNA was extracted from the indicated organs of a transgenic mouse (line 90; Tg) and its TRX-negative littermate (non-Tg). 31 and 27 cycles of the PCR procedure were performed to detect the mRNA of human TRX (hTRX, top) and hprt (bottom), respectively. Samples were resolved by electrophoresis on 2% agarose gels and stained with an ethidium bromide solution.
Figure 3.

Top, Western blot analysis of human TRX expression in pancreatic islets. Pancreatic islets were isolated from 8-wk-old (NOD × B6) F1 mice and their nontransgenic littermates. Approximately 15 μg of islet protein was loaded in each well. The amounts of human TRX in the lysates of transgenic islets from lines 90 and 21 mice were densitometrically estimated to be 25 ng on the imaging film exposed using an ECL detection kit. Prolonged exposure of a film revealed a faint band of human TRX in line 48 (data not shown). Bottom, Immunoblot analysis of the endogenous TRX expression in islets and pancreas. The lysates of islets were prepared from (NOD × B6) F1 transgenic mice (line 90), their TRX-negative littermates, and BALB/c mice. The lysates of the pancreas were prepared from a (NOD × B6) F1 transgenic mouse. These lysates were subjected to Western blot analysis using mouse TRX–specific serum. 2.5 μg (a) and 5 μg (b) of protein were loaded for each sample. The imaging film was exposed overnight.
Insulin Secretory Capacities of Transgenic Islets.
We next examined whether transgenic TRX expression in β cells affects glucose-responsive insulin secretion. The insulin secretory capacities of (NOD × B6) F1 transgenic islets at three glucose concentrations (5, 10, and 25 mM) were 0.40 ± 0.05, 1.02 ± 0.08, and 2.76 ± 0.15 ng/h/islet (mean ± SD), respectively. The insulin secretory capacities of their TRX-negative littermates were 0.43 ± 0.03, 1.04 ± 0.07, and 2.83 ± 0.13 ng/h/islet, respectively. There was no significant difference between the results from the islets of transgenic mice and those of their TRX-negative littermates at any glucose concentration (unpaired t test, NS for all), indicating that the overexpression of TRX did not affect the insulin secretory capacity of β cells.
Incidence of Diabetes.
To examine the effect of TRX expression on the development of diabetes, female NOD transgenic mice and their female TRX-negative littermates were monitored for glucosuria up to 32 wk of age. In transgenic mice of lines 21 and 90, the cumulative incidence of diabetes was markedly reduced at 32 wk of age (Fig. 4). Statistical analysis demonstrated a significant difference between NOD transgenic mice and their TRX-negative littermates (Kaplan-Meier method, P < 0.01 for both lines). The incidence of diabetes was not significantly reduced in transgenic mice of line 48, in which the TRX expression was very low.
Figure 4.
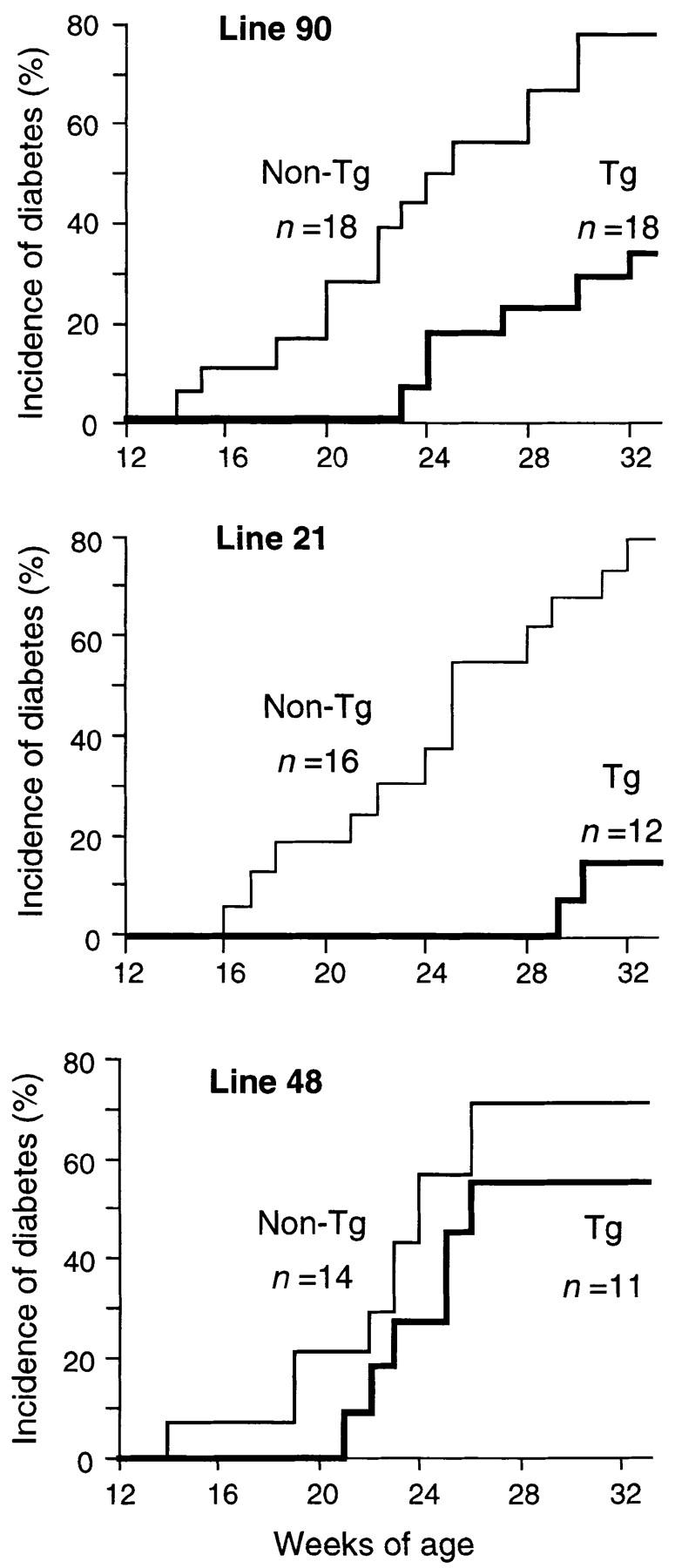
Cumulative incidence of diabetes in each NOD transgenic strain. Mice were monitored with weekly measurements of urine glucose. The number (n) of animals tested is shown in each figure. Statistical analysis demonstrated a significant difference between transgenic mice and their nontransgenic littermates in lines 90 and 21 (Kaplan-Meier method, P < 0.01 for both), but not in line 48 (NS).
Insulitis Score.
To determine whether TRX overexpression affects the development of insulitis in pancreatic islets, we performed histochemical analyses to determine the degree of the insulitis in NOD transgenic mice and their TRX-negative littermates before the onset of overt diabetes. The average insulitis scores of 12-wk-old female NOD transgenic mice (line 90) and their female TRX-negative littermates were 1.63 ± 0.32 and 1.57 ± 0.26 (mean ± SEM), respectively, indicating that the transgenic TRX overexpression does not attenuate the development of insulitis.
Blood Glucose Levels and Insulin Content after STZ Injection.
We next examined whether TRX overexpression confers resistance to STZ-induced diabetes. In this experiment, we used (NOD × B6) F1 transgenic mice and their nontransgenic littermates to avoid the complications in interpretation that could arise from the autoimmune diabetes accompanying the NOD background. No evidence of insulitis was observed in these mice. Before STZ treatment, fasting blood glucose levels were 80.0 ± 5.7 mg/dl for transgenic mice (n = 9) and 77.6 ± 9.7 mg/dl for their TRX-negative littermates (n = 9). 7 d after STZ injection, blood glucose levels were >500 mg/dl in most of the nontransgenic mice, but were only moderately elevated in TRX transgenic mice, as shown in Fig. 5 a. There was a significant difference in blood glucose levels between the transgenic mice and their TRX-negative littermates. We next examined the insulin content of the pancreas of STZ-treated transgenic and nontransgenic littermates after subjecting them to a 6-h fast (Fig. 5 b). Pancreatic insulin content was significantly higher in the transgenic mice (21.8 ± 9.4 ng/mg pancreatic weight; n = 5) than in their TRX-negative littermates (8.2 ± 4.2 ng/mg; n = 5). Without STZ treatment, insulin content was the same for the transgenic mice (66 ± 7.0 ng/mg; n = 5) and their TRX-negative littermates (63.0 ± 8.6 ng/mg; n = 5). These results clearly indicate that human TRX overexpression protects β cells from the cytotoxicity caused by STZ administration.
Figure 5.
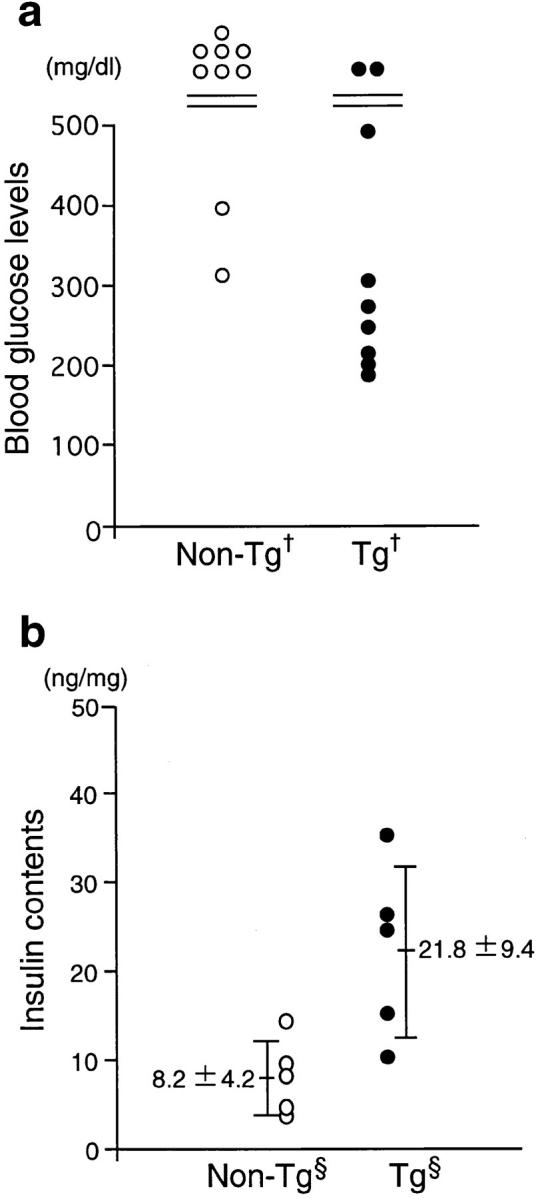
Alteration of blood glucose levels and insulin content after STZ treatment. STZ (250 mg/kg) was injected intraperitoneally into 8-wk-old (NOD × B6) F1 mice after an 18-h starvation period. (a) 7 d after injection, the blood glucose concentration was measured from mice fed ad libitum. When the concentration surpassed 500 mg/dl, the upper limit of measurement, its value was indicated as >500 mg/dl. There was a significant difference between (NOD × B6) F1 transgenic mice (line 90, male; n = 9) and their TRX-negative littermates (male, n = 9) (Mann-Whitney U test, † P < 0.05). (b) Pancreata were excised from 6-h-starved (NOD × B6) F1 transgenic mice (line 21, female; n = 5) and their TRX-negative littermates (female, n = 5) 10 d after STZ injection. Pancreatic insulin contents of transgenic mice were significantly higher than those of their TRX-negative littermates (unpaired t test, § P < 0.05).
Discussion
IDDM is characterized by the infiltration of lymphocytes into the pancreatic islets followed by the destruction of β cells, which leads to overt diabetes. However, it is not well understood how β cells are destroyed by the immunocytes in islets. The coexistence of various cell types and their complex interactions in the inflammatory islets make it very difficult to study the mechanism of β cell destruction in vivo. To overcome this complexity, we adopted a strategy in which molecules that are assumed to affect β cell destruction are directly tested by overexpressing them specifically in β cells in vivo. In this study, we have generated NOD transgenic mice that express human TRX, which has strong antioxidative and antiapoptotic activity (20, 26). Analysis by RT-PCR demonstrated that human TRX was expressed exclusively in the islets of our transgenic mice. Immunoblot analysis showed that the ratio of human TRX to total protein of the transgenic islets was ∼1.7 ng/μg (Fig. 3, top). The endogenous TRX levels of normal organs are less than ∼0.3 ng/μg (J. Yodoi, unpublished data). Therefore, the TRX levels in transgenic islets are considerably higher than those of normal tissues. An excessive expression of TRX may be toxic to cells, as pointed out by Baker et al. (44). However, we consider this an unlikely problem in our transgenic mice because endogenous TRX expression is often enhanced to 0.3–2 ng/μg or more (28, 45) in immature, transformed, or neoplastic cells (26–29, 44, 45).
The targeted overexpression of TRX in pancreatic β cells remarkably reduced the incidence of diabetes (Fig. 4), indicating a protective effect of TRX against the autoimmune destruction of β cells. Destruction of β cells induced by the ROI-generating agent STZ was also attenuated by TRX overexpression, as evidenced by lower blood glucose levels and higher insulin content in TRX transgenic mice than in their TRX-negative littermates. These results indicate that the overexpression of the antioxidative molecule TRX in β cells confers protective effects against spontaneous autoimmune diabetes as well as experimental diabetes induced by STZ. Recently, Kubisch et al. (46) reported that the overexpression of Drosophila Cu/Zn superoxide dismutase, a superoxide-scavenging enzyme, in β cells confers resistance to experimental diabetes induced by alloxan. Taken together, these results strongly suggest that ROIs play major roles in the β cell destruction that accompanies not only experimental diabetes induced by the ROI-generating drugs STZ and alloxan, but also the spontaneous autoimmune diabetes of the NOD mouse.
Our study revealed that the expression of endogenous TRX is very low in islet cells compared with pancreatic exocrine cells (Fig. 3, bottom). Previously, Hansson et al. (47) suggested that a lower expression of TRX occurs in islet cells than in pancreatic exocrine cells, although their immunohistochemical analysis was not quantitative. They also showed that TRX expression was suppressed in recently fed mice. This attenuation of TRX expression in insulin-secreting β cells may be one of the reasons β cells are highly susceptible to oxidative stress. Recent studies have indicated a stepwise development of autoimmune diabetes, with insulitis the first phase and massive destruction of β cells the second (48). We showed that there was no significant difference in the severity of insulitis between NOD transgenic mice and their TRX-negative littermates, indicating that overexpression of TRX did not prevent insulitis, but rather attenuated the cytotoxicity of the infiltrating immunocytes in islets. This result is consistent with the function of TRX as an antioxidative protein. It has also been reported that IDDM develops without insulitis in transgenic mice overexpressing inducible NO synthase (iNOS) in β cells (49), probably through the increased oxidative stress due to NO production. This report and our present study together suggest that oxidative stress is one of the major effector mechanisms of β cell destruction by infiltrating inflammatory cells.
Recently, apoptosis has also been suggested as a pivotal mechanism of β cell destruction in autoimmune (10–13) and drug-induced diabetes (14, 50). The Fas/FasL system has been implicated in β cell apoptosis. NOD transgenic mice that express FasL in their β cells were reported to be highly susceptible to autoimmune diabetes (12), whereas Fas-deficient NOD-lpr/lpr mice were resistant to autoimmune diabetes (12, 13). Recombinant TRX has been reported to protect cells against apoptosis mediated by both TNF and anti-Fas agonistic Ab (51). Very recently, mammalian TRX has been shown to function as a direct inhibitor of apoptosis signal-regulating kinase (ASK)-1 (52). These results suggest that the antiapoptotic function of TRX may also participate in the protection of β cells after immunocytic infiltration.
Although β cell–targeted overexpression of TRX effectively suppressed the incidence of autoimmune diabetes in NOD mice, a low percentage of transgenic mice still developed diabetes with significantly later onset. At present, we can only speculate on the molecular mechanisms causing diabetes in these mice. It is probable that TRX could not confer complete resistance to the oxidative stress and β cell death caused by infiltrating cells. Alternatively, other secondary mechanisms for β cell destruction, such as the perforin-dependent cytotoxicity of CD8+ T cells (53), might have led to the autoimmune diabetogenic β cell loss.
In human IDDM, overt diabetes develops when β cells are almost completely destroyed. Subjects at high risk for the future development of IDDM can only be identified by the presence of autoantibodies, which indicate the presence of autoimmunity and probable ongoing β cell destruction. The prevention of and intervention in IDDM is only possible during the second phase in the development of the autoimmune process. Therefore, these effects of TRX overexpression suggest a new approach to protecting subjects in the second phase of IDDM or to delaying the development of overt diabetes.
This study provides the first direct evidence that an antioxidative and antiapoptotic protein has protective effects on autoimmune diabetes. Our results strongly suggest that oxidative stress plays an essential role in β cell destruction caused by inflammatory cells in islets. The results also suggest an antioxidative and antiapoptotic strategy as a promising approach for the prevention of and intervention in IDDM in particular, as well as other autoimmune diseases in general, in subjects with ongoing autoimmunity.
Acknowledgments
The authors thank Ms. R. Takano and Drs. H. Aihara, J. Kawagishi, T. Kobayashi, and Y. Nitta for the generation of transgenic mice. We are grateful to Drs. Y. Takagi, Y. Ueda, and E. Yamato for helpful advice.
This work was supported by grants-in-aid from the Japanese Ministry of Education, Science, Sports, and Culture.
Abbreviations used in this paper
- B6
C57BL/6J
- IDDM
insulin-dependent diabetes mellitus
- NO
nitric oxide
- NOD
nonobese diabetic
- ROI
reactive oxygen intermediate
- RT
reverse transcription
- STZ
streptozotocin
- TRX
thioredoxin
- hprt
hypoxanthine-guanine phosphoribosyl transferase
References
- 1.Gepts W. Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes. 1965;14:619–633. doi: 10.2337/diab.14.10.619. [DOI] [PubMed] [Google Scholar]
- 2.Makino S, Kunimoto K, Muraoka Y, Mizushima Y, Katagiri K, Tochino Y. Breeding of a non-obese, diabetic strain of mice. Jikken Dobutsu. 1980;29:1–13. doi: 10.1538/expanim1978.29.1_1. [DOI] [PubMed] [Google Scholar]
- 3.Kikutani H, Makino S. The murine autoimmune diabetes model: NOD and related strains. Adv Immunol. 1992;51:285–322. doi: 10.1016/s0065-2776(08)60490-3. [DOI] [PubMed] [Google Scholar]
- 4.Grankvist K, Marklund S, Sehlin J, Taljedal IB. Superoxide dismutase, catalase and scavengers of hydroxyl radical protect against the toxic action of alloxan on pancreatic islet cells in vitro. Biochem J. 1979;182:17–25. doi: 10.1042/bj1820017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kroncke KD, Kolb-Bachofen V, Berschick B, Burkart V, Kolb H. Activated macrophages kill pancreatic syngeneic islet cells via arginine-dependent nitric oxide generation. Biochem Biophys Res Commun. 1991;175:752–758. doi: 10.1016/0006-291x(91)91630-u. [DOI] [PubMed] [Google Scholar]
- 6.Corbett JA, Wang JL, Sweetland MA, Lancaster JR, Jr, McDaniel ML. Interleukin-1 beta induces the formation of nitric oxide by beta-cells purified from rodent islets of Langerhans. Evidence for the beta-cell as a source and site of action of nitric oxide. J Clin Invest. 1992;90:2384–2391. doi: 10.1172/JCI116129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mandrup-Poulsen T. The role of interleukin-1 in the pathogenesis of IDDM. Diabetologia. 1996;39:1005–1029. doi: 10.1007/BF00400649. [DOI] [PubMed] [Google Scholar]
- 8.Eizirik DL, Flodstrom M, Karlsen AE, Welsh N. The harmony of the spheres: inducible nitric oxide synthase and related genes in pancreatic beta cells. Diabetologia. 1996;39:875–890. doi: 10.1007/BF00403906. [DOI] [PubMed] [Google Scholar]
- 9.Rabinovitch A, Suarez-Pinzon WL, Sorensen O, Bleackley RC. Inducible nitric oxide synthase (iNOS) in pancreatic islets of nonobese diabetic mice: identification of iNOS-expressing cells and relationships to cytokines expressed in the islets. Endocrinology. 1996;137:2093–2099. doi: 10.1210/endo.137.5.8612552. [DOI] [PubMed] [Google Scholar]
- 10.Kurrer MO, Pakala SV, Hanson HL, Katz JD. Beta cell apoptosis in T cell-mediated autoimmune diabetes. Proc Natl Acad Sci USA. 1997;94:213–218. doi: 10.1073/pnas.94.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O'Brien BA, Harmon BV, Cameron DP, Allan DJ. Apoptosis is the mode of beta cell death responsible for the development of IDDM in the nonobese diabetic (NOD) mouse. Diabetes. 1997;46:750–757. doi: 10.2337/diab.46.5.750. [DOI] [PubMed] [Google Scholar]
- 12.Chervonsky AV, Wang Y, Wong FS, Visintin I, Flavell RA, Janeway CA, Jr, Matis LA. The role of Fas in autoimmune diabetes. Cell. 1997;89:17–24. doi: 10.1016/s0092-8674(00)80178-6. [DOI] [PubMed] [Google Scholar]
- 13.Itoh N, Imagawa A, Hanafusa T, Waguri M, Yamamoto K, Iwahashi H, Moriwaki M, Nakajima H, Miyagawa J, Namba M, et al. Requirement of Fas for the development of autoimmune diabetes in nonobese diabetic mice. J Exp Med. 1997;186:613–618. doi: 10.1084/jem.186.4.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaneto H, Fujii J, Seo HG, Suzuki K, Matsuoka T, Nakamura M, Tatsumi H, Yamasaki Y, Kamada T, Taniguchi N. Apoptotic cell death triggered by nitric oxide in pancreatic beta-cells. Diabetes. 1995;44:733–738. doi: 10.2337/diab.44.7.733. [DOI] [PubMed] [Google Scholar]
- 15.Grankvist K, Marklund SL, Taljedal IB. CuZn-superoxide dismutase, Mn-superoxide dismutase, catalase and glutathione peroxidase in pancreatic islets and other tissues in the mouse. Biochem J. 1981;199:393–398. doi: 10.1042/bj1990393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gandy SE, III, Galbraith RA, Crouch RK, Buse MG, Galbraith GM. Superoxide dismutase in human islets of Langerhans. N Engl J Med. 1981;304:1547–1548. doi: 10.1056/NEJM198106183042518. [DOI] [PubMed] [Google Scholar]
- 17.Malaisse WJ, Malaisse-Lagae F, Sener A, Pipeleers DG. Determinants of the selective toxicity of alloxan to the pancreatic beta cell. Proc Natl Acad Sci USA. 1982;79:927–930. doi: 10.1073/pnas.79.3.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cornelius JG, Luttge BG, Peck AB. Antioxidant enzyme activities in IDD-prone and IDD-resistant mice: a comparative study. Free Radic Biol Med. 1993;14:409–420. doi: 10.1016/0891-5849(93)90090-h. [DOI] [PubMed] [Google Scholar]
- 19.Lenzen S, Drinkgern J, Tiedge M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic Biol Med. 1996;20:463–466. doi: 10.1016/0891-5849(96)02051-5. [DOI] [PubMed] [Google Scholar]
- 20.Holmgren A. Thioredoxin. Annu Rev Biochem. 1985;54:237–271. doi: 10.1146/annurev.bi.54.070185.001321. [DOI] [PubMed] [Google Scholar]
- 21.Luthman M, Holmgren A. Rat liver thioredoxin and thioredoxin reductase: purification and characterization. Biochemistry. 1982;21:6628–6633. doi: 10.1021/bi00269a003. [DOI] [PubMed] [Google Scholar]
- 22.Laurent TC, Moore EC, Reichard P. Enzymatic synthesis of deoxyribonucleotides. IV. Isolation and characterization of thioredoxin, the hydrogen donor from Escherichia coliB. J Biol Chem. 1964;239:3436–3444. [PubMed] [Google Scholar]
- 23.Huber HE, Russel M, Model P, Richardson CC. Interaction of mutant thioredoxins of Escherichia coliwith the gene 5 protein of phage T7. The redox capacity of thioredoxin is not required for stimulation of DNA polymerase activity. J Biol Chem. 1986;261:15006–15012. [PubMed] [Google Scholar]
- 24.Hirota K, Matsui M, Iwata S, Nishiyama A, Mori K, Yodoi J. AP-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1. Proc Natl Acad Sci USA. 1997;94:3633–3638. doi: 10.1073/pnas.94.8.3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steitz TA. A mechanism for all polymerases. Nature. 1998;391:231–232. doi: 10.1038/34542. [DOI] [PubMed] [Google Scholar]
- 26.Nakamura H, Nakamura K, Yodoi J. Redox regulation of cellular activation. Annu Rev Immunol. 1997;15:351–369. doi: 10.1146/annurev.immunol.15.1.351. [DOI] [PubMed] [Google Scholar]
- 27.Berggren M, Gallegos A, Gasdaska JR, Gasdaska PY, Warneke J, Powis G. Thioredoxin and thioredoxin reductase gene expression in human tumors and cell lines, and the effects of serum stimulation and hypoxia. Anticancer Res. 1996;16:3459–3466. [PubMed] [Google Scholar]
- 28.Sasada T, Iwata S, Sato N, Kitaoka Y, Hirota K, Nakamura K, Nishiyama A, Taniguchi Y, Takabayashi A, Yodoi J. Redox control of resistance to cis-diamminedichloroplatinum (II) (CDDP): protective effect of human thioredoxin against CDDP-induced cytotoxicity. J Clin Invest. 1996;97:2268–2276. doi: 10.1172/JCI118668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yokomizo A, Ono M, Nanri H, Makino Y, Ohga T, Wada M, Okamoto T, Yodoi J, Kuwano M, Kohno K. Cellular levels of thioredoxin associated with drug sensitivity to cisplatin, mitomycin C, doxorubicin, and etoposide. Cancer Res. 1995;55:4293–4296. [PubMed] [Google Scholar]
- 30.Sonenshein GE. Rel/NF-kappa B transcription factors and the control of apoptosis. Semin Cancer Biol. 1997;8:113–119. doi: 10.1006/scbi.1997.0062. [DOI] [PubMed] [Google Scholar]
- 31.Karin M Z.g. Liu, and E. Zandi. AP-1 function and regulation. Curr Opin Cell Biol. 1997;9:240–246. doi: 10.1016/s0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- 32.Schenk H, Klein M, Erdbrugger W, Droge W, Schulze-Osthoff K. Distinct effects of thioredoxin and antioxidants on the activation of transcription factors NF-kappa B and AP-1. Proc Natl Acad Sci USA. 1994;91:1672–1676. doi: 10.1073/pnas.91.5.1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qin J, Clore GM, Kennedy WM, Huth JR, Gronenborn AM. Solution structure of human thioredoxin in a mixed disulfide intermediate complex with its target peptide from the transcription factor NF kappa B. Structure (Lond) 1995;3:289–297. doi: 10.1016/s0969-2126(01)00159-9. [DOI] [PubMed] [Google Scholar]
- 34.Qin J, Clore GM, Kennedy WP, Kuszewski J, Gronenborn AM. The solution structure of human thioredoxin complexed with its target from Ref-1 reveals peptide chain reversal. Structure (Lond) 1996;4:613–620. doi: 10.1016/s0969-2126(96)00065-2. [DOI] [PubMed] [Google Scholar]
- 35.Miyazaki J, Araki K, Yamato E, Ikegami H, Asano T, Shibasaki Y, Oka Y, Yamamura K. Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology. 1990;127:126–132. doi: 10.1210/endo-127-1-126. [DOI] [PubMed] [Google Scholar]
- 36.Gordon JW, Ruddle FH. Gene transfer into mouse embryos: production of transgenic mice by pronuclear injection. Methods Enzymol. 1983;101:411–433. doi: 10.1016/0076-6879(83)01031-9. [DOI] [PubMed] [Google Scholar]
- 37.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 38.Gotoh M, Maki T, Kiyoizumi T, Satomi S, Monaco AP. An improved method for isolation of mouse pancreatic islets. Transplantation (Baltimore) 1985;40:437–438. doi: 10.1097/00007890-198510000-00018. [DOI] [PubMed] [Google Scholar]
- 39.Mitsui A, Hirakawa T, Yodoi J. Reactive oxygen-reducing and protein-refolding activities of adult T cell leukemia-derived factor/human thioredoxin. Biochem Biophys Res Commun. 1992;186:1220–1226. doi: 10.1016/s0006-291x(05)81536-0. [DOI] [PubMed] [Google Scholar]
- 40.Okamoto Y, Ishida H, Taminato T, Tsuji K, Kurose T, Tsuura Y, Kato S, Imura H, Seino Y. Role of cytosolic Ca2+in impaired sensitivity to glucose of rat pancreatic islets exposed to high glucose in vitro. Diabetes. 1992;41:1555–1561. doi: 10.2337/diab.41.12.1555. [DOI] [PubMed] [Google Scholar]
- 41.Kurasawa K, Koike T, Matsumura R, Takabayashi K, Tomioka H, Ito I, Yoshida S. The immunosuppressant FK-506 prevents progression of diabetes in nonobese diabetic mice. Clin Immunol Immunopathol. 1990;57:274–279. doi: 10.1016/0090-1229(90)90041-n. [DOI] [PubMed] [Google Scholar]
- 42.Junod A, Lambert AE, Stauffacher W, Renold AE. Diabetogenic action of streptozotocin: relationship of dose to metabolic response. J Clin Invest. 1969;48:2129–2139. doi: 10.1172/JCI106180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McEvoy, R.C. 1984. Tissue culture of fetal rat pancreatic islets: quantification of changes in the number of islet cells during culture. In Methods in Diabetes Research I, part A. J. Larner and S.L. Pohl, editors. John Wiley & Sons, Inc., New York. 227–237.
- 44.Baker A, Payne CM, Briehl MM, Powis G. Thioredoxin, a gene found overexpressed in human cancer, inhibits apoptosis in vitro and in vivo. Cancer Res. 1997;57:5162–5167. [PubMed] [Google Scholar]
- 45.Gallegos A, Berggren M, Gasdaska JR, Powis G. Mechanisms of the regulation of thioredoxin reductase activity in cancer cells by the chemopreventive agent selenium. Cancer Res. 1997;57:4965–4970. [PubMed] [Google Scholar]
- 46.Kubisch HM, Wang J, Bray TM, Phillips JP. Targeted overexpression of Cu/Zn superoxide dismutase protects pancreatic beta-cells against oxidative stress. Diabetes. 1997;46:1563–1566. doi: 10.2337/diabetes.46.10.1563. [DOI] [PubMed] [Google Scholar]
- 47.Hansson HA, Holmgren A, Rozell B, Taljedal IB. Immunohistochemical localization of thioredoxin and thioredoxin reductase in mouse exocrine and endocrine pancreas. Cell Tissue Res. 1986;245:189–195. doi: 10.1007/BF00218100. [DOI] [PubMed] [Google Scholar]
- 48.Andre I, Gonzalez A, Wang B, Katz J, Benoist C, Mathis D. Checkpoints in the progression of autoimmune disease: lessons from diabetes models. Proc Natl Acad Sci USA. 1996;93:2260–2263. doi: 10.1073/pnas.93.6.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Takamura T, Kato I, Kimura N, Nakazawa T, Yonekura H, Takasawa S, Okamoto H. Transgenic mice overexpressing type 2 nitric-oxide synthase in pancreatic beta cells develop insulin-dependent diabetes without insulitis. J Biol Chem. 1998;273:2493–2496. doi: 10.1074/jbc.273.5.2493. [DOI] [PubMed] [Google Scholar]
- 50.Morgan NG, Cable HC, Newcombe NR, Williams GT. Treatment of cultured pancreatic B-cells with streptozotocin induces cell death by apoptosis. Biosci Rep. 1994;14:243–250. doi: 10.1007/BF01209729. [DOI] [PubMed] [Google Scholar]
- 51.Matsuda M, Masutani H, Nakamura H, Miyajima S, Yamauchi A, Ynehara S, Uchida A, Irimajiri K, Horiuchi A, Yodoi J. Protective activity of adult T cell leukemia-derived factor (ADF) against tumor necrosis factor-dependent cytotoxicity on U937 cells. J Immunol. 1991;147:3837–3841. [PubMed] [Google Scholar]
- 52.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK)-1. EMBO (Eur Mol Biol Organ) J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kagi D, Odermatt B, Seiler P, Zinkernagel RM, Mak TW, Hengartner H. Reduced incidence and delayed onset of diabetes in perforin-deficient nonobese diabetic mice. J Exp Med. 1997;186:989–997. doi: 10.1084/jem.186.7.989. [DOI] [PMC free article] [PubMed] [Google Scholar]