

Abstract
We explored the role of Gi protein signaling in the regulation of interleukin (IL)-12 production and T helper cell type 1 (Th1) T cell differentiation. In initial studies, we showed that treatment of normal mice with pertussis toxin (PT), which inhibits Gi protein signaling, enhanced the capacity of splenocytes to produce IL-12 in response to both microbial and nonmicrobial stimuli. In addition, PT treatment increased the production of tumor necrosis factor (TNF)-α and IL-10 by stimulated cells. These findings were corroborated by the fact that untreated Gi2α2/− mice exhibited enhanced production of IL-12 and TNF-α by splenocytes, and of IL-12 p40 by purified spleen CD8α+ lymphoid dendritic cells. Finally, we showed that while normal BALB/c mice infected with Leishmania major exhibited a nonhealing phenotype, those treated with PT when infection was initiated exhibited a healing phenotype along with an enhancement of leishmania-specific Th1 responses in draining lymph nodes. Further, healing was prevented by coadministration of anti–IL-12 and PT. These data demonstrate that endogenous Gi protein signaling has a primary role in the regulation of IL-12 production and the induction of Th1 responses in vivo.
Keywords: G protein, interleukin 12, T helper cell type 1, pertussis toxin, leishmaniasis
Introduction
Prior studies from this and other laboratories have demonstrated that signaling via Gi protein–coupled seven transmembrane domain receptors (7TDR) can inhibit the production of IL-12 by APCs and the subsequent induction of Th1 responses 1 2 3 4 5. Thus, it has been shown that the CC chemokine macrophage chemoattractant protein (MCP)-1–4, and the natural chemoattractant C5a, when added exogenously, can suppress IL-12 production from APCs in vitro 1 2. In addition, treatment of mice with MCP-1 has been shown to inhibit the induction of septic shock after exposure to LPS, and to enhance the induction of oral tolerance 3 5. These effects of MCP-1 are consistent with its ability to inhibit IL-12 production, as IL-12 has been proposed to have enhancing and inhibitory roles, respectively, in these processes. Finally, tetrahydrocannabinol and morphine, which act via Gi protein–coupled cannabinoid, and opioid receptors have been shown to inhibit the induction of Th1 responses in vivo 6 7.
Although these studies demonstrated that exogenous application of 7TDR ligands can result in the suppression of IL-12 production and Th1 responses, they did not address the role of endogenously produced 7TDR ligands in the regulation of normally occurring immune responses in vivo. In this report, we first show that treatment of normal mice with pertussis toxin (PT), which acts to inhibit Gi protein signaling in leukocytes 8 9, results in an enhanced capacity of splenocytes to produce IL-12 in response to both microbial and nonmicrobial stimuli. To rule out the possibility that PT acts via a non–Gi-dependent pathway 10, we show that splenocytes and dendritic cells (DCs) from mice deficient in Gi2α, a member of the Gi protein family, exhibit enhanced production of IL-12 similar to that seen with cells from PT-treated wild-type (WT) mice. Finally, we show that PT treatment of BALB/c mice infected with Leishmania major results in a healing phenotype and the enhancement of leishmania-specific Th1 responses in draining LNs. Taken together, these data strongly support the view that Gi protein signaling plays a central role in the regulation of IL-12 production and the induction of Th1 responses in vivo.
Materials and Methods
Mice.
Gi2α-deficient (Gi2α2/−) mice on the C57BL/6 background were bred from homozygous breeding pairs 11 originally provided by Baylor College of Medicine (Houston, TX). Age- and sex-matched WT C57BL/6 control mice as well as female BALB/c mice were obtained from the National Cancer Institute, National Institutes of Health. All mice used were between 8 and 13 wk of age and conventionally housed.
Reagents.
PT was purchased from List Biological Laboratories. Soluble leishmania antigen (SLA) was prepared as described previously 12. Staphylococcus aureus, Cowan's strain I (SAC; Pansorbin®), was supplied by Calbiochem. Recombinant murine IFN-γ was purchased from BD PharMingen. LPS (from Escherichia coli, serotype 0127:B8) was purchased from Sigma-Aldrich. Immunostimulatory CpG-containing oligodeoxynucleotide 1826 (TCCATGACGTTCCTGACGTT; ODN) 13 was synthesized with a nuclease-resistant phosphorothioate backbone by Operon Technologies. Recombinant trimerized murine CD40 ligand (CD40L) was provided by Immunex.
Treatment of Animals and Parasite Challenge.
8–12-wk-old female BALB/c mice were treated with 400 ng of PT in 100 μl of PBS intravenously on days 0 and 2. For antibody treatment, mice were given 2 mg of anti–murine IL-12 (clone C17.8) or control antibody on days 0, 2, 4, and 8. The initial treatment with PT and/or anti–IL-12 was followed by inoculation of 105 L. major (WHOM/IR/−/173) metacyclic promastigotes into the right hind footpad 14. Footpad swelling was measured weekly using a metric caliper. 6–7 wk after infection, mice were killed and draining LNs were removed for analysis of antigen-specific cytokine responses (see below). In addition, feet from representative animals were removed and fixed in 10% buffered formalin. Paraffin sections were made and stained with Giemsa stain according to established procedures.
Cell Culture Conditions and Measurement of Cytokine Production.
Splenocytes were obtained from Gi2α−/− mice and WT control mice and cultured at 2 × 106 cells/ml in RPMI 1640 (Biosource International) supplemented with 10% fetal bovine serum (Biosource International), 100 μg/ml penicillin, 10 μg/ml streptomycin, 50 μg/ml gentamicin (Life Technologies), 5% Medium NCTC-109 (Life Technologies), 15 mM Hepes buffer, 0.005 mM 2-ME, and 2 mM l-glutamine (cRPMI) at 37°C and 6% CO2. Cells were cultured with the indicated stimuli for 24 h, at which time supernatants were removed and frozen at −20°C until measurement of cytokines. Transiently adherent DCs were isolated by plating splenocytes on tissue culture dishes and incubating for 1 h at 37°C and 6% CO2. The plates were then washed with warmed PBS. Transiently adherent DC-enriched cells were then harvested after an additional 24 h of incubation at 37°C in cRPMI and stimulated at 6 × 105 cells/ml. Highly purified lymphoid DCs were prepared as described previously 15. In brief, spleens were digested with collagenase D (400 U/ml; Roche Molecular Biochemicals) and DNase I (15 μg/ml; Roche Molecular Biochemicals), treated with EDTA (5 mM), and CD11c+ cells were positively selected with anti–mouse CD11c-coated magnetic beads (Miltenyi Biotec). Selected cells were then stained with PE-labeled anti-CD8α and FITC-conjugated anti-B220 antibodies, and B220−CD8α+ cells were isolated by flow cytometric sorting (FACStar™; Becton Dickinson). Sorted DCs (98% for CD11c+ and CD8α+) were plated at 105 cells/200 μl and stimulated as indicated. For measurement of leishmania-specific cytokine responses, single cell preparations from draining popliteal LNs taken from mice 6 wk after parasite infection were plated in triplicate in a 96-well microtiter plate at 3 × 105 cells/200 μl. SLA was added to cultures at 2.5 μg/ml, and culture supernatants were analyzed for the presence of cytokines 14. IFN-γ was assessed by ELISA using antibody pairs from BD PharMingen. The lower limit of detection was 50 pg/ml for IFN-γ. All other cytokines were assayed by ELISA using the OptEIA™ set reagents (BD PharMingen) according to the manufacturer's instructions. The lower limit of sensitivity for the IL-12p70, IL-12p40, and TNF-α ELISAs was 30 pg/ml, for the IL-10 ELISA was 10 pg/ml, and for the IL-4 ELISA was 5 pg/ml.
Statistical Analysis.
Results represent the mean ± SD where applicable. Statistical significance of differences was determined by the Student's t test.
Results and Discussion
PT is an exotoxin produced by Bordetella pertussis with a hexameric structure similar to cholera toxin and E. coli heat-labile toxin 8. The pentameric B subunit mediates binding of the toxin to glycoprotein receptors on many eukaryotic cells. After binding, the A subunit of PT enters the cell and mediates ADP-ribosylation of the α subunits of Gi proteins. ADP-ribosylation results in inactivation of signaling. Thus, in initial studies to address our hypothesis that Gi protein signaling by endogenously produced 7TDR ligands will inhibit IL-12 production and Th1 differentiation in vivo, we treated mice with PT and assessed the ability of splenocytes from treated mice to produce IL-12 upon stimulation in vitro.
As shown in Fig. 1, we found that splenocytes from BALB/c mice pretreated with PT produced significantly higher amounts of IL-12 p40 and IL-12 p70 after exposure in vitro to the well-established IL-12 inducer SAC and IFN-γ (for a review, see reference 16) compared with splenocytes from non–PT-treated mice. This was also shown using unmethylated bacterial DNA sequences containing CpG motifs (ODNs), which are capable of stimulating IL-12 production in the absence of IFN-γ 17. Interestingly, both TNF-α and IL-10 production were also increased by PT treatment. Because IL-10 has been shown to be capable of suppressing IL-12 production 18, the fact that IL-10 levels were increased along with IL-12 suggested that the inhibitory effect of Gi protein signaling on IL-12 production is not due to stimulation of IL-10. In the absence of stimulation in vitro, isolated splenocytes cultured with PT alone did not produce any of the measured cytokines, suggesting that PT itself does not directly induce the cells to produce cytokines. Several prior reports are consistent with the ability of PT to enhance IL-12 production in vivo. Thus, it has been shown that natural infection by B. pertussis, as well as vaccination with a whole cell vaccine containing PT, induces a Th1-predominant T cell response 19 20. In addition, PT can enhance delayed-type hypersensitivity reactions 21 and has been shown to increase the severity of disease in animal models of Th1-mediated autoimmunity, such as experimental autoimmune encephalomyelitis 22 and experimental autoimmune uveitis 16.
Figure 1.
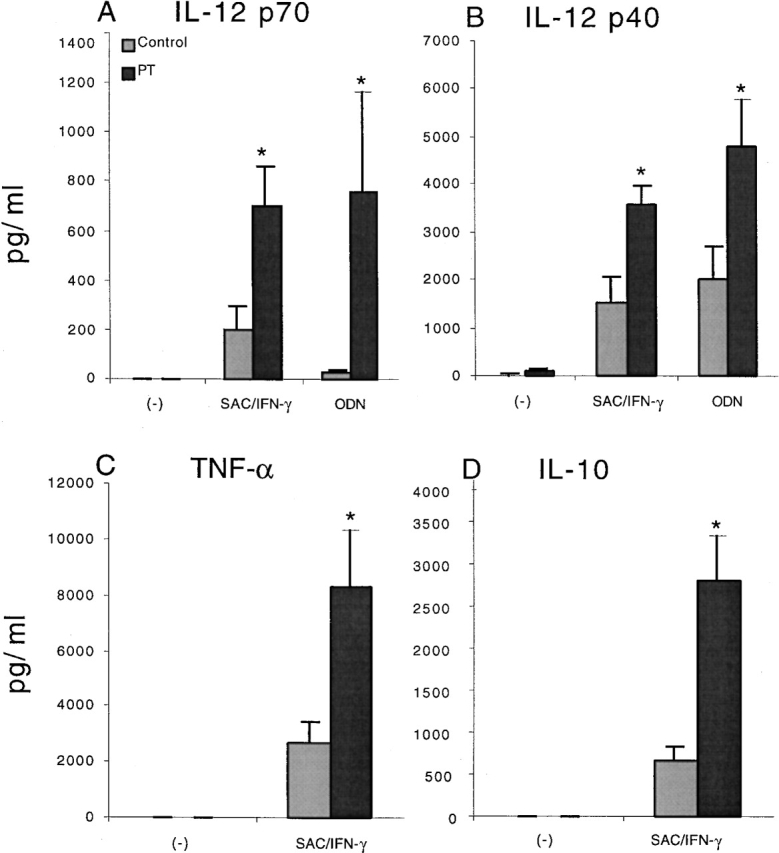
Treatment of mice with PT enhances IL-12 and TNF-α production by cultured splenocytes. This enhancement is seen with T cell–dependent and –independent stimuli and is not due to the suppression of IL-10 production. BALB/c mice were given 400 ng PT in 100 μl PBS. Controls were sex- and age-matched untreated BALB/c mice. Splenocytes from individual mice (n = 5) were prepared 20 h after PT treatment and stimulated with SAC (0.02%) and IFN-γ (10 ng/ml), or with ODNs (5 μg/ml). (A) IL-12 p70 production. (B) IL-12 p40 production. (C) TNF-α production. (D) IL-10 production. Data are presented as mean value ± SD and are representative of three separate experiments producing similar results. *P < 0.008.
To demonstrate that the effect of PT on enhancing cytokine production from splenocytes was due to the ability of PT to ADP-ribosylate and inactivate Gi proteins, and not to cellular activation by the pentameric B subunit 10 or to non-Gi protein–mediated signaling, we next examined cytokine production from cells from untreated Gi2α2/− mice, i.e., mice with defective Gi2 protein signaling due to targeted disruption of the Gi2α gene. A role for Gi2 in regulating immune responses was suggested by studies demonstrating that conventionally housed Gi2α2/− mice on certain genetic backgrounds spontaneously develop a Th1-mediated inflammatory colitis, with high local levels of IL-12 11 23. As shown in Fig. 2 A, we found that whole splenocytes from Gi2α2/− mice produced significantly higher levels of IL-12 p40, IL-12 p70, and TNF-α, and similar levels of IL-10 after stimulation with SAC and IFN-γ or with ODNs, compared with splenocytes from age- and sex-matched control mice. Interestingly, the degree of increase in levels of IL-12p40, IL-12p70, and TNF-α was virtually identical to that seen for the PT-treated WT mice. Splenocytes from Gi2α2/− mice were not enriched for cells capable of producing IL-12, i.e., monocyte/macrophages, DCs, or B cells, as determined by flow cytometry, compared with WT mice (data not shown). In addition, cell viability both before and after in vitro stimulation was identical for cells from both WT and Gi2α2/− mice (data not shown), so neither differences in starting cell populations nor differences in viability were factors influencing the level of cytokines produced by the Gi2α2/− cells.
Figure 2.
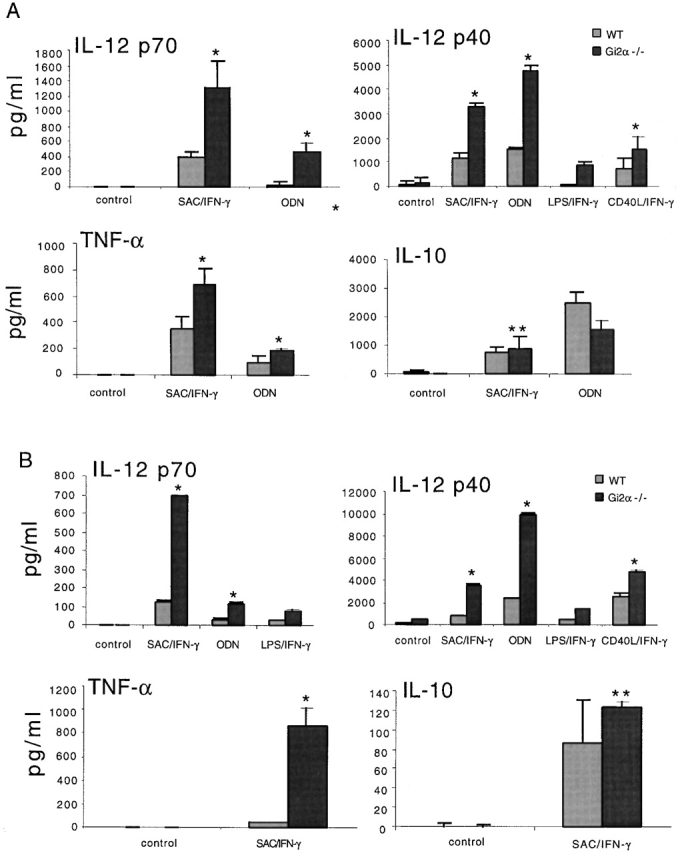
Increased IL-12 and TNF-α production by whole splenocytes and transiently adherent DC-enriched spleen cells from Gi2α2/− mice. Splenocytes and DC-enriched spleen cells from individual Gi2α2/− (n = 3) and WT (n = 3) mice were prepared and stimulated with SAC (0.02%), LPS (1 μg/ml), or CD40LT (4 μg/ml), together with IFN-γ (10 ng/ml), or with ODNs (5 μg/ml) alone. (A) Cytokine levels from whole splenocytes. (B) Cytokine levels from DC-enriched spleen cells. Data are presented as mean value ± SD and are representative of four separate experiments producing similar results. *P < 0.005; **P > 0.5.
Since it is now clear that DCs are an important source of IL-12 and are potent inducers of primary T cell responses 24, we also tested the ability of DCs from Gi2α2/− mice to produce IL-12, as well as other cytokines in response to bacterial and T cell stimuli. As shown in Fig. 2 B, stimulation of transiently adherent DC-enriched (40–60%) spleen cells with SAC, LPS, or a trimerized form of recombinant CD40L (to stimulate DCs via surface-expressed CD40) and IFN-γ or ODNs alone resulted in levels of IL-12 p70 and IL-12 p40 that were two- to fivefold higher than from control cells. In addition, DC-enriched cells from Gi2α2/− mice produced higher levels of TNF-α and similar amounts of IL-10 compared with cells from control mice.
It has recently been shown that the subpopulation of murine spleen DCs that expresses CD8α, and is thought to be derived from a lymphoid precursor, has an enhanced capacity to produce IL-12 and to induce the differentiation of Th1 cells compared with CD8α2CD11b+ myeloid-derived spleen DCs 25 26. Therefore, another possible explanation for why whole spleen DC populations from Gi2α2/− mice produce more IL-12 is that DC populations in such mice are skewed to a lymphoid phenotype. To test this possibility, we analyzed freshly isolated CD11c+ spleen DCs from Gi2α2/− and control mice for the expression of CD8α. We found no difference in the proportions of CD8α+ and CD8α2 CD11c+ DCs in the spleens of Gi2α2/− and WT mice (data not shown).
Next, we isolated highly purified CD8α+ DCs from spleens of Gi2α2/− and normal mice, and tested their ability to produce IL-12. As shown in Table , consistent with the other findings from this study, the CD8α+ DCs from Gi2α2/− mice produced significantly higher levels of IL-12 p40 than CD8α+ DCs from WT mice. These data support the hypothesis that PT, when given to WT mice, acts primarily by its ability to inhibit Gi protein signaling. In addition, they support a primary role for the specific Gi protein, Gi2, in immune regulation.
Table 1.
In Vitro Production of IL-12 p40 by Highly Purified CD8α+ DCs from Gi2α2/− and WT Mice
| IL-12 p40 | ||
|---|---|---|
| CD8α+ DCs | WT | Gi2α−/− |
| pg/ml | ||
| Unstimulated | <30 | 208.24 (± 59.71) |
| SAC/IFN-γ | 1,130.93 (± 119.42) | 6,669.02 (± 345.70) |
In a final series of studies, we determined the biological significance of Gi signaling pathways on the development of Th1 responses in vivo by assessing the effect of PT on the immune response to L. major, a well-studied infectious disease model whose resolution depends on an adequate Th1 response 27 28. In using this model, we took advantage of the fact that L. major infection of BALB/c mice results in progressive disease that correlates with the lack of a significant IFN-γ response to leishmanial antigens in draining LNs. This in turn has been shown to be due to an early Th2 (IL-4) response that can be negated by the administration of IL-12 and redirection of the response down the Th1 pathway 29. Thus, if PT induces Th1 responses, as suggested by the data presented above, it should have the same effect on L. major infection as IL-12. To examine this hypothesis, we treated BALB/c mice with PT at the time of infection and 48 h later. As shown in Fig. 3 and Fig. 4, such treatment resulted in a healing phenotype, which was accompanied by enhanced IFN-γ and TNF-α production by cells from the draining LNs after stimulation in vitro with L. major–specific antigens. In addition, IL-10 production by these cells was enhanced, again supporting the probability that PT does not act to increase IL-12 and IFN-γ production by inhibiting IL-10. In contrast, IL-4 production was low and not significantly different between cells from PT-treated and untreated mice. The latter finding is consistent with prior studies demonstrating that the induction of increased IFN-γ–producing T cells, rather than a reduction of IL-4–producing T cells, correlates with protection in this model 30. As expected, when anti–IL-12 was coadministered with PT, the enhanced production of IFN-γ, IL-10, and TNF-α by stimulated LN cells was not seen, and a nonhealing phenotype was again seen, as in the untreated mice.
Figure 3.



In vivo treatment with PT protects BALB/c mice from L. major infection. BALB/c mice were given 400 ng PT intravenously on days 0 and 2, and 2 mg anti–IL-12 or control antibody (controlAb) intraperitoneally on days 0, 2, 4, and 8. Mice were challenged in the hind footpad with 105 live L. major metacyclic promastigotes 2 h after the first dose of PT. (A) Footpad thickness was measured weekly. Data are presented as mean value ± SD for five individual mice, and are representative of two experiments producing similar results. (B) Giemsa-stained paraffin sections of footpads from representative control or PT-treated mice. An arrow indicates an amastigote inside a macrophage (original magnification: ×100).
Figure 4.
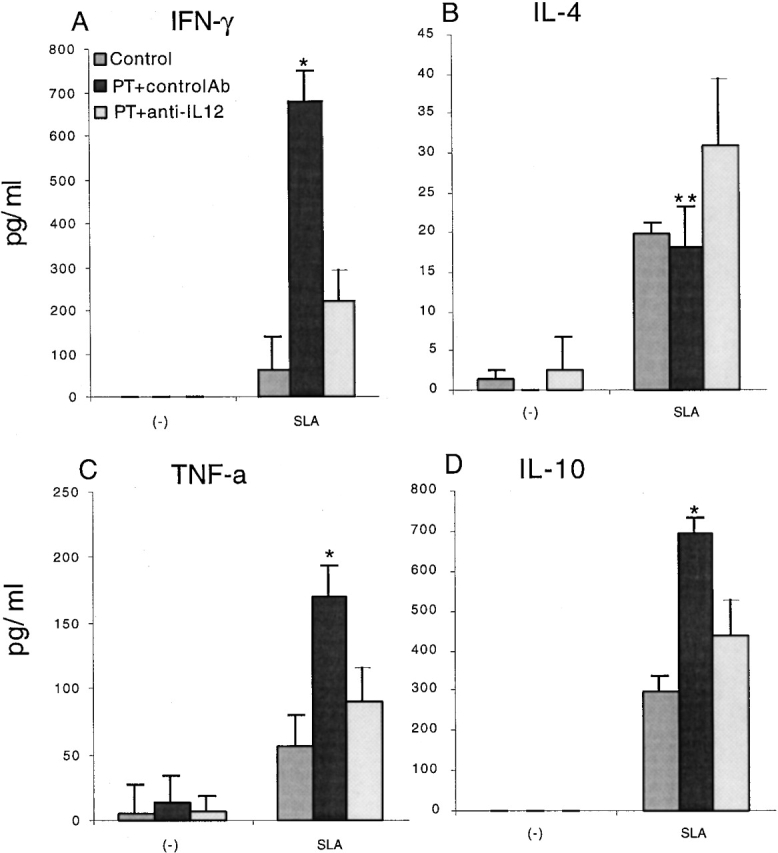
Cytokine production from draining LN cells after infection with L. major. BALB/c mice that were untreated (Control), treated with PT and control antibody (controlAb), or treated with PT and anti–IL-12 (n = 5 for each group) were infected in the footpad with L. major. Cells from draining popliteal LNs were isolated 6 wk after infection and cultured with or without SLA (2.5 μg/ml) for 24 h (for measuring IL-10 and TNF-α production) or 48 h (for measuring INF-γ and IL-4 production). (A) IFN-γ production. (B) IL-4 production. (C) TNF-α production. (D) IL-10 production. Data are presented as mean value ± SD for five individual mice, and are representative of two separate experiments producing similar results. *P < 0.005; **P > 0.5.
To confirm the lack of footpad swelling seen in the PT-treated mice was due to the elimination of parasites and not simply to a lack of influx of immune or inflammatory cells, we sectioned the feet of representative mice 6 wk after infection. As shown in Fig. 3 B, feet of control mice were significantly infected, as shown by the presence of visible amastigotes throughout the section. In contrast, feet from PT-treated mice had few, if any, visible amastigotes and had modest infiltration by lymphocytes.
Taken together, the data presented here demonstrate a major role for Gi protein signaling in the regulation of proinflammatory cytokine production from APCs, including CD8α+ DCs in mice. They also show that PT can enhance the Th1 response to L. major in susceptible BALB/c mice, resulting in protection against progressive disease. The fact that reversal of disease susceptibility was prevented by the coadministration of anti–IL-12 is consistent with the hypothesis that PT directly enhances the production of IL-12, which then drives increased Th1 development and protection in vivo. However, it is also likely that the increased production of TNF-α after PT treatment contributes to protection from disease by driving IFN-γ production by NK cells and by enhancing the effects of IFN-γ on nitric oxide–mediated parasite killing by host cells. In contrast, the effect of PT is unlikely to be due to an inhibitory effect on immune cell trafficking, since this should result in the inhibition rather than the enhancement of host resistance.
An implication of these data is that there is a constitutive suppression of proinflammatory cytokine production in vivo which is mediated by 7TDR ligands that activate Gi proteins, particularly Gi2. Among the candidate molecules that mediate such negative signals are chemokines, since we have demonstrated that selective chemokines (MCP-1–4, FMLP, and C5a, but not regulated upon activation, normal T cell expressed and secreted chemokine [RANTES], macrophage inflammatory protein [MIP]-1α, MIP-1β, or stromal cell–derived factor 1) suppress IL-12 production from human monocytes in vitro 1. One intriguing possibility in this regard is that endogenous production of MCP proteins by monocyte/macrophages upon stimulation with bacterial products results in an autocrine regulation of proinflammatory cytokine production after binding and signaling via surface-expressed CC chemokine receptor 2 (CCR2).
Acknowledgments
We thank Drs. W. Strober and S. Strauss for reviewing the manuscript and for helpful comments.
References
- Braun M.C., Lahey E., Kelsall B.L. Selective suppression of IL-12 production by chemoattractants. J. Immunol. 2000;164:3009–3017. doi: 10.4049/jimmunol.164.6.3009. [DOI] [PubMed] [Google Scholar]
- Wittmann M., Zwirner J., Larsson V.A., Kirchhoff K., Begemann G., Kapp A., Gotze O., Werfel T. C5a suppresses the production of IL-12 by IFN-gamma-primed and lipopolysaccharide-challenged human monocytes. J. Immunol. 1999;162:6763–6769. [PubMed] [Google Scholar]
- Karpus W.J., Kennedy K.J., Kunkel S.L., Lukacs N.W. Monocyte chemotactic protein 1 regulates oral tolerance induction by inhibition of T helper cell 1–related cytokines. J. Exp. Med. 1998;187:733–741. doi: 10.1084/jem.187.5.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chensue S.W., Warmington K.S., Ruth J.H., Sanghi P.S., Lincoln P., Kunkel S.L. Role of monocyte chemoattractant protein-1 (MCP-1) in Th1 (mycobacterial) and Th2 (schistosomal) antigen-induced granuloma formationrelationship to local inflammation, Th cell expression, and IL-12 production. J. Immunol. 1996;157:4602–4608. [PubMed] [Google Scholar]
- Zisman D.A., Kunkel S.L., Strieter R.M., Tsai W.C., Bucknell K., Wilkowski J., Standiford T.J. MCP-1 protects mice in lethal endotoxemia. J. Clin. Invest. 1997;99:2832–2836. doi: 10.1172/JCI119475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein T.W., Newton C., Zhu W., Daaka Y., Friedman H. delta 9-Tetrahydrocannabinol, cytokines, and immunity to Legionella pneumophila . Proc. Soc. Exp. Biol. Med. 1995;209:205–212. doi: 10.3181/00379727-209-43897b. [DOI] [PubMed] [Google Scholar]
- Pellis N.R., Harper C., Dafny N. Suppression of the induction of delayed hypersensitivity in rats by repetitive morphine treatments. Exp. Neurol. 1986;93:92–97. doi: 10.1016/0014-4886(86)90148-2. [DOI] [PubMed] [Google Scholar]
- Kaslow H.R., Burns D.L. Pertussis toxin and target eukaryotic cellsbinding, entry, and activation. FASEB J. 1992;6:2684–2690. doi: 10.1096/fasebj.6.9.1612292. [DOI] [PubMed] [Google Scholar]
- Kehrl J.H. Heterotrimeric G protein signalingroles in immune function and fine-tuning by RGS proteins. Immunity. 1998;8:1–10. doi: 10.1016/s1074-7613(00)80453-7. [DOI] [PubMed] [Google Scholar]
- Wong W.S., Rosoff P.M. Pharmacology of pertussis toxin B-oligomer. Can. J. Physiol. Pharmacol. 1996;74:559–564. doi: 10.1139/cjpp-74-5-559. [DOI] [PubMed] [Google Scholar]
- Rudolph U., Finegold M.J., Rich S.S., Harriman G.R., Srinivasan Y., Brabet P., Boulay G., Bradley A., Birnbaumer L. Ulcerative colitis and adenocarcinoma of the colon in G alpha i2-deficient mice. Nat. Genet. 1995;10:143–150. doi: 10.1038/ng0695-143. [DOI] [PubMed] [Google Scholar]
- Afonso L.C.C., Scharton T.M., Vieira L.Q., Wysocka M., Trinchieri G., Scott P. The adjuvant effect of interleukin-12 in a vaccine against Leishmania major . Science. 1994;263:235–237. doi: 10.1126/science.7904381. [DOI] [PubMed] [Google Scholar]
- Davis H.L., Weeratna R., Waldschmidt T.J., Tygrett L., Schorr J., Krieg A.M., Weeranta R. CpG DNA is a potent enhancer of specific immunity in mice immunized with recombinant hepatitis B surface antigen J. Immunol. 160 1998. 870 876[published erratum at 162:3103] [PubMed] [Google Scholar]
- Gurunathan S., Sacks D.L., Brown D.R., Reiner S.L., Charest H., Glaichenhaus N., Seder R.A. Vaccination with DNA encoding the immunodominant LACK parasite antigen confers protective immunity to mice infected with Leishmania major . J. Exp. Med. 1997;186:1137–1147. doi: 10.1084/jem.186.7.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A., Kelsall B.L. Freshly isolated Peyer's patch, but not spleen, dendritic cells produce interleukin 10 and induce the differentiation of T helper type 2 cells. J. Exp. Med. 1999;190:229–239. doi: 10.1084/jem.190.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver P.B., Chan C.C., Wiggert B., Caspi R.R. The requirement for pertussis to induce EAU is strain-dependentB10.RIII, but not B10.A mice, develop EAU and Th1 responses to IRBP without pertussis treatment. Invest. Ophthalmol. Vis. Sci. 1999;40:2898–2905. [PubMed] [Google Scholar]
- Klinman D.M., Yi A.K., Beaucage S.L., Conover J., Krieg A.M. CpG motifs present in bacteria DNA rapidly induce lymphocytes to secrete interleukin 6, interleukin 12, and interferon gamma. Proc. Natl. Acad. Sci. USA. 1996;93:2879–2883. doi: 10.1073/pnas.93.7.2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X., Riemann H., Gri G., Trinchieri G. Positive and negative regulation of interleukin-12 gene expression Eur. Cytokine Netw. 9Suppl.1998. 54 64 [PubMed] [Google Scholar]
- Brady M.T., Mahon B.P., Mills K.H. Pertussis infection and vaccination induces Th1 cells. Immunol. Today. 1998;19:534. doi: 10.1016/s0167-5699(98)01359-0. [DOI] [PubMed] [Google Scholar]
- Mahon B.P., Ryan M.S., Griffin F., Mills K.H. Interleukin-12 is produced by macrophages in response to live or killed Bordetella pertussis and enhances the efficacy of an acellular pertussis vaccine by promoting induction of Th1 cells. Infect. Immun. 1996;64:5295–5301. doi: 10.1128/iai.64.12.5295-5301.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sewell W.A., Munoz J.J., Vadas M.A. Enhancement of the intensity, persistence, and passive transfer of delayed-type hypersensitivity lesions by pertussigen in mice. J. Exp. Med. 1983;157:2087–2096. doi: 10.1084/jem.157.6.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz J.J., Bernard C.C., Mackay I.R. Elicitation of experimental allergic encephalomyelitis (EAE) in mice with the aid of pertussigen. Cell. Immunol. 1984;83:92–100. doi: 10.1016/0008-8749(84)90228-4. [DOI] [PubMed] [Google Scholar]
- Hornquist C.E., Lu X., Rogers F.P., Rudolph U., Shappell S., Birnbaumer L., Harriman G.R. G(alpha)i2-deficient mice with colitis exhibit a local increase in memory CD4+ T cells and proinflammatory Th1-type cytokines. J. Immunol. 1997;158:1068–1077. [PubMed] [Google Scholar]
- Banchereau J., Steinman R.M. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Maldonado-Lopez R., De Smedt T., Michel P., Godfroid J., Pajak B., Heirman C., Thielemans K., Leo O., Urbain J., Moser M. CD8α1 and CD8α− subclasses of dendritic cells direct the development of distinct T helper cells in vivo. J. Exp. Med. 1999;189:587–592. doi: 10.1084/jem.189.3.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulendran B., Smith J.L., Caspary G., Brasel K., Pettit D., Maraskovsky E., Maliszewski C.R. Distinct dendritic cell subsets differentially regulate the class of immune response in vivo. Proc. Natl. Acad. Sci. USA. 1999;96:1036–1041. doi: 10.1073/pnas.96.3.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott P., Natovitz P., Coffman R.L., Pearce E., Sher A. Immunoregulation of cutaneous leishmaniasis. T cell lines that transfer protective immunity or exacerbation belong to different T helper subsets and respond to distinct parasite antigens. J. Exp. Med. 1988;168:1675–1684. doi: 10.1084/jem.168.5.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzel F.P., Sadick M.D., Holaday B.J., Coffman R.L., Locksley R.M. Reciprocal expression of interferon gamma or interleukin 4 during the resolution or progression of murine leishmaniasis. Evidence for expansion of distinct helper T cell subsets. J. Exp. Med. 1989;169:59–72. doi: 10.1084/jem.169.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzel F.P., Schoenhaut D.S., Rerko R.M., Rosser L.E., Gately M.K. Recombinant interleukin-12 cures mice infected with Leishmania major . J. Exp. Med. 1993;177:1505–1509. doi: 10.1084/jem.177.5.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris L., Troutt A.B., Handman E., Kelso A. Changes in the precursor frequencies of IL-4 and IFN-gamma secreting CD4+ cells correlate with resolution of lesions in murine cutaneous leishmaniasis. J. Immunol. 1992;149:2715–2721. [PubMed] [Google Scholar]