

Abstract
We have previously shown that the obligate intracellular pathogen chlamydia can suppress interferon (IFN)-γ–inducible major histocompatibility complex (MHC) class II expression in infected cells by degrading upstream stimulation factor (USF)-1. We now report that chlamydia can also inhibit both constitutive and IFN-γ–inducible MHC class I expression in the infected cells. The inhibition of MHC class I molecule expression correlates well with degradation of RFX5, an essential downstream transcription factor required for both the constitutive and IFN-γ–inducible MHC class I expression. We further demonstrate that a lactacystin-sensitive proteasome-like activity identified in chlamydia-infected cell cytosolic fraction can degrade both USF-1 and RFX5. This proteasome-like activity is dependent on chlamydial but not host protein synthesis. Host preexisting proteasomes may not be required for the unique proteasome-like activity. These observations suggest that chlamydia-secreted factors may directly participate in the proteasome-like activity. Efforts to identify the chlamydial factors are underway. These findings provide novel information on the molecular mechanisms of chlamydial evasion of host immune recognition.
Keywords: MHC class I suppression, RFX5 degradation, IFN-γ induction, chlamydial infection, proteasomal activity
Introduction
CD8+ T cell–mediated immunity plays an important role in controlling intracellular pathogen infections 1 2, and MHC class I expression on infected cells is a prerequirement for CD8+ T cell recognition of the infected cells 3. The CD8+ T cell recognition of the infected cells may lead to both cell killing 4 that may enable the infected hosts to eliminate the intracellularly sequestrated pathogens and secretion of cytokines 5 that may be able to restrict the intracellular pathogen growth. Therefore, to avoid CD8+ T cell recognition, many intracellular pathogens such as cytomegalovirus 6, Toxoplasma gondii 7, Listeria monocytogenes 8, and Leishmania donovani 9 have evolved strategies for inhibiting MHC class I expression on the infected cells. The natural target cells of intracellular pathogens such as epithelial cells can express MHC class I molecules constitutively, and the expression level of MHC class I molecules on these cells can be significantly upregulated with IFN-γ stimulation 10. The IFN-γ induction of MHC class I expression may greatly enhance the ability of the infected cells to present the intracellular pathogen-derived peptide on cell surface for CD8+ T cell recognition 11. At transcription level, both constitutive and IFN-γ–inducible MHC class I expression require a common downstream transcription complex, RFX. The RFX complex, consisting of multiple subunits including RFX5, RFXAP, and p41 12, can directly bind to a conserved X1 regulatory element upstream of MHC class I heavy chain and β2-microglobulin (β2M) genes 13 14. Mutations in RFX5 gene abolish binding of RFX to the X1 regulatory element 15, and human cells carrying RFX5 mutations can not properly express MHC class I even in the presence of IFN-γ stimulation 16, demonstrating the importance of RFX5 in both constitutive and IFN-γ–inducible MHC class I expression. Therefore, the RFX transcription complex can be a vulnerable (or effective) target for microbial pathogens to suppress both constitutive and IFN-γ–inducible MHC class I expression. However, although many viruses have been found to evolve strategies for interrupting MHC class I expression by acting at the posttranslation level 17 18 19 20, so far there is no report on microbial interruption of MHC expression by targeting at the transcription complex RFX.
Chlamydia is an obligate intracellular pathogen of humans and animals and has adapted so successfully that it can persist in hosts for a long period of time after an acute infection 21 22. Chlamydial immune evasion may contribute to the chlamydial persistence. We have previously demonstrated that chlamydia can suppress IFN-γ–inducible MHC class II expression by degrading upstream stimulation factor (USF)-1 23, which may allow the infected cells to avoid CD4+ T cell recognition. To escape from CD8+ T cell–mediated immunity, chlamydia has been found to possess a strong antiapoptotic activity for potentially evading the killing mechanisms of CD8+ T cells 24. However, even without effective killing of the target cells, CD8+ T cell recognition of the infected cells may still lead to secretion of lymphokines that are able to restrict intracellular pathogen growth. This selection pressure may further drive intracellular pathogens to evolve strategies for preventing CD8+ T cell recognition by altering MHC class I antigen presentation 25 26 27 28. Since the intracellular chlamydia organisms can persist in a wide range of host species for a long period of time, chlamydia may have also evolved strategies for evading CD8+ T cell recognition. To test this hypothesis, we evaluated the effect of chlamydial infection on MHC class I expression with or without IFN-γ stimulation. We found that chlamydia can inhibit both constitutive and IFN-γ–inducible MHC class I expression by degrading a critical transcription factor, RFX5, that is essential for MHC class I expression. Furthermore, a chlamydia-dependent proteasome-like activity (CPA) was identified in the chlamydia-infected cell cytosolic fraction. CPA was found to be responsible for both USF-1 and RFX5 degradation. We hypothesize that a chlamydial factor(s) may directly participate in CPA, as the CPA required chlamydial but not host protein synthesis and was likely to be independent of preexisting host proteasomes.
Materials and Methods
Chlamydial Infection and IFN-γ Stimulation.
The following human cell lines were used: HeLa (cervical epithelium; American Type Culture Collection [ATCC]); HL (airway epithelium; Washington Research Foundation; reference 29); MRC-5 (fibroblast; ATCC), and WSI (fibroblast; provided by Dr. P.J. van den Elsen, Leiden University Medical Center, Leiden, The Netherlands). Cells were infected with Chlamydia trachomatis LGV2 strain at a multiplicity of infection (MOI) of 5 or as indicated and for 24–30 h or as indicated in individual experiments 23. Cells with or without infection were stimulated with human IFN-γ (PharMingen) at 200 U/ml or as indicated for another 10 h (for reverse transcriptase [RT]-PCR analysis) or 24–30 h (for flow cytometry and Western blot analysis).
Flow Cytometry.
Cell samples were stained with mouse anti–HLA-A, -B, and -C (HB94; ATCC), mouse anti–human β2M (32271A; PharMingen), or normal mouse IgG (Zymed Labs., Inc.). Primary antibody binding was detected using goat anti–mouse IgG + IgM conjugated with FITC (Caltag Labs.) and analyzed with a FACSCalibur™ equipped with CELLQuest™ software (Becton Dickinson). Dead cells were excluded by propidium iodide staining.
Western Blot Assay.
Western blot assay was carried out as we previously described 23 24. Rabbit antibodies were used to detect RFX5 (Rockland Immunochemicals), USF-1 (SC-229; Santa Cruz Biotechnology, Inc.), USF-2 (SC-862; Santa Cruz), and 11S regulatory subunit PA28α (PW8185; AFFINITI Research Products Ltd.). Mouse antibodies were used to detect HLA-A and -B (HB296; ATCC), β2M (HB149; ATCC), β1 integrin (provided by Dr. J. Wilkins, University of Manitoba, Manitoba, Canada), 20S proteasome α subunit HC2 (MCP20; AFFINITI), 20S proteasome α subunit HC3 (MCP21; AFFINITI), 20S proteasome subunits α1, 2, 3, 5, 6, and 7 (MCP231; AFFINITI), 20S proteasome subunit β7 (MCP205; AFFINITI), and a chlamydial major outer membrane protein (MOMP; clone MC22; our unpublished data). Primary antibody binding was detected with horseradish peroxidase–conjugated goat anti–mouse IgG or –rabbit IgG, depending on the source of the primary antibodies, and visualized using an ECL kit (Amersham).
RT-PCR Assay.
Cell samples were collected for RNA extraction using the Rneasy™ Mini Kit from QIAGEN Inc. 2 μg of total RNA was used for each cDNA synthesis with random primers and the 1st Strand cDNA synthesis kit from Boehringer Mannheim. Aliquots of the cDNA samples were used as template for amplifying specific gene fragments by PCR reactions 23. The primers used for gene amplification were: for β2M amplification, 5′-TCTCGCTCCGTGGCCTTAG (forward) and 5′-ATGTCTCGATCCCACTTAACT (reverse); for HLA class I heavy chain amplification, 5′-GTGGGCTAGGTGGACGAC (forward) and 5′-TTCTCCAGGTATCTGCGG (reverse); for USF-1 amplification, 5′-TGGCACTGGTCAATTCTTTGTG (forward) and 5′-GTTGCTGTCATTCTTGATGAC (reverse); for RFX5, 5′-TCCTTCAGTTCCATCGTTGAG (forward) and 5′-TTCAGCTGTCCTCTTGACACC (reverse); and for β-actin, 5′-GTGGGGCGCCCCAGGCACCA (forward) and 5′-CTCCTTAATGTCACGCACGATTTC (reverse). β-actin mRNA detection was used as an internal control for the amount of cDNA synthesized. To ensure the specificity of the mRNA detection, all primers were designed to cover at least two exons, and parallel samples without RT were run as negative controls. The amplified DNA products were run on an agarose gel and visualized with ethidium bromide staining.
Immunoprecipitation.
Immunoprecipitation was carried out as described elsewhere 30 with some modifications. For antibody depletion experiments, cell samples were dounced to make the cytosolic fraction S100 as we previously described 24, and the proteasome complexes in S100 were precipitated with the mAb MCP21 (specific to 20S proteasome α subunit HC3; reference 31) previously bound to protein G–Sepharose beads. The S100 supernatants after depletion of the proteasome complexes were compared with the S100 without prior antibody depletion for their ability to degrade RFX5 in a cell-free assay (see below). For two-dimensional PAGE analysis, cells with or without chlamydial infection were continuously labeled with S35–methionine/cysteine (ICN) for 24 h, and the radiolabeled cells were lysed with a RIP buffer containing 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM PMSF, 1 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 μg/ml pepstatin A 24. The lysates were precipitated with the MCP21 antibody, and the precipitated pellets were run on two-dimensional PAGE. The first dimension was nonequilibrium pH gradient electrophoresis toward the cathode for 3 h at 400 V. The second dimension run was in Laemmli SDS-PAGE gels with 12% polyacrylamide. The gels were then fixed and subjected to autoradiography. For pulse–chase labeling experiments, cells with or without chlamydial infection were pulsed with S35–methionine/cysteine for 30 min, and the pulsed cells were washed, detached, and aliquoted into four portions. One portion was immediately lysed in RIP buffer as the zero chase time point sample. The other three portions were chased in normal growth medium at 37°C for an additional 30 min, 2 or 6 h before they were lysed. The cell lysates were precipitated with an anti–HLA-A, -B, and -C (HB94; ATCC) and anti-β2M (HB149; ATCC) antibody, respectively. Portions of the anti–HLA-A, -B, and -C antibody–precipitated pellets were treated with endoglycosidase H (EndoH; catalog no. 100119; Boehringer Mannheim) in 60 mM sodium acetate, pH 5.4, as previously described 30. The precipitated pellets with or without the EndoH treatment were run on SDS-PAGE gels for autoradiography analysis.
Cell-free Proteolysis Assays.
The S100 fractions made from chlamydia-infected cells were used as the source of enzyme and were incubated with cellular extracts containing target proteins for 1 h at 37°C. The incubated mixtures were then analyzed for specific target protein degradation on Western blot. For inhibition experiments, inhibitors were added to the enzyme preparations before mixing with the target proteins. Inhibitors used in this study include: lactacystin (catalog no. 426102; Calbiochem Corp.), MG115 (carbobenzoxy-l-leucyl-l-leucyl-l-norvalinal; catalog no. 474780; Calbiochem), MG-132 (carbobenzoxy-l-leucyl-l-leucyl-l-leucinal; catalog no. 474790; Calbiochem), PSI (Z-Ile-Glu(OBu)-Ala-Leu-H [aldehyde]; Peptide Institute Inc.), AAF (Ala-Ala-Phe chloromethylketone; Sigma Chemical Co.), PMSF (Sigma), pepstatin A (Sigma), and aprotinin (Sigma).
Column Chromatography.
The S100 fractions from chlamydia-infected cells were applied to a Mono Q-anion exchange or a Superdex 200 size exclusion column (Pharmacia Biotech Inc.). A linear gradient from 0 to 1 M NaCl of eluent B was used for anion exchange, and a phosphate buffer, pH 7.4, was used for size exclusion chromatography with an AKTA purifier 10 instrument (Pharmacia Biotech Inc.). Fractions were assayed for degradation of the nuclear transcription factor RFX5 and for the presence of host cell proteasome subunits.
Results
Both Constitutive and IFN-γ–inducible MHC Class I Expressions Are Inhibited in Chlamydia-infected Cells.
To test our hypothesis that chlamydia may suppress MHC class I expression, we evaluated both constitutive and IFN-γ–inducible MHC class I expression in chlamydia-infected cells. A flow cytometry approach was first used to compare the surface expression of MHC class I in normal HeLa and chlamydia-infected HeLa cells with or without IFN-γ stimulation (Fig. 1 A). Normal HeLa cells expressed a basal level of MHC class I heavy chain (HLA-A, -B, and -C) and β2M, and the expression of these molecules was significantly upregulated by IFN-γ stimulation. However, HeLa cells infected with chlamydia displayed remarkably reduced levels of both HLA-A, B, and -C and β2M regardless of IFN-γ stimulation. These observations suggest that chlamydial infection completely blocked the IFN-γ induction of MHC class I molecules and also reduced the constitutive expression level.
Figure 1.
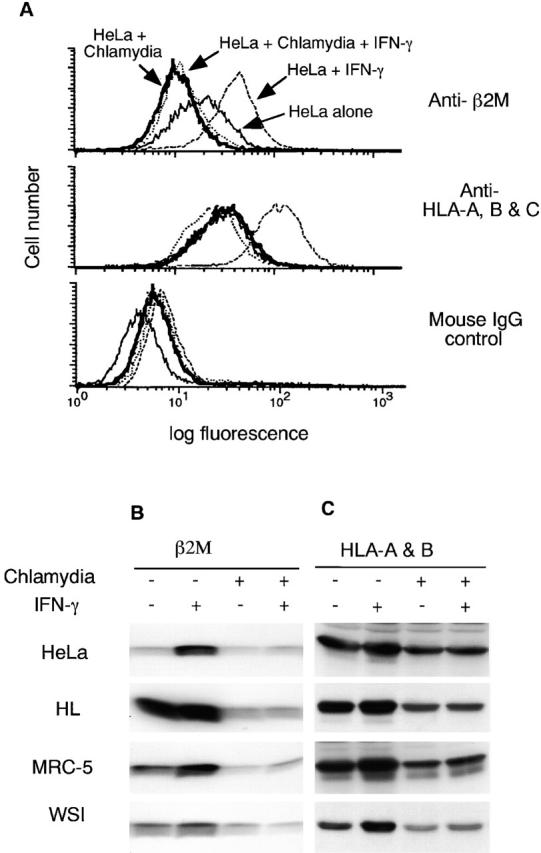


Chlamydia inhibits both constitutive and IFN-γ–inducible MHC class I expression in infected cells. HeLa cells with or without chlamydial infection were stimulated with IFN-γ or unstimulated and collected for flow cytometry (A), Western blot (B and C), and RT-PCR (E) analysis. Chlamydia prevents both constitutive and IFN-γ–inducible HLA-A, -B, and -C heavy chain and β2M surface expression (A). Chlamydia suppresses the total cellular protein level of both constitutive and IFN-γ–inducible β2M (B) and HLA-A and -B heavy chains (C) in various human cell lines. Chlamydia inhibits the mRNA expression of both MHC class I heavy chains and β2M (E). For pulse–chase labeling experiment (D), HL cells with or without chlamydial infection were metabolically labeled with S35–methionine/cysteine for 30 min, and the pulsed cell samples were aliquoted and chased for various times as indicated. Mature or immature bands correspond to proteins resistant or sensitive to EndoH digestion as determined in a separate experiment (data not shown).
The chlamydial suppression of MHC class I expression was further evaluated in multiple cell lines on Western blot (Fig. 1B and Fig. C). All four human cell lines tested (two epithelial cells, HeLa and HL, and two fibroblast cells, MRC-5 and WSI) displayed a basal level of MHC class I heavy chain (HLA-A and -B; Fig. 1 C) and β2M molecule (Fig. 1 B). The basal level was significantly reduced after chlamydial infection, suggesting that chlamydial infection can suppress the constitutive expression of MHC class I molecules. Although IFN-γ stimulation greatly upregulated the MHC class I heavy chain and β2M expression in all four cell lines, chlamydial infection completely inhibited the IFN-γ induction. These results confirm that chlamydial infection can inhibit both constitutive and IFN-γ–inducible expression of MHC class I molecules. The fact that total cellular level of MHC class I was detected in the Western blot assay suggests that the chlamydia-suppressed cell surface expression of MHC class I molecules shown in Fig. 1 A may not be due to an alternation of MHC class I intracellular trafficking. To further determine whether chlamydial infection affects intracellular trafficking of MHC class I molecules, we compared the maturation process of both MHC class I heavy chains and β2M molecule in mock- and chlamydia-infected cells (Fig. 1 D). Although the overall levels of class I heavy chains and β2M molecules in the chlamydia-infected cells was lower than that in mock-infected cells, the maturation rate (comparing samples chased up to 2 h) and half-life (comparing 6 h–chased samples) of class I heavy chains and β2M molecule were similar between the mock- and chlamydia-infected cells. The MHC class I heavy chains gained partial and full resistance to EndoH digestion at 30 min and 2 h after pulse, respectively, in both mock- and chlamydia-infected cells (data not shown). This observation is consistent with a previous finding that chlamydial infection did not affect the trafficking of host cell glycoproteins including MHC class I heavy chains 32. As chlamydial infection did not alter MHC class I heavy chain and β2M intracellular trafficking but significantly reduced the total protein level, we hypothesize that chlamydia may inhibit MHC class I expression at either the transcription or translation level.
We evaluated the mRNA levels of both the MHC class I heavy chain and β2M in cells with or without chlamydial infection and with or without IFN-γ stimulation using a semiquantitative RT-PCR assay (Fig. 1 E). Chlamydial infection significantly inhibited both MHC class I heavy chain and β2M mRNA expression, especially that induced by IFN-γ, which suggests that the chlamydial suppression of MHC class I occurred at the transcription level. We next evaluated the effects of chlamydial infection on transcription factors required for MHC class I expression.
Chlamydial Inhibition of MHC Class I Expression Correlates with Loss of Host Transcription Factor RFX5 in Chlamydia-infected Cells.
It has been demonstrated that RFX5 is an essential component of transcription complex RFX required for both constitutive and IFN-γ–inducible MHC class I expression 16. We measured the level of RFX5 protein in chlamydia-infected cells (Fig. 2 A). HeLa cells displayed normal levels of RFX5, HLA-A and -B heavy chains, β2M, and β1 integrin molecules, and the expression of HLA-A and -B heavy chains and β2M was further upregulated upon IFN-γ stimulation. However, HeLa cells infected with chlamydia regardless of IFN-γ stimulation showed a significant reduction in HLA-A and -B and β2M and complete loss of RFX5. The level of β1 integrin (as a control membrane protein) in chlamydia-infected cells was largely unchanged. The chlamydial MOMP was only detected in chlamydia-infected cells. These observations demonstrated a correlation between the inhibition of MHC class I expression and the loss of the transcription factor RFX5 in chlamydia-infected cells.
Figure 2.

Inhibition of MHC class I expression correlates with RFX5 degradation. (A) HeLa cells with or without chlamydial infection and with or without IFN-γ stimulation were analyzed on Western blot for the levels of RFX5, HLA-A and -B heavy chains, β2M, β1 integrin (as a control membrane protein), and a chlamydial MOMP. Chlamydia-infected cell samples showed reduced levels of HLA heavy chain and β2M and complete loss of RFX5. (B) Cell-free assay analysis of HLA-A and -B heavy chain, β2M, and RFX5 degradation. Cytosolic S100 fractions from either normal HeLa (HeLa S100) or chlamydia-infected HeLa cells (chlamydia S100) were incubated with the corresponding target proteins (either a membrane protein extract as the source of HLA-A and -B heavy chains and β2M or a nuclear protein extract as the source of RFX5). The incubated mixtures were analyzed on Western blot for the levels of the residual target proteins with the corresponding antibodies. Chlamydia S100 selectively degrades RFX5 but not HLA-A and -B heavy chains and β2M.
We next tried to understand how RFX5 was completely lost in chlamydia-infected cells. RFX5 mRNA levels in normal and chlamydia-infected HeLa cells were compared using RT-PCR, and no difference was found (data not shown), suggesting that RFX5 mRNA was normally transcripted in chlamydia-infected cells and that the loss of RFX5 protein may occur at or downstream of translation level. We have previously identified a CPA in chlamydia-infected cells. This CPA selectively degraded transcription factor USF-1, and the CPA-mediated degradation was sensitive to lactacystin inhibition 23. We tested whether the lactacystin-sensitive CPA could also degrade RFX5 (Fig. 2 B). In this experiment, a nuclear protein extract was used as the source of RFX5. To exclude the possibility of direct degradation of MHC class I molecules, the MHC class I molecules were also used as degradation targets in the same assay. Since we have determined that a cytosolic soluble protein fraction after 100,000 g centrifugation (designated as S100) contains the highest CPA (data not shown), the S100 from chlamydia-infected HeLa cells (chlamydia S100) was used as the source of the CPA, and the S100 from normal HeLa cells (HeLa S100) was used as control. Neither chlamydia S100 nor HeLa S100 alone showed any significant amounts of target proteins HLA-A and -B, β2M (both membrane proteins), or RFX5 (nuclear protein), suggesting that the S100 fractions used in this experiment had minimal contamination with proteins from other cellular compartments. When HeLa S100 was incubated with the target proteins, all target proteins detected were still intact (Fig. 2 B). However, incubation with chlamydia S100 completely degraded RFX5 but not HLA-A and -B and β2M. Furthermore, RFX5 degradation was inhibited by lactacystin. These observations demonstrated that the lactacystin-sensitive CPA in chlamydia S100 selectively degraded the transcription factor RFX5, which further strengthens a causal relationship between chlamydia-induced degradation of RFX5 and chlamydial suppression of MHC class I expression. Combining this data with our previous observation on chlamydial suppression of IFN-γ–inducible MHC class II expression 23, we conclude that the same CPA was responsible for the degradation of both RFX5 and USF-1. The next step was to further characterize the CPA.
Degradation of RFX5 Is Dissociated from Host Cell Proteasomes.
Various inhibitors known to be able to inhibit different eukaryotic proteases and proteasomes were used for characterizing the CPA by measuring RFX5 degradation in a cell-free assay (Fig. 3 A). Chlamydia S100 completely degraded RFX5, whereas HeLa S100 failed to do so. The RFX5 degradation was inhibited by the proteasome inhibitor lactacystin at a final concentration of 20 μM. These observations confirmed that the typical CPA was responsible for the RFX5 degradation in this assay. Interestingly, the CPA-mediated RFX5 degradation was not inhibitable by other proteasome inhibitors, including PSI, MG115, and MG132 (carbobenzoxy-l-leucyl-l-leucyl-l-leucinal), suggesting that the CPA is different from the conventional 20S or 26S proteasome activity. It was recently demonstrated that a tripeptidyl peptidase II (TPP2) and some other AAF-inhibitable proteases can substitute the function of the conventional proteasomes in stressed cells 33 34. We evaluated the role of TPP2 in RFX5 degradation. The potent TPP2 inhibitor AAF, even at a final concentration of 100 μM, failed to block the RFX5 degradation, suggesting that AAF-inhibitable protease activities including TPP2 were not involved in the CPA. We also evaluated the effects of many other protease inhibitors such as phenylmethylsulfonyl fluoride, pepstatin A, and aprotinin on the CPA, and no inhibition was found with any of these inhibitors (data not shown). Together, these observations suggest that the CPA represents a distinct proteasome-like activity.
Figure 3.




Degradation of RFX-5 is dissociated from host proteasomes. (A) Inhibition of RFX5 degradation by proteasome inhibitors in a cell-free assay. Chlamydia S100 or HeLa S100 cells were incubated with a nuclear protein extract (as a source of RFX5) in the presence or absence of various inhibitors (proteasome inhibitors: lactacystin, PSI, MG115, and MG132; TPP2 inhibitor: AAF). The incubated mixtures were then analyzed on Western blot for residual RFX5 levels. ns denotes nonspecific binding. (B) Comparison of 20S proteasome subunit distribution patterns between normal and chlamydia-infected HeLa cell samples. HeLa cells with or without chlamydial infection were metabolically labeled with S35–methionine/cysteine for 24 h, and the cell lysates were precipitated with antibody MCP21 (recognizing one of the 20S proteasome subunits). The precipitates were subjected to a two-dimensional PAGE, and the protein patterns were visualized by autoradiography. The 20S proteasome subunits were numbered based on the description in reference 31. (C) Ion exchange column separation of RFX5 cleavage activity from host proteasome subunits. Chlamydia S100 was applied to a Mono Q-anion exchange column, which was eluted with a linear gradient of 0–1 M NaCl, and the eluent protein concentration was monitored using 280 nm absorbence. The eluted fractions with even numbers were measured for RFX5 cleavage in a cell-free assay and detected for the presence of host proteins, including various α subunits, β7 subunit of the 20S proteasome, and 11S regulatory PA28α on Western blot. (D) Size exclusion column separation of RFX5 cleavage activity from host proteasome subunits. Chlamydia S100 was applied to a Superdex 200 size exclusion column, which was eluted with PBS. The eluted fractions were measured as described in the legend for panel C. (E) Effect of 20S proteasome depletion on RFX5 cleavage activity. HeLa or chlamydia S100 was precipitated with antibody MCP21 to deplete the 20S proteasome complexes. The supernatants after depletion were compared for RFX5 cleavage activity with the intact S100 (S100 without prior antibody depletion). The RFX5 cleavage activity was measured in a cell-free assay, and the residual RFX5 was detected on Western blot (top panel). The efficiency of the 20S proteasome depletion was evaluated by monitoring residual 20S proteasome subunits in the S100 with prior antibody depletion on Western blot (bottom panel). Although both heavy and light chains of the antibody MCP21 were detected, the proteasome subunit HC3 was no longer detectable in the depleted supernatant.
We next compared the 20S proteasome precipitation patterns between normal HeLa and chlamydia-infected HeLa cells (Fig. 3 B). The two-dimensional gel distribution pattern of the radioisotope-labeled 20S proteasome subunits precipitated from the chlamydia-infected HeLa cells was similar to that from normal HeLa cells, suggesting that chlamydial infection did not significantly alter the 20S proteasome subunit composition. It is, therefore, unlikely that the CPA is caused by chlamydia-induced modification of host proteasomes.
A column chromatography approach was used to further differentiate the CPA (measured using the RFX5 degradation assay) from the normal host cell proteasomes (Fig. 3c and Fig. d). The chlamydia S100 was subjected to an ion exchange (Fig. 3 C) or size exclusion (Fig. 3 D) column separation, and the separated fractions were measured for both the CPA and the presence of host proteasome components. Maximal CPA was found in fraction 12 of the ion exchange column, while most of the detected proteasome subunits 31 35 existed in fraction 14. Similarly, there was no obvious overlapping between the CPA and any of the detected 20S proteasome subunits in fractions from the size exclusion column (Fig. 3 D). These observations suggest that host proteasomes may not be required for the CPA. However, there are still some overlaps between the host proteasomes and the CPA in both column separation assays, although at a minimal level, which may suggest a role of the overlapping minority of the host proteasomes in the CPA. The potential contribution of host proteasomes to the CPA was further evaluated using the following antibody depletion experiment (Fig. 3 E). Either HeLa S100 or chlamydia S100 was subjected to precipitation with the mAb MCP21, and the supernatants after precipitation (designated as S100-de) were compared with intact S100 for their ability to degrade RFX5 (Fig. 3 E, top panel). Chlamydia S100 completely degraded RFX5 (Fig. 3 E), and, more importantly, the chlamydia S100-de still caused efficient degradation of RFX5 (Fig. 3 E), suggesting that the 20S host proteasomes were not required for the CPA, as depletion of the 20S proteasome did not affect RFX5 degradation. The removal of host 20S proteasomes was sufficient, as the proteasome subunits were no longer detectable in S100 with prior antibody depletion (Fig. 3 E, bottom panel).
Both the Inhibition of MHC Class I Molecule Expression and the Degradation of RFX5 Are Dependent on Chlamydial but Not Host Protein Synthesis.
We have demonstrated that the degradation of RFX5 was dissociated from preexisting host proteasomes, which suggests that the CPA responsible for RFX5 degradation may be produced by newly synthesized proteins. To test whether chlamydial protein synthesis is required for the CPA, we first correlated the degradation of RFX5 with chlamydial infection dose (Fig. 4 A) and infection time course (Fig. 4 B). As MOI (ratio of number of organisms/number of host cells) increased, more chlamydial MOMP protein was produced and more RFX5 was degraded, suggesting a correlation between the amount of chlamydial protein synthesis and severity of the RFX5 degradation. The time course study also revealed a similar correlation: as infection prolonged, more chlamydial MOMP protein was synthesized and more RFX5 was degraded (Fig. 4 B). USF-1 and -2 levels were also monitored in the dose titration and time course experiments using an antibody that can bind to both USF isoforms. USF-1 was used as positive control, since we have previously demonstrated the degradation of USF-1 in chlamydia-infected cells 23. USF-2 was used as negative control, as we found that the level of USF-2 was not affected by chlamydial infection 23. Consistent with our previous observation 23, USF-1 degradation correlated with the infection severity, whereas USF-2 was maintained at a similar level in both the time course and dose titration experiments. The correlation between the chlamydial infection severity and both RFX5 and USF-1 degradation suggests that chlamydial protein synthesis may be required for the CPA. We used antibiotics specifically inhibiting either prokaryotic or eukaryotic protein synthesis to further confirm whether chlamydial or host protein synthesis is required for the CPA (Fig. 4 C). Normal host cells expressed a basal level of MHC class I heavy chains, β2M (both upregulated by IFN-γ), RFX5, and USF-1, whereas chlamydia-infected cells displayed a remarkably reduced level or even complete loss of these molecules regardless of IFN-γ stimulation (Fig. 4 C). The chlamydial MOMP was detected only in chlamydia-infected cell samples. Treatment with rifampin (inhibiting prokaryotic transcription) or chloramphenicol (inhibiting prokaryotic translation) completely inhibited chlamydial MOMP protein synthesis, prevented degradation of the transcription factors RFX5 and USF-1, and restored MHC class I expression (Fig. 4 C), suggesting that chlamydial protein synthesis is necessary for both the inhibition of MHC class I and degradation of RFX5 and USF-1. Treatment with penicillin (only inhibiting chlamydial particle assembly without affecting chlamydial protein synthesis) was unable to block chlamydial MOMP protein synthesis and failed to prevent degradation of RFX5 and USF-1 and inhibition of MHC class I expression, suggesting that chlamydial particle assembly is not required for the chlamydial effects. Finally, cycloheximide treatment (inhibiting eukaryotic protein synthesis) did not affect chlamydial protein synthesis and various chlamydia-induced effects, suggesting that host newly synthesized proteins are not required for the suppression of MHC class I and degradation of RFX5 in chlamydia-infected cells.
Figure 4.
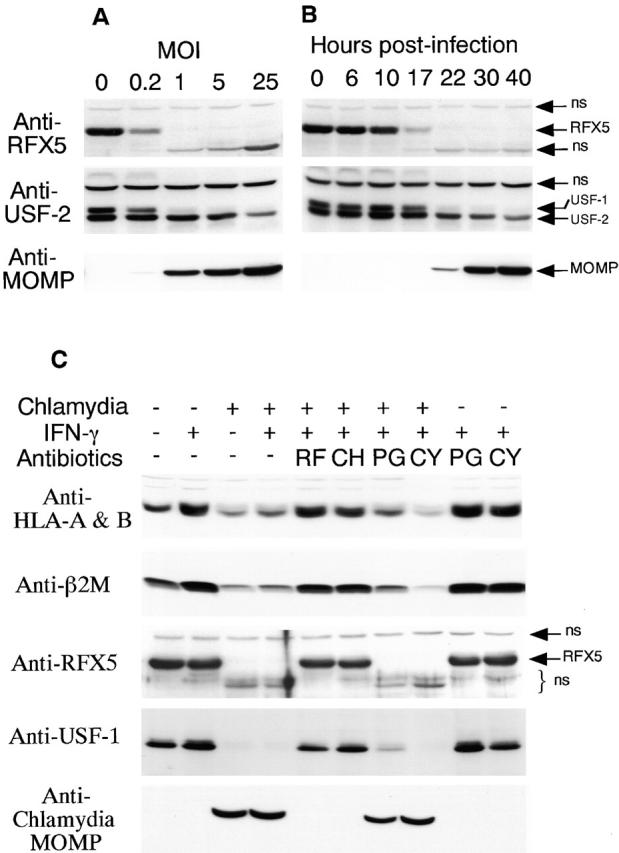
Chlamydial but not host protein synthesis is required for both the degradation of RFX5 and suppression of HLA-A and -B heavy chains and β2M. (A) Correlation between infection dose and RFX5 degradation. 30 h after chlamydial infection at various MOI, HeLa cells were analyzed for the levels of chlamydial MOMP and host RFX5, USF-1, and USF-2 proteins on Western blot. The anti–USF-2 antibody used in this experiment can bind to both USF-1 and -2. These two USF isoforms can be separated on blots if gels are run far enough. Since we have previously shown that USF-1 is degraded in chlamydia-infected cells 23, USF-1 is used as a positive control. Since USF-2 protein level is usually not altered by chlamydial infection, it serves as a negative control. ns denotes nonspecific binding. (B) Time course relationship between chlamydial growth and RFX5 degradation. At various time points after infection, HeLa cell samples were analyzed on Western blot as described in A. (C) Inhibition of chlamydial but not host protein synthesis prevents RFX5 degradation and restores HLA-A and -B heavy chain and β2M expression. Rifampin (RF; final concentration = 0.1 μg/ml), chloramphenicol (CH; 60 μg/ml) and penicillin (PG; 100 μg/ml) were added at the beginning of chlamydial infection and maintained throughout the culture. Cycloheximide (CY; 10 μg/ml) was added to the culture 10 h before IFN-γ treatment and maintained during the IFN-γ stimulation. The treated HeLa cells were analyzed for protein levels of HLA-A and -B heavy chains, β2M, RFX5, USF-1, and chlamydial MOMP on Western blot. ns denotes nonspecific binding.
Discussion
Animal hosts have evolved multiple, sometimes seemingly wasteful, defense responses for controlling microbial infections, which, in turn, select for microbial pathogens with the ability to evade multiple host defense mechanisms. We have found that, like many other successful intracellular pathogens, chlamydia has evolved varied strategies for counteracting both immune recognition and immune effector mechanisms 23 24. The chlamydial immune evasion may contribute to the success of chlamydial persistence, a major cause of chlamydia-induced diseases in humans 36. To avoid CD4+ T cell recognition, chlamydia can efficiently inhibit IFN-γ–inducible MHC class II expression by degrading transcription factor USF-1 23. USF-1 degradation may also affect MHC class I expression, as USF-1 is required for the expression of class II transactivator (CIITA) and CIITA may participate in the transcription of IFN-γ–inducible MHC class I genes 37. However, our current finding on chlamydial suppression of both constitutive and IFN-γ–inducible MHC class I expression cannot be explained by the USF-1 degradation alone. This is because (a) many USF-1/CIITA-independent pathways can also mediate IFN-γ induction of MHC class I expression (reference 10) and (b) neither CIITA nor USF-1 has been found to play any significant role in the constitutive expression of MHC class I molecules. Many epithelial cells lacking CIITA can constitutively express MHC class I molecules. Therefore, the chlamydial suppression of both constitutive and IFN-γ–inducible MHC class I expression is likely to depend on a mechanism independent of USF-1 degradation.
We have correlated the chlamydial inhibition of MHC class I expression with the degradation of RFX5, a critical downstream transcription factor essential for both constitutive and IFN-γ–inducible MHC class I expression 16. RFX5 is a key component of the transcription complex RFX 16 38. Human cells carrying RFX5 mutations cannot properly express MHC class I even in the presence of IFN-γ stimulation, and transfection of these mutant cells with plasmid encoding competent RFX5 can restore the normal expression of MHC class I molecules 16. Therefore, RFX5 degradation may represent an effective strategy for suppressing MHC class I expression by the chlamydia organisms. Interestingly, MHC class II molecule expression also requires the RFX complex 13 14. In fact, RFX complex is a shared downstream transcription complex essential for both MHC class I and class II gene transcription 14. Humans carrying RFX5 mutations develop type C bare lymphocyte syndrome because of deficiency in MHC class II expression 15. Therefore, besides the degradation of USF-1 23, the chlamydia-induced RFX5 degradation also contributes to the suppression of MHC class II expression in chlamydia-infected cells. Degradation of both RFX5 and USF-1 may allow chlamydia to ensure that both MHC class I and class II expression are efficiently suppressed in the infected cells.
Blocking the function of transcription factors required for mounting host defense responses may represent an effective immune evasion strategy used by intracellular pathogens. It has been recently shown that adenoviral E1A can directly bind to signal transducer and activator of transcription 1 (Stat1) and block Stat1-dependent gene activation 39. It has also been demonstrated that human cytomegalovirus can induce degradation of Janus tyrosine kinase (JAK)1 and block IFN-γ JAK/STAT signaling pathways 40. Here, we report that chlamydia, an obligate intracellular pathogen, has evolved the strategy for directly degrading host transcription factors USF-1 23 and RFX5.
What is the functional outcome of the chlamydia-induced suppression of MHC class I expression? This study has demonstrated that chlamydial infection caused RFX5 degradation 17 h after infection (Fig. 4 B) and induced a significant suppression of cell surface MHC class I molecules 24 h or later after infection (Fig. 1 A). Therefore, at an early stage during chlamydial infection, chlamydial early proteins 41 and even structural proteins 42 can be processed and presented by host MHC molecules, which may allow antigen-specific T cells to recognize and attack the infected cells. This scenario may provide an explanation for some in vitro observations that CD8+ T cells were able to lyse chlamydia-infected target cells 43 44. However, once the chlamydial antiapoptotic activity 24 and chlamydia-induced suppression of MHC class I molecules are developed, the chlamydia-specific T cells may not be able to detect and attack the infected cells anymore, which is consistent with the observations that chlamydia-immunized CTLs failed to lyse chlamydia-infected cells in some cases 45 46. More importantly, the chlamydial suppression of MHC class I may play a significant role in facilitating chlamydial survival during natural infection. It has been shown that chlamydia-infected cells can only release a significant amount of inflammatory cytokines 24 h after infection 47, suggesting that the circulating chlamydia-specific T cells are most effectively recruited to the infection sites only after the middle cycle of chlamydial growth (24 h after infection). Therefore, the chlamydial suppression of MHC class I expression, occurring at a time when host T cell attack on the chlamydia-infected cells is most likely to happen, may be beneficial for chlamydial survival in the infected hosts. This hypothesis is further supported by various previous findings that mice deficient in β2M 48 or perforin 49 did not show enhanced susceptibility to chlamydial infection.
We have demonstrated that a CPA identified in chlamydia-infected cell cytosolic fraction is responsible for the degradation of both USF-1 and RFX5. The CPA is dependent on chlamydial but not host protein synthesis. Although we have presented considerable evidence showing that preexisting host 20S proteasomes may not be required for the CPA, we cannot completely exclude the involvement of host proteasomes in CPA. This is because there is a possibility that only small amounts of proteasome subunits, after the chlamydia-dependent modification, may be sufficient for participating in the observed CPA. Indeed, a minimal overlap was observed between the host proteasomes and the CPA in the column separation experiments (Fig. 3d and Fig. e). It is also possible that the chlamydia-secreted factor(s) may be responsible for specifically binding to certain host transcription factors such as RFX5 and targeting the bound molecules to host proteasomes for degradation. This scenario may explain both the substrate specificity of CPA and sensitivity of CPA to lactacystin inhibition.
Regardless of the precise mechanisms involved in the CPA, the fact that we were able to measure the CPA by detecting RFX5 degradation using a cell-free assay in fractions separated by various types of columns should lead us to purify and identify the CPA factors encoded by either chlamydia or host alone or both. On a size exclusion column, a CPA was separated in the fractions following those of 20S proteasomes (Fig. 3 D), suggesting that the factor(s) is smaller or forms a smaller complex than the 20S proteasomes. On an anion exchange column, a CPA was eluted before various 20S proteasome subunits but after an 11S regulatory PA28α subunit (Fig. 3 C), suggesting that the factor(s) has a pI higher than 20S proteasomes but lower than the 11S regulatory subunit. If a multicomponent or multistep mechanism is involved in the CPA and the multiple components can be separated into different fractions as the purification proceeds, a mix and match approach will be employed to define the essential factors required for the CPA.
Acknowledgments
This work was supported by the Medical Research Council (MRC) of Canada. G. Zhong was the recipient of an MRC scholarship award when this work was carried out.
Footnotes
Abbreviations used in this paper: β2M, β2-microglobulin; CPA, chlamydia-dependent proteasome-like activity; CIITA, class II transactivator; EndoH, endoglycosidase H; MOI, multiplicity of infection; MOMP, major outer membrane protein; RT, reverse transcriptase; STAT, signal transducer and activator of transcription; TPP2, tripeptidyl peptidase II; USF, upstream stimulation factor.
References
- Harty J.T., Bevan M.J. Responses of CD8(+) T cells to intracellular bacteria. Curr. Opin. Immunol. 1999;11:89–93. doi: 10.1016/s0952-7915(99)80016-8. [DOI] [PubMed] [Google Scholar]
- Bouwer H.G., Barry R.A., Hinrichs D.J. Acquired immunity to an intracellular pathogenimmunologic recognition of L. monocytogenes-infected cells. Immunol. Rev. 1997;158:137–146. doi: 10.1111/j.1600-065x.1997.tb01000.x. [DOI] [PubMed] [Google Scholar]
- Germain R.N. MHC-dependent antigen processing and peptide presentationproviding ligands for T lymphocyte activation. Cell. 1994;76:287–299. doi: 10.1016/0092-8674(94)90336-0. [DOI] [PubMed] [Google Scholar]
- Moretta A. Molecular mechanisms in cell-mediated cytotoxicity. Cell. 1997;90:13–18. doi: 10.1016/s0092-8674(00)80309-8. [DOI] [PubMed] [Google Scholar]
- Tascon R.E., Stavropoulos E., Lukacs K.V., Colston M.J. Protection against Mycobacterium tuberculosis infection by CD8+ T cells requires the production of gamma interferon. Infect. Immun. 1998;66:830–834. doi: 10.1128/iai.66.2.830-834.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones T.R., Hanson L.K., Sun L., Slater J.S., Stenberg R.M., Campbell A.E. Multiple independent loci within the human cytomegalovirus unique short region down-regulate expression of major histocompatibility complex class I heavy chains. J. Virol. 1995;69:4830–4841. doi: 10.1128/jvi.69.8.4830-4841.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luder C.G., Lang T., Beuerle B., Gross U. Down-regulation of MHC class II molecules and inability to up-regulate class I molecules in murine macrophages after infection with Toxoplasma gondii . Clin. Exp. Immunol. 1998;112:308–316. doi: 10.1046/j.1365-2249.1998.00594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller S., Kugler S., Goebel W. Suppression of major histocompatibility complex class I and class II gene expression in Listeria monocytogenes-infected murine macrophages. FEMS Immunol. Med. Microbiol. 1998;20:289–299. doi: 10.1111/j.1574-695X.1998.tb01139.x. [DOI] [PubMed] [Google Scholar]
- Reiner N.E., Ng W., McMaster W.R. Parasite-accessory cell interactions in murine leishmaniasis. II. Leishmania donovani suppresses macrophage expression of class I and class II major histocompatibility complex gene products. J. Immunol. 1987;138:1926–1932. [PubMed] [Google Scholar]
- Gobin S.J., Peijnenburg A., Keijsers V., van den Elsen P.J. Site alpha is crucial for two routes of IFN gamma-induced MHC class I transactivationthe ISRE-mediated route and a novel pathway involving CIITA. Immunity. 1997;6:601–611. doi: 10.1016/s1074-7613(00)80348-9. [DOI] [PubMed] [Google Scholar]
- Gaczynska M., Rock K.L., Goldberg A.L. Gamma-interferon and expression of MHC genes regulate peptide hydrolysis by proteasomes. Nature. 1993;365:264–267. doi: 10.1038/365264a0. [DOI] [PubMed] [Google Scholar]
- Masternak K., Barras E., Zufferey M., Conrad B., Corthals G., Aebersold R., Sanchez J.C., Hochstrasser D.F., Mach B., Reith W. A gene encoding a novel RFX-associated transactivator is mutated in the majority of MHC class II deficiency patients. Nat. Genet. 1998;20:273–277. doi: 10.1038/3081. [DOI] [PubMed] [Google Scholar]
- van den Elsen P.J., Gobin S.J.P., van Eggermond M.C., Peijnenburg A. Regulation of MHC class I and II gene transcriptiondifferences and similarities. Immunogenetics. 1998;48:208–221. doi: 10.1007/s002510050425. [DOI] [PubMed] [Google Scholar]
- van den Elsen P.J., Peijnenburg A., van Eggermond M.C., Gobin S.J. Shared regulatory elements in the promoters of MHC class I and class II genes. Immunol. Today. 1998;19:308–312. doi: 10.1016/s0167-5699(98)01287-0. [DOI] [PubMed] [Google Scholar]
- Steimle V., Durand B., Barras E., Zufferey M., Hadam M.R., Mach B., Reith W. A novel DNA-binding regulatory factor is mutated in primary MHC class II deficiency (bare lymphocyte syndrome) Genes Dev. 1995;9:1021–1032. doi: 10.1101/gad.9.9.1021. [DOI] [PubMed] [Google Scholar]
- Gobin S.J., Peijnenburg A., van Eggermond M., van Zutphen M., van den Berg R., van den Elsen P.J. The RFX complex is crucial for the constitutive and CIITA-mediated transactivation of MHC class I and beta2-microglobulin genes. Immunity. 1998;9:531–541. doi: 10.1016/s1074-7613(00)80636-6. [DOI] [PubMed] [Google Scholar]
- Lehner P.J., Karttunen J.T., Wilkinson G.W., Cresswell P. The human cytomegalovirus US6 glycoprotein inhibits transporter associated with antigen processing-dependent peptide translocation. Proc. Natl. Acad. Sci. USA. 1997;94:6904–6909. doi: 10.1073/pnas.94.13.6904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengel H., Flohr T., Hammerling G.J., Koszinowski U.H., Momburg F. Human cytomegalovirus inhibits peptide translocation into the endoplasmic reticulum for MHC class I assembly. J. Gen. Virol. 1996;77:2287–2296. doi: 10.1099/0022-1317-77-9-2287. [DOI] [PubMed] [Google Scholar]
- Fruh K., Ahn K., Peterson P.A. Inhibition of MHC class I antigen presentation by viral proteins. J. Mol. Med. 1997;75:18–27. doi: 10.1007/s001090050082. [DOI] [PubMed] [Google Scholar]
- Hengel H., Reusch U., Gutermann A., Ziegler H., Jonjic S., Lucin P., Koszinowski U.H. Cytomegaloviral control of MHC class I function in the mouse. Immunol. Rev. 1999;168:167–176. doi: 10.1111/j.1600-065x.1999.tb01291.x. [DOI] [PubMed] [Google Scholar]
- Patton D.L., Askienazy-Elbhar M., Henry-Suchet J., Campbell L.A., Cappuccio A., Tannous W., Wang S.P., Kuo C.C. Detection of Chlamydia trachomatis in fallopian tube tissue in women with postinfectious tubal infertility. Am. J. Obstet. Gynecol. 1994;171:95–101. doi: 10.1016/s0002-9378(94)70084-2. [DOI] [PubMed] [Google Scholar]
- Cappuccio A.L., Patton D.L., Kuo C.C., Campbell L.A. Detection of Chlamydia trachomatis deoxyribonucleic acid in monkey models (Macaca nemestrina) of salpingitis by in situ hybridizationimplications for pathogenesis. Am. J. Obstet. Gynecol. 1994;171:102–110. doi: 10.1016/s0002-9378(94)70085-0. [DOI] [PubMed] [Google Scholar]
- Zhong G., Fan T., Liu L. Chlamydia inhibits interferon γ–inducible major histocompatibility complex class II expression by degradation of upstream stimulatory factor 1. J. Exp. Med. 1999;189:1931–1938. doi: 10.1084/jem.189.12.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan T., Lu H., Hu H., Shi L., McClarty G.A., Nance D.M., Greenberg A.H., Zhong G. Inhibition of apoptosis in chlamydia-infected cellsblockade of mitochondrial cytochrome c release and caspase activation. J. Exp. Med. 1998;187:487–496. doi: 10.1084/jem.187.4.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossendorp F., Eggers M., Neisig A., Ruppert T., Groettrup M., Sijts A., Mengede E., Kloetzel P.M., Neefjes J., Koszinowski U. A single residue exchange within a viral CTL epitope alters proteasome-mediated degradation resulting in lack of antigen presentation. Immunity. 1996;5:115–124. doi: 10.1016/s1074-7613(00)80488-4. [DOI] [PubMed] [Google Scholar]
- Bertoletti A., Sette A., Chisari F.V., Penna A., Levrero M., De Carli M., Fiaccadori F., Ferrari C. Natural variants of cytotoxic epitopes are T-cell receptor antagonists for antiviral cytotoxic T cells Nature. 369 1994. 407 410[see comments] [DOI] [PubMed] [Google Scholar]
- Wiertz E.J., Jones T.R., Sun L., Bogyo M., Geuze H.J., Ploegh H.L. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell. 1996;84:769–779. doi: 10.1016/s0092-8674(00)81054-5. [DOI] [PubMed] [Google Scholar]
- Ahn K., Gruhler A., Galocha B., Jones T.R., Wiertz E.J., Ploegh H.L., Peterson P.A., Yang Y., Fruh K. The ER-luminal domain of the HCMV glycoprotein US6 inhibits peptide translocation by TAP. Immunity. 1997;6:613–621. doi: 10.1016/s1074-7613(00)80349-0. [DOI] [PubMed] [Google Scholar]
- Cles L.D., Stamm W.E. Use of HL cells for improved isolation and passage of Chlamydia pneumoniae . J. Clin. Microbiol. 1990;28:938–940. doi: 10.1128/jcm.28.5.938-940.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong G., Castellino F., Romagnoli P., Germain R.N. Evidence that binding site occupancy is necessary and sufficient for effective major histocompatibility complex (MHC) class II transport through the secretory pathway redefines the primary function of class II–associated invariant chain peptides (CLIP) J. Exp. Med. 1996;184:2061–2066. doi: 10.1084/jem.184.5.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendil K.B., Kristensen P., Uerkvitz W. Human proteasomes analysed with monoclonal antibodies. Biochem. J. 1995;305:245–252. doi: 10.1042/bj3050245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scidmore M.A, Fischer E.R., Hackstadt T. Sphingolipids and glycoproteins are differentially trafficked to the Chlamydia trachomatis inclusion. J. Cell Biol. 1996;134:363–374. doi: 10.1083/jcb.134.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glas R., Bogyo M., McMaster J.S., Gaczynska M., Ploegh H.L. A proteolytic system that compensates for loss of proteasome function. Nature. 1998;392:618–622. doi: 10.1038/33443. [DOI] [PubMed] [Google Scholar]
- Geier E., Pfeifer G., Wilm M., Lucchiari-Hartz M., Baumeister W., Eichmann K., Niedermann G. A giant protease with potential to substitute for some functions of the proteasome. Science. 1999;283:978–981. doi: 10.1126/science.283.5404.978. [DOI] [PubMed] [Google Scholar]
- Hendil K.B., Khan S., Tanaka K. Simultaneous binding of PA28 and PA700 activators to 20 S proteasomes. Biochem. J. 1998;332:749–754. doi: 10.1042/bj3320749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward E.M. Mechanisms of chlamydia-induced diseases. In: Stephens R.S., editor. ChlamydiaIntracellular Biology, Pathogenesis, and Immunity. ASM Press; Washington, DC: 1999. pp. 171–210. [Google Scholar]
- Martin B.K., Chin K.C., Olsen J.C., Skinner C.A., Dey A., Ozato K., Ting J.P. Induction of MHC class I expression by the MHC class II transactivator CIITA. Immunity. 1997;6:591–600. doi: 10.1016/s1074-7613(00)80347-7. [DOI] [PubMed] [Google Scholar]
- Moreno C.S., Rogers E.M., Brown J.A., Boss J.M. Regulatory factor X, a bare lymphocyte syndrome transcription factor, is a multimeric phosphoprotein complex. J. Immunol. 1997;158:5841–5848. [PubMed] [Google Scholar]
- Look D.C., Roswit W.T., Frick A.G., Gris-Alevy Y., Dickhaus D.M., Walter M.J., Holtzman M.J. Direct suppression of Stat1 function during adenoviral infection. Immunity. 1998;9:871–880. doi: 10.1016/s1074-7613(00)80652-4. [DOI] [PubMed] [Google Scholar]
- Miller D.M., Rahill B.M., Boss J.M., Lairmore M.D., Durbin J.E., Waldman J.W., Sedmak D.D. Human cytomegalovirus inhibits major histocompatibility complex class II expression by disruption of the Jak/Stat pathway. J. Exp. Med. 1998;187:675–683. doi: 10.1084/jem.187.5.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scidmore M.A, Rockey D.D., Fischer E.R., Heinzen R.A., Hackstadt T. Vesicular interactions of the Chlamydia trachomatis inclusion are determined by chlamydial early protein synthesis rather than route of entry. Infect. Immun. 1996;64:5366–5372. doi: 10.1128/iai.64.12.5366-5372.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.K., Angevine M., Demick K., Ortiz L., Rudersdorf R., Watkins D., DeMars R. Induction of HLA class I-restricted CD8+ CTLs specific for the major outer membrane protein of Chlamydia trachomatis in human genital tract infections. J. Immunol. 1999;162:6855–6866. [PubMed] [Google Scholar]
- Starnbach M.N., Bevan M.J., Lampe M.F. Murine cytotoxic T lymphocytes induced following Chlamydia trachomatis intraperitoneal or genital tract infection respond to cells infected with multiple serovars. Infect. Immun. 1995;63:3527–3530. doi: 10.1128/iai.63.9.3527-3530.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty P.R., Rasmussen S.J., Stephens R.S. Cross-reactive cytotoxic T-lymphocyte-mediated lysis of Chlamydia trachomatis- and Chlamydia psittaci-infected cells. Infect. Immun. 1997;65:951–956. doi: 10.1128/iai.65.3.951-956.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qvigstad E., Hirschberg H. Lack of cell-mediated cytotoxicity towards Chlamydia trachomatis infected target cells in humans. Acta Pathol. Microbiol. Immunol. Scand. 1984;92:153–159. doi: 10.1111/j.1699-0463.1984.tb00067.x. [DOI] [PubMed] [Google Scholar]
- Pavia C.S., Schachter J. Failure to detect cell-mediated cytotoxicity against Chlamydia trachomatis-infected cells. Infect. Immun. 1983;39:1271–1274. doi: 10.1128/iai.39.3.1271-1274.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen S.J., Eckmann L., Quayle A.J., Shen L., Zhang Y.X., Anderson D.J., Fierer J., Stephens R.S., Kagnoff M.F. Secretion of proinflammatory cytokines by epithelial cells in response to Chlamydia infection suggests a central role for epithelial cells in chlamydial pathogenesis. J. Clin. Invest. 1997;99:77–87. doi: 10.1172/JCI119136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison, R.P., K. Feilzer, and D.B. Tumas. Gene knockout mice establish a primary protective role for major histocompatibility complex class II-restricted responses in Chlamydia trachomatis genital tract infection. Infect. Immun. 63:4661–4668. [DOI] [PMC free article] [PubMed]
- Perry L.L., Feilzer K., Hughes S., Caldwell H.D. Clearance of Chlamydia trachomatis from the murine genital mucosa does not require perforin-mediated cytolysis or Fas-mediated apoptosis. Infect. Immun. 1999;67:1379–1385. doi: 10.1128/iai.67.3.1379-1385.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]