

Abstract
Cadherins are expressed in tissue-restricted patterns and typically mediate homophilic adhesion. Cadherins also mediate lymphocyte adhesion, providing the opportunity for lymphocyte attachment to parenchymal cells. The best characterized example of lymphocyte adhesion to a tissue-specific cell adhesion molecule, as opposed to a vascular endothelial adhesion molecule, is the interaction between integrin αEβ7 on intraepithelial lymphocytes and E-cadherin on epithelial cells. However, the molecular basis for an integrin–cadherin interaction is not well defined. Realization that the cadherin domain adopts a topology similar to the immunoglobulin (Ig) fold suggested that integrin recognition of E-cadherin might be similar to recognition of Ig superfamily ligands. Thus, we modeled domain 1 of human E-cadherin and studied the role of solvent-exposed loops that connect Ig-like core-forming β strands. Mutational analyses localized the integrin αEβ7 recognition site to the top of domain 1 at the face formed by the BC and FG loops, a site distinct from the region recognized in intercellular adhesion molecule (ICAM)-1, -2, and -3, mucosal addressin cell adhesion molecule 1 (MAdCAM-1), vascular cell adhesion molecule 1 (VCAM-1), and fibronectin by their integrin ligands. Moreover, the integrin αEβ7 binding site is distinct from the homophilic binding site on E-cadherin. These studies provide a conceptual basis for integrin–cadherin binding and extend the model that an Ig-like fold can serve as a scaffold for recognition.
Keywords: cadherins, integrins, cell adhesion, T lymphocytes, protein binding
Introduction
Although several specific adhesion molecules mediate lymphocyte adhesion to the vascular endothelium, few molecular interactions have been identified that are involved in the specific retention of lymphocytes in tissues. The best characterized tissue-specific interaction is that between integrin αEβ7 on mucosal T cells, and epithelial (E)-cadherin expressed on epithelial cells. E-cadherin has been shown to mediate T cell–epithelial cell adhesion via αEβ7 in cell–cell and cell–fusion protein adhesion assays in vitro 1 2. Moreover, as the number of intraepithelial lymphocytes (IELs) is markedly reduced in αE-deficient mice, the E-cadherin–αEβ7 interaction is important in lymphocyte localization in the epithelium 3. Yet, the molecular basis for the binding of an integrin and a cadherin is not well understood.
Integrins are heterodimeric integral membrane proteins composed of noncovalently associated α and β subunits. The integrin αE chain contains an inserted domain (I or A) within its NH2-terminal region 4. The crystal structures of the A domains of the integrin αL, αM, and α2 chains have been determined and reveal a metal ion binding site located at the top of the A domain, 5 6 7 8 9. Five of the six coordination sites for the divalent cation are contributed directly or indirectly via water by conserved residues in the A domain that include a linear D-X-S-X-S motif and discontinuous threonine and aspartate residues 5 6 7 8 9. In one crystal structure, the sixth coordination site was provided by a glutamate residue from an adjacent, symmetry-related A domain, suggesting a direct role for an acidic residue in the ligand in coordination of the metal ion 6. It has been proposed that all integrin β chains contain a similar A-like domain 6. Certain integrin ligands contain acidic residues that are critical for integrin adhesion and that are present in solvent-exposed locations accessible for integrin recognition, generally on loops connecting the strands of Ig-like domains. For example, recognition of the first Ig domains of vascular cell adhesion molecule 1 (VCAM-1) and mucosal addressin cell adhesion molecule 1 (MAdCAM-1) requires an acidic residue at the tip of the CD loop on the lower side of the domain 1 10 11 12 13, and integrin αLβ2 recognition of intracellular adhesion molecule (ICAM)-1, ICAM-2, and ICAM-3 requires an acidic residue that is the last residue of the C strand on the lower side of domain 1 14 15.
The αEβ7–E-cadherin interaction is unique among integrin ligands. Cadherins are type 1 integral membrane proteins with five extracellular homologous “cadherin” domains, a transmembrane region, and a highly conserved cytoplasmic tail, and are classically described as calcium-dependent homophilic cell adhesion molecules important in morphogenesis and tissue architecture 16. Although cadherins do not share detectable sequence homology with Ig superfamiy (IgSF) members, the structural determination of the NH2-terminal domains of murine E- and neural (N)-cadherin by NMR and x-ray crystallography reveals that the overall topology of the cadherin domain consists of seven β strands that form a β barrel and is similar to the Ig fold 17 18 19 20 21. This realization led us to hypothesize that αEβ7 might recognize E-cadherin in a manner related to how integrins recognize their IgSF counterreceptors.
Here, we model the first domain of human E-cadherin on the murine E-cadherin crystal structure. Through site-directed mutagenesis, we elucidate the face and specific residues that are the adhesion site for binding the integrin αEβ7 and show that this site is distinct from that mediating homophilic cadherin adhesion. These molecular interactions provide a conceptual basis for the adhesion of leukocytes to parenchymal cells based on the expression of tissue-specific cadherins.
Materials and Methods
mAbs.
The following mouse anti–human mAbs were used: αE7-1 (anti-αEβ7, IgG2a) 22, αE7-2 (anti-αEβ7, IgG1) 22, αE7-3 (anti-αEβ7, IgG1) 22, E4.6 (anti–E-cadherin, IgG1) 23, HECD-1 (anti–E-cadherin, IgG1; Zymed) 24, and SHE78-7 (anti–E-cadherin, IgG2a; Zymed).
Cell Cultures.
Human IEL-496 T cells were derived from intestinal IELs and maintained as described previously 2 25. Human embryonic kidney (HEK)-293-EBNA cells (Invitrogen) were maintained in DMEM (GIBCO BRL) with 10% fetal bovine serum (FBS) at 10% CO2. Transfectants of the human chronic myelogenous leukemia cell line K562 that express αEβ7 (K562-αEβ7) were provided by Dr. David Erle (University of California San Francisco, San Francisco, CA) and maintained as described previously 26. Human breast epithelial cells MCF-7 (American Type Culture Collection) were grown in DMEM with 10% FBS at 10% CO2.
Construction of Mutated E-Cadherin-Fc Expression Vectors.
Human E-cadherin-Fc (five extracellular domains of E-cadherin fused to the Fc portion of human IgG1) 2 was excised from pCDM8 with EcoRV and NotI and ligated into pBluescript SK II (pBS; Stratagene). Mutations were introduced by PCR using pBS-E-cadherin-Fc as a template and Pfu polymerase (Stratagene) according to the manufacturer's recommendations with 25 cycles of 94°C for 1 min, 52°C for 2 min, 72°C for 2 min, and one cycle at 72°C for 10 min. For mutations with a nearby restriction site, PCR was performed with one primer complementary to the wild-type sequence and a second primer containing the mutation and a restriction site. Mutations to the BC loop were made with a forward primer complementary near the 5′ end of E-cadherin (hEprimV 5′-AAG TCA GTT CAG ACT CCA GCC C-3′) and a reverse primer incorporating the mutation and the MscI site (hEprimH 5′-AGC TCC TTG GCC AGT GAT GCT GTA GAA AAC CTT GCC TGC TTT GTC-3′ for E31A, hEprimAL2 5′-AGC TCC TTG GCC AGT GAT GCT GTA GAA AAC CTT GCC ATC TTT GTC-3′ for E31D, hEprimT 5′-GCT CCT TGG CCA GTG ATG CGT TAG AAA ACC GCG CCT TC-3′ for K33A, and hEprimU 5′-GCT CCT TGG CCA GTG ATG CTG TAG AAA ACC TTG GTA TCT CTG TCT TT-3′ for KEG30-32RDT). The D44A mutation in the CD loop was made using a forward primer containing the MscI site, the D44A mutation (hEprimA 5′-ATC ACT GGC CAA GGA GCT GCC ACA CCC CCT GT-3′), and a reverse wild-type primer (hEprimB 5′-CTC CAT TGG ATC CTC AAC TGC-3′). The E56A mutation in the DE loop was made using a forward wild-type primer (hEprimG 5′-TAC GGT TTC ATA ACC CAA CAG ATC CAT TTC TT-3′) and a reverse primer containing the E56A mutation and the Eco57I site (hEprimJ 5′-GTT CTC TAT CCA GAG GCT CTG TCA CCT TCA GCC ATC CTG TTG CTC TTT C-3′).
The other mutations were introduced by overlap extension PCR 27. First, PCR was performed with a forward primer complementary to the wild-type sequence near the 5′ end of E-cadherin (hEprimV) and a reverse primer containing the mutation (hEprimO 5′-TGT AGA AAA CCT TGC CTT CTT TGT CTT TGT TGG ATG CGA TCT G-3′ for K25A, hEprimP 5′-TGT AGA AAA CCT TGC CTT CTT TGT CTT TGG CGG ATT TGA TCT G-3′ for N27A, hEprimQ 5′-TGT AGA AAA CCT TGC CTT CTT TGT CTG CGT TGG ATT TGA TCT G-3′ for K28A, hEprimR 5′-TGT AGA AAA CCT TGC CTT CTG CGT CTT TGT TGG ATT TGA TCT G-3′ for K30A, hEprimAA 5′-CTC CAT TGG ATC CTC AAC TGC ATT CCC GTT GGC TGA CAC-3′ for S83A, hEprimAC 5′-CTC CAT TGG ATC CTC AAC TGC AGC CCC GTT GGA TGA CAC-3′ for N86A, and hEprimAE 5′-GGT TAC CGT GAT CAA AAT CTC CAT TGG ATC CGC AAC TGC-3′ for E89A). Concurrently, PCR was performed with a forward primer complementary to the 5′ end of the primer containing the mutation (hEprimS 5′-AGA AGG CAA GGT TTT CTA CAG CAT CAC TGG CCA AGG AGC-3′ for K25A, N27A, K28A, and K30A; hEprimAD 5′-GCA GTT GAG GAT CCA ATG GAG ATT-3′ for S83A and N86A; and hEprimAH 5′-ACT GGC CAA GGA GCT GAC ACA CCC CCT-3′ for E89A) and reverse primer located downstream (hEprimW 5′-TGT TGT CGT TAA CCC CTC ACC-3′). A subsequent PCR reaction was performed using these two overlapping PCR products as the template and primers at the 5′ (hEprimV) and 3′ (hEprimW) ends. The PCR products were cleaved with restriction enzymes (NcoI and MscI for K25A, N27A, K28A, K30A, E31A, E31D, KEG30-32RDT, and K33A; MscI and BamHI for D44A; PflMI and Eco57I for E56A; and NcoI and SnaBI for S83A, N86A, and E89A) and ligated into pBS-E-cadherin-Fc. The PCR-amplified region of each clone was sequenced. The pBS-E-cadherin-Fc mutants were serially digested, first with NotI and ScaI, and then with HindIII. The mutated E-cadherin-Fc insert was ligated into pCEP4 (Invitrogen) and cleaved with HindIII and NotI. DNA for transfections was prepared using a Qiagen Plasmid Maxi Kit.
Production of E-Cadherin-Fc Fusion Proteins.
Purified wild-type E-cadherin-Fc fusion protein was produced as described previously 2. Mutated and wild-type E-cadherin-Fc fusion protein was produced by transient transfection of HEK-293-EBNA cells with 10 μg of pCEP4-E-cadherin-Fc plasmid DNA using the CaPO4 transfection protocol provided in the Stratagene Mammalian Transfection kit. To produce the cadherin-Fc proteins, transfected cells were grown in DMEM containing 10% ultra-low Ig FBS (GIBCO BRL). After 5–10 d, the culture supernatants were harvested, filtered through a 0.2-μm membrane, and stored at 4°C.
ELISA.
Recombinant soluble E-cadherin-Fc fusion proteins were quantitated in culture supernatants by ELISA. The wells of Linbro Titertek 96-well plates (ICN Flow Laboratories) were coated with goat anti–human IgG polyclonal antisera (Zymed) at 10 μg/ml in 35 mM NaHCO3, 15 mM Na2CO3, pH 9.6 for 16 h at 4°C. The wells were washed three times with 200 μl of Tris-buffered saline (TBS), 1 mM CaCl2, pH 7.4. Subsequently, the wells were blocked with TBS, 1 mM CaCl2, 1% BSA for 2 h at room temperature. The wells were washed three times with TBS, 1 mM CaCl2, 0.05% Tween 20. Supernatants from HEK-293-EBNA cell transfectants were incubated for 2 h at room temperature, and purified wild-type E-cadherin-Fc was used as the standard. Bound fusion protein was detected with alkaline phosphatase–conjugated goat anti–human IgG Fc-specific (Sigma Chemical Co.) antibody and Sigma 104® phosphatase substrate (Sigma Chemical Co.). The E-cadherin-Fc fusion proteins were produced in the range of 10 to 75 μg/ml. To determine the reactivity of the E-cadherin-Fc mutants to E-cadherin mAbs, ELISA was performed as described above except that 96-well plates were coated with 1 μg/ml of purified anti–human E-cadherin mAbs, E4.6 or HECD-1.
Western Blot.
200 ng of wild-type and mutated E-cadherin-Fc fusion proteins were resolved by SDS-PAGE (7% acrylamide) under reducing conditions. Proteins were transferred to polyvinylidene difluoride membranes (Immobilon-P; Millipore). Membranes were blocked with 2% gelatin, washed, and then probed with peroxidase-conjugated goat anti–human IgG Fc (Sigma Chemical Co.) at 1:100,000. After washing, blots were developed using enhanced chemiluminescence (NEN Life Science Products).
Adhesion Assays.
Heterophilic adhesion assays were performed as described previously 1 2. In brief, 96-well plates were coated with goat anti–human IgG antibody and blocked as described for ELISA. The wells were washed and subsequently coated with culture supernatants containing the E-cadherin-Fc proteins or purified human IgG1 (Calbiochem-Novabiochem Corp.). IEL-496 or K562-αEβ7 cells were labeled with BCECF-AM (Molecular Probes). The adhesion assay was performed in Hepes-buffered saline (HBS) with 1 mM MgCl2, 1 mM MnCl2, 1 mM CaCl2, 50 mM dextrose, and 0.1% BSA with 50,000 cells/well for 10 min at 37°C. Labeled cells were detected using a Fluorescence Concentration Analyzer plate reader (IDEXX Laboratories, Inc.), and the percentage of cells bound was calculated.
The homophilic adhesion assay was performed as above with the following modifications. MCF-7 epithelial cells were released from Corning T75 culture flasks and labeled with 10 μg of BCECF-AM in 5 ml of 0.02% (wt/vol) bovine pancreas trypsin (Sigma Chemical Co.) in HBS 1 mM CaCl2 for 15 min at 37°C. 10 ml of 0.04% (wt/vol) soybean trypsin inhibitor type I-S (Sigma Chemical Co.) in HBS, 1 mM CaCl2 was added, and the cells were washed twice with cold HBS without CaCl2. Homophilic adhesion was carried out in HBS with 1 mM CaCl2, 0.1% BSA, and 50 mM dextrose.
Antibody blocking experiments were performed by preincubation of cells or E-cadherin–Fc-coated wells with 20–40 μg/ml purified mAb for 10 min at 4°C. Adhesion was carried out in final concentrations of 10–20 μg/ml purified mAbs.
Statistical Analysis.
Using a two-sided t test and controlling for plate to plate variability, average values of cell adhesion to mutated E-cadherin-Fc fusion protein were compared with average values of cell adhesion to wild-type E-cadherin-Fc to determine whether adhesion to the mutant was significantly different from adhesion to wild-type. Using the Bonferroni conservative adjustment for a confidence limit of 95% with 13 tests, a P value of 0.05/13 or P ≤ 0.0038 was considered statistically significant. The analysis was performed using the program “SAS” for UNIX.
Modeling of Human E-Cadherin.
Sequence alignments were performed using the Genetics Computer Group program PileUp. Human E-cadherin was modeled based on the murine E-cadherin crystal structure (available from the Protein Data Bank, http://www.rcsb.org/pdb, under accession no. 1EDH) 19. Using the program “O” (T.A. Jones, Uppsala University, and M. Kjelgaard, Aahus University), sequence substitution of human E-cadherin residues into the murine E-cadherin structure was performed. The side chain conformation of human E-cadherin residues was chosen to be similar to that of the murine E-cadherin residues while potential close contact was avoided.
Results
Sequence Analysis and Modeling of Human E-Cadherin.
The amino acid sequence of the first domain of human E-cadherin was aligned with that of murine E- and N-cadherin, and there did not appear to be any deletions or insertions (Fig. 1 A). Domain 1 of human E-cadherin shares 89% amino acid sequence identity with domain 1 of murine E-cadherin. Interestingly, all 11 substituted residues are solvent-exposed based on the crystal structure of the two NH2-terminal domains of murine E-cadherin 19. Therefore, the structure of human E-cadherin is predicted to be very similar to that of murine E-cadherin. As the core structure of the β barrel is predicted to be highly conserved, we developed a model of human E-cadherin based on the murine E-cadherin structure (Fig. 1 B).
Figure 1.

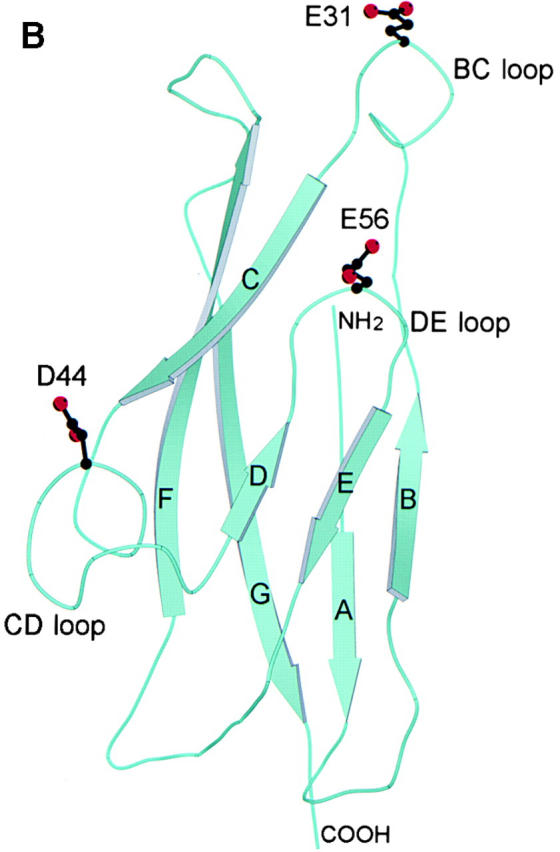
Structural analysis of human E-cadherin. (A) Amino acid alignment of domain 1 of human E-cadherin with human P-cadherin and murine E- and N-cadherin. The positions of the β strands were determined by the Definition of Secondary Structure of Proteins program (reference 51) and are indicated by arrows above the alignment. The PENE sequence in the AB loop, the LDRE sequence in the EF loop, and the DQNDN sequence in the connecting strand, which are predicted to be involved in the binding calcium, are indicated by Ca2+. Residues E31, D44, and E56 are shown in boldface. (B) Ribbon diagram of the human E-cadherin domain 1 model. Human E-cadherin was modeled based on the previously described murine E-cadherin crystal structure. Domain 1 of E-cadherin has seven β strands labeled A through G. The side chains of E31 on the BC loop at the top of domain 1, of D44 on the CD loop on the lower side of domain 1, and of E56 on the DE loop pointing upward in the center of the domain are represented as stick diagrams with carbon atoms, shown in black, and oxygen atoms, shown in red.
This model of human E-cadherin was used to consider possible interaction sites in cadherin–integrin binding with special reference to solvent-exposed acidic residues on loop structures. The seven β strands in the cadherin domain form two antiparallel β sheets, one formed by β strands D, E, and B and the other by β strands A, G, F, and C. Without the conserved intersheet disulfide bond present in Ig domains, the β strands in cadherin domains have a more cylindrical arrangement that has been termed a β barrel. Loops extend from and connect the β strands, and the majority of the solvent-exposed residues are located on the loops. The BC loop is exposed at the top of domain 1 of E-cadherin, contains a single turn of 310 helix, and as a whole has a high atomic mobility 18 19. The BC loop contains two acidic residues, D29 and E31. Residue D29 is conserved among cadherin domains (Fig. 1 A). Based on our model, the side chain of D29 is predicted to point into the core of the structure and hydrogen bond to Y36. As D29 may be important in preserving the conformation of the BC loop, it was not mutated. The side chain of E31 is solvent-exposed and highly accessible at the tip of the BC loop, and thus a good candidate for integrin recognition (Fig. 1 B). The CD loop that protrudes from the lower side of the domain contains two conserved proline residues (Fig. 1 A) and assumes a helical structure termed a quasi–β helix (Fig. 1 B; 18, 19). The central residues of the CD loop of human E-cadherin, GAD44TP, share a striking similarity to the integrin binding motif, G/Q I/L E/D S/T S/P, found in the C strands of ICAM-1, ICAM-2 and ICAM-3, and in the CD loops of VCAM-1 and MAdCAM-1 28. Based on the similarity to other integrin binding sites, the solvent-exposed D44 was considered a good candidate for integrin αEβ7 recognition. The DE loop in E-cadherin points upward and is solvent-exposed in the central portion of the domain. The side chain of E56 is also solvent-exposed and located at the tip of the DE loop, and thus also has the potential to be involved in integrin αEβ7 recognition (Fig. 1 B). The FG loop is located at the top of the molecule adjacent to the BC loop (Fig. 1 B), and there are no negatively charged residues on the tip of the FG loop of human E-cadherin (Fig. 1 A). The AB and EF loops are located at the bottom of the cadherin domain (Fig. 1 B). The acidic residues, E11 and E13, in the AB loop and acidic residues, D67 and E69, in the EF loop are predicted to be involved in binding calcium along with residues from the second cadherin domain (Fig. 1 A; 17). These residues are predicted to be important in domain–domain interactions, and thus were not selected for mutation as potential integrin binding residues.
Effect of Mutation of the Central Acidic Residues in the BC, CD, and DE Loops of E-Cadherin on Heterophilic Adhesion to IELs.
We used a recombinant fusion protein consisting of the five extracellular domains of human E-cadherin fused to the Fc portion of human IgG1 in order to study E-cadherin loop interactions with integrin αEβ7 2. Mutants of the E-cadherin-Fc fusion protein were produced by transient transfection of HEK-293 cells as described in Materials and Methods. The concentration of E-cadherin-Fc in the culture supernatants was determined by ELISA using purified wild-type E-cadherin-Fc as a standard. Western blot analysis of the E-cadherin-Fc mutants using an anti–human IgG Fc antibody demonstrated that the products were the correct size and confirmed the results of quantitation by ELISA (data not shown). To ensure that the E-cadherin-Fc mutants were properly folded, each of the mutants was tested for the ability to support homophilic adhesion, a function that is localized to domain 1 (16; and see below). Additionally, all of the mutants were recognized by two anti–E-cadherin mAbs in ELISA (data not shown): E4.6, which blocks heterophilic adhesion 23, and HECD-1, which blocks homophilic adhesion 29.
Cell–fusion protein adhesion assays were performed to determine the function of site-directed mutants of E-cadherin-Fc in heterophilic adhesion to integrin αEβ7–bearing cells. E-cadherin-Fc fusion protein was bound to the wells of 96-well plates, and fluorescently labeled T cell adhesion to E-cadherin-Fc was determined by quantitating the fluorescence bound. Adhesion of the human intestinal IEL–derived T cell line IEL-496 to immobilized wild-type E-cadherin-Fc was dose dependent (Fig. 2, ♦). Maximal adhesion (60%) to wild-type E-cadherin-Fc was achieved with 0.02 μg of E-cadherin-Fc added per well, whereas human IgG1, which contains the same Fc portion as the fusion protein, did not support adhesion (Fig. 2, compare ♦ to ▪). The specificity of adhesion in this assay was confirmed by mAb blocking with anti-αE mAb (αE7-2) and anti–E-cadherin mAb (E4.6), both of which blocked IEL adhesion to E-cadherin-Fc (data not shown).
Figure 2.
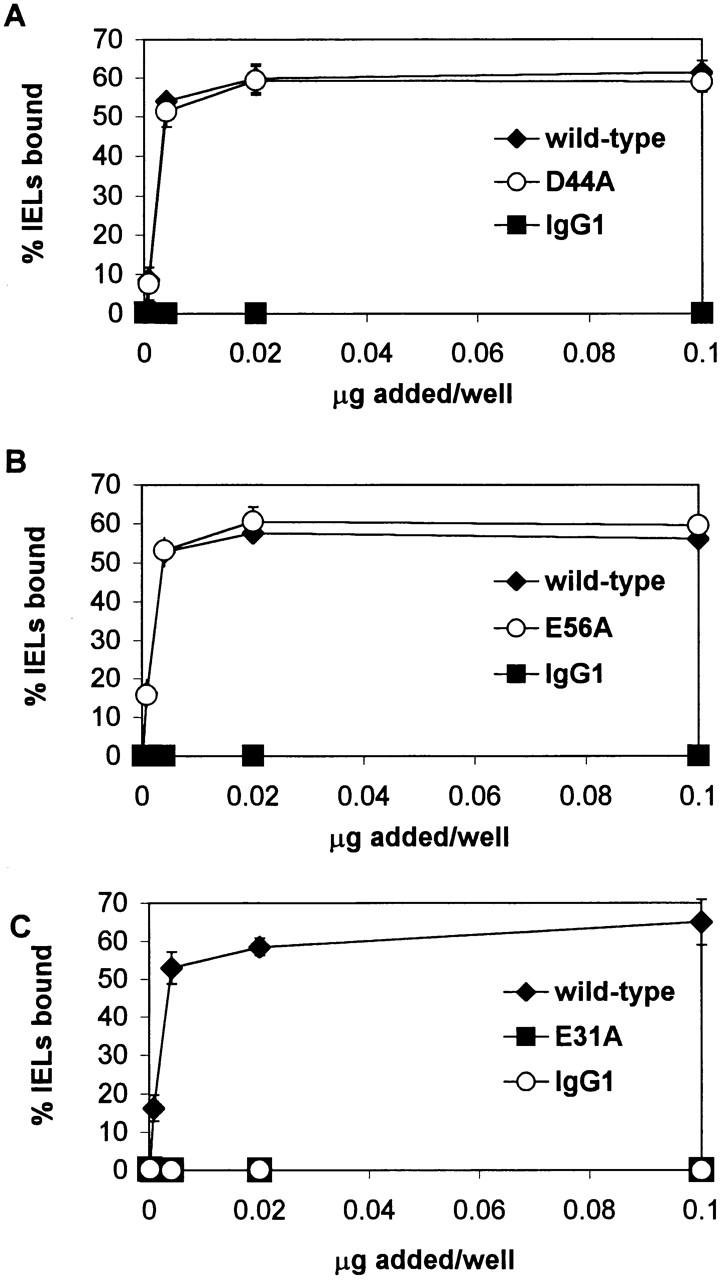
IEL adhesion to E-cadherin-Fc containing mutations of the central acidic residue in the BC, CD, and DE loops. Serial dilutions of culture supernatants from transient transfection with wild-type or mutated E-cadherin-Fc were immobilized on microtiter plate wells coated with goat anti–human IgG antiserum. Purified human IgG1 diluted in culture media was used as a negative control. Adhesion of IELs was determined in the presence of HBS with 1 mM MnCl2, 1 mM MgCl2, 1 mM CaCl2, 50 mM glucose, and 0.1% BSA, as described in Materials and Methods. Adhesion was determined in at least two independent experiments with each condition performed in triplicate. Dose–response curves are shown as the mean percentage of IELs bound ± 1 SD for a representative experiment. (A) Effect of D44A mutation in the CD loop. (B) Effect of the E56A mutation in the DE loop. (C) Effect of E31A mutation in the BC loop.
To delineate the critical loop residues of E-cadherin that mediate adhesion to αEβ7, acidic loop residues in E-cadherin-Fc were selected for alanine substitution based on the human E-cadherin model (above), and mutants were tested for their ability to support adhesion of IEL-derived T cells (Fig. 2). D44A and wild-type E-cadherin-Fc have similar dose–response curves, and above saturating levels of fusion protein, 59% of IELs adhered to the D44A mutant of E-cadherin-Fc fusion protein compared with 61% of IELs that adhered to wild-type E-cadherin-Fc (Fig. 2 A, ○ and ♦ are overlapping in figure, P ≤ 0.71). Thus, there was no significant difference between IEL adhesion to the E-cadherin-Fc CD loop mutant D44A and wild-type. Similarly, the DE loop mutant E56A and wild-type E-cadherin-Fc share similar dose–response curves, and at saturating levels E56A supported 60% of IEL adhesion compared with 57% for wild-type (Fig. 2 B, P ≤ 0.67). Thus, there was no significant difference between IEL adhesion to E56A and wild-type E-cadherin-Fc. In contrast to the CD and DE loop mutations that preserved αEβ7 adhesion to E-cadherin, the E31A mutation at the tip of the BC loop completely abolished detectable adhesion of IELs. No IELs bound to the E31A mutant, compared with 56% of IELs that bound to wild-type (Fig. 2 C, P ≤ 0.0001).
Refinement of the Heterophilic Adhesion Site.
Although these studies pointed to a critical role for E31 in E-cadherin adhesion to αEβ7, it is unlikely that the specificity of the integrin αEβ7–E-cadherin interaction is determined by a single residue. The contact between the two molecules most likely involves multiple residues that determine specificity and stabilize the interaction. Using the human E-cadherin model (Fig. 1 B), we selected additional residues with solvent-exposed side chains in the proximity of E31 for site-directed mutagenesis. In the BC loop, residues K25, N27, K28, K30, and K33 have solvent-exposed side chains in close proximity to the side chain of E31 and may be important in recognition of integrin αEβ7. Thus, E-cadherin-Fc fusion proteins containing mutations of these residues to alanine were produced and tested in adhesion to αEβ7-expressing cells. Beginning at the NH2 terminus of the BC loop, we found that 69% of the input IELs adhered to E-cadherin-Fc containing the K25A mutation compared with 72% of IELs that adhered to wild-type E-cadherin-Fc (Fig. 3 A, P ≤ 0.35). Continuing along the BC loop, mutants N27A, K28A, and K30A supported adhesion of 58% of the IELs compared with wild-type, which supported adhesion of 62% of the IELs (Fig. 3B–D; P ≤ 0.086, 0.11, 0.014, respectively). At the COOH-terminal end of the BC loop, the K33A mutant supported adhesion of 60% of the IELs, which was the same percentage of IEL adhesion supported by wild-type E-cadherin-Fc (Fig. 3 E, P ≤ 0.46). Thus, there were no significant differences between IEL adhesion to wild-type E-cadherin-Fc and E-cadherin-Fc mutants K25A, N27A, K28A, K30, and K33A.
Figure 3.

IEL adhesion to E-cadherin-Fc BC loop mutants. Adhesion was determined in at least two independent experiments in which each condition was performed in triplicate, as described in the legend to Fig. 2. Results are shown as the mean percentage of IELs bound ± 1 SD for a representative experiment. (A) Dose–response of IEL adhesion to K25A. (B) Dose–response of IEL adhesion to N27A. (C) Dose–response of IEL adhesion to K28A. (D) Dose–response of IEL adhesion to K30A. (E) Dose–response of IEL adhesion to K33A. (F) Dose–response of IEL adhesion to KEG30-32RDT. (G) Dose–response of IEL adhesion to E31D.
Recognizing that >100-fold more human placental (P)-cadherin-Fc than E-cadherin-Fc fusion protein is required to support IEL adhesion 2, we compared the BC loops of these two ligands. The only differences between the BC loop of E-cadherin and P-cadherin are residues 30, 31, and 32 (Fig. 1 A). Thus, we substituted the central three residues of the BC loop of P-cadherin (RD31T) in place of KE31G in E-cadherin (mutant termed KEG30-32RDT) to determine the effect on IEL adhesion. Even though both wild-type E-cadherin and the KEG30-32RDT mutant contain an acidic residue at position 31, the KEG30-32RDT mutation completely abolished adhesion of IELs (Fig. 3 F, P ≤ 0.0001). To distinguish the role of E31 from the flanking residues, we also constructed the conservative E31D mutation. Surprisingly, the E31D mutation also completely abolished detectable adhesion of IELs (Fig. 3 G, P ≤ 0.0001).
The FG loop is located adjacent to the BC loop at the top of the cadherin domain, suggesting that side chains of this loop might also participate in integrin binding (Fig. 1 B). Analysis of the human E-cadherin model revealed that the side chains of residues S83, N86, and E89 in the FG loop are in close proximity to the critical E31 residue in the BC loop. E-cadherin-Fc fusion protein containing the FG loop S83A mutation was found to support adhesion of 68% of the IELs compared with adhesion of 72% of the IELs to wild-type E-cadherin-Fc (Fig. 4 A, P ≤ 0.55). E-cadherin-Fc with the N86A mutation supported adhesion of 57% of the IELs compared with adhesion of 53% of the IELs supported by wild-type E-cadherin-Fc (Fig. 4 B, P ≤ 0.068). These results indicated that the S83A and N86A mutations did not significantly affect αEβ7-dependent adhesion of IELs to E-cadherin. E-cadherin-Fc containing the E89A mutation supported adhesion of 12% of the IELs compared with adhesion of 72% of the IELs to wild-type (Fig. 4 C), suggesting that the E89A mutation substantially diminished αEβ7-dependent IEL adhesion to E-cadherin (P ≤ 0.0001). For easier comparison, the effect of the mutations on adhesion to IELs is summarized as the percentage of adhesion compared with wild-type (Fig. 5 A). The three mutants that revealed the greatest effect were all located in the BC loop and involved residue E31. These mutations completely abrogated adhesion to <2% of wild-type adhesion. In addition, E89A in the FG loop reduced heterophilic adhesion to 20% of wild-type. In contrast, the other mutations located in the remainder of the BC and FG loop as well as the CD and DE loops supported >82% of wild-type adhesion.
Figure 4.
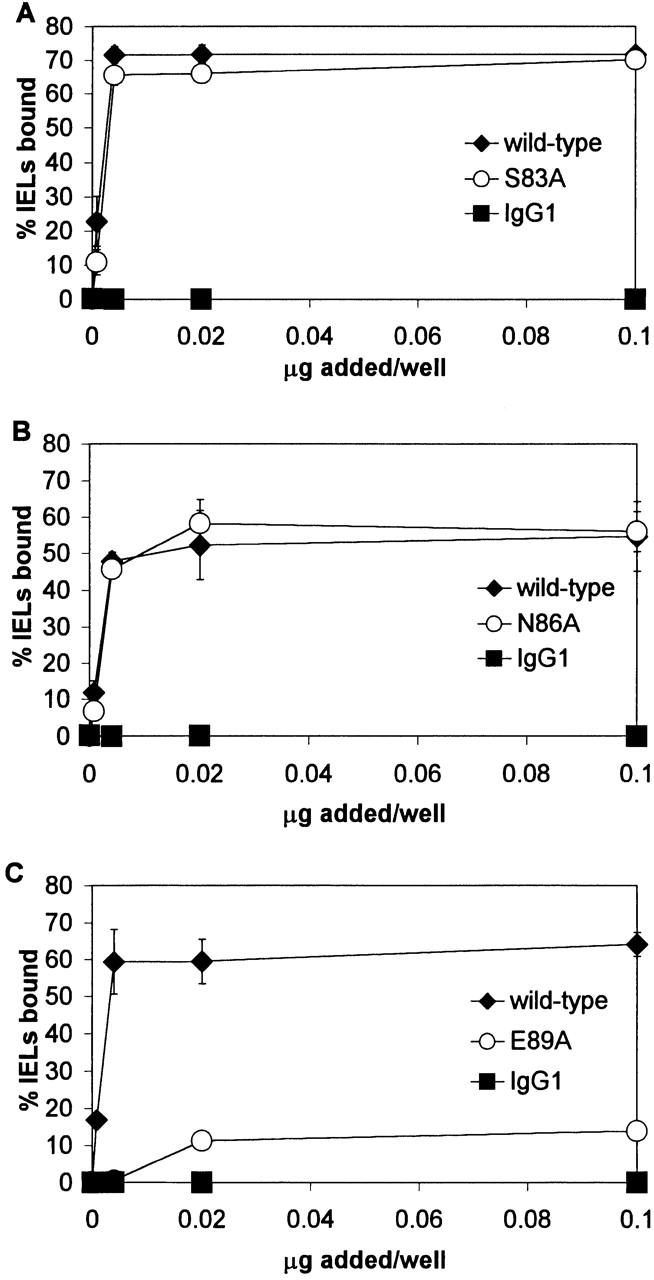
IEL adhesion to E-cadherin-Fc FG loop mutants. Adhesion was determined in at least two independent experiments in which each condition was performed in triplicate, as described in the legend to Fig. 2. Results are shown as the mean percentage of IELs bound ± 1 SD for a representative experiment. (A) Dose–response of IEL adhesion to S83A. (B) Dose–response of IEL adhesion to N86A. (C) Dose–response of IEL adhesion to E89A.
Figure 5.
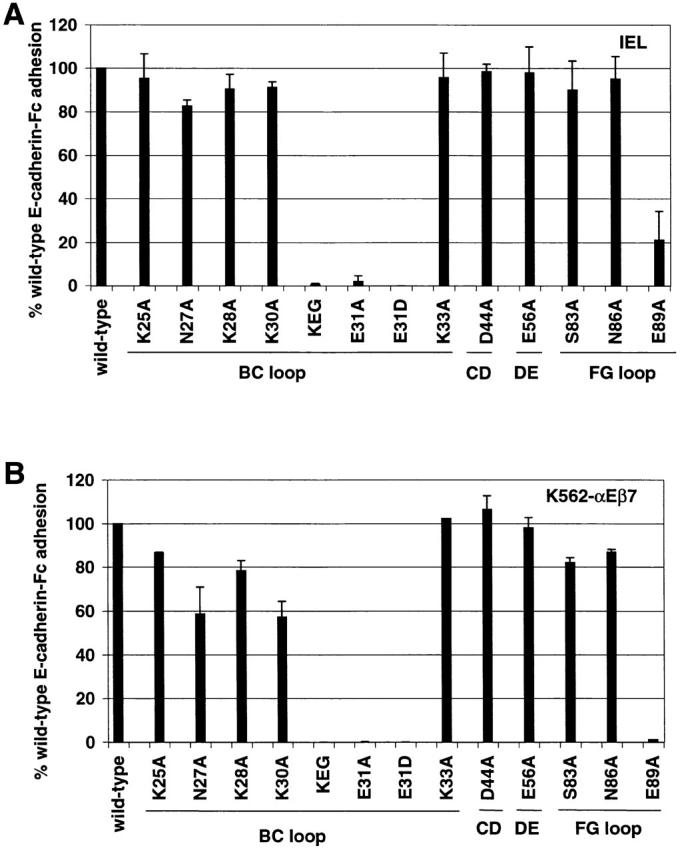
Heterophilic adhesion of IELs (A) and K562-αEβ7 (B) to E-cadherin-Fc mutants. Using adhesion to saturating amounts of E-cadherin-Fc, the mean adhesion to each mutant is expressed relative to adhesion to wild-type E-cadherin-Fc (% cells bound to mutant E-cadherin-Fc/% cells bound to wild-type E-cadherin-Fc × 100). Adhesion to wild-type E-cadherin-Fc is shown as 100%. Adhesion above the saturating dose was chosen to minimize the effect of any errors in quantitation of the fusion protein. Mutation KEG30-32RDT is abbreviated KEG. The loop containing the mutated residues is marked below the lines. Results are shown as the mean ± 1 SD of at least two independent experiments in which each condition was performed in triplicate. A two-sided t test was used to compare the mean value of IEL adhesion to each E-cadherin-Fc mutant with the mean value of IEL adhesion to wild-type E-cadherin-Fc. (A) IEL adhesion to the panel of E-cadherin-Fc mutants. Statistically significant differences are observed for KEG30-32RDT (P ≤ 0.0001), E31A (P ≤ 0.0001), E31D (P ≤ 0.0001), and E89A (P ≤ 0.0001). (B) K562-αEβ7 adhesion to the section of E-cadherin-Fc mutants. Statistically significant differences are observed for N27A (P ≤ 0.0001), K28A (P ≤ 0.0001), K30A (P ≤ 0.0001), KEG30-32RDT (P ≤ 0.0001), E31A (P ≤ 0.0001), E31D (P ≤ 0.0001), N86A (P ≤ 0.0017), and E89A (P ≤ 0.0001).
Additional Mutations Reduce Heterophilic Adhesion of K562-αEβ7 Transfectants to E-Cadherin.
To confirm the importance of E31 on the top of the BC loop and E89 on the FG loop in IEL adhesion to E-cadherin-Fc and to identify additional residues involved in heterophilic adhesion, we examined the adhesion of K562-αEβ7 transfectants to the panel of E-cadherin-Fc mutants under the same conditions used to study heterophilic adhesion of IEL T cells. We hypothesized that K562-αEβ7 transfectants might identify other residues involved in heterophilic adhesion, as K562-αEβ7 transfectants adhere less strongly to E-cadherin-Fc under the same conditions. Similar to IEL adhesion, heterophilic adhesion of K562-αEβ7 transfectants to immobilized E-cadherin-Fc is dose dependent, with maximal adhesion achieved with 0.02 μg of E-cadherin-Fc added per well. Adhesion is specific, as <1% of K562 mock transfectants bound to E-cadherin-Fc–coated wells, and <1% of K562-αEβ7 transfectants bound to IgG1-coated wells (data not shown). However, mean maximal adhesion of K562-αEβ7 transfectants to wild-type E-cadherin-Fc is 30% compared with mean maximal adhesion of IEL to wild-type E-cadherin-Fc, which is 58% (data not shown).
Heterophilic adhesion of K562-αEβ7 transfectants to mutants E31A, E31D, and KEG30-32RDT was not detectable, confirming the findings in adhesion to in vitro–cultured IEL T cells expressing αEβ7 (Fig. 5 B). E-cadherin-Fc containing K25A and K33A mutations in the BC loop supported adhesion similar to wild-type: 87% (P ≤ 0.0113) and 104% (P ≤ 0.36), respectively, as a percentage of adhesion to wild-type E-cadherin (Fig. 5 B). BC loop mutants N27A, K28A, and K30A only partially supported adhesion of K562-αEβ7 transfectants: 57% (P ≤ 0.0001), 79% (P ≤ 0.0001), and 53% (P ≤ 0.0001), respectively, as a percentage of adhesion to wild-type E-cadherin (Fig. 5 B). Thus, K562-αEβ7 transfectants identified additional BC loop mutations with statistically significant differences in the ability to support heterophilic adhesion compared with wild-type, namely N27A, K28A, and K30A. Mutants containing the D44A change in the CD loop and the E56A change in DE loop supported heterophilic adhesion to K562-αEβ7 similar to wild-type: 105% (P ≤ 0.62) and 102% (P ≤ 0.40), respectively, like the situation for these mutations in adhesion to IELs. FG loop mutations S83A and N86A partially supported heterophilic adhesion to K562-αEβ7: 80% (P ≤ 0.0001) and 86% (P ≤ 0.0017), respectively, as a percentage of adhesion to wild-type E-cadherin-Fc (Fig. 5 B). In contrast, FG loop mutant E89A did not support detectable adhesion, confirming the finding with adhesion to IELs (Fig. 5 B). In summary, the K562-αEβ7 transfectants confirmed the role of mutations noted for adhesion of IELs and identified several additional mutants with smaller but statistically significant differences in the ability to support heterophilic adhesion compared with wild-type (N27A, K28A, K30A, S83A, and N86A). For a conservative interpretation, we emphasize mutations N27A and K30A, as they resulted in the largest (>40%) reduction in αEβ7-dependent adhesion (Fig. 5 B).
Location of αEβ7 Adhesion Site on E-Cadherin.
We analyzed the human E-cadherin model examining the location of residues affecting or not affecting adhesion of αEβ7-expressing cells to E-cadherin-Fc. As shown in the ribbon diagram of domain 1 of E-cadherin, the side chains of residues K25 and K33 on the BC loop and E56 on the DE loop point in approximately the same direction away from the sheet formed by the D, E, and B strands (Fig. 6 A). Mutation of residues K25, K33, and E56 to alanine in E-cadherin-Fc did not affect heterophilic adhesion to IELs or K562-αEβ7 transfectants, suggesting that the integrin does not approach E-cadherin at the DEB sheet. The side chains of residues D44 on the CD loop and S83 and N86 on the FG loop are located on the other side of E31 and point away from the CFG face (Fig. 6 A). Mutations D44A, S83A, and N86A did not have a substantial effect on heterophilic adhesion to IELs or K562-αEβ7 transfectants, suggesting that the integrin does not approach E-cadherin at the CFG face. In contrast, the side chains of residues N27, K28, K30, and E31 on the BC loop as well as E89 on the FG loop are highly solvent-exposed at the top of the molecule on the face formed by the BC and FG loops (Fig. 6 A). Mutation of E31 and E89 severely diminished heterophilic adhesion of both IEL and K562-αEβ7 transfectants. Mutations N27A, K28A, and K30A had statistically significant reductions in adhesion to K562-αEβ7 transfectants, and N27A and K30A reduced K562-αEβ7 transfectant adhesion >40%.
Figure 6.

Ribbon and surface diagrams of domain 1 of E-cadherin demonstrating the location of mutated residues. (A) Ribbon diagram of E-cadherin domain 1. The side chains of residues involved in adhesion to αEβ7 are shown with stick diagrams in red. Mutation of side chains shown in blue did not diminish adhesion to αEβ7 or diminished adhesion to αEβ7 <20%. (B) Surface diagram of E-cadherin domain 1 in the same orientation as A. Residues involved in adhesion to αEβ7 are shown in red, and residues that did not affect adhesion to αEβ7 are shown in blue. (C) Surface diagram of E-cadherin domain 1 rotated ∼180° on a vertical axis from B.
A surface diagram of E-cadherin in the same orientation as the ribbon diagram in Fig. 6 A more clearly demonstrates the location of the side chains that were mutated (Fig. 6 B). The side chains of residues N27, K30, E31, and E89, which are involved in adhesion to αEβ7, are shown in red. Despite the fact the implicated residues come from different loops, they are located on the same face (Fig. 6 B). Residues that do not substantially affect adhesion to αEβ7 are shown in blue. Rotation of the surface diagram 180° on a vertical axis demonstrates that residues on the opposite side are not involved in adhesion to αEβ7 (Fig. 6 C). Therefore, it is likely that the integrin αEβ7 approaches domain 1 of human E-cadherin at the face formed by the BC and FG loops.
Analysis of the three-dimensional human E-cadherin model suggests that the side chains of residues N27, K30, and E31 protrude upward and could directly interact with the integrin (partially illustrated in Fig. 6 A). Residue E89 likely forms interactions with one or both residues, S26 and K28. E89 has the potential to form a salt bridge with the main chain amide of K28, similar to the E89/R28 pair observed in murine E-cadherin 19. E89 might also form a hydrogen bond with residue S26. These interactions would serve to stabilize the BC loop that presents residues N27, K30, and E31 for integrin recognition. In summary, mutational analyses of the NH2-terminal domain of human E-cadherin identified a localized group of exposed residues at the top of the domain that are likely to be part of the integrin αEβ7 binding site either by directly binding to the integrin or by supporting the conformation of the integrin binding residues.
Homophilic Adhesion of E-Cadherin-Fc Mutants.
We tested the ability of E-cadherin-Fc and its mutants to support homophilic adhesion of E-cadherin–expressing MCF-7 breast epithelial cells. Adhesion of MCF-7 cells to E-cadherin-Fc was dose dependent, with maximal adhesion of 40% of cells bound with 0.02 μg of E-cadherin-Fc added per well (Fig. 7, ♦), whereas adhesion to human IgG1 was not observed (Fig. 7, ▪). Confirming the specificity of this adhesion assay, we found that the adhesion of MCF-7 cells to E-cadherin-Fc was blocked by anti–E-cadherin mAbs HECD-1 and SHE78-7, but not by isotype-matched control mAbs against αEβ7, αE7-1, and αE7-2 (data not shown), and the interaction did not require the presence of magnesium or manganese.
Figure 7.
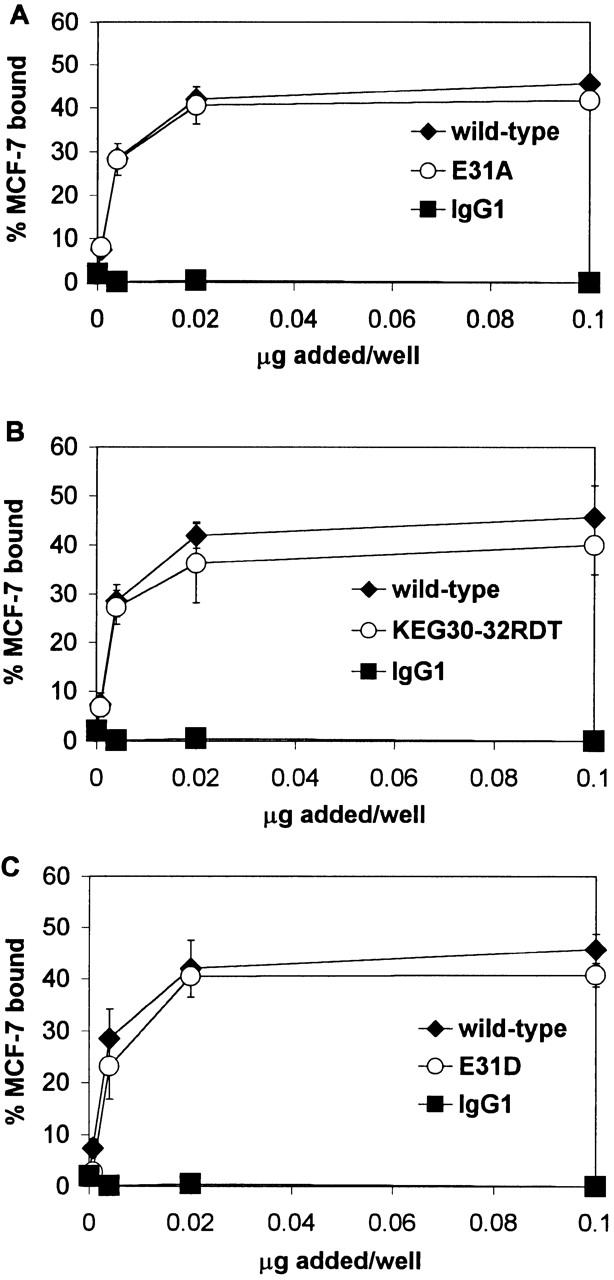
Homophilic adhesion of MCF-7 cells to E-cadherin-Fc mutants E31A, KEG30-32RDT, and E31D. Serial dilutations of wild-type and mutated E-cadherin and IgG1 were immobilized on plates coated with goat anti–human IgG antiserum. Homophilic adhesion assays with E-cadherin–expressing MCF-7 cells were performed in HBS with 1 mM CaCl2, 50 mM glucose, and 0.1% BSA, as described in Materials and Methods. Results are shown as the mean percentage of MCF-7 cells bound ± 1 SD and are representative of at least two independent experiments in which each condition was performed in triplicate. (A) Homophilic adhesion to KEG30-32RDT (KEG). (B) Homophilic adhesion to E31A. (C) Homophilic adhesion to E31D.
The E-cadherin-Fc mutants were tested in homophilic adhesion to MCF-7 epithelial cells to demonstrate that the mutants were functionally folded and to determine whether the heterophilic adhesion site for αEβ7 is distinct from the homophilic site for E-cadherin. E-cadherin-Fc mutations that completely abrogated IEL adhesion did not affect homophilic adhesion (Fig. 7). E31A E-cadherin-Fc supported homophilic adhesion of 41% of the MCF-7 cells (Fig. 7 A), KEG30-32RDT E-cadherin-Fc supported homophilic adhesion of 38% of the MCF-7 cells (Fig. 7 B), and E31D E-cadherin-Fc supported homophilic adhesion of 41% of the MCF-7 cells (Fig. 7 C), compared with adhesion of 44% of the MCF-7 cells to wild-type E-cadherin-Fc. Thus, there were no significant differences between MCF-7 homophilic adhesion to wild-type E-cadherin-Fc and E-cadherin-Fc mutants E31A, KEG30-32RDT, and E31D, despite the effect these mutations revealed in heterophilic adhesion. The ability of these E-cadherin mutants to function in homophilic adhesion confirms that these loop mutations did not affect proper folding of the cadherin domain and that the heterophilic αEβ7 adhesion site is distinct from the homophilic E-cadherin adhesion site.
Fig. 8 summarizes the effect of the mutations on homophilic adhesion to saturating amounts of mutated E-cadherin-Fc fusion proteins relative to adhesion to wild-type E-cadherin-Fc. Mutants K25A and N27A located at the beginning of the BC loop exhibit a mild reduction in homophilic adhesion compared with wild-type E-cadherin-Fc. Mutant K25A supports 76% of the homophilic adhesion observed to wild-type E-cadherin-Fc (P ≤ 0.0003), and mutant N27A supports 62% of the homophilic adhesion observed to wild-type E-cadherin-Fc (P ≤ 0.0001). There were no significant differences between the adhesion of the other E-cadherin-Fc BC loop mutants (K28A, K30A, and K33A) and wild-type. Mutants K28A, K30A, and K33A supported homophilic adhesion 94, 99, and 91% as well as wild-type E-cadherin, respectively. Mutants D44A in the CD loop and E56A in the DE loop supported homophilic adhesion 94 and 90% as well as wild-type E-cadherin-Fc. Mutants S83A and N86A on the FG loop and N86A also supported wild-type levels of homophilic adhesion: 105 and 93%, respectively. Homophilic adhesion supported by mutants D44A, E56A, S83A, and N86A is not significantly different from adhesion supported by wild-type. In contrast, E-cadherin-Fc containing the E89A mutation on the G strand supported only 2% of the homophilic adhesion observed for wild-type E-cadherin (P ≤ 0.0001), similar to its inability to support heterophilic adhesion. As the E89A mutant was efficiently secreted and recognized by two anti-E-cadherin mAbs, it remains likely that it is properly folded and has a role in both heterophilic and homophilic adhesion.
Figure 8.

Homophilic adhesion of MCF-7 cells to E-cadherin-Fc mutants. Adhesion of MCF-7 cells to saturating amounts of mutated E-cadherin-Fc is expressed relative wild-type E-cadherin-Fc, as described in the legend to Fig. 5. Results are shown as the mean ± 1 SD of two independent experiments in which each condition was performed in triplicate. Using a two-sided t test, statistically significant differences were observed between homophilic adhesion to wild-type E-cadherin-Fc and K25A (P ≤ 0.0003), N27A (P ≤ 0.0001), and E89A (P ≤ 0.0001).
Although we have not specifically addressed the homophilic adhesion site, several of the residues we mutated have been implicated in homophilic adhesion in previous studies. Mutational analyses localize the homophilic binding site to domain 1 of E-cadherin and homophilic adhesion specificity determining residues to serine residues on either side of the HAV sequence at the end of the F strand: S78 and S83 16. Simultaneous mutation of both S78 and S83 in E-cadherin to the corresponding residues in P-cadherin does not abolish calcium-dependent homophilic cell aggregation, but allows cadherin transfectants to aggregate with both E- and P-cadherin 16. In agreement, our results showed that point mutation of S83A does not diminish homophilic adhesion. The homophilic adhesion interface suggested by the adhesion dimer in the N-cadherin domain 1 crystal structure includes interactions between the C strand and C′ strand (prime indicates that the structure comes from the adjacent molecule), reciprocal contact between the DE loop to FG′ loop, and the CD loop to CD′ loop interactions 18. D44 is the only residue we mutated that is present at the homophilic adhesion interface. Mutation of D44A did not diminish homophilic adhesion, suggesting that either the loss of D44's contributions is not significant enough to diminish homophilic adhesion because of the complexity of the homophilic adhesive interaction or the adhesion dimer seen in the crystal structure may not be the physiological interface.
Previous studies suggest that homophilic adhesion requires dimerization 30 31. However, controversy exists over which part of the cadherin domain is involved in cis-dimerization 18 19 20. In our study, mutation of residues K25, N27, and E89 significantly diminished homophilic adhesion. As E-cadherin-Fc mutants K25A and N27A exhibit near wild-type adhesion to IELs, they are most likely functionally folded. As can be observed in Fig. 6 B, the residues that disrupt homophilic adhesion are located on the same face. These residues could affect homophilic adhesion by directly being part of the homophilic binding site or indirectly by contributing to cis-dimerization of E-cadherin. Residues K25, N27, and E89 are located at the cis-dimer interface observed in the murine N-cadherin domain 1 crystal structure 18 and have the potential to support cis-dimerization. Thus, we propose that mutations K25, N27, and E89 indirectly affect homophilic adhesion by interfering with cis-dimerization. This interpretation supports a physiological role for the cis-dimer in homophilic adhesion.
Discussion
A well-defined feature of integrin ligands is the presence of an aspartic acid or glutamic acid residue that is essential for integrin adhesion. The critical acidic residue in the ligand may provide a coordination site for the divalent cation that is essential for integrin adhesion 6. Our first approach was to mutate solvent-exposed acidic residues on the various loops of human E-cadherin to localize integrin recognition to a particular loop. We selected potential residues from our model of human E-cadherin based on the crystal structure of murine E-cadherin 19. Mutational analyses of the exposed acidic residues on the BC, CD, and DE loops localized the αEβ7 binding site to E31 in the BC loop of domain 1. This result is consistent with a previous analysis of acidic loop residues in murine E-cadherin using αEβ7-expressing cells adhering to L cell transfectants expressing murine E-cadherin mutants 32. We further analyzed the role of the BC loop in determining the specificity of the E-cadherin interaction with αEβ7 with the KEG30-32RDT mutation that substitutes P- for E-cadherin residues at the tip of the BC loop. Failure of this mutant to support heterophilic cadherin adhesion indicates that the central residues in the BC loop of P-cadherin cannot support adhesion of αEβ7. In contrast, substitution of the CD loops of MAdCAM-1 and VCAM-1 does not dramatically affect integrin recognition. Previous studies demonstrated that the CD loop residues of ICAM-1, GIET, can substitute for the CD loop of VCAM-1, QIDS, in a cell–cell α4β1-dependent adhesion assay 33. Additionally, previous studies demonstrated that the CD loop residues of MAdCAM-1, GLDTS, can substitute for the CD loop of VCAM-1, QIDSP, in mediating α4β1-mediated cell spreading on and α4β7-cell adhesion to VCAM-Fc 34. The difference between this study and previous work emphasizes that the integrin αEβ7 displays extreme specificity for the specific acidic residue at the adhesion site and for the specific residues flanking the acidic residue.
To determine whether the lack of adhesion to the KEG30-32RDT mutant was due to the conservative change in the acidic residue or due to the side chains of the flanking residues, we made the E31D mutation. Relevant mutations of other integrin ligands have been tested in adhesion. Mutation of the critical acidic residue D40 in CD loop of domain 1 of soluble, recombinant VCAM-1 to glutamate did not diminish cell adhesion via α4β1 35 and resulted in a <50% reduction in cell spreading via α4β1 34. However, the D40E mutation in recombinant VCAM-1 and the homologous D41E mutation in recombinant murine MAdCAM-1 resulted in an almost complete loss of cell adhesion via α4β7 34. Mutation of the critical acidic residue on the C strand of domain 1 of ICAM-1 E34D almost completely abrogated binding to αLβ2 in a cell-free system 36. These previous studies suggest that changing the critical acidic residue to another acidic residue has little affect on α4β1 recognition, but almost entirely abrogates recognition by α4β7 and αLβ2. Here, the E31D E-cadherin-Fc mutation abrogated adhesion of IELs and K562-αEβ7 transfectants, suggesting that αEβ7 has remarkably fine specificity for the length of the side chain of glutamic acid residue 31 in E-cadherin (Fig. 5). Analysis of the murine E-cadherin crystal structure reveals that there is one hydrogen bond between the main chain carbonyl group of residue 28 (R28 in murine, and K28 in human E-cadherin) and the main chain amide group of residue 32 (T32 in murine, and G32 in human E-cadherin). It is likely that this hydrogen bond will make the tip of the BC loop assume a rigid conformation so that the key residue E31 can be posed in the correct configuration for binding to the integrin, potentially the MIDAS metal center. Moreover, we have also noticed a minihydrophobic core between the BC and FG loops, centered around V34, which should further stabilize the BC loop. This minihydrophobic core includes V88, A80, and Y36, as well as those aliphatic portions of other residues like K28; all of these residues are conserved. There is one more hydrogen bond in this region between the hydroxyl group of Y36 and the main chain amide group of S26. Although the overall atomic mobility of BC loop in the murine E- and N-cadherin crystal structure is high with respect to the core 18 19, these interactions serve to make the long BC loop rigid with respect to itself. From an energetic point of view, a rigid loop is more favorable during the ligand binding interaction. This may explain why the conservative E31D mutation abolishes adhesion. If the key binding motif is in a very delicate conformation designed for integrin binding, even the size of the one methyl group difference between glutamate and aspartate could be significant. Similar hydrogen bonding systems that surround key acidic integrin binding residues and help rigidify the binding motif have been reviewed for other IgSF ligands of both A domain and non–A domain containing integrins 37.
Our mutational analyses of selected residues with solvent-exposed side chains towards the top of E-cadherin domain 1 revealed that the side chains of N27, K30, E31, and E89 are important for integrin αEβ7 recognition. Modeling of the position of these side chains in human E-cadherin revealed that they are all located on the same face formed by the BC and FG loops at the top of domain 1 (Fig. 6 B). As previously discussed, these residues may directly interact with the integrin αEβ7 or may be important in maintaining the conformation of the residues that directly contact the integrin.
The integrin ligands ICAM-1, -2, -3, VCAM-1, and MAdCAM-1 belong to the IgSF. Although the tenth type III repeat of fibronectin and E-cadherin lack the sequence homology and conserved disulfide bonds to be considered an Ig fold, both of these domains share an Ig-like β sandwich topology consisting of two β sheets made up of three or four antiparallel β strands 18 19 38. This β sandwich topology appears to be a conserved scaffold in integrin recognition. However, our studies emphasize that integrins use different exposed surfaces for recognition of different ligands. In MAdCAM-1 and VCAM-1, the critical acidic residue is present at the tip of the CD loop that protrudes from the side on the lower half of domain 1 (Fig. 9; 10–13, 28, 33, 34, 39 40 41). In contrast, in the ICAMs the acidic residue critical for integrin recognition is located at the end of the C strand on a flat recognition surface located on the lower side of domain 1 (Fig. 9; 14, 15, 36, 42 43 44 45 46). The binding site for integrins α5β1, αIIbβ3, and αVβ3 on fibronectin is the RGD sequence on an extended FG loop of the tenth type III repeat (Fig. 9; 38, 47). In contrast, αEβ7 recognizes E-cadherin at the top of the Ig-like cadherin first domain (Fig. 9). The critical acidic residue E31 is located at the very tip of the outwardly extended BC loop, and the integrin recognition surface contains residues from the BC and the adjacent FG loop. The local conformation around the critical acidic residue E31 in E-cadherin is most similar to the conformation of the corresponding FG loop in fibronectin. In conclusion, integrin recognition of E-cadherin extends the general paradigm that Ig-like folds serve as a scaffold for integrin recognition, but is unique in using the BC loop at the top of the domain (Fig. 9).
Figure 9.

Comparison of integrin recognition sites in E-cadherin, fibronectin, MAdCAM-1, and ICAM-1. Ribbon diagrams of domain 1 of murine E-cadherin (Protein Database accession no. 1EDH; reference 19), the tenth type III repeat of human fibronectin (accession no. 1FNF; reference 36), domain 1 of human MAdCAM-1 (accession no. 1BQS; reference 10), and domain 1 of human ICAM-1 (accession no. 1IC1; reference 41) are shown. The location of the side chains of acidic residues critical for integrin adhesion are shown with stick diagrams using black for carbon atoms, red for oxygen atoms, and blue for nitrogen atoms. Where applicable, the loop involved in integrin recognition is highlighted in orange. The location of the integrin recognition sites in MAdCAM-1 and ICAM-1 is also representative of VCAM-1 and ICAM-2/ICAM-3, respectively. ICAM-1, ICAM-2, VCAM-1, and MAdCAM-1 all have an extra disulfide bond, not characteristic of the Ig fold, between the BC loop and the FG loop or F strand. The conserved disulfide bond of Ig folds and the extra disulfide bond between the BC and FG loops are shown in yellow in the MAdCAM-1 and ICAM-1 structures.
Importantly, while the three mutants involving E31 (E31A, KEG30-32RDT, and E31D) did not support detectable heterophilic adhesion, they did support homophilic adhesion of cells expressing wild-type E-cadherin. These results are the first direct evidence that the heterophilic adhesion site on E-cadherin for αEβ7 is distinct from the homophilic E-cadherin–E-cadherin adhesion site. This finding is consistent with previous studies examining the homophilic adhesion site and localizing it to the face formed by the C strand, D strand, and FG loop and in particular to residues at the COOH terminus of the F strand and in the FG loop 16 18 48. As the E-cadherin residues critical for heterophilic adhesion to αEβ7 are distinct from those required for homophilic adhesion, it is possible to block heterophilic adhesion selectively while preserving E-cadherin's homophilic function. The analyses of αEβ7 recognition of E-cadherin suggest that blockade of this interaction may be similar in molecular terms to that of other integrin–IgSF interactions. The current data provide both the conceptual basis and suitable assay systems to identify therapeutic inhibitors. Our data suggest that the potential exists to design therapeutics that selectively block the E-cadherin–αEβ7 interaction and do not disrupt E-cadherin homophilic adhesion. Integrin αEβ7 is expressed on nearly all T cells localized in the epithelium, as well as on certain mast cells and dendritic cells 22 49 50. Agents that block the interaction of αEβ7-expressing cells with E-cadherin–expressing epithelium could be useful therapeutics in mucosal inflammatory conditions such as inflammatory bowel disease.
Acknowledgments
We thank Dr. Lawren Daltroy for his guidance with the statistical analysis, and Dr. Ulrike Strauch for her help in maintaining the K562 transfectants (both of the Division of Rheumatology, Immunology and Allergy, Brigham and Women's Hospital).
This work was supported by National Institutes of Health grants to M.B. Brenner, HL48675 (to J.H. Wang), and AI01212 (to D.A. Mandelbrot), and by a Wellcome Trust International Prize Travelling Research Fellowship and a Crohn's and Colitis Foundation of America Fellowship (to J.M.G. Higgins).
Footnotes
Abbreviations used in this paper: E, epithelial; FBS, fetal bovine serum; HBS, Hepes-buffered saline; HEK, human embryonic kidney; IEL, intraepithelial lymphocyte; IgSF, Ig superfamily; ICAM, intercellular adhesion molecule; K562-αEβ7, K562 cells stably transfected with αEβ7; MAdCAM-1, mucosal addressin cell adhesion molecule 1; N, neural; P, placental; TBS, Tris-buffered saline; VCAM-1, vascular cell adhesion molecule 1.
References
- Cepek K.L., Parker C.M., Madara J.L., Brenner M.B. Integrin αΕβ7 mediates adhesion of T lymphocytes to epithelial cells. J. Immunol. 1993;150:3459–3470. [PubMed] [Google Scholar]
- Higgins J.M.G., Mandelbrot D.A., Shaw S.K., Russell G.J., Murphy E.A., Chen Y.T., Nelson W.J., Parker C.M., Brenner M.D. Direct and regulated interaction of integrin αEβ7 with E-cadherin. J. Cell Biol. 1998;140:197–210. doi: 10.1083/jcb.140.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoen M.P., Arya A., Murphy E.A., Adams C.A., Strauch U.G., Agace W., Marsal J., Donohue J.P., Her J., Deier D.R. Mucosal T lymphocyte numbers are selectively reduced in integrin αE (CD103)-deficient mice. J. Immunol. 1999;162:6641–6649. [PubMed] [Google Scholar]
- Shaw S.K., Cepek K.L., Murphy E.A., Russell G.J., Brenner M.B., Parker C.M. Molecular cloning of the human mucosal lymphocyte integrin αE subunit. Unusual structure and restricted RNA distribution. J. Biol. Chem. 1994;269:6016–6025. [PubMed] [Google Scholar]
- Qu A., Leahy D.J. Crystal structure of the I-domain from the CD11a/CD18 (LFA-1, αLβ2) integrin. Proc. Natl. Acad. Sci. USA. 1995;92:10277–10281. doi: 10.1073/pnas.92.22.10277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.O., Rieu P., Arnaout M.A., Liddington R. Crystal structure of the A domain from the α subunit of integrin CR3 (CD11b/CD18) Cell. 1995;80:631–638. doi: 10.1016/0092-8674(95)90517-0. [DOI] [PubMed] [Google Scholar]
- Qu A., Leahy D.J. The role of the divalent cation in the structure of the I domain from the CD11a/CD18 integrin. Structure. 1996;4:931–942. doi: 10.1016/s0969-2126(96)00100-1. [DOI] [PubMed] [Google Scholar]
- Emsley J., King S.L., Bergelson J.M., Liddington R.C. Crystal structure of the I domain from integrin α2β1 . J. Biol. Chem. 1997;272:28512–28517. doi: 10.1074/jbc.272.45.28512. [DOI] [PubMed] [Google Scholar]
- Baldwin E.T., Sarver R.W., Bryant G.L., Curry K.A., Fairbanks M.B., Finzel B.C., Garlick R.L., Heinrikson R.L., Horton N.C., Kelley L.L. Cation binding to the integrin CD11b I domain and activation model assessment. Structure. 1998;6:923–935. doi: 10.1016/s0969-2126(98)00093-8. [DOI] [PubMed] [Google Scholar]
- Tan K., Casasnovas J.M., Liu J.H., Briskin M.J., Springer T.A., Wang J.H. The structure of immunoglobulin superfamily domains 1 and 2 of MAdCAM-1 reveals novel features important for integrin recognition. Structure. 1998;6:793–801. doi: 10.1016/s0969-2126(98)00080-x. [DOI] [PubMed] [Google Scholar]
- Viney J.L., Jones S., Chiu H.H., Lagrimac B., Renz M.E., Presta L.G., Jackson D., Hillan K.J., Lew S., Fong S. Mucosal addressin cell adhesion molecule-1a structural and functional analysis demarcates the integrin binding motif. J. Immunol. 1996;157:2488–2497. [PubMed] [Google Scholar]
- Jones E.Y., Harlos K., Bottomley M.J., Robinson R.C., Driscoll P.C., Edward R.M., Clements J.M., Dudgeon T.J., Stuart D.I. Crystal structure of an integrin-binding fragment of vascular cell adhesion molecule1 at 1.8 Å resolution. Nature. 1995;373:539–544. doi: 10.1038/373539a0. [DOI] [PubMed] [Google Scholar]
- Vonderheide R.H., Tedder T.F., Springer T.A., Staunton D.E. Residues within a conserved amino acid motif of domains 1 and 4 of VCAM-1 are required for binding to VLA-4. J. Cell Biol. 1994;125:215–222. doi: 10.1083/jcb.125.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casasnovas J.M., Springer T.A., Liu J.H., Harrison S.C., Wang J.H. Crystal structure of ICAM-2 reveals a distinctive integrin recognition surface. Nature. 1997;387:312–315. doi: 10.1038/387312a0. [DOI] [PubMed] [Google Scholar]
- Staunton D.E., Dustin M.L., Erickson H.P., Springer T.A. The arrangement of the immunoglobulin-like domains of ICAM-1 and the binding sites for LFA-1 and rhinovirus. Cell. 1990;61:243–254. doi: 10.1016/0092-8674(90)90805-o. [DOI] [PubMed] [Google Scholar]
- Nose A., Tsugi K., Takeichi M. Localization of the specificity determining sites in cadherin cell adhesion molecules. Cell. 1990;61:147–155. doi: 10.1016/0092-8674(90)90222-z. [DOI] [PubMed] [Google Scholar]
- Overduin M., Harvey T.S., Bagby S., Tong K.I., Yau P., Takeichi M., Ikura M. Solution structure of the epithelial cadherin domain responsible for selective cell adhesion. Science. 1995;267:386–389. doi: 10.1126/science.7824937. [DOI] [PubMed] [Google Scholar]
- Shapiro L., Fannon A.M., Kwong P.D., Thompson A., Lehmann M.S., Gruebel G., Legrand J.F., Als-Nielsen J., Colman D.R., Hendrickson W. Structural basis of cell-cell adhesion by cadherins. Nature. 1995;374:327–337. doi: 10.1038/374327a0. [DOI] [PubMed] [Google Scholar]
- Nagar B., Overduin M., Ikura M., Rini J.M. Structural basis of calcium-induced E-cadherin rigidification and dimerization. Nature. 1996;380:360–364. doi: 10.1038/380360a0. [DOI] [PubMed] [Google Scholar]
- Tamura K., Shan W.S., Hendrickson W.A., Colman D.R., Shapiro L. Structure-function analysis of cell adhesion by neural (N-) cadherin. Neuron. 1998;20:1152–1163. doi: 10.1016/s0896-6273(00)80496-1. [DOI] [PubMed] [Google Scholar]
- Pertz O., Bozic D., Koch A.W., Fasuer C., Brancaccio A., Engel J. A new crystal structure, Ca2+ dependence and mutational analysis reveal molecular details of E-cadherin homoassociation. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:1738–1747. doi: 10.1093/emboj/18.7.1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell G.J., Parker C.M., Cepek K.L., Mandelbrot D.A., Sood A., Mizoguchi E., Ebert E.C., Brenner M.B., Bhan A.K. Distinct structural and functional epitopes of the αEβ7 integrin. Eur . J. Immunol. 1994;24:2832–2841. doi: 10.1002/eji.1830241138. [DOI] [PubMed] [Google Scholar]
- Cepek K.L., Shaw S.K., Parker C.M., Russell G.J., Morrow J.S., Rimm D.L., Brenner M.B. Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the αEβ7 integrin. Nature. 1994;372:190–193. doi: 10.1038/372190a0. [DOI] [PubMed] [Google Scholar]
- Shimoyama Y., Yoshida T., Terada M., Shimosato Y., Abe O., Hirohashi S. Molecular cloning of a human Ca2+-dependent cell–cell adhesion molecule homologous to mouse placental cadherinits low expression in human placental tissues. J. Cell Biol. 1989;109:1787–1794. doi: 10.1083/jcb.109.4.1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell G.J., Parker C.M., Sood A., Mizogushi E., Ebert E.C., Bhan A.K., Brenner M.B. P126 (CDw101), a costimulatory molecule preferentially expressed on mucosal T lymphocytes. J. Immunol. 1996;157:3366–3374. [PubMed] [Google Scholar]
- Abitorabi M.A., Pachynski R.K., Ferrando R.E., Tidswell M., Erle D.J. Presentation of integrins on leukocyte microvillia role for the extracellular domain in determining membrane localization. J. Cell Biol. 1997;139:563–571. doi: 10.1083/jcb.139.2.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho S.N., Hunt H.D., Horton R.M., Pullen J.K., Pease L.R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- Briskin M.J., Rott L., Butcher E.C. Structural requirements for mucosal vascular addressin binding to its lymphocyte receptor α4β7. Common themes among integrin-Ig family interactions. J. Immunol. 1996;156:719–726. [PubMed] [Google Scholar]
- Watabe J., Nagafuchi A., Tsukita S., Takeichi M. Induction of polarized cell–cell association and retardation of growth by activation of the E-cadherin-catenin adhesion system in a dispersed carcinoma line. J. Cell Biol. 1994;127:247–256. doi: 10.1083/jcb.127.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomschy A., Fasuer C., Landwehr R.E.J. Homophilic adhesion of E-cadherin occurs by a co-operative two-step interaction of N-terminal domains. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:3507–3514. [PMC free article] [PubMed] [Google Scholar]
- Brieher W.M., Yap A.S., Gumbiner B.M. Lateral dimerization is required for the homophilic binding activity of C-cadherin. J. Cell Biol. 1996;135:487–496. doi: 10.1083/jcb.135.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karecla P.I., Green S.J., Bowden S.J., Coadwell J., Kilshaw P.J. Identification of the binding site for integrin αEβ7 in the N-terminal domain of E-cadherin. J. Biol. Chem. 1996;271:30909–30915. doi: 10.1074/jbc.271.48.30909. [DOI] [PubMed] [Google Scholar]
- Osborn L., Vassallo C., Browning B.G., Tizard R., Haskard D.O., Benjamin C.D., Dougas I., Kirchhausen T. Arrangement of domains, and amino acid residues required for binding of vascular cell adhesion molecule-1 to its counter-receptor VLA-4 (α4β1) J. Cell Biol. 1994;124:601–608. doi: 10.1083/jcb.124.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newham P., Craig S.E., Seddon G.N., Schofield N.R., Ress A., Edwards R.M., Jones E.Y., Humphries M.J. α4 integrin binding interfaces on VCAM-1 and MAdCAM-1. Integrin binding footprints identify accessory binding sites that play a role in integrin specificity. J. Biol. Chem. 1997;272:19429–19440. doi: 10.1074/jbc.272.31.19429. [DOI] [PubMed] [Google Scholar]
- Renz M.E., Chiu H.H., Jones S., Fox J., Kim K.J., Presta L.G., Fong S. Structural requirements for adhesion of soluble recombinant murine vascular cell adhesion molecule-1 to α4β1 . J. Cell Biol. 1994;125:1395–1406. doi: 10.1083/jcb.125.6.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher K.L., Lu J., Kim K.J., Presta L.G., Bodary S.C. Identification of the binding site in intercellular adhesion molecule 1 for its receptor, leukocyte function-associated antigen 1. Mol. Biol. Cell. 1997;8:501–515. doi: 10.1091/mbc.8.3.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Springer T.A. Structural specializations of immunoglobulin superfamily members for adhesion to integrins and viruses. Immunol. Rev. 1998;163:197–215. doi: 10.1111/j.1600-065x.1998.tb01198.x. [DOI] [PubMed] [Google Scholar]
- Leahy D.J., Aukhil I., Erickson H.P. 2.0 Å crystal structure of a four-domain segment of human fibronectin encompassing the RGD loop and synergy region. Cell. 1996;84:155–164. doi: 10.1016/s0092-8674(00)81002-8. [DOI] [PubMed] [Google Scholar]
- Wang J.H., Pepinsky R.B., Stehle T., Liu J.H., Karpusas M., Browning B., Osborn L. The crystal structure of an N-terminal two-domain fragment of vascular cell adhesion molecule 1 (VCAM-1)a cyclic peptide based on the domain 1 C-D loop can inhibit VCAM-α4 integrin interaction. Proc. Natl. Acad. Sci. USA. 1995;92:5714–5718. doi: 10.1073/pnas.92.12.5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn L., Vassallo C., Benjamin C.D. Activated endothelium binds lymphocytes through a novel binding site in the alternately spliced domain of vascular cell adhesion molecule-1. J. Exp. Med. 1992;176:99–107. doi: 10.1084/jem.176.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu H.H., Crowe D.T., Renz M.E., Presta L.G., Jones S., Weissman I.L., Fong S. Similar but nonidentical amino acid residues on vascular cell adhesion molecule-1 are involved in the interaction with α4β1 and α4β7 under different activity states. J. Immunol. 1995;155:5257–5267. [PubMed] [Google Scholar]
- Bella J., Kolatkar P.R., Marlor C.W., Greve J.M., Rossman M.G. The structure of the two amino-terminal domains of human ICAM-1 suggest how it functions as a rhinovirus receptor and as an LFA-1 integrin ligand. Proc. Natl. Acad. Sci. USA. 1998;95:4140–4145. doi: 10.1073/pnas.95.8.4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casasnovas J.M., Stehle T., Lie J.H., Wang J.H., Springer T.A. A dimeric crystal structure for the N-terminal two domains of intercellular adhesion molecule-1. Proc. Natl. Acad. Sci. USA. 1998;95:4134–4139. doi: 10.1073/pnas.95.8.4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell E.D., May A.P., Simmons D.L. The leukocyte function-associated antigen-1 (LFA-1)-binding site on ICAM-3 comprises residues on both faces of the first immunoglobulin domain. J. Immunol. 1998;161:1363–1370. [PubMed] [Google Scholar]
- Klickstein L.B., York M.R., de Fougerolles A.R., Springer T.A. Localization of the binding site of intercellular adhesion molecule-3 (ICAM-3) for lymphocyte function-associated antigen-1 (LFA-1) J. Biol. Chem. 1996;271:23920–23927. doi: 10.1074/jbc.271.39.23920. [DOI] [PubMed] [Google Scholar]
- Holness C.L., Bates P.A., Little A.J., Buckley C.D., McDowall A., Bossy D., Hogg N., Simmons D.L. Analysis of the binding site on intercellular adhesion molecule 3 for the leukocyte integrin lymphocyte function-associated antigen 1. J. Biol. Chem. 1995;270:877–884. doi: 10.1074/jbc.270.2.877. [DOI] [PubMed] [Google Scholar]
- Hynes R.O. Integrinsversatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- Blaschuk O., Sullivan R., David S., Pouliot Y. Identification of a cadherin cell adhesion recognition sequence. Dev. Biol. 1990;139:227–229. doi: 10.1016/0012-1606(90)90290-y. [DOI] [PubMed] [Google Scholar]
- Smith T.J., Ducharme L.A., Shaw S.K., Parker C.M., Brenner M.B., Kilshaw P.J., Weis J.H. Murine M290 integrin expression modulated by mast cell activation. Immunity. 1994;1:393–403. doi: 10.1016/1074-7613(94)90070-1. [DOI] [PubMed] [Google Scholar]
- Kilshaw P.J. Expression of the mucosal T cell integrin αM290β7 by a major subpopulation of dendritic cells in mice. Eur. J. Immunol. 1993;23:3365–3368. doi: 10.1002/eji.1830231246. [DOI] [PubMed] [Google Scholar]
- Kabsch W., Sandler C. Dictionary of protein secondary structurepattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]