

Abstract
The M88.7 T cell clone recognizes an antigen presented by HLA B*1302 on the melanoma cell line M88. A cDNA encoding this antigen (NA88-A) was isolated using a library transfection approach. Analysis of the genomic gene's sequence identified it is a processed pseudogene, derived from a retrotranscript of mRNA coding for homeoprotein HPX42B. The NA88-A gene exhibits several premature stop codons, deletions, and insertions relative to the HPX42B gene. In NA88-A RNA, a short open reading frame codes for the peptide MTQGQHFLQKV from which antigenic peptides are derived; a stop codon follows the peptide's COOH-terminal Val codon. Part of the HPX42B mRNA's 3′ untranslated region codes for a peptide of similar sequence (MTQGQHFSQKV). If produced, this peptide can be recognized by M88.7 T cells. However, in HPX42B mRNA, the peptide's COOH-terminal Val codon is followed by a Trp codon. As a result, expression of HPX42B mRNA does not lead to antigen production. A model is proposed for events that participated in creation of a gene coding for a melanoma antigen from a pseudogene.
Keywords: peptide, epitope, processing, tumor immunity, CTL
Introduction
CTLs can recognize antigens presented by human tumor cells. In the hope that manipulating this CTL response to combat cancer will prove possible, a number of groups have started to identify the antigens in question (for review see reference 1). The results of several clinical trials of both adoptive immunotherapy 2 and vaccination 3 4 5 based on this work suggest that the effort is worthwhile.
Most of the antigens characterized to date are HLA class I restricted and presented by melanoma cells. Many of them are derived from normal or mutated gene products obtained by classical pathways of gene expression 1. However, an increasing number of reports describe antigens resulting from the use of nonclassical pathways of gene expression, when unexpected events take place during transcription, splicing, or translation 6. Thus, the NA17-A antigen 7 is translated from a transcript obtained by activation of a cryptic promoter within intron sequences of the N-acetylglucosaminyltransferase-V gene. Reverse strand transcription of a housekeeping gene gives rise to an antigen recognized by CTLs on a human kidney tumor 8. Intron retention during splicing is responsible for generation of some antigens from the gp100 9 and tyrosinase-related protein (TRP)-2 10 genes. Use of alternative open reading frames (ORFs) for translation can also create antigens, as described for a breast and melanoma-shared antigen 11, a gp75TRP-1 antigen 12, a renal cell carcinoma antigen derived from the intestinal carboxyl esterase gene 13, and an antigen derived from the LAGE-1 gene 14.
We describe here a new tumor antigen generating an HLA class I–restricted CTL response against melanoma. Surprisingly, the antigen is coded for by a processed pseudogene. Expression of the “living” counterpart of the pseudogene does not lead to a corresponding CTL response. This difference between the two genes provides insights into how a gene coding for a tumor antigen was created.
Materials and Methods
Cell Lines.
Melanoma cell lines (M) were established from metastatic tumor fragments from different patients. IPC 277/5 and IGR 1/54 melanoma cell lines were gifts from C. Aubert (INSERM U119, Marseille, France). The FM2.29 cell line was a gift from J. Zeuthen (Danish Cancer Society Research Center, Copenhagen, Denmark). DAUV melanoma cell line and mouse fibrosarcoma WEHI 164 clone 13 used for TNF assays was obtained from T. Boon (Ludwig Institute for Cancer Research, Brussels, Belgium). T cell clone M88.7 was derived from tumor infiltrating lymphocytes of patient M88 (A*0201, A*0301, B*4002, B*1302, Cw*02022, Cw*0602) as described previously 15.
HLA Transfection in Tumor Cell Lines.
Cells were seeded in 96-well plates and incubated until 80% confluent before addition of expression vector (100 ng) and Lipofectamine (0.5 μl) in serum-free medium. After 12–15 h, the transfection mixture was replaced with complete medium. Transfected tumor cell lines were used for stimulation of T cell clone M88.7 36–48 h after transfection.
Library Screening.
Poly A+ RNA was obtained from M88 cells using the FastTrack 2.0 mRNA extraction kit (Invitrogen Corp.). mRNA was converted to cDNA using a cDNA synthesis kit (Stratagene Inc.). The cDNA obtained was inserted into pCI-neo (Promega). Recombinant plasmids were electroporated into Escherichia coli XL2-Blue MRF′ (Stratagene Inc.). For screening, 1,500 pools of 100 ampicillin-resistant bacteria per pool were constituted. Plasmid DNA was extracted by the alkaline lysis method from each pool 16. For genomic gene cloning, a human cosmid library (provided by A. Haunauer, IGBMC, Strasbourg, France) was hybridized with the 2.7-kb NA88-A cloned cDNA. Selected regions of the positive cosmid identified were sequenced using the Applied Biosystems 373 DNA sequencer with the DNA sequencing Kit Dye terminator (Perkin Elmer).
Transfection of COS-7 Cells.
DEAE–dextran-chloroquine transfection was as described 17. In brief, 1.5 × 104 COS-7 cells were transfected with 100 ng of the HLA B*1302 expression vector and 100 ng of a pool of the cDNA library. Transfectants were tested after 48 h for their ability to stimulate TNF production by T cells. One positive pool was identified, and 500 individual plasmids obtained from it tested for the ability to trigger T cell clone TNF release as described for pools.
T Cell Stimulation Assay.
T cells (2.5 × 103 per well) were added to COS-7 cells or tumor cells 24–48 h after transfection. Culture supernatants were harvested 6 h later and tested for TNF content by measuring culture supernatant cytotoxicity to WEHI 164 clone 13 in a colorimetric assay 18.
Production of Modified cDNAs.
Truncated NA88-A cDNAs were generated using standard techniques 16 and cloned into pcDNA3 (Invitrogen Corp.) or into pCI-neo. pNA88-A Trp, pNA88-A Ser, and pHPX42B Stop were generated using the QuikChange™ Site-Directed Mutagenesis Kit (Stratagene Inc.).
Reverse Transcriptase–PCR Assays.
Total RNA was extracted by the guanidinium–cesium chloride procedure 16 and used for cDNA synthesis 19. Some samples of tumor and normal tissue cDNA were provided by F. Brasseur (Ludwig Cancer Institute, Brussels, Belgium). To test for genomic DNA contamination, cDNA samples were used for PCR with primers designed to amplify the MAGE-1 genomic gene. None of the samples used here gave a positive result in this test. For amplification of NA88-A cDNA, PCR assays were performed with primers 5′-ccttgtggttcgagtccc-3′ and 5′-GGACACGGCCACAGGACC-3′ for 35 cycles (1 min at 94°C, 1 min at 62°C, and 1 min 30 s at 72°C). Under these conditions, primers will not amplify HPX42B cDNA. PCR products were size fractionated and blotted to Hybond-N+ (Amersham Pharmacia Biotech) before hybridization with the cloned NA88-A cDNA. Samples were normalized for RNA and cDNA integrity and quantity by PCR amplification of human β-actin cDNA 19.
Peptide Stimulation Assays.
T cell clone M88.7 was stimulated with different concentrations of peptide (Leiden University Medical Center) in the presence or absence of 10 μg/ml brefeldin-A (Sigma-Aldrich). For stimulation without brefeldin-A, culture supernatants were harvested 6 h later and tested for TNF content. For stimulation with brefeldin-A, after 6 h cells were fixed for 10 min with PBS containing 4% paraformaldehyde, washed twice, and stored at 4°C. Fixed, stimulated cells were stained for cytokine. In brief, cells were stained with the PE-conjugated anti–IL-2 mAb (MQ1-17H12; BD PharMingen) at a concentration of 5 μg/ml for 30 min at room temperature. Reagent dilutions and washes were made with PBS containing 0.1% BSA (Sigma-Aldrich) and 0.1% saponin (Sigma-Aldrich). After staining and washes, cells were resuspended in PBS and analyzed on a FACScan™ (Becton Dickinson).
Results
The M88.7 T Cell Clone Recognizes an Antigen Presented by HLA B*1302.
The CD8+ T cell clone M88.7, isolated from a tumor-invaded lymph node of melanoma patient M88, has no lytic activity but is able to secrete TNF and IL-2 on stimulation by autologous M88 melanoma cells (data not shown). To identify the restricting molecule, a panel of 12 melanoma cell lines not recognized by M88.7 T cells was transiently transfected with expression vectors for the different HLA molecules of the M88 patient. After transfection with the HLA B*1302 vector (pHLA B*1302), 6 of the 12 lines stimulated TNF release by M88.7 T cells (Fig. 1 A).
Figure 1.
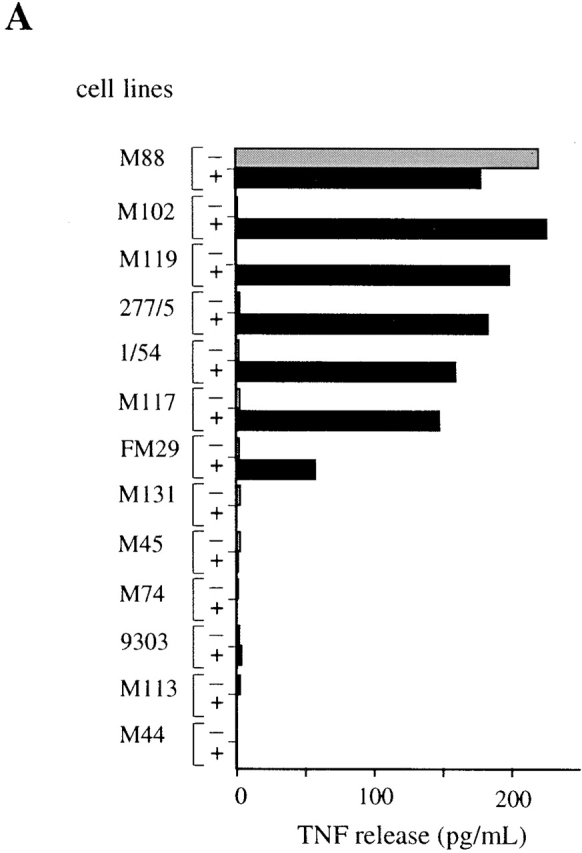

Characterization of the antigen recognized by M88.7 T cells. (A) Restriction element determination. Melanoma cell lines were tested for their ability to induce TNF release by M88.7 T cells before (−) and after (+) transfection with an HLAB*1302 expression vector. (B) Cloning of the cDNA. COS-7 cells were transfected with plasmids as indicated and tested for their ability to induce TNF release by M88.7 T cells.
Identification of a cDNA Coding for the Antigen.
A cDNA library was made from M88 RNA. Plasmid DNA extracted from pools of bacterial colonies from the library was cotransfected with pHLA B*1302 into COS-7 cells. After 48 h, M88.7 T cells were added and TNF production was assayed. Plasmid pool 651 proved positive in this test (Fig. 1 B), and the individual plasmid (pNA88-A) coding for the antigen recognized by M88.7 T cells recovered from it. Antigenic peptide production directed by pNA88-A is not limited to transfected COS-7 cells: an HLA B*1302 melanoma cell line not normally recognized by M88.7 T cells became recognizable after transfection with pNA88-A (data not shown).
The NA88-A cDNA was sequenced (EMBL/GenBank/DDBJ accession no. AF164963). Expression of the NA88-A gene was then studied by reverse transcriptase (RT)-PCR using primers 400 and 970 (Fig. 2 A) chosen from the sequence. All six melanoma cell lines known to produce antigen (i.e., those that stimulated TNF production by M88.7 T cells after transfection with pHLA B*1302; see Fig. 1 A) yielded an RT-PCR product of the size expected for the NA88-A product. These RT-PCR products hybridized to NA88-A cDNA (data not shown). The remaining six cell lines known to not produce antigen (i.e., those unable to stimulate TNF production by M88.7 T cells, even after transfection with pHLA B*1302; Fig. 1 A) yielded no such RT-PCR product (data not shown).
Figure 2.
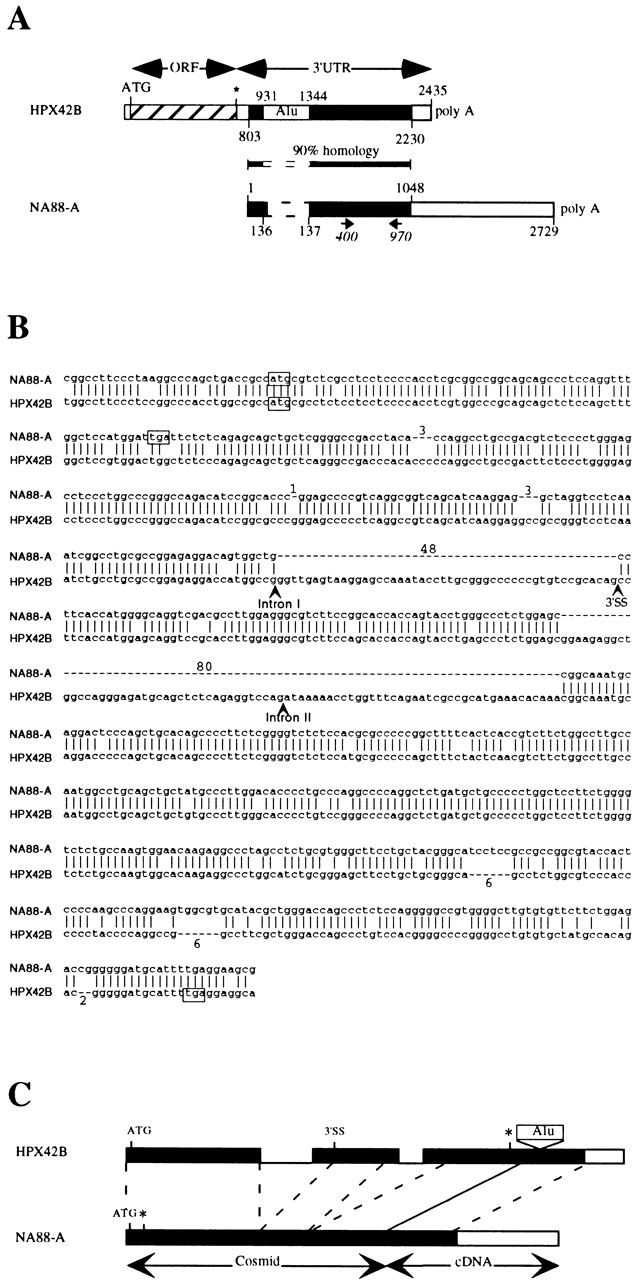
Schematic representations of HPX42B and NA88-A sequences. Drawings are not to scale. (A) cDNAs; EMBL/GenBank/DDBJ accession nos. AF164963 (NA88-A) and AF068006 (HPX42B). Homologous regions of the two cDNAs are marked in black. For HPX42B cDNA, the open reading frame coding for the homeoprotein (ORF) and the 3′ UTR containing an Alu family member (Alu) are marked. For NA88-A cDNA, the positions of primers used for RT-PCR are marked (400 and 970). (B) Alignment of HPX42B and NA88-A genomic sequences, EMBL/GenBank/DDBJ accession nos. AF164964 and AC006177, respectively. The positions of two HPX42B gene introns are marked, but not their sequences. Dashes represent bases missing in one of the two sequences, and the number of bases concerned is marked. The alternative 3′ splice site of the HPX42B second exon is marked 3′SS. For the NA88-A sequence, the last base shown (g) corresponds to the first base of the cloned cDNA whose sequence is represented in Fig. 2 A. (C) Schematic alignment of the genomic sequences. For the HPX42B gene, exons are marked as boxes, and introns separating them are shown as lines. The alternative 3′ splice site is marked 3′SS. The positions of initiation codons (ATG) and stop codons (*) discussed in the text and boxed in A are marked. The NA88-A gene representation is a composite using information derived from the cosmid and the cDNA as indicated.
Using the RT-PCR approach validated by the above results, we went on to search for NA88-A RNA in a variety of normal and tumor tissues. We were unable to detect NA88-A RNA in any of the normal tissues tested, including normal melanocytes in culture, with the possible exception of testis (only two samples were tested, though both proved positive; data not shown). Among the tumor samples we tested, expression of NA88-A RNA was limited to some metastatic melanomas (7 of the 66 tested; data not shown).
NA88-A RNA Is Similar to the Homeoprotein-encoding HPX42B mRNA.
There was no long ORF on the 2.7-kb NA88-A cDNA, although it did contain an AATAAA polyadenylation signal and terminal poly A tail. While the sequence was new, its first 1,048 nucleotides proved to be ∼90% identical to two noncontiguous parts (separated by an Alu family member) of the 3′ untranslated region (UTR) of the previously reported HPX42B cDNA (Fig. 2 A; similar sequences are shown in black). HPX42B cDNA was originally obtained in a search for new homeobox genes, using degenerate PCR on cDNA derived from a purified population of CD34+ human hemopoietic cells 20. The similarity between the NA88-A and HPX42B sequences suggested that the cloned NA88-A cDNA could be part of the 3′ UTR of a homeoprotein-encoding gene related to HPX42B. To test this hypothesis, we decided to isolate the genomic NA88-A gene by screening a human cosmid library.
A Processed Pseudogene Encodes NA88-A RNA.
We isolated one NA88-A cosmid and sequenced part of it to search for sequences coding for a HPX42B-like protein. The NA88-A sequence determined is shown in Fig. 2 B, aligned with the corresponding HPX42B gene sequence. (A region of chromosome 10 including the HPX42B has been sequenced. Comparison of this sequence with the HPX42B cDNA shows that the gene contains two introns. Their positions, but not their sequences, are marked in Fig. 2 B.) The NA88-A and HPX42B sequences are very similar. However, relative to the HPX42B gene, the NA88-A gene contains a number of mutations. One point mutation converts codon 22 of the HPX42B gene (tgg) into a premature stop codon (tga; boxed in Fig. 2 B). Relative to the HPX42B gene, the NA88-A gene also contains a number of deletions and insertions. While several of these remove or add codons without changing the reading frame, two of the deletions (of 1 and 80 bp) and one insertion (of 2 bp) do change it. As a result of all these mutations, the NA88-A gene no longer codes for a homeoprotein.
Further comparison of the genes shows that the NA88-A gene lacks equivalents of the HPX42B gene's introns (Fig. 2B and Fig. C). These observations strongly suggest that the NA88-A gene is a processed pseudogene, resulting from chromosomal integration of part of a retrotranscript of HPX42B mRNA. For splicing of the first HPX42B intron, the HPX42B mRNA employed to generate the NA88-A gene appears to have used an alternative 3′ splice site (3′SS in Fig. 2B and Fig. C). We cannot tell how the second intron was removed, as the NA88-A gene contains a large deletion covering the region corresponding to the junction between HPX42B exons 2 and 3. One hallmark of processed pseudogenes is the presence of an A-rich sequence corresponding to the poly A tail of the original mRNA. However, as the NA88-A gene lacks sequences corresponding to the end of the HPX42B mRNA's 3′ UTR (Fig. 2 A), this A-rich sequence is not expected to be present in the NA88-A pseudogene. In conclusion, our data show that the NA88-A antigen is coded for (a) by a processed pseudogene and (b) by sequences derived from the 3′ UTR of the HPX42B mRNA.
Identification of the Peptide Recognized by M88.7 T Cells.
To identify the region of the 2,729-bp NA88-A cDNA coding for the antigenic peptide, we tested a number of subfragments for their ability to direct antigen production. TNF was produced by M88.7 T cells exposed to COS-7 cells cotransfected with pHLA B*1302 and vectors containing the NA88-A cDNA region 1–722, but not by M88.7 T cells exposed to COS-7 cells cotransfected with pHLA B*1302 and vectors containing the NA88-A cDNA region 1–700 (Fig. 3 A). These results positioned the end of the sequence coding for the antigenic peptide between nucleotides 700 and 722. Peptides covering all three reading frames encoded by the cDNA region 684–730 were synthesized (Fig. 3 A) and tested for their capacity to induce TNF production by M88.7 T cells. Only one of them led to significant TNF production (peptide 1, RMTQGQHFLQKV; Fig. 3 B).
Figure 3.

Identification of the antigenic peptide. (A) Translation products of nucleotides 674–730 of NA88-A cDNA in all three reading frames. 1, 2, and 3: peptides synthesized. (B) Production of TNF by M88.7 T cells in response to M88 cells or one of the three synthetic peptides shown in A as marked. (C and D) Determination of the minimal antigenic peptide. Production of TNF (C) and IL-2 (D) by the M88.7 T clone in response to different synthetic peptides derived from peptide 1. For A, T cell clone M88.7 was stimulated with different concentrations of each peptide. For B, T cell clone M88.7 was stimulated with 50 μM peptide. Fixed, stimulated cells were stained for cytokine and analyzed on a FACScan™. Percentages of positive cells are indicated in the dot plots.
To characterize the antigenic peptide further, two 10-mers and two 9-mers representing different but overlapping regions of the 12-mer peptide 1 were tested in parallel for their capacity to induce secretion of TNF by M88.7 T cells (Fig. 3 C). Results obtained show (a) that removal of the COOH-terminal valine abolishes peptide activity and (b) that the 10-mer (TQGQHFLQKV) and the 9-mer (QGQHFLQKV) with this valine present are both about as active as the parent 12-mer. A similar conclusion was reached when IL-2 production by M88.7 T cells exposed to the peptides was measured (Fig. 3 D).
HPX42B mRNA Expression Does Not Lead to T Cell Stimulation.
An alignment of the HPX42B and NA88-A cDNA sequences covering the region of the NA88-A sequence coding for the antigenic peptide is shown in Fig. 4 A. Part of the 3′ UTR of the HPX42B cDNA can potentially encode the peptide MTQGQHFSQKV, which differs from the NA88-A antigenic peptide by a single substitution, serine (underlined) for leucine. To test if HPX42B mRNA expression leads to stimulation of M88.7 T cells, COS-7 cells were cotransfected with pHLAB*1302 and pHPX42B (Fig. 4 B), an expression vector containing the entire HPX42B cDNA. Transfected cells were unable to stimulate TNF production by M88.7 T cells (Fig. 4 C). However, COS-7 cells cotransfected with pHLAB*1302 and a point-mutated NA88-A expression vector coding for the serine-containing HPX42B peptide (pNA88-A Ser; Fig. 4 B) did stimulate TNF production by M88.7 T cells (Fig. 4 C), albeit less well than cells transfected with wild-type pNA88-A. Thus, although both NA88-A RNA and HPX42B mRNA code for a peptide capable of stimulating M88.7 T cells, only NA88-A RNA expression leads to peptide presentation.
Figure 4.
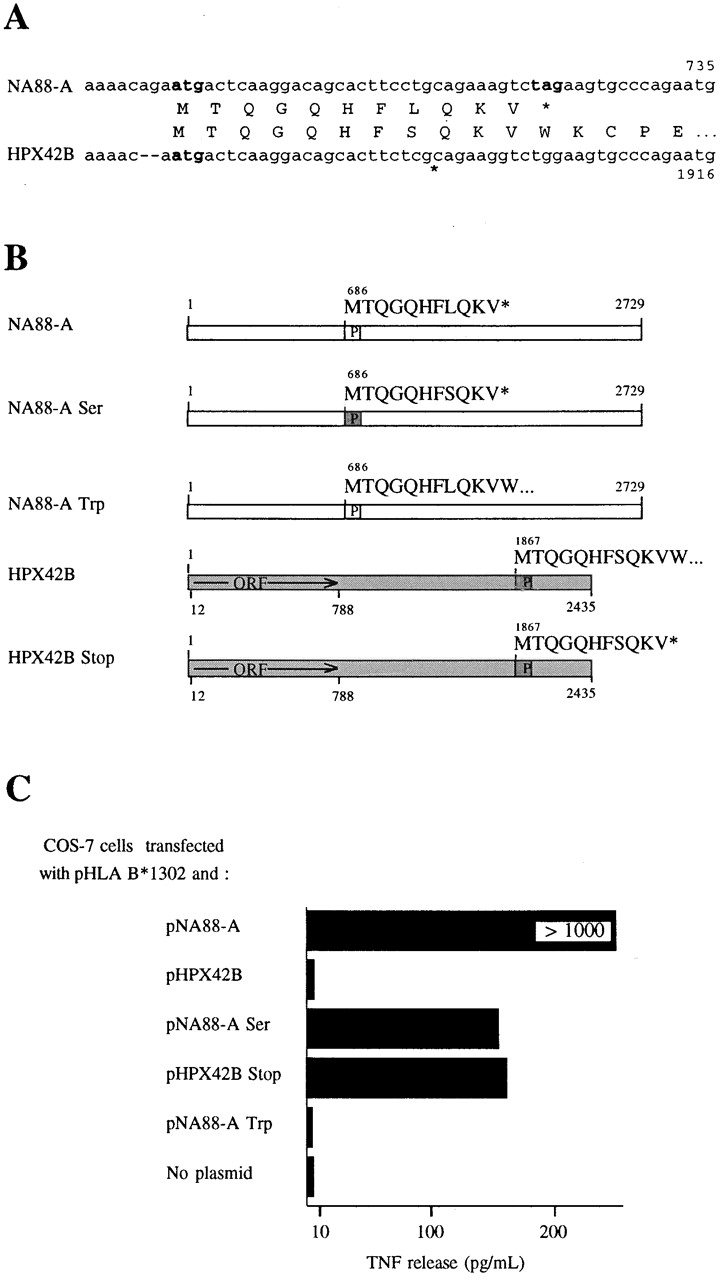
Identification of differences between HPX42B and NA88-A cDNAs important for their differential ability to generate a signal stimulating TNF release by M88.7 T cells. (A) Comparison of NA88-A and HPX42B nucleotide and encoded protein sequences in the region coding for antigenic peptide. Potential initiation and stop codons are indicated in bold characters. (B) Schematic representations of normal and mutated NA88-A and HPX42B cDNA sequences. Boxes marked P code for antigenic peptides and are white for the NA88-A peptide and grey for the HPX42B peptide (as marked above each box). For NA88-A Ser, a Leu codon, CTG, has been changed to a Ser codon, TCG, within the peptide-coding region. For NA88-A Trp, a stop codon tag immediately following the antigenic peptide's terminal Val codon has been changed to a Trp codon, tgg. For HPX42B Stop, a Trp codon, tgg, immediately following a Val codon has been changed to a stop codon, tag. (C) pcDNA3-based plasmids containing cDNAs as described in B were cotransfected into COS-7 cells with pHLA B*1302. The ability of transfected cells to induce TNF release by M88.7 T cells was measured. Note that all results shown here are from transfections carried out simultaneously in parallel, allowing comparison of results between samples. TNF release values for pNA88-A are higher than those shown in the separate experiment of Fig. 1 and Fig. 3, as results depend on COS-7 cell transfection efficiencies and the condition of the WEHI cells used for TNF assays, both of which vary between individual experiments.
A Stop Codon in the NA88-A Gene Is Vital for the T Cell Response.
In NA88-A RNA, the codon coding for the COOH-terminal Val of the antigenic peptide is followed by a stop codon, whereas in HPX42B mRNA, the corresponding Val codon is followed by a Trp codon (Fig. 4 A). This difference is important. COS-7 cells cotransfected with pHLAB*1302 and pNA88-A Trp (Fig. 4 B), in which the stop codon has been replaced by the Trp codon found in HPX42B mRNA, were unable to stimulate significant TNF production by M88.7 T cells (Fig. 4 C). However, COS-7 cells cotransfected with pHLAB*1302 and pHPX42B Stop (Fig. 4 B), in which the Trp codon has been mutated to a stop codon, were able to stimulate TNF production by M88.7 T cells (Fig. 4 C). TNF levels obtained were similar to those obtained using pNA88-A Ser, which codes for the same serine-containing peptide.
Discussion
The NA88-A gene is a processed pseudogene, which no longer codes for any likely functional protein. The NA88-A antigen is encoded by a very short ORF, which was derived from part of a functional mRNA's 3′ UTR. Our results thus extend the possible sources of tumor antigen coding sequences to “junk” DNA and seriously raise the possibility that any DNA sequence can lead to antigen production, the only limitation being that it must be transcribed. As is not surprising for junk DNA, transcription of the NA88-A gene is limited. Significant expression is found only in a fraction of metastatic melanomas. Transcription of the gene may occur only at a late stage in melanoma, after multiple genetic changes have accumulated facilitating aberrant transcription.
In contrast, the NA88-A's “parent” gene, the HPX42B gene, is transcribed in a variety of normal tissues (our unpublished observations). The 3′ UTR of HPX42B mRNA has codons for a peptide that can be recognized by M88.7 T cells. However, unlike for NA88-A RNA, expression of HPX42B mRNA does not lead to T cell stimulation. One reason is that antigenic peptides must be excised from a precursor of 125 amino acids (which begins MTQGQHFSQKVWKCPEWE…; antigenic peptide in bold). Thus, expression of a mutated HPX42B mRNA, in which the Trp codon immediately following the peptide's terminal Val codon, is replaced by a stop codon, will lead to T cell stimulation. The absence of M88.7 T cell stimulation after wild-type HPX42B mRNA expression can be explained if downstream flanking residues stop appropriate peptides from being produced in quantities sufficient for presentation. Proteolytic cleavage COOH-terminal to the peptide's Val residue may be inefficient. Other cleavages may be favored that lead to peptides containing MTQGQHFSQKV but that cannot be presented productively to M88.7 T cells, due to a problem with peptide stability, TAP (transporter associated with antigen processing) transport, or MHC loading. Alternatively, the downstream residues could provoke cleavage within the MTQGQHFSQKV sequence itself. In favor of these suggestions, a number of reports exist 21 22 23 24 25 26 27 28 describing the influence of flanking residues on epitope production and/or presentation.
None of the above problems is likely to adversely affect production of NA88-A antigenic peptide. The peptide MTQGQHFLQKV is the direct translation product of a short ORF (a stop codon follows the Val codon), and antigenic peptides can be derived from it after at most minimal NH2 terminal trimming. In addition to avoiding potential problems with precursor processing (see above), the lack of any significant processing requirement should limit the level of transcription and translation required to produce sufficient levels of peptide for T cell stimulation. This could be important, as the NA88-A RNA contains no primary ORF, but rather many short ORFs, and there is no obvious reason why the ORF coding for the antigenic peptide should be chosen for translation rather than any of the others. Translation of the antigenic peptide's ORF is thus likely to be inefficient.
In conclusion, the NA88-A gene codes for a melanoma-specific antigen today, in large part because, during its evolution, a point mutation transformed a Trp codon into a stop codon. This significantly augmented production of antigenic peptide.
Acknowledgments
We thank P. Coulie for testing whether the antigen recognized by M88.7 T cells was already known. We thank T. Boon, C. Aubert, and J. Zeuthen for gifts of cell lines and F. Brasseur and Anne Moreau for samples of tumor- and normal tissue–derived cDNA. We thank A. Haunauer for help screening the cosmid library and I. Berard for help in sequencing. We thank P. Moretti and C. D'Andrea for HPX42B cDNA fragments.
This work was supported in part by the Ligue Nationale contre le Cancer (axe “Immunologie des Tumeurs”), the Ligue des Deux Sèvres, the Ligue de Loire Atlantique, and the Association pour la Recherche contre le Cancer.
Footnotes
A. Moreau-Aubry and S. Le Guiner contributed equally to this work.
References
- Van den Eynde B.J., Van der Bruggen P. T cell defined tumor antigens. Curr. Opin. Immunol. 1997;9:684–693. doi: 10.1016/s0952-7915(97)80050-7. [DOI] [PubMed] [Google Scholar]
- Rosenberg S.A., Yannelli J.R., Yang J.C., Topalian S.L., Schwartzentruber D.J., Weber J.S., Parkinson D.R., Seipp C.A., Einhorn J.H., White D.E. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J. Natl. Cancer Inst. 1994;865:1159–1166. doi: 10.1093/jnci/86.15.1159. [DOI] [PubMed] [Google Scholar]
- Marchand M., Weynants P., Rankin E., Arienti F., Belli F., Parmiani G., Cascinelli N., Bourlond A., Vanwijck R., Humblet Y. Tumor regression responses in melanoma patients treated with a peptide encoded by gene MAGE-3. Int. J. Cancer. 1995;63:883–885. doi: 10.1002/ijc.2910630622. [DOI] [PubMed] [Google Scholar]
- Rosenberg S.A., Yang J.C., Schwartzentruber D.J., Hwu P., Marincola F.M., Topalian S.L., Restifo N.P., Dudley M.E., Schwarz S.L., Spiess P.J. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat. Med. 1998;4:321–327. doi: 10.1038/nm0398-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestle F.O., Alijagic S., Gilliet M., Sun Y., Grabbe S., Dummer R., Burg G., Schadendorf D. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat. Med. 1998;4:328–332. doi: 10.1038/nm0398-328. [DOI] [PubMed] [Google Scholar]
- Mayrand S.M., Green W.R. Non-traditionally derived CTL epitopesexceptions that prove the rules? Immunol. Today. 1998;19:551–556. doi: 10.1016/s0167-5699(98)01342-5. [DOI] [PubMed] [Google Scholar]
- Guilloux Y., Lucas S., Brichard V.G., Van P.A., Viret C., De Plaen E., Brasseur F., Lethe B., Jotereau F., Boon T. A peptide recognized by human cytolytic T lymphocytes on HLA-A2 melanomas is encoded by an intron sequence of the N-acetylglucosaminyltransferase V gene. J. Exp. Med. 1996;183:1173–1183. doi: 10.1084/jem.183.3.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Eynde B.J., Gaugler B., Probst-Kepper M., Michaux L., Devuyst O., Lorge F., Weynants P., Boon T. A new antigen recognized by cytolytic T lymphocytes on a human kidney tumor results from reverse strand transcription. J. Exp. Med. 1999;190:1793–1800. doi: 10.1084/jem.190.12.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins P.F., El Gamil M., Li Y.F., Fitzgerald E.B., Kawakami Y., Rosenberg S.A. The intronic region of an incompletely spliced gp100 gene transcript encodes an epitope recognized by melanoma-reactive tumor-infiltrating lymphocytes. J. Immunol. 1997;159:303–308. [PubMed] [Google Scholar]
- Lupetti R., Pisarra P., Verrecchia A., Farina C., Nicolini G., Anichini A., Bordignon C., Sensi M., Parmiani G., Traversari C. Translation of a retained intron in tyrosinase-related protein (TRP)2 mRNA generates a new cytotoxic T lymphocyte (CTL)-defined and shared human melanoma antigen not expressed in normal cells of the melanocytic lineage. J. Exp. Med. 1998;188:1005–1016. doi: 10.1084/jem.188.6.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.F., Johnston S.L., Zeng G., Topalian S.L., Schwartzentruber D.J., Rosenberg S.A. A breast and melanoma-shared tumor antigenT cell responses to antigenic peptides translated from different open reading frames. J. Immunol. 1998;161:3598–3606. [PubMed] [Google Scholar]
- Wang R.F., Parkhurst M.R., Kawakami Y., Robbins P.F., Rosenberg S.A. Utilization of an alternative open reading frame of a normal gene in generating a novel human cancer antigen. J. Exp. Med. 1996;183:1131–1140. doi: 10.1084/jem.183.3.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronsin C., Chung-Scott V., Poullion I., Aknouche N., Gaudin C., Triebel F. A non-AUG-defined alternative open reading frame of the intestinal carboxyl esterase mRNA generates an epitope recognized by renal cell carcinoma-reactive tumor-infiltrating lymphocytes in situ. J. Immunol. 1999;163:483–490. [PubMed] [Google Scholar]
- Aarnoudse C.A., van den Doel P.B., Heemskerk B., Schrier P.I. Interleukin-2-induced, melanoma-specific T cells recognize CAMEL, an unexpected translation product of LAGE-1. Int. J. Cancer. 1999;82:442–448. doi: 10.1002/(sici)1097-0215(19990730)82:3<442::aid-ijc19>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Pandolfino M.C., Viret C., Gervois N., Guilloux Y., Davodeau F., Diez E., Jotereau F. Specificity, T cell receptor diversity and activation requirements of CD4+ and CD8+ clones derived from human melanoma-infiltrating lymphocytes. Eur. J. Immunol. 1992;22:1795–1802. doi: 10.1002/eji.1830220719. [DOI] [PubMed] [Google Scholar]
- Maniatis T., Fritsh E.F., Sambrook J. Molecular Cloning 2nd ed 1989. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: pp. 1,659 [Google Scholar]
- Brichard V., Van Pel A., Wolfel T., Wolfel C., De Plaen E., Lethe B., Coulie P., Boon T. The tyrosinase gene codes for an antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J. Exp. Med. 1993;178:489–495. doi: 10.1084/jem.178.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M.B., Nielsen S.E., Berg K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J. Immunol. Methods. 1989;119:203–210. doi: 10.1016/0022-1759(89)90397-9. [DOI] [PubMed] [Google Scholar]
- Labarriere N., Diez E., Pandolfino M.C., Viret C., Guilloux Y., Le G.S., Fonteneau J.F., Dreno B., Jotereau F. Optimal T cell activation by melanoma cells depends on a minimal level of antigen transcription. J. Immunol. 1997;158:1238–1245. [PubMed] [Google Scholar]
- Moretti P., Simmons P., Thomas P., Haylock D., Rathjen P., Vadas M., D'Andrea R. Identification of homeobox genes expressed in human haemopoietic progenitor cells. Gene. 1994;144:213–219. doi: 10.1016/0378-1119(94)90380-8. [DOI] [PubMed] [Google Scholar]
- Theobald M., Ruppert T., Kuckelkorn U., Hernandez J., Haussler A., Ferreira E.A., Liewer U., Biggs J., Levine A.J., Huber C. The sequence alteration associated with a mutational hotspot in p53 protects cells from lysis by cytotoxic T lymphocytes specific for a flanking peptide epitope. J. Exp. Med. 1998;188:1017–1028. doi: 10.1084/jem.188.6.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann C.C., Tong L., Cua R., Sensintaffar J., Stohlman S. Differential effects of flanking residues on presentation of epitopes from chimeric peptides. J. Virol. 1994;68:5306–5310. doi: 10.1128/jvi.68.8.5306-5310.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Val M., Schlicht H.J., Ruppert T., Reddehase M.J., Koszinowski U.H. Efficient processing of an antigenic sequence for presentation by MHC class I molecules depends on its neighboring residues in the protein. Cell. 1991;66:1145–1153. doi: 10.1016/0092-8674(91)90037-y. [DOI] [PubMed] [Google Scholar]
- Eisenlohr L.C., Yewdell J.W., Bennink J.R. Flanking sequences influence the presentation of an endogenously synthesized peptide to cytotoxic T lymphocytes. J. Exp. Med. 1992;175:481–487. doi: 10.1084/jem.175.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedermann G., Butz S., Ihlenfeldt H.G., Grimm R., Lucchiari M., Hoschutzky H., Jung G., Maier B., Eichmann K. Contribution of proteasome-mediated proteolysis to the hierarchy of epitopes presented by major histocompatibility complex class I molecules. Immunity. 1995;2:289–299. doi: 10.1016/1074-7613(95)90053-5. [DOI] [PubMed] [Google Scholar]
- Ossendorp F., Eggers M., Neisig A., Ruppert T., Groettrup M., Sijts A., Mengede E., Kloetzel P.M., Neefjes J., Koszinowski U. A single residue exchange within a viral CTL epitope alters proteasome-mediated degradation resulting in lack of antigen presentation. Immunity. 1996;5:115–124. doi: 10.1016/s1074-7613(00)80488-4. [DOI] [PubMed] [Google Scholar]
- Shastri N., Serwold T., Gonzalez F. Presentation of endogenous peptide/MHC class I complexes is profoundly influenced by specific C-terminal flanking residues. J. Immunol. 1995;155:4339–4346. [PubMed] [Google Scholar]
- Yellen-Shaw A.J., Wherry E.J., Dubois G.C., Eisenlohr L.C. Point mutation flanking a CTL epitope ablates in vitro and in vivo recognition of a full-length viral protein. J. Immunol. 1997;158:3227–3234. [PubMed] [Google Scholar]