

Abstract
Receptors for immunoglobulin (Ig)G (FcγRs) are important for the antibody-mediated effector functions of the immune system. FcγRI and FcγRIII trigger cell activation through a common γ chain, whereas FcγRII acts as a negative regulator of antibody production and immune complex–triggered activation. Here we describe the in vivo consequences of FcγR deficiency in a mouse model of human rheumatoid arthritis. FcRγ chain–deficient mice on arthritis-susceptible DBA/1 background were immunized with collagen for induction of collagen-induced arthritis. The DBA/1 mice lacking FcRγ chain were protected from collagen-induced arthritis in contrast to wild-type mice, although both groups produced similar levels of IgG anticollagen antibodies. In comparison, DBA/1 mice lacking FcγRII developed an augmented IgG anticollagen response and arthritis. These observations suggest a crucial role of FcγRI and FcγRIII in triggering autoimmune arthritis.
Keywords: autoimmunity, mice, knock-outs, immunoglobulin receptor, antibodies
Introduction
IgG immune complexes (ICs) are of central importance in the humoral immune system and are strongly implicated in promoting inflammation and autoimmune diseases. Part of the inflammatory response is attributed to the binding of ICs to Fc receptors for IgG (FcγRs) on leukocytes. By cross-linking FcγRs, a variety of cellular responses are triggered including phagocytosis, antibody-dependent cellular cytotoxicity, release of inflammatory mediators, IC clearance, and regulation of antibody production. In this way, FcγRs form a molecular link between the humoral and cellular branches of the immune system. In the mouse, there are three types of Fcγ receptors, the high-affinity receptor FcγRI, capable of binding monomeric IgG, and the two low-affinity receptors FcγRII and FcγRIII, which bind IgG in the form of ICs. Both FcγRI and FcγRIII are multimeric, whereas FcγRII is a single chain receptor. FcγRI and FcγRIII trigger cell activation through a common γ chain that contains an immunoreceptor tyrosine-based activation motif (ITAM). In contrast, FcγRII contains an immunoreceptor tyrosine-based inhibitory motif (ITIM) that via cocross-linking inhibits activation signals through receptors containing ITAMs (for a review, see reference 1). The specific contribution of each of the FcγR classes to normal and pathological immune responses is still not fully understood. Mice deficient in the γ subunit (FcRγ−/−) are unable to phagocytose IgG-opsonized particles or to mediate antibody-dependent cytotoxicity by NK cells 2 and respond very poorly to IC-mediated enhancement of antibody responses 3. Furthermore, FcRγ−/− mice show a grossly diminished Arthus reaction 4 and are resistant to autoantibody-dependent experimental hemolytic anemia, thrombocytopenia, and glomerulonephritis 5 6. These results suggest that a wide range of inflammatory and autoimmune diseases may be mediated by FcγRs and not, as previously thought, primarily by complement factors (for a review, see reference 7). In contrast, targeted disruption of FcγRII in the mouse results in elevated Ig levels in response to antigen challenge, augmented IgG-mediated anaphylaxis, and IC-mediated alveolitis 3 8 9, indicating that FcγRII acts as a negative regulator of antibody responses and IC-triggered activation.
Collagen-induced arthritis (CIA), a model of rheumatoid arthritis (RA), is induced in certain susceptible strains of mice with injection of collagen type II (CII) in CFA 10. This gives rise to a polyarthritis, characterized by synovial hyperplasia, infiltration of mononuclear cells, pannus formation, and destruction of cartilage and bone 11. It has been previously well documented that antibodies to CII are a prerequisite for CIA. B cell-deficient mice do not develop arthritis 12, and arthritis can be transferred with hyperimmune anti-CII serum concentrate 13 or polyclonal IgG anti-CII antibodies 14 15. Moreover, strain susceptibility to CIA correlates well with high antibody responders to CII 11 and high levels of IgG anti-CII antibodies. B cells producing IgG anti-CII have been found in several RA patients 16 17 as well, where presence of serum IgG anti-CII early in disease is predictive of rapidly progressive RA 18. However, the effector mechanism by which antibodies contribute to arthritis development has not been well understood. In the last few years, Fc receptors have been proposed as candidate molecules for induction of inflammation, and in a recent report, we showed that CIA is suppressed in mice lacking the low-affinity receptor for IgE, FcεRII (CD23) 19. It has also been shown that deletion of FcγRII can render arthritis-resistant 129/SvJ and C57BL/6 hybrid mice susceptible to CIA 20.
In this study, we have investigated the role of FcγRs in CIA by studying FcRγ chain–deficient mice on arthritis-susceptible DBA/1 background. We show here that mice lacking the FcRγ chain are almost completely resistant to CIA, although they develop similar immune reactivity to CII as wild-type mice. In contrast, DBA/1 mice lacking FcγRII develop an augmented CIA with elevated serum IgG anti-CII antibodies.
Materials and Methods
Mice.
FcRγ chain–deficient mice 2 and FcγRII-deficient mice 8 were backcrossed into DBA/1 background (H-2q) (Bomholtgaard Ltd.) for five generations. The backcrossed mice were then intercrossed to generate mice homozygous for the disrupted FcRγ chain allele (FcRγ2/2) or the disrupted FcγRII allele (FcγRII−/−). Littermates homozygous for the wild-type FcRγ chain allele (FcRγ1/1) or the FcγRII allele (FcγRII+/+) were used as controls. The FcRγ chain and the FcγRII genotypes were determined by PCR performed on isolated tail DNA. PCR for the FcRγ genotype was carried out using primer sets described elsewhere 2, and for the FcγRII genotype three different primers were used: Neo (5′-CTG GTG CTT TAC GGT ATC GCC-3′), 5′EC1 (5′-AAA CTC GAC CCC CCG TGG ATC-3′), and 3′EC1 (5′-TTG ACT GTG TTA AAC GTG TAG-3′). PCR was performed in 20-μl volumes using 10 mM Tris-HCl, pH 8.3, 50 mM KCl, 4.4 mM MgCl2, 0.2 mM dNTPs, 0.22 μM of primers, and 1 U of AmpliTaq DNA polymerase (Perkin-Elmer Cetus) for 35 cycles.
All mice were backcrossed, bred, and maintained at the Animal Units of the Biomedical Centre and at the Unit of Pathology, Uppsala University. The animals were fed rodent chow and water ad libitum. Experiments were performed in age-matched mutant and wild-type male mice.
Collagen Preparation.
Bovine type II collagen (BCII) was prepared from nasal cartilage by pepsin digestion and subsequent purification as described previously 21. BCII was solubilized to a concentration of 2 mg/ml in 0.01 M acetic acid (HAc) at 4°C with constant mixing overnight.
Induction of CIA.
For induction of CIA, BCII was emulsified with an equal volume (1:1) of CFA (Difco), and 50 μl of the emulsion was injected intradermally, under light ether anesthesia, at the base of the tail of each mouse.
Arthritis development was assessed by inspection three times a week. Clinical severity of arthritis was quantified according to a graded scale from 0 to 3 as follows: 0, normal; 1, detectable swelling in one joint; 2, swelling in more than one but not in all joints; and 3, severe swelling of the entire paw and/or ankylosis. Each paw was graded, and each mouse could achieve a maximum score of 12. A mean arthritic score value among only arthritic mice was calculated.
Histologic Assessment of CIA.
At termination of the experiments, the hind paws of four FcRγ+/+ and four FcRγ−/− mice were removed. The paws were fixed in phosphate buffer containing 4% formaldehyde, decalcified in EDTA, and paraffin embedded as described previously 22. Sagital sections (5 μm) were stained with hematoxylin and eosin and evaluated “blindly” (without knowledge of the treatment groups).
Measurement of Anti-CII Antibodies.
Mice were bled from the tails at different time points after immunization, and individual sera were analyzed for CII-specific IgG antibodies by ELISA. Microtiter plates (Immunolon 2; Dynex Technologies) were coated overnight at 4°C with 50 μl of native BCII in PBS at 200 μg/ml. Plates were washed with PBS containing 0.05% Tween 20 (PBS/Tween), and serum samples were added in serial dilution with PBS/Tween and incubated for 2 h at room temperature (rt). The plates were then washed and incubated for 2 h at rt with 50 μl of sheep anti–mouse IgG conjugated to alkaline phosphatase (Jackson ImmunoResearch Laboratories) diluted 1:1,000 in PBS/Tween. After additional washings, 50 μl of p-nitrophenyl phosphate substrate (Sigma Chemical Co.) diluted in diethanolamine buffer at 1 mg/ml was applied. Absorbances were read after 20 min at 405 nm. A polyclonal anti-BCII standard with known concentration was included on every microtiter plate to allow calculation of the antibody content by using Softmax software (Molecular Devices). The standard was purified by affinity chromatography from pooled sera obtained from BCII hyperimmunized mice.
ELISA to detect CII-specific IgG isotypes was performed with a modified protocol of the assay described above. After incubation of serum samples overnight at 4°C, 50 μl of biotinylated rat anti–mouse IgG1 (diluted 1:2,000), IgG2a (diluted 1:10,000), IgG2b (diluted 1:10,000), or IgG3 (diluted 1:5,000) (all from Southern Biotechnology Associates, Inc.) was applied to the plates for 5 h at rt. After washings 50 μl of streptavidin–alkaline phosphatase (diluted 1:1,000; Serotec) was added and incubated for 1 h at rt. The plates were washed and p-nitrophenyl phosphate substrate was added. The concentration of antibodies was calculated by comparison with the polyclonal anti-BCII standard.
Proliferation Assay.
FcRγ2/2 and FcRγ+/+ mice were killed 14 d after BCII/CFA immunization and inguinal, popliteal, and axillary LNs were removed. Individual single cell suspensions were made in DMEM supplemented with 2-ME (50 μM), Hepes (10 mM), glutamine (20 mM), penicillin (100 U/ml), streptomycin (100 μg/ml), and 5% FCS. The LN cells (LNCs, 5 × 105) were plated in 96-well round-bottomed microtiter plates and stimulated in triplicate in the absence or presence of 5, 50, or 100 μg/ml of heat-denatured BCII (dCII) in 0.01 M HAc. The cells were incubated at 37°C in 5% CO2 for 4 d, and 1 μCi/well of [3H]TdR was added to the culture for the last 18 h. [3H]TdR incorporation was measured using a β-scintillation counter, and the results were expressed as the mean cpm ± SD of the LNC preparations derived from four FcRγ2/2 or four FcRγ+/+ mice.
Statistics.
The severity of arthritis was analyzed using the Mann-Whitney U-test and the frequency of arthritis by Fisher's exact test. The antibody levels and proliferation assay were analyzed with Student's t test.
Results
DBA/1 Mice Lacking FcRγ Chain Are Highly Protected from CIA.
To investigate the involvement of the FcγRs in the development of CIA, FcRγ chain–deficient mice and their littermate controls, each on DBA/1 background, were immunized with CII. Clinical arthritis was observed in FcRγ1/1 mice from day 21 onward (Fig. 1A and Fig. B). The disease progressed to severe arthritis, and by the termination of the experiment 80% of the FcRγ+/+ mice were arthritic (Fig. 1 A) with a mean arthritic score of 7 (Fig. 1 B). In contrast, only one FcRγ−/− mouse developed clinical signs of arthritis within the first few weeks after immunization (Fig. 1A and Fig. B). This mouse had swelling in a single digit that went into spontaneous remission after 10 d. Around days 50 and 70 after immunization, another two FcRγ−/− mice developed mild arthritis (Fig. 1A and Fig. B). The arthritis manifestations in these mice were similar to the previously arthritic FcRγ−/− mouse, with clinical arthritis restricted to the swelling of only a single digit. To confirm the clinical assessments, at killing the clinically positive hind paws of the two responding FcRγ−/− mice as well as hind limbs of two nonarthritic FcRγ−/− mice and those of four clinically positive FcRγ+/+ mice were subjected to histopathology. Arthritis in wild-type mice included synovial hyperplasia, increased vascularization, and extensive infiltration of periarticular tissue by mononuclear cells and granulocytes. Frequently seen was pannus formation and severe erosion of cartilage and bone (Fig. 2 A). By comparison, the joints of the two FcRγ−/− mice that developed arthritis exhibited synovial hyperplasia and synovial villi formation (Fig. 2 B), whereas inflammatory cell infiltrates and erosions of cartilage and bone were absent. Joints of nonarthritic FcRγ−/− mice showed no pathological changes. The synovial tissue was normal, and cartilage and underlying bone were intact (Fig. 2 C).
Figure 1.
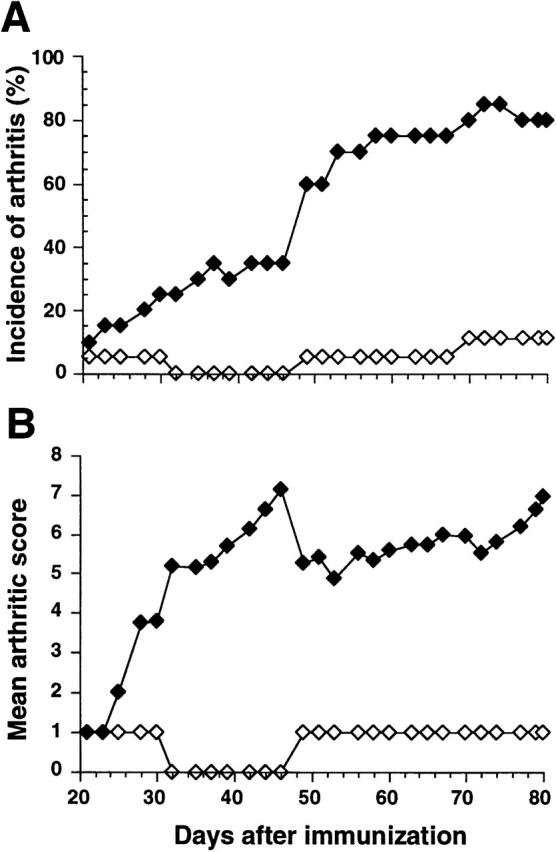
Protection from CIA in FcRγ-deficient DBA/1 mice. CII-immunized FcRγ+/+ mice (filled symbols, n = 20) and FcRγ−/− mice (open symbols, n = 18) were observed for arthritic lesions, and the percentage of mice that developed disease (A) and the mean severity of arthritis in diseased animals (B) are shown. The figure shows results from one representative experiment out of two performed.
Figure 2.
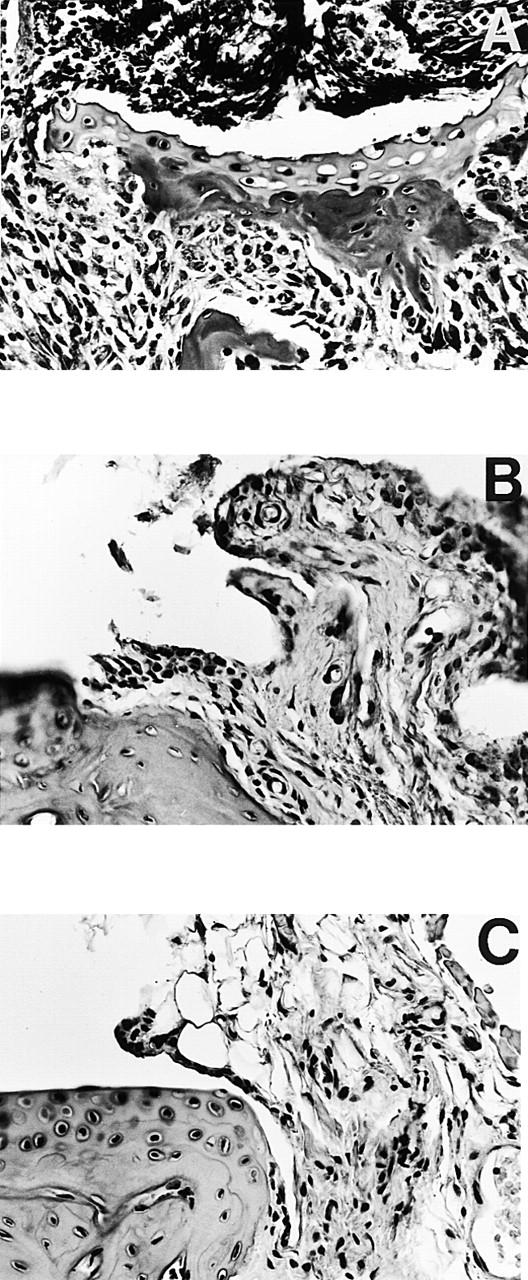
Histopathology of tarsal joints from FcRγ+/+ and FcRγ−/− DBA/1 mice 80 d after CII immunization. Severe arthritis was seen in FcRγ+/+ mice (A) with inflammatory cellular infiltrate, invasive pannus, and erosions of cartilage and bone clearly detectable. The few FcRγ−/− mice that developed disease (B) showed proliferation of synovial lining layer, synovial villi formation, but absence of cellular infiltrate and erosions. Joints of nonaffected FcRγ−/− mice (C) were normal in appearance, with normal synovia and smooth intact cartilage. Representative sagittal paraffin sections with hematoxylin-eosin stain; original magnifications: (A) ×20; (B and C) ×50.
The Anti-CII Response Is Not Altered in FcRγ−/− Mice.
To investigate if the immune response against CII was different in FcRγ−/− compared with FcRγ+/+ mice, we analyzed cellular and humoral immunity to CII. BCII-primed LNCs from FcRγ−/− and FcRγ+/+ mice had a low proliferative response to antigenic stimulation with dCII (Fig. 3). No significant differences of the CII-specific proliferation were found between the groups.
Figure 3.

Proliferation of CII-primed LNCs in response to CII. LNCs from BCII-immunized FcRγ+/+ (black bars, n = 4) and FcRγ−/− mice (hatched bars, n = 4) were stimulated in vitro with different antigen doses of heat-denatured CII (dCII). Proliferative responses were determined after 4 d of culturing by uptake of [3H]TdR. No significant difference between the groups was found.
In sera taken from mice periodically during the experiment, it was shown that the total IgG anti-CII levels did not differ between FcRγ−/− and FcRγ+/+ mice (Fig. 4 A). However, FcRγ−/− mice developed significantly higher IgG1 anti-CII levels at all time points, whereas IgG2a, IgG2b, and IgG3 levels were not significantly different between the two groups (Fig. 4 B).
Figure 4.
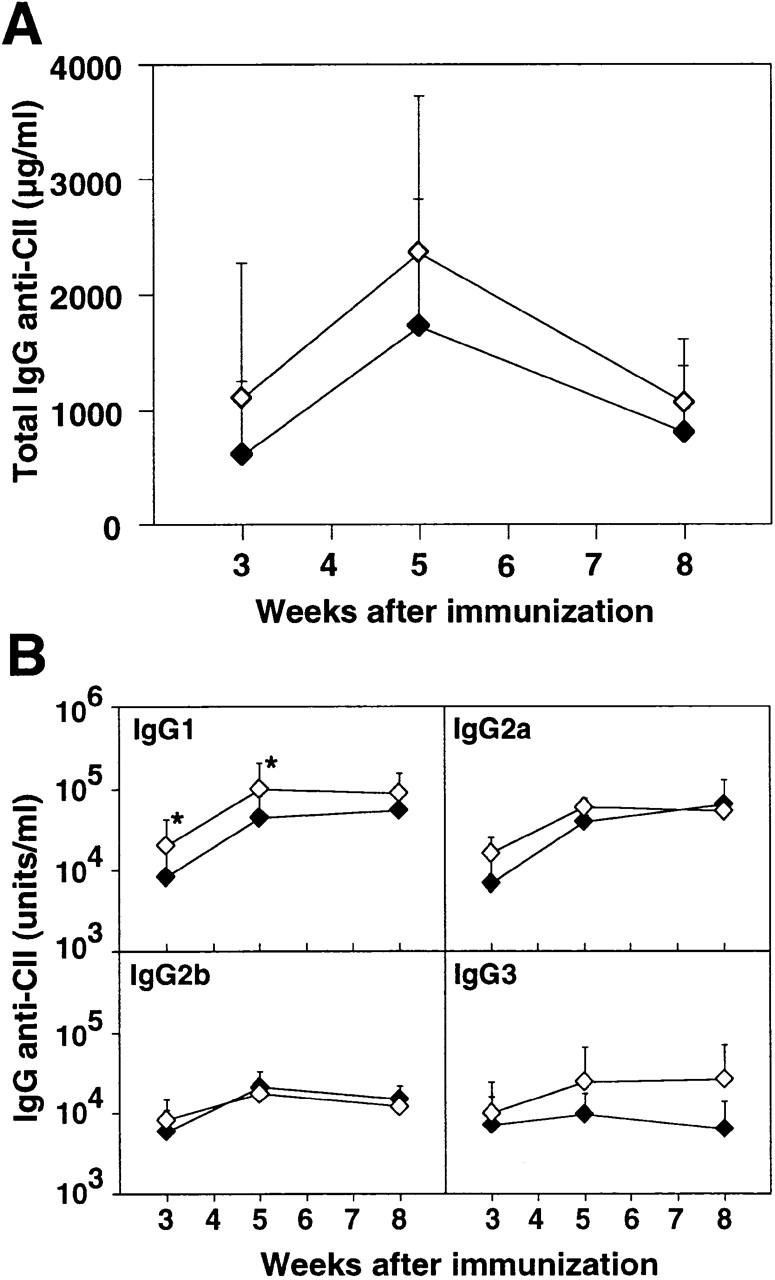
Anti-CII antibodies in FcRγ-deficient DBA/1 mice. Circulating CII-specific antibodies were determined periodically after BCII immunization in individual sera of FcRγ+/+ (filled symbols) and FcRγ−/− mice (open symbols). The mean ± SD antibody levels of total IgG anti-CII (A) and subclass-specific IgG anti-CII (B) are shown. *P < 0.05 compared with the FcRγ+/+ group.
Augmented CIA in DBA/1 Mice Lacking FcγRII.
In two independent experiments, FcγRII−/− mice on DBA/1 background proved to be more susceptible for induction of arthritis than FcγRII+/+ littermates. As early as 30 d after immunization, 75% of the FcγRII−/− mice had developed arthritis, whereas only 8% of the FcγRII+/+ mice were arthritic (P < 0.01; Fig. 5 A). The FcγRII−/− mice developed not only a more rapidly progressing arthritis, but also a clinically more severe disease; by day 42 after immunization, FcγRII−/− mice exhibited a mean clinical score of 9.36 ± 3.0 in contrast to 3.7 ± 2.3 in FcγRII+/+ mice (P < 0.05; Fig. 5 B). Furthermore, the FcγRII−/− mice developed very high serum IgG anti-CII levels; at 5 wk, a mean of 4.75 mg/ml total IgG anti-CII was found in the FγRII-deficient mice compared with 0.82 mg/ml in the FγRII+/+ mice (Fig. 6 A). The CII-specific antibody response in the FcγRII−/− mice was not restricted to a particular subclass: all subclasses were significantly increased at 3 and 5 wk after immunization compared with FcγRII+/+ mice (Fig. 6 B).
Figure 5.
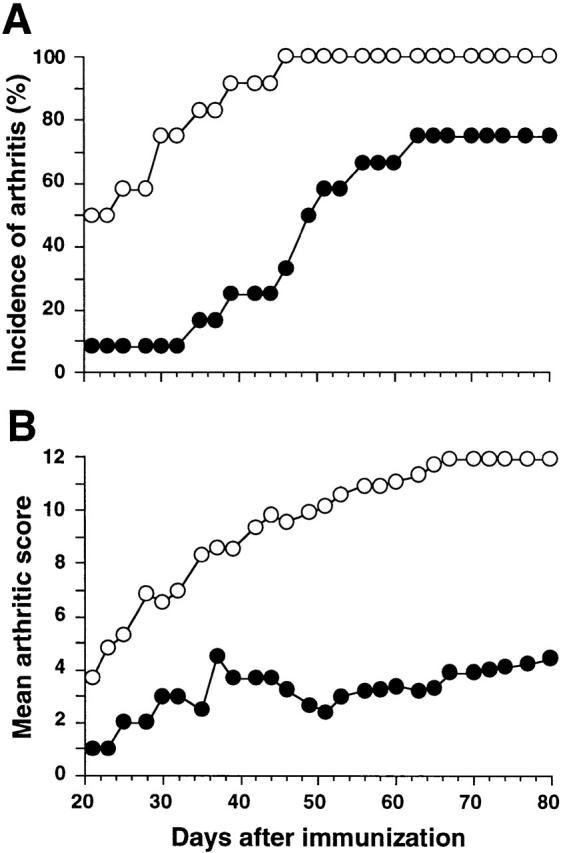
Augmented CIA in FcγRII-deficient DBA/1 mice. CII-immunized FcγRII+/+ (filled symbols, n = 12) and FcγRII−/− mice (open symbols, n = 12) were observed for arthritic lesions, and the percentage of mice that developed disease (A) and the mean severity of arthritis in diseased animals (B) are depicted. The figure shows results from one representative experiment out of two performed.
Figure 6.
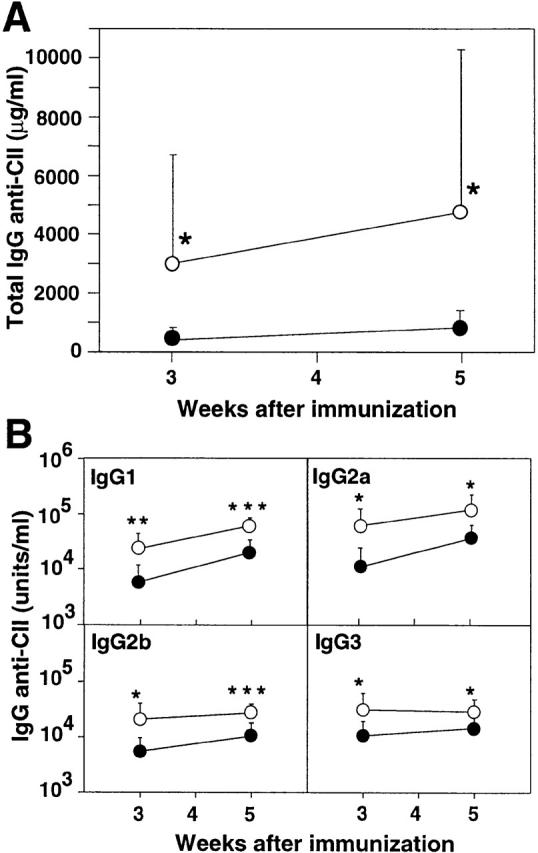
Anti-CII antibodies in FcγRII-deficient DBA/1 mice. Circulating CII-specific antibodies were determined periodically after BCII immunization in individual sera of FcγRII+/+ (filled symbols) and FcγRII−/− mice (open symbols). The mean ± SD antibody levels of total IgG anti-CII (A) and subclass-specific IgG anti-CII (B) are shown. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with the FcγRII+/+ group.
Discussion
The CIA model has been shown to be dependent on B cells 12, promoting the important IgG anti-CII response. Thus, high amounts of IgG anti-CII antibodies have proven to be pathogenic when transferred to naive recipients 14 15. Here we show that mice lacking the FcRγ chain are almost completely resistant to CIA, although equal levels of IgG anti-CII antibodies were demonstrated in FcRγ−/− mice as in wild-type mice during the course of the experiment. The proliferative response to CII in FcRγ−/− mice was similar to that in wild-type animals, suggesting that the T and B cell compartments function normally in FcRγ−/− mice. Absence of arthritis in spite of this indicates that the FcRγ chain is linked to a crucial “downstream” effector arm in the development of arthritis. As IgG antibodies are important for this process, as also shown in other arthritis models 23, the most likely receptors implicated are FcγRI and/or FcγRIII, both shown to be functionally impaired in FcRγ−/− animals 2. However, the definite identity of the receptor(s) involved cannot be established until mouse strains selectively lacking the various FcRγ chain–associated receptors are available on an arthritis-susceptible background. Previous reports have shown that inflammation triggered by ICs depends primarily on engagement and activation of γ chain–associated Fc receptors 4 5 6. This study adds to this, also highlighting the absolute requirement for γ chain activation in the complex autoimmune disease model, CIA. In addition, our results show that neither FcγRII nor the complement system, both present in the FcRγ−/− animals, are sufficient to trigger CIA.
There are several, not mutually exclusive, effector pathways that could be used by the FcRγ chain in the development of arthritis. FcRγ chain–containing receptors were recently shown to play a crucial role for the ability of ICs to trigger strong antibody responses in vivo 3. This enhancement of antibody responses by ICs may be operative in the early phases of CIA, resulting in accumulation of IgG autoantibodies that may precipitate and bind to joint constituents. Uptake by resident FcγRIII and/or FcγRI inflammatory cells may trigger inflammation and recruitment of circulating monocytes and neutrophils. In fact, the lack of infiltrating leukocytes in the synovium of FcRγ−/− mice could be linked to a decreased chemotactic activity in the joints due to FcγRIII deficiency. Cross-linking of FcγRIII on human monocytes from peripheral blood with immobilized IgG induces monocyte chemoattractant protein 1 24, a chemokine also detected in joints of RA patients 25.
In this study, we find diverse susceptibility to CIA by studying two different FcγR-deficient strains on the arthritis-susceptible DBA/1 background. Thus, DBA/1 mice lacking FcγRII developed a clinically more severe arthritis with an earlier onset than wild-type mice. We propose that these divergent results are due to the fact that FcRγ−/− mice lack FcγRI and FcγRIII, whereas in FcγRII−/− mice these FcγRs are present. FcγRII has been shown to function as a negative regulator of antibody production and IC-triggered activation, where mice lacking FcγRII show a greatly enhanced IgG-mediated passive cutaneous anaphylactic response 8. FcγRII has also been demonstrated to have an inhibitory role on macrophages by regulating phagocytic and calcium flux responses 9. Thus, we interpret the augmented CIA in FcγRII−/− mice as a result of elevated antibody levels and an amplified effector response to ICs. The increased susceptibility of arthritis in FcγRII−/− DBA/1 mice compared with FcγRII+/+ mice is in line with a previous finding where a CIA-resistant mouse strain carrying the H-2b haplotype was rendered susceptible to CIA through deletion of FcγRII 20.
Our findings clearly demonstrate the important role of distinct FcγRs for induction and suppression of arthritis. IgG-triggered activation of the ITAM-associated FcγRI and FcγRIII are crucial for arthritis development, whereas ITIM-associated FcγRII downregulates autoimmune responses and arthritis. The balance of FcRγ-stimulatory and FcγRII-inhibitory signals will most likely determine the threshold of IC stimulation and the outcome of arthritis after immunization with CII in a potent adjuvant. Depending on which receptors are available for ligation, disease can either be completely prevented or dramatically enhanced. The data presented suggest that a future possibility for treating RA could be to find ways to specifically inhibit signaling through the FcRγ chain.
Acknowledgments
We thank Dr. Steven Applequist for his helpful discussions and critical reading of the manuscript.
This work was supported by the Swedish Medical Research Council, the Swedish Rheumatism Association, King Gustav V's 80-Year Foundation, the Swedish Society of Medicine, Åke Wiberg's Foundation, Börje Dahlin's Foundation, Anna-Greta Crafoord's Foundation, and the Nanna Svartz Foundation.
References
- Gessner J.E., Heiken H., Tamm A., Schmidt R.E. The IgG Fc receptor family. Ann. Hematol. 1998;76:231–248. doi: 10.1007/s002770050396. [DOI] [PubMed] [Google Scholar]
- Takai T., Li M., Sylvestre D., Clynes R., Ravetch J.V. FcR γ chain deletion results in pleiotrophic effector cell defects. Cell. 1994;76:519–529. doi: 10.1016/0092-8674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- Wernersson S., Karlsson M.C.I., Dahlström J., Mattsson R., Verbeek J.S., Heyman B. IgG-mediated enhancement of antibody responses is low in Fc receptor γ chain-deficient mice and increased in FcγRII-deficient mice. J. Immunol. 1999;163:618–622. [PubMed] [Google Scholar]
- Sylvestre D.L., Ravetch J.V. Fc receptors initiate the Arthus reactionredefining the inflammatory cascade. Science. 1994;265:1095–1098. doi: 10.1126/science.8066448. [DOI] [PubMed] [Google Scholar]
- Clynes R., Ravetch J. Cytotoxic antibodies trigger inflammation through Fc receptors. Immunity. 1995;3:21–26. doi: 10.1016/1074-7613(95)90155-8. [DOI] [PubMed] [Google Scholar]
- Park S.Y., Ueda S., Ohno H., Hamano Y., Tanaka M., Shiratori T., Yamazaki T., Arase H., Arase N., Karasawa A. Resistance of Fc receptor-deficient mice to fatal glomerulonephritis. J. Clin. Invest. 1998;102:1229–1238. doi: 10.1172/JCI3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravetch J.V., Clynes R.A. Divergent roles for Fc receptors and complement in vivo. Annu. Rev. Immunol. 1998;16:421–432. doi: 10.1146/annurev.immunol.16.1.421. [DOI] [PubMed] [Google Scholar]
- Takai T., Ono M., Hikida M., Ohmori H., Ravetch J.V. Augmented humoral and anaphylactic response in FcγRII-deficient mice. Nature. 1996;379:346–349. doi: 10.1038/379346a0. [DOI] [PubMed] [Google Scholar]
- Clynes R., Maizes J.S., Guinamard R., Ono M., Takai T., Ravetch R.V. Modulation of immune complex–induced inflammation in vivo by the coordinate expression of activation and inhibitory Fc receptors. J. Exp. Med. 1999;189:179–185. doi: 10.1084/jem.189.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtenay J.S., Dallman M.J., Dayan A.D., Martin A., Mosedale B. Immunisation against heterologous type II collagen induces arthritis in mice. Nature. 1980;283:666–668. doi: 10.1038/283666a0. [DOI] [PubMed] [Google Scholar]
- Wooley P.H., Luthra H.S., Stuart J.M., David C.S. Type II collagen–induced arthritis in mice. I. Major histocompatibility complex (I region) linkage and antibody correlates. J. Exp. Med. 1981;154:688–700. doi: 10.1084/jem.154.3.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson L., Jirholt J., Holmdahl R., Jansson L. B cell-deficient mice do not develop type II collagen-induced arthritis (CIA) Clin. Exp. Immunol. 1998;111:521–526. doi: 10.1046/j.1365-2249.1998.00529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart J.M., Dixon F.J. Serum transfer of collagen-induced arthritis in mice. J. Exp. Med. 1983;158:378–392. doi: 10.1084/jem.158.2.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooley P.H., Luthra H.S., Krco C.J., Stuart J.M., David C.S. Type II collagen-induced arthritis in mice. II. Passive transfer and suppression by intravenous injection of anti-type II collagen antibody or free native type II collagen. Arthritis Rheum. 1984;27:10–17. doi: 10.1002/art.1780270907. [DOI] [PubMed] [Google Scholar]
- Holmdahl R., Jansson L., Larsson A., Jonsson R. Arthritis in DBA/1 mice induced with passively transferred type II collagen immune serum. Scand. J. Immunol. 1990;31:147–157. doi: 10.1111/j.1365-3083.1990.tb02754.x. [DOI] [PubMed] [Google Scholar]
- Clague R.B., Moore L.J. IgG and IgM antibody to native type II collagen in rheumatoid arthritis serum and synovial fluid. Evidence for the presence of collagen-anticollagen immune complexes in synovial fluid. Arthritis Rheum. 1984;27:1370–1377. doi: 10.1002/art.1780271207. [DOI] [PubMed] [Google Scholar]
- Rönnelid J., Lysholm J., Engström-Laurent A., Klareskog L., Heyman B. Local anti-collagen type II antibody production in rheumatoid arthritis synovial fluid; evidence for an HLA-DR4-restricted IgG response. Arthritis Rheum. 1994;37:1023–1029. doi: 10.1002/art.1780370707. [DOI] [PubMed] [Google Scholar]
- Cook A.D., Rowley M.J., Mackay I.R., Gough A., Emery P. Antibodies to type II collagen in early rheumatoid arthritis. Correlation with disease progression. Arthritis Rheum. 1996;39:1720–1727. doi: 10.1002/art.1780391015. [DOI] [PubMed] [Google Scholar]
- Kleinau S., Martinsson P., Gustavsson S., Heyman B. Importance of CD23 for collagen-induced arthritisdelayed onset and reduced severity in CD23-deficient mice. J. Immunol. 1999;162:4266–4270. [PubMed] [Google Scholar]
- Yuasa T., Kubo S., Yoshino T., Ujike A., Matsumura K., Ravetch J.V., Takai T. Deletion of Fcγ receptor IIB renders H-2b mice susceptible to collagen-induced arthritis. J. Exp. Med. 1999;189:187–194. doi: 10.1084/jem.189.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller E.J. Structural studies on cartilage collagen employing limited cleavage and solubilization with pepsin. Biochemistry. 1972;11:4903–4909. doi: 10.1021/bi00776a005. [DOI] [PubMed] [Google Scholar]
- Kleinau S., Erlandsson H., Holmdahl R., Klareskog L. Adjuvant oils induce arthritis in the DA rat. I. Characterization of the disease and evidence for an immunological involvement. J. Autoimmun. 1991;4:871–880. doi: 10.1016/0896-8411(91)90050-M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korganow A.-S., Ji H., Mangialaio S., Duchatelle V., Pelanda R., Martin T., Degott C., Kikutani H., Rajewsky K., Pasquali J.-L. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity. 1999;10:451–461. doi: 10.1016/s1074-7613(00)80045-x. [DOI] [PubMed] [Google Scholar]
- Marsh C.B., Wewers M.D., Tan L.C., Rovin B.H. Fcγ receptor cross-linking induces peripheral blood mononuclear cell monocyte chemoattractant protein-1 expression. J. Immunol. 1997;158:1078–1084. [PubMed] [Google Scholar]
- Loetscher P., Dewald B., Baggiolini M., Seitz M. Monocyte chemoattractant protein 1 and interleukin 8 production by rheumatoid synoviocyteseffects of anti-rheumatic drugs. Cytokine. 1994;6:162–170. doi: 10.1016/1043-4666(94)90038-8. [DOI] [PubMed] [Google Scholar]