

Abstract
M11L, a novel 166–amino acid membrane-associated protein expressed by the poxvirus, myxoma virus, was previously found to modulate apoptosis after infection of rabbit leukocytes. Furthermore, infection of rabbits with an M11L knockout virus unexpectedly produced lesions with a profound proinflammatory phenotype. We show here that M11L is antiapoptotic when expressed independently of other viral proteins, and is directed specifically to mitochondria by a short COOH-terminal region that is necessary and sufficient for targeting. This targeting region consists of a hydrophobic domain flanked by basic amino acid residues, adjacent to a positively charged tail. M11L blocks staurosporine-induced apoptosis by preventing mitochondria from undergoing a permeability transition, and the mitochondrial localization of this protein is essential for this function. We show that M11L is specifically required to inhibit the apoptotic response of monocytes/macrophages during virus infection, as cells of this lineage undergo apoptosis when infected with the M11L knockout virus. As monocyte apoptosis is uniquely proinflammatory, we propose that this observation reconciles the paradoxical proapoptotic and proinflammatory phenotypes of the M11L knockout virus. We suggest that apoptosis of tissue macrophages represents an important antiviral defense, and that the inhibition of apoptosis by viral proteins can be directed in a cell-specific fashion.
Keywords: apoptosis, inflammation, mitochondria, monocyte, Poxviridae infection
Introduction
Virus infection of a host triggers numerous cellular defensive responses designed to limit viral replication and contain the infection. To ensure continued virion production in the face of this immune response, viruses have, in turn, developed strategies to counteract cellular defenses and maintain a suitable environment for their own replication. A key innate cellular response to infection is apoptosis 1 2 3, and a growing number of viral antiapoptotic proteins continue to be characterized 1 2 4 5 6.
In certain cases, the roles of these viral antiapoptotic proteins have been defined. Some, including the viral Fas-associated death domain–like IL-1β–converting enzyme (FLICE) inhibitory proteins (vFLIPs), encoded by many gammaherpesviruses as well as by the molluscum contagiosum poxvirus 2 7, target the initial signal transduction events leading to apoptosis. The vFLIPs specifically disrupt signaling by the Fas/TNF family of death receptor proteins by preventing complete assembly of the death-inducing signaling complex on the cytoplasmic domains of these receptors after activation. Viruses have also devised strategies to target the caspases, the family of apoptotic proteases that play key roles as initiators and effectors of apoptotic events. Viral proteins able to counteract the caspases include p35 of baculovirus, the poxviral serpin CrmA/Spi2, and the E3-14.7K protein of adenovirus 2 5 8. The baculovirus inhibitors of apoptosis proteins (IAPs) are another class of proteins implicated in the regulation of caspase activation, a function that may be linked to their ability to counter the effects of the proapoptotic proteins RPR, GRIM, HID, and DOOM in insect cells 9. Other viral proteins modulate the cell death checkpoint mediated by the Bcl-2 family of apoptotic regulators. Viral proteins such as the Bcl-2 homologues encoded by the lymphotropic gammaherpesviruses, the E1B-19K protein of adenovirus, and the 5-HL protein of African swine fever virus have sequence and/or functional homology to cellular Bcl-2 proteins and are able to abrogate the activity of the proapoptotic Bcl-2 family members, in some cases by direct physical interaction 2 5 7.
Recent experimental findings have indicated a pivotal role for mitochondria in the “decision to die” checkpoint regulated by Bcl-2 proteins 10 11 12 13. In this context, Bcl-2 proteins are believed to regulate the mitochondrial permeability transition (PT) pore and thereby control the release of cytochrome c and other proteins from within mitochondria into the cytoplasm. Once these proteins are able to interact with cytoplasmic components, they become proapoptotic and mediate the activation of key downstream effectors such as caspase-3. Like their cellular counterparts, certain viral Bcl-2 family members have been associated with the mitochondrial checkpoint. In particular, stable expression of the herpesvirus saimiri Bcl-2 protein in Jurkat lymphocytes has been shown to prevent loss of mitochondrial membrane potential, cytochrome c release, and caspase-3 activation after ligation of the Fas receptor 14. Recently, an antiapoptotic human cytomegalovirus protein, vMIA, has been described that has no homology to other known proteins and localizes to mitochondria. This protein inhibits mitochondrial changes typically associated with apoptosis, such as the release of cytochrome c into the cytoplasm and, significantly, binds the adenine nucleotide carrier subunit of the permeability transition (PT) pore 15. The characterization of novel viral proteins such as vMIA demonstrates how analysis of viral protein function can provide valuable insight into normal cellular processes.
Several poxvirus-encoded proteins have been implicated in regulating apoptotic cascades, but presently most of these proteins have an undefined mechanism of action. Included among the poxvirus apoptotic modulators are several proteins encoded by myxoma virus 16 17, a Leporipoxvirus which is the causative agent of a lethal disease, myxomatosis, in the European or laboratory rabbit 18. Myxoma virus apoptotic modulators include M-T2, M-T4, M-T5, and M11L, as revealed by the finding that expression of these proteins is required during infection of RL-5 rabbit lymphocytes to prevent apoptosis and allow efficient virus replication 19 20 21.
M11L is a novel myxoma virus–encoded protein that currently has no database homologues outside the poxvirus family 22. It is 166 amino acids in length and has no distinct structural motifs apart from a hydrophobic stretch of 18 amino acids near the COOH terminus that constitutes a putative transmembrane region. M11L plays an important role in the virulence of myxoma virus during host infection. This was demonstrated during characterization of a myxoma virus variant unable to express the M11L protein as a result of a targeted gene disruption. In marked contrast to the parental virus, which gives rise to the lethal symptoms of myxomatosis, the M11L deletion mutant elicited a highly attenuated, nonlethal disease phenotype in laboratory rabbits. Despite its reduced virulence, however, the lesions produced by the M11L knockout virus were unusual, and histological analysis revealed signs of vigorous inflammatory activity. The knockout virus was also shown to be impaired in its ability to replicate in primary rabbit splenocytes 23. The attributes of M11L suggested a model in which this protein could act as a virulence factor by preventing apoptosis of leukocytes during host infection, thus compromising the effectiveness of cellular protective mechanisms designed to limit viral propagation.
In this paper, we demonstrate that M11L is antiapoptotic when expressed independently from other viral proteins. Significantly, M11L localizes to mitochondria, and we provide evidence that the protection afforded by M11L influences the mitochondrial checkpoint. Finally, we show that M11L is required to maintain the viability of primary rabbit monocytes infected with myxoma virus and suggest a key role for M11L linking the inhibition of apoptosis with inflammation suppression during infection.
Materials and Methods
Cells.
RL-5 rabbit CD4+ lymphocytes (National Institutes of Health AIDS Reagent Program) and HeLa cells (American Type Culture Collection) were maintained in RPMI medium (GIBCO BRL) supplemented with 10% fetal bovine serum (FBS; GIBCO BRL). Primary rabbit monocytes were cultured in the same medium supplemented with 20% FBS. Rat2 fibroblasts, COS-7 monkey fibroblasts, and HepG2 human hepatocellular carcinoma cells (American Type Culture Collection) were maintained in DMEM (GIBCO BRL) supplemented with 10% FBS. BGMK monkey kidney cells (obtained from Dr. S. Dales, University of Western Ontario) were maintained in DMEM supplemented with 10% newborn calf serum (GIBCO BRL). All media contained 200 U/ml penicillin and 200 μg/ml streptomycin.
Virus Infections.
Recombinant viruses used in this study have been described previously. These include vMyxlac, a control myxoma virus that produces M11L, vMyxM11L− which fails to produce M11L owing to a targeted gene disruption 23, and vMyxM11LR, a revertant virus in which the gene disruption has been repaired 20. A vaccinia virus construct that overexpresses M11L, VVM11L 22, and a control vaccinia virus, VV601, were also employed. Other myxoma virus constructs used in this study included the M-T2 24, M-T4 19, and Serp-1 25 targeted disruption mutants. Cells were infected at a multiplicity of infection (MOI) of 10 as described 19.
Plasmid Constructs.
The M11L coding sequence was amplified by PCR using the primers 5′Eco (GCTAGAATTCATGATGTCTCGTTTAAAGAC) or 5′Xho (GGATCTCGAGATGATGTCTCGTTTAAAGAC) and 3′Sal (CGTAGTCG-ACTAGGTCCCTCGGTACC), and cloned into the T-tailed vector pT7blue (Novagen). For retrovirus-directed expression of M11L, the gene was subcloned into the murine leukemia virus–based vector pBabePuro 26 to produce the vector pBabePuroM11L. To allow the expression of GFP-M11L, a fusion protein consisting of green fluorescent protein (GFP) appended to the NH2 terminus of M11L, the M11L coding sequence was cloned into the GFP expression vector pS65T-C1 (Clontech) encoding the S65T variant of GFP so that the M11L sequence was inserted downstream of and in frame with the GFP coding sequence. A mutated form of M11L was created using a PCR-based approach. The M11L coding sequence was amplified using the 5′Xho primer detailed above and a 3′ primer M11Lstop (AACTGCCGCGGTTAGATAGACGGATCATTT) incorporating a stop codon in place of the codon specifying isoleucine at position 143, the first amino acid of the hydrophobic region. A restriction fragment containing the mutated codon was excised from this amplified PCR product and used to replace the corresponding fragment of the wild-type M11L gene. These constructs were subcloned into the pEGFP-C1 vector (Clontech) to allow expression of the same M11L constructs appended to the COOH terminus of the EGFP (enhanced) variant of GFP, which has a higher fluorescence intensity. To identify a minimal mitochondrial targeting signal contained in M11L, a restriction fragment containing the coding sequence for the last 25 amino acids of the protein was cloned into pEGFP-C1. As a positive control, the Bcl-2 coding sequence (provided by Dr. S. Farrow, Glaxo Wellcome Research and Development, Stevenage, UK) was cloned into pEGFP-C1 to allow expression of an EGFP–Bcl-2 chimeric protein. Correct construction of all GFP chimeras was verified by DNA sequencing analysis.
Ectopic Expression of M11L.
Control pBabePuro and pBabePuroM11L vectors were transiently transfected into BOSC 23 cells and packaged into ecotropic virus particles as described 27. After infection of Rat2 fibroblasts, pooled clones of cells that had stably incorporated the respective proviruses were selected on the basis of ability to grow in the presence of 2.5 μg/ml puromycin, and designated Rat2puro and Rat2M11L. Expression of the M11L protein by Rat2M11L cells was verified by immunoblot analysis (data not shown). Transient expression of GFP chimeric proteins for flow cytometry was accomplished by transfecting HeLa cells with GFP plasmid constructs using the Lipofectin Plus reagent (GIBCO BRL) as described 28. Analysis was conducted 24 h after transfection.
Induction and Measurement of Apoptosis.
Apoptosis was induced by addition of staurosporine (Sigma Chemical Co.) to the cell culture medium at a final concentration of 2 μM. Apoptotic cells were identified by measuring the characteristic elevation in levels of nicked DNA using the previously described terminal deoxynucleotidyl transferase–mediated dUTP-fluorescein nick end labeling (TUNEL) reaction 19 29.
Measurement of Caspase-3 Activation.
Rat2puro and Rat2M11L cells (106) were incubated with 5 μM staurosporine for up to 4 h. Caspase-3 activation was detected by SDS-PAGE and immunoblot analysis as described 30 using an antibody directed against the large subunit of the active enzyme (provided by Dr. D. Nicholson, Merck-Frosst Center for Therapeutic Research, Montreal, Quebec, Canada).
Confocal Microscopy.
To study M11L localization in myxoma virus–infected cells, BGMK cells grown on coverslips were treated 20 h after infection with the mitochondrial-specific fluorescent marker Mitotracker Red CXMRos (Molecular Probes) at a final concentration of 30 ng/ml. Accumulation of the dye was allowed to occur for 20 min at 37°C. The cells were fixed with 2% paraformaldehyde/PBS for 30 min at room temperature and permeabilized for 2 min with cold 0.1% Triton X-100/0.1% sodium citrate buffer. Cells were then incubated for 20 min at room temperature with a polyclonal rabbit anti-M11L antibody 22 diluted 1:50 in PBS followed by incubation for 20 min at room temperature with a secondary FITC-conjugated anti–rabbit antibody (Jackson ImmunoResearch Laboratories) at a dilution of 1:200 in PBS. Coverslips were mounted using 50% PBS/50% glycerol solution, and confocal images were obtained using an LSM510 laser scanning confocal microscope mounted on a Zeiss Axiovert 100M microscope equipped with a 63× 1.4 oil immersion Plan-Apochromat objective. FITC excitation was induced by illumination at 488 nm, and the fluorescent signal was collected using a 505–530-nm band pass filter. Mitotracker Red fluorescence was induced by illumination at 543 nm and was detected using a 560-nm long pass filter.
Images of live cells were obtained by growing cells in 3.5-cm-diameter cell culture dishes modified so that a section of the base was replaced by a glass No. 1 coverslip (Fisher Scientific). Partially confluent monolayers of COS-7 or HeLa cells were each transfected with 4 μg DNA using the Lipofectin reagent and OptiMEM medium (GIBCO BRL) according to the manufacturer's specifications. The transfected cells were incubated for 24 h, and mitochondria were stained with Mitotracker Red CXMRos (Molecular Probes) by addition of the dye to the culture medium at a concentration of 15 ng/ml. Cells were replenished with RPMI medium lacking phenol red (GIBCO BRL) and examined by confocal microscopy using the same filter settings described above. Red and green signals were collected sequentially to eliminate bleed-through. Neutral density filters were set at levels of 80% or higher to minimize photobleaching.
Analysis of Protease Sensitivity.
HepG2 cells (5 × 105) were infected at an MOI of 10 with the M11L-overexpressing vaccinia virus VVM11L, or with the control virus VV601 for 12 h. The cells were harvested and resuspended in 200 μl digitonin lysis buffer (75 mM NaCl, 1 mM NaH2PO4, 8 mM Na2HPO4, 250 mM sucrose, and 95 μg/ml digitonin) at 4°C for 5 min to permit selective permeabilization of the plasma membrane without disrupting intracellular membranes 31. Both samples were divided into four aliquots of 50 μl each and centrifuged at 15,000 g for 15 min at 4°C, and the supernatants were retained. The pellets were resuspended in 50 μl digitonin lysis buffer. Duplicate samples were then treated with the protease inhibitor PMSF (Sigma Chemical Co.) at a final concentration of 1 mM immediately or treated with 2.5 μg/μl proteinase K (Boehringer Mannheim) at 4°C for 20 min before the reaction was quenched by PMSF addition. One of the duplicate samples was then resuspended in SDS sample buffer, and the whole cell lysate was used to detect cytochrome c oxidase subunit IV (COX IV). The other sample was used to immunoprecipitate M11L. This was achieved by dilution of the sample to 1 ml in NP-40 lysis buffer (50 mM Tris-HCl, pH 8, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 2 mM EDTA, and 1 mM PMSF) and agitation at 4°C for 30 min. Insoluble material was pelleted at 4°C by centrifugation at 15,000 g for 10 min. The supernatant was removed, and 10 μl of rabbit polyclonal antibody 22 was added. The samples were incubated at 4°C with constant agitation for 2 h, and 20 μl of a 50% slurry of protein A–Sepharose beads (Amersham Pharmacia Biotech) was added before further incubation for 1 h. The beads were then pelleted by low speed centrifugation, washed in lysis buffer, and boiled in SDS sample buffer for 5 min. For COX IV and M11L detection, proteins were separated by SDS-PAGE on a 15% gel and electroblotted onto Immobilon-P membrane (Millipore). COX IV was detected using a primary mouse mAb (Molecular Probes) at a concentration of 0.4 μg/ml and a secondary anti–mouse-horseradish peroxidase conjugate (Bio-Rad) at 1:6,000 dilution followed by enhanced chemiluminescence detection (ECL; Amersham Pharmacia Biotech). M11L was detected using the primary rabbit polyclonal antibody at a dilution of 1:500 and a secondary protein A–horseradish peroxidase conjugate (Pierce Chemical Co.) at a dilution of 1:10,000 followed by ECL detection.
Measurement of Mitochondrial Membrane Potential in Cells Expressing M11L Constructs.
Rat2puro or Rat2M11L cells were cultured in 12-well plates (5 × 105 cells/well) and treated with 2 μM staurosporine for 4 h. As the staurosporine stock was prepared in DMSO, control cells were treated with an equivalent amount of DMSO alone. Thereafter, the cells were stained with the green fluorescent dye 3,3′dihexyloxacarbocyanine iodide (DiOC6; Molecular Probes). The fluorescence intensity of this dye has been found to correlate with the mitochondrial membrane potential and provide an accurate measure of loss of this potential during apoptosis 32. Cells were incubated with 0.5 nM DiOC6 at 37°C for 10 min, harvested with trypsin, and then washed and resuspended in RPMI lacking phenol red (GIBCO BRL). To verify that a decrease in the fluorescence signal intensity did accompany a loss of mitochondrial membrane potential, control cells were treated with the protonophore carbonyl cyanide m-chlorophenyl hydrazone (CCCP; Molecular Probes), which causes dissipation of the proton gradient across the mitochondria inner membrane 32. CCCP was added to the cell culture medium at a final concentration of 50 μM during and after dye addition. Flow cytometric analysis was conducted using a Becton Dickinson FACSCalibur™ flow cytometer equipped with an argon ion laser with 15 mW of excitation at 488 nm. Data were acquired at 10,000 cells per sample, and the fluorescent signal due to excitation of DiOC6 at 488 nm was detected through the FL1 channel equipped with a 530-nm filter (30-nm band pass). Light scatter signals were acquired at linear gain, and fluorescence signals were acquired at logarithmic gain.
HeLa cells transiently expressing GFP plasmid constructs were treated with 2.5 μM staurosporine, and control samples were treated with DMSO alone. Measurement of changes in mitochondrial membrane potential in this system was conducted using the ethyl ester of tetramethylrhodamine (TMRE; Molecular Probes), as the accumulation of this dye in the mitochondria of HeLa cells correlates directly with the magnitude of the negative mitochondrial membrane potential. In addition, this dye displays no appreciable nonspecific binding, self-quenching, or cellular toxicity 33 34. TMRE was added to cells at a final concentration of 0.1 μM, and CCCP was added to a control sample. Flow cytometric analysis to detect green fluorescence due to GFP and orange fluorescence due to TMRE was conducted using a FACSCalibur™ instrument specified above. GFP fluorescence was collected after a 530/30-nm band pass filter, and TMRE fluorescence was acquired through a 585/42-nm band pass filter. Electronic fluorescence compensation was set to eliminate any spectral overlap of the emitted signals. Data were acquired at 10,000 cells per sample with fluorescence signals at logarithmic gain. The percentage of GFP-positive cells that were also TMRE-positive was then calculated for each gated cell population.
Isolation, Infection, and Analysis of Primary Rabbit Monocytes.
For each experiment, heparinized blood was obtained by cardiac puncture from a healthy New Zealand White laboratory rabbit and subjected to Ficoll-Paque (Amersham Pharmacia Biotech) density gradient separation. The buffy coat containing white blood cells was collected, and cells were cultured in RPMI medium containing 20% FBS for 4 h; the monocyte population was enriched as a result of adherence to the plastic culture dish. The adherent cells were detached from the plastic support using warm 1× SSC (150 mM sodium chloride, 15 mM sodium citrate, pH 7.5) and infected with appropriate viruses at an MOI of 10. Typically, infection rates exceeded 70% as verified by staining with X-gal 23. The infected cells were cultured in medium containing 20% FBS for 12 h, fixed in 2% paraformaldehyde/PBS, and apoptotic cells were detected using the TUNEL reaction. The monocyte population in each sample was identified by indirect immunofluorescence by incubation with a mouse anti–rabbit CD11b antibody (Spring Valley) at a dilution of 1:50 at room temperature for 20 min, followed by incubation with a PE-conjugated F(ab′)2 goat anti–mouse secondary antibody (Dako) at a dilution of 1:20 at room temperature for 20 min. Data were acquired using a Becton Dickinson FACScan™ flow cytometer using the same settings described for the FACSCalibur™ instrument. The fluorescein-dUTP signal from TUNEL-positive cells and PE fluorescence from CD11b-positive cells was analyzed for the gated cell population.
Results
The M11L Protein of Myxoma Virus Is Antiapoptotic.
In view of the experimental findings that several different targeted gene disruptions in the myxoma virus genome yielded virus variants with proapoptotic phenotypes 19 20 21, it followed that myxoma virus encodes more than one protein able to directly modulate apoptotic cascades. First, however, it was of interest to ascertain whether myxoma virus infection provided cells with protection from exogenous apoptotic stimuli in addition to the process of virus infection itself. RL-5 rabbit T lymphocytes were mock-infected or infected with myxoma virus (vMyxlac), and 12 h after infection were treated with the apoptosis inducer staurosporine at a concentration of 2 μM for up to 4 h. Apoptotic cells were identified by measuring the characteristic elevation in the levels of nicked DNA using TUNEL analysis. After staurosporine treatment, the percentage of TUNEL-positive (apoptotic) cells was found to be substantially higher in the mock-infected cell population than was the case with infected cells (Fig. 1 A). Thus, myxoma virus infection counteracts the proapoptotic effects of staurosporine.
Figure 1.
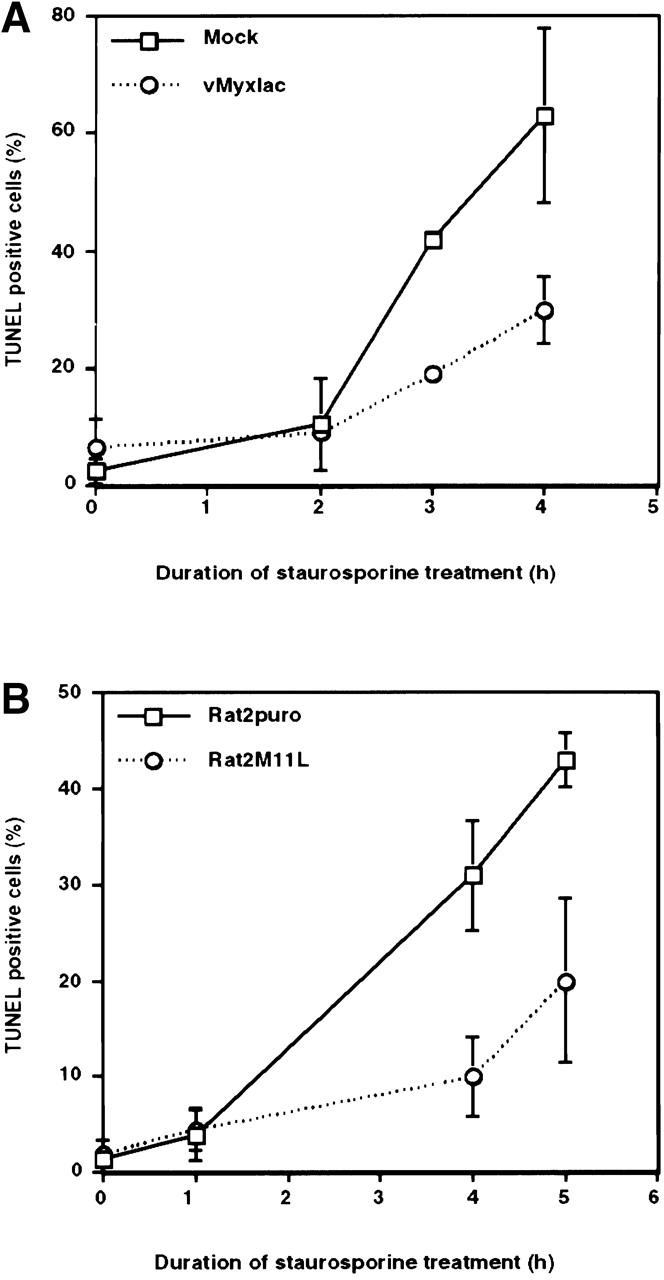

M11L is antiapoptotic. (A) Mock- or myxoma virus (vMyxlac)-infected RL-5 rabbit T lymphocytes were treated with 2 μM staurosporine, and apoptosis was measured using TUNEL analysis at the time intervals indicated. Levels of TUNEL-positive (apoptotic) cells were elevated in the mock-infected cell population compared with myxoma virus–infected cells, indicating that virus infection protects RL-5 cells from apoptosis after staurosporine treatment. (B) Rat2 fibroblasts ectopically expressing M11L (Rat2M11L) or containing the vector alone (Rat2puro) were treated with 2 μM staurosporine, and apoptosis was monitored at the times indicated using TUNEL analysis. Apoptosis levels were elevated in Rat2puro cells compared with Rat2M11L cells, indicating that M11L expression alone protects Rat2 cells from the proapoptotic effects of staurosporine. (C) Rat2puro and Rat2M11L cells were treated with 5 μM staurosporine for the times indicated, and caspase-3 was detected in whole cell lysates by SDS-PAGE and immunoblot analysis using an antibody directed against the large subunit of the active caspase. Cleavage of the 32-kD procaspase-3 to produce the detectable 19-kD component of the active caspase was observed in Rat2puro cells (top) but was considerably reduced in Rat2M11L cells (bottom). Hence, M11L expression impedes caspase-3 activation after treatment of Rat2 cells with staurosporine.
In contrast to parental myxoma virus, the M11L knockout virus has a distinct proapoptotic phenotype when infecting RL-5 cells 20. This finding suggested that, during the normal course of myxoma virus infection, M11L is one of the factors that provides an antiapoptotic function 16. Therefore, we investigated the possibility that M11L has protective, antiapoptotic properties that are able to extend beyond the context of virus infection. To achieve this, we expressed M11L in Rat2 fibroblasts using a retrovirus-based approach. The ability of staurosporine to induce apoptosis in M11L-expressing Rat2M11L cells and control Rat2puro cells, transfected with empty vector alone, was monitored by measuring DNA fragmentation using the TUNEL assay. Whereas 2 μM staurosporine treatment over a 5-h duration produced a steady increase in the levels of TUNEL-positive Rat2puro cells, the levels of TUNEL-positive Rat2M11L cells were considerably reduced under the same conditions, indicating that M11L expression alone confers resistance to staurosporine-induced apoptosis (Fig. 1 B). This result provided the first evidence that M11L is antiapoptotic when expressed independently of other myxoma virus proteins and is able to function in cells derived from a species other than rabbit.
To confirm the idea that M11L directly impacts an apoptotic cascade, we asked whether M11L interferes with end-stage apoptotic processes, such as the cleavage and activation of the effector caspase, caspase-3. To address this question, we monitored caspase-3 processing in Rat2puro and Rat2M11L cells by immunoblot analysis using an antibody directed against the large subunit of the active caspase. Treatment of Rat2puro cells with 5 μM staurosporine over a duration of 4 h resulted in activation of caspase-3 as revealed by reduction in the amount of the 32-kD proenzyme and appearance of the p19 cleavage product, a component of the active enzyme (Fig. 1 C, top). In contrast, caspase-3 activation in Rat2M11L cells after the same treatment was substantially reduced (Fig. 1 C, bottom), with only a low level of activation being apparent after 4-h treatment (compare lane 4, top and bottom). In additional experiments (data not shown), M11L expression in Rat2 fibroblasts was found to inhibit staurosporine-induced cleavage of poly(ADP-ribose) polymerase, a substrate of effector caspases such as caspase-3 35. These findings suggest that M11L impedes apoptotic signaling events upstream of caspase-3 activation.
M11L Localizes to Mitochondria in Infected Cells.
Our studies demonstrated that M11L is antiapoptotic, but database searches failed to identify similarities between M11L and any other protein of known function. However, sequence analysis of M11L revealed the presence of a COOH-terminal putative transmembrane domain. This observation prompted us to investigate the subcellular localization of M11L during myxoma virus infection. BGMK cells were infected with M11L-expressing myxoma virus (vMyxlac) or the M11L knockout virus (vMyxM11L−), and 20 h after infection, M11L was visualized by indirect immunofluorescence and confocal microscopy. As expected, M11L was detected in vMyxlac-infected cells (Fig. 2 A, panel a) and not in cells infected by the knockout virus (Fig. 2 A, panel d). In addition, M11L was observed to have a punctate, cytoplasmic distribution reminiscent of mitochondrial targeting. To determine whether M11L did, indeed, localize to mitochondria, cells were treated with the mitochondrion-specific dye Mitotracker Red (Fig. 2 A, panels b and e). When the fluorescent signals due to M11L and Mitotracker Red were superimposed, a uniform yellow image was produced, indicating that the two signals were coincident (Fig. 2 A, panel c). No such signal was observed in the case of cells infected with the knockout virus (Fig. 2 A, panel f). This result provides evidence that, within myxoma virus–infected cells, intracellular M11L localizes predominantly to mitochondria.
Figure 2.


M11L localizes to mitochondria in infected cells. (A) BGMK cells infected for 20 h with M11L-expressing myxoma virus (vMyxlac) or the M11L knockout virus (vMyxM11L−) were treated with Mitotracker Red to identify mitochondria, and M11L was detected by indirect immunofluorescence. Cells were visualized by confocal microscopy. As expected, M11L was detected in cells infected with vMyxlac (a) but not in cells infected with the M11L knockout virus (d), and Mitotracker Red produced punctate mitochondrial staining (b and e). Superimposed Mitotracker Red and M11L signals (c) yielded a yellow image, indicating that M11L localizes to mitochondria. This was not observed in cells infected with the knockout virus (f). Bar, 10 nm. (B) The proteinase K (PK) sensitivity of the 18-kD M11L (top) or 17-kD COX IV (bottom) proteins was assessed. Digitonin lysates of HepG2 cells infected with M11L-expressing VVM11L or the control virus VV601 (CNTL) were prepared 12 h after infection. Pellet (lanes 1, 2, 5, and 6) and supernatant (sup; lanes 3, 4, 7, and 8) fractions were isolated. Samples were subjected to proteinase K treatment for 20 min (PK 20 min; lanes 1–4) or left untreated (PK 0 min; lanes 5–8), and M11L or COX IV were detected by SDS-PAGE and immunoblotting. M11L (top) but not COX IV (bottom) in the pellet fraction (lane 1) was sensitive to proteinase K treatment, indicating that although M11L is membrane associated, it is orientated towards the cytosol.
To investigate the topology of M11L within mitochondria, we conducted experiments to determine whether M11L is proteinase K sensitive. For these experiments, we used the human hepatocyte-derived HepG2 cell line, as these cells contain a high proportion of mitochondria. Digitonin extracts of HepG2 cells were separated into pellet and supernatant fractions after infection for 12 h with the M11L-overexpressing virus vector VVM11L or the control virus VV601 (which does not express M11L). Samples were treated with proteinase K or left untreated before immunoblot analysis of M11L or COX IV, a protein that resides in the inner mitochondrial membrane. As can be seen from Fig. 2 B (top), the majority of the M11L protein is present in the pellet fraction (lanes 1 and 5), which contains membrane-associated components. A small proportion of M11L can be detected in the soluble fraction (lanes 3 and 7), as would be expected for a protein produced by a cytoplasmic virus. M11L protein was not present in cells infected with the control virus, as expected (lanes 2, 4, 6, and 8). COX IV was only detected in the pellet fractions (Fig. 2 B, bottom, lanes 1, 2, 5, and 6). However, when the protease sensitivity of M11L was assessed, it was evident that M11L present in the pellet fraction was proteinase K sensitive (Fig. 2 B, top, lane 1). M11L present in the supernatant fraction was also protease sensitive, as revealed after overexposure of the immunoblot (data not shown). In contrast, COX IV, present in the pellet fraction, was protease resistant (Fig. 2 B, bottom, lanes 1 and 2). These data provide evidence that M11L, although associated with mitochondria, is exposed on the cytoplasmic face of the organelle.
M11L Expressed in Uninfected Cells Localizes to Mitochondria and Contains a COOH-terminal Mitochondrial Targeting Signal.
We next sought to determine the spatial distribution of M11L in live, transfected cells. A chimeric form of M11L bearing an NH2-terminal GFP tag was expressed in both COS-7 and HeLa cells. As was the case for M11L detected in virus-infected BGMK cells, GFP-tagged M11L displayed a punctate cytoplasmic distribution both in COS-7 cells (Fig. 3 d) and HeLa cells (data not shown) which was suggestive of association with intracellular membranes. Upon comparison of the GFP-M11L signal fluorescence pattern with that of Mitotracker Red in the same cells (Fig. 3 e), the two signals were found to be coincident (Fig. 3 f), indicating that GFP-M11L localizes to mitochondria. In contrast, GFP alone produced a diffuse signal throughout COS-7 cells (Fig. 3, a and c), reflecting its lack of intracellular targeting and lack of correspondence with Mitotracker Red staining (Fig. 3 b). These experiments therefore show that a GFP tag, when added to the NH2 terminus of M11L, does not alter the mitochondrial localization of the protein, and that this localization is consistent in cells having different species of origin.
Figure 3.

GFP-tagged M11L localizes to mitochondria. COS-7 cells expressing GFP alone (a), M11L bearing an NH2-terminal GFP tag (d), or a GFP-tagged, truncated form of M11L (GFP-M11Lstop) lacking the last 24 amino acids including the hydrophobic region (g) were visualized by confocal microscopy. Mitochondria were identified by Mitotracker Red staining (b, e, and h). When the Mitotracker Red fluorescence signal was merged with that of GFP-M11L, a yellow image was produced (f), indicating that GFP-M11L localizes to mitochondria in live, transfected cells. In contrast, no colocalization was observed in the case of GFP alone (c) or truncated M11L (i). Failure of truncated M11L to localize to mitochondria indicates that the last 24 amino acids are necessary for targeting. Bar, 10 nm.
Since the COOH-terminal 24 amino acids of M11L include a stretch of 18 amino acids that constitute a putative transmembrane domain, we wished to determine whether this domain was important for targeting. To address this question, the localization of a GFP-tagged, truncated form of M11L lacking the COOH-terminal 24 amino acids was investigated. Interestingly, this truncated form of M11L was distributed diffusely throughout the cytoplasm and nucleus in COS-7 cells (Fig. 3g and Fig. i) and showed no correspondence with the staining pattern due to Mitotracker Red (Fig. 3 h). The same result was obtained using HeLa cells (data not shown). This shows that the COOH-terminal 24 amino acids are necessary for M11L targeting to mitochondria, and removal of this domain, which includes the putative transmembrane region and short 6–amino acid positively charged tail, prevents mitochondrial localization.
We next sought to determine whether the 25–amino acid COOH-terminal region of M11L alone was sufficient for mitochondrial targeting. The coding sequence for this minimal region (designated mt, Fig. 4) was appended to GFP and was found to direct GFP to punctate cytoplasmic structures in both COS-7 (Fig. 4 a) and HeLa (Fig. 4 d) cells. When the distribution of this fluorescent signal was compared with that of Mitotracker Red (Fig. 4b and Fig. e) in the same cells, the two signals were found to be coincident (Fig. 4c and Fig. f). Hence, the COOH-terminal 25 amino acids of M11L comprise a signal that is sufficient for mitochondrial targeting. This targeting motif includes an 18–amino acid putative transmembrane domain flanked by positively charged lysine residues adjacent to a short 6–amino acid COOH-terminal tail with a net positive charge (Fig. 5). A truncated form of this sequence consisting of only the last 19 amino acids of M11L failed to localize GFP to mitochondria (data not shown), indicating the requirement for a hydrophobic stretch sufficiently long to form a transmembrane segment within the targeting signal. The M11L COOH-terminal targeting signal conforms to a newly described consensus for directing proteins to mitochondria. This consensus consists of a hydrophobic region flanked by positively charged residues adjacent to a short, positively charged tail and was initially identified in vesicle-associated membrane protein 1B (VAMP-1B), monoamine oxidase A and B, and Bcl-2 36. We found this consensus to be additionally present in diverse Bcl-2 family members (Fig. 5).
Figure 4.

M11L contains a COOH-terminal mitochondrial targeting signal. COS-7 or HeLa cells expressing GFP-mt, a construct consisting of GFP tagged with the COOH-terminal 25 amino acids of M11L (mt) containing the putative transmembrane domain (underlined), were visualized by confocal microscopy. The distribution of the GFP-mt (a and d) and Mitotracker red (b and e) was found to be coincident (c and f). Hence, the COOH-terminal 25 amino acids of M11L are sufficient for mitochondrial targeting. Bars, 10 nm.
Figure 5.

The M11L mitochondrial targeting signal belongs to a consensus found in other proteins. Proposed COOH-terminal consensus for targeting Bcl-2 family members to the mitochondrial outer membrane. The COOH-terminal sequences shown include those of the antiapoptotic Bcl-2 family members Bcl-2, Bcl-XL, Boo/Diva, and CED-9, the viral antiapoptotic proteins M11L, BHRF-1, and KSbcl-2, as well as the proapoptotic proteins Nip3 and Nix. aa, amino acid.
M11L Prevents Mitochondria from Undergoing a Permeability Transition after Apoptosis Induction.
Since M11L inhibits apoptosis and localizes to mitochondria, it was of interest to determine whether M11L could function by protecting mitochondria from apoptotic changes, notably a permeability transition revealed by loss of electrical potential difference across the inner membrane. The mitochondrial membrane potential in Rat2puro and Rat2M11L cells was measured as a function of DiOC6 fluorescence in the presence or absence of staurosporine treatment. Representative results from one of three experiments shown in Fig. 6 A reveal that the mitochondria of control cells from both cell lines stained brightly (Fig. 6 A, panels a and c). Fluorescence was diminished in the presence of the protonophore CCCP, an uncoupler of the electron transport chain that induces collapse of the mitochondrial inner membrane potential (Fig. 6 A, panel a insert). However, when subjected to the proapoptotic effects of staurosporine, mitochondria in Rat2puro cells were found to be considerably more sensitive to loss of membrane potential than were Rat2M11L cells (Fig. 6 A, panels b and d). These results indicate that M11L can protect mitochondria from undergoing a permeability transition after receipt of an apoptotic signal induced by staurosporine.
Figure 6.

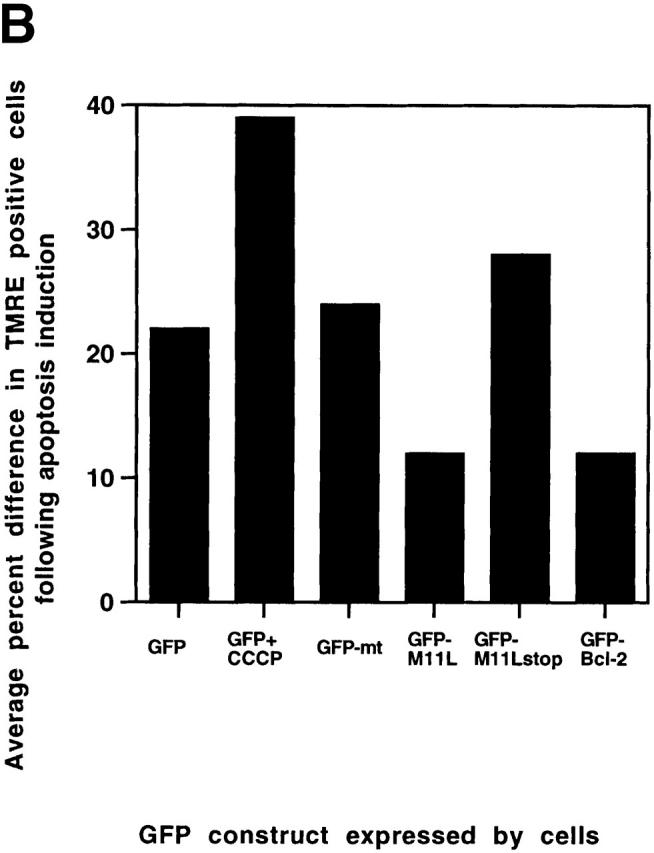
M11L prevents the mitochondrial permeability transition. (A) Rat2puro (top) or Rat2M11L (bottom) cells were maintained as controls or treated with staurosporine for 4 h and stained with the fluorescent dye DiOC6 to obtain a measure of the mitochondrial membrane potential. Representative results of one of three separate experiments are shown. Control cells (a and c) displayed intense staining with the dye, indicating normal mitochondrial function. The protonophore CCCP markedly attenuated the fluorescent signal, as expected (a, insert). A similar reduction in the fluorescent signal was seen in Rat2puro cells after apoptosis induction by staurosporine (b). In contrast, signal intensity and, therefore, mitochondrial function was retained in Rat2M11L cells after the same treatment (d). This shows that M11L plays a role in preserving mitochondrial function after apoptosis induction. (B) HeLa cells were transiently transfected to allow expression of GFP, mitochondria-targeted GFP (GFP-mt), or the fusion proteins GFP-M11L, GFP-M11Lstop, or GFP–Bcl-2. The percentage of GFP-expressing cells that also displayed TMRE fluorescence was determined with or without staurosporine treatment. The reduction in the percentage of TMRE-positive cells after staurosporine treatment is represented here graphically and is the average of two separate experiments. The results show that a large percentage of cells expressing GFP, GFP-mt, and the GFP-M11Lstop chimera (which is not localized to mitochondria) failed to retain TMRE fluorescence after staurosporine treatment. CCCP also produced a loss of TMRE fluorescence in GFP-expressing cells, as expected. In contrast, GFP-M11L and GFP–Bcl-2 expressing cells maintained TMRE fluorescence after staurosporine treatment. This indicates that M11L, like Bcl-2, can protect the mitochondria of HeLa cells from undergoing loss of membrane potential after apoptosis induction.
Similarly, we investigated the ability of transiently expressed GFP constructs to protect HeLa cells from loss of mitochondrial membrane potential after staurosporine treatment. TMRE was used to detect mitochondrial changes in this series of experiments because, like DiOC6, it displays decreased fluorescence intensity as mitochondrial membrane potential diminishes but it emits a signal in the orange spectral range and therefore does not interfere with GFP fluorescence. TMRE has been successfully used to monitor mitochondrial function in HeLa cells by confocal microscopy 33 and, in this study, was also found to be suitable for flow cytometric analysis, as is the related methyl ester TMRM 32. The percentage of cells expressing GFP constructs that were also TMRE-positive was determined in control and staurosporine-treated cells. The reduction in the percentage of TMRE-positive cells after staurosporine treatment was then calculated. The average results for two separate experiments are graphically represented in Fig. 6 B. As shown by this figure, expression of GFP alone, GFP targeted to mitochondria (GFP-mt), and the nontargeted GFP-M11Lstop truncation mutant all failed to prevent a loss of TMRE fluorescence. This suggests that these constructs cannot protect mitochondria from undergoing a loss of membrane potential after staurosporine treatment. Treatment of GFP-expressing cells with CCCP also induced a marked loss of TMRE fluorescence. In contrast, expression of the GFP-M11L chimera and the positive control GFP–Bcl-2 construct resulted in TMRE fluorescence being retained in HeLa cells after the same treatment. This indicates that M11L, like Bcl-2, can protect mitochondria in HeLa cells from undergoing a permeability transition induced by the proapoptotic effects of staurosporine. This property is dependent on M11L being correctly localized to mitochondria.
M11L Is Required to Prevent Apoptosis during Infection of Primary Rabbit Monocytes.
On the basis of the experimental findings described in this paper, it would be predicted that infection of rabbit leukocytes by the M11L knockout virus would result in elevated levels of apoptosis in these cells. Apoptosis is normally an immunologically silent event 37, and therefore the strongly proinflammatory disease phenotype elicited in rabbits infected with the knockout virus is a seemingly conflicting observation. However, it has been reported that monocyte apoptosis has the unusual property of promoting inflammation 37 38. Therefore, we decided to test whether infection of monocytes with the M11L knockout virus was able to induce apoptosis in these cells, a situation that could explain the proinflammatory phenotype of this virus.
Primary rabbit monocytes were isolated from peripheral rabbit blood and infected with several myxoma virus constructs. After 12 h of infection, apoptotic cells were detected by means of the TUNEL reaction, and cells of the monocyte lineage were identified by the presence of the CD11b surface marker. Populations of dual-positive cells representing apoptotic monocytes were quantitated by flow cytometry. Fig. 7 shows representative results from one of four separate experiments and reveals that only the M11L knockout virus induced notable levels of apoptosis in primary rabbit monocytes. In contrast, when monocytes were infected with virus expressing M11L, including the control vMyxlac virus, the M11L revertant virus, and myxoma virus constructs with other targeted gene disruptions, no elevation in levels of apoptosis was observed. This finding is of interest, as the M11L, M-T2, and M-T4 proteins are all required to prevent apoptosis during infection of the RL-5 lymphocytes, and disruption of any one of these genes produces a virus with a proapoptotic phenotype in this cell line. In contrast, there appears to be a unique requirement for M11L production during primary rabbit monocyte infection. Virus constructs unable to produce either the M-T2 or M-T4 protein, but still able to produce M11L, do not have a proapoptotic phenotype in primary monocytes. This result identifies a cell lineage where M11L production is functionally significant in preventing apoptosis during infection, and provides an explanation for the seemingly conflicting observations that the M11L knockout virus induces apoptosis but also produces lesions with extensive infiltrations of inflammatory cells in infected animals.
Figure 7.

M11L is required to prevent apoptosis during myxoma virus infection of primary rabbit monocytes. Primary rabbit monocytes were infected with the M11L knockout virus (vMyxM11L−), a revertant of this knockout virus (vMyxM11LR), control vMyxlac, or three other myxoma virus constructs with targeted gene disruptions (vMyxT2−, vMyxT4−, and vMyxSerp−). Apoptosis was measured by TUNEL analysis (horizontal axis), and the CD11b-positive cells of monocyte origin were identified by indirect immunofluorescence (vertical axis). Apoptotic monocytes are represented in the second quadrant (percentage of total cells shown). Apoptosis levels were elevated in cells infected with the M11L knockout virus (vMyxM11L−) but not in cells infected with the other virus variants, all of which express M11L. The data shown are representative of four separate experiments and demonstrate a distinct role for M11L in preventing apoptosis of infected monocytes.
Discussion
The unique role of M11L in the virulence of myxoma virus was indicated by two previous experimental observations. First, in contrast to control virus, an M11L knockout myxoma virus elicited a markedly attenuated disease phenotype associated with unusual tumor-like lesions containing large numbers of infiltrating inflammatory cells 23. Second, M11L was identified as a factor required to prevent apoptosis during myxoma virus infection of RL-5 lymphocytes in vitro 20. These observations suggested a model in which M11L acts as a virulence factor by virtue of its ability to prevent infected leukocytes from initiating a protective apoptotic response, thereby promoting viral replication.
These results suggested that M11L exerts a host cell–protective effect when the process of infection itself serves as the apoptotic trigger. In this paper, we show for the first time that M11L, when expressed independently from other viral proteins, can protect these cells from the proapoptotic effects of another inducer, namely staurosporine. The finding that M11L can prevent caspase-3 activation and poly(ADP-ribose) polymerase cleavage suggests that this protein has a direct effect on a strategic step in an apoptotic cascade upstream of caspase-3.
In this study, we investigated the intracellular localization of M11L and demonstrated that this protein is primarily targeted to mitochondria. A previous study 22 based on indirect immunofluorescence analysis of nonpermeabilized cells revealed that M11L can be detected on the surface of infected cells. However, we have shown that surface M11L probably represents only a minor proportion of the total amount of protein produced in myxoma virus–infected cells. The current study used the wild-type protein expressed in the context of myxoma virus infection as well as a plasmid-encoded GFP-M11L chimera visualized in live, transfected cells to investigate the protein's intracellular localization. We demonstrated that the majority of the M11L protein within cells is targeted to mitochondria and remains accessible to proteinase K digestion. This indicates that M11L is associated with the outer mitochondrial membrane and is oriented towards the cytoplasm. Hence, M11L has an intracellular distribution similar to many Bcl-2–like antiapoptotic proteins of both cellular and viral origin 11 13. M11L contains a signal within the COOH-terminal 25 amino acids that is necessary and sufficient for mitochondrial targeting. This mitochondrial COOH-terminal targeting signal conforms to a newly proposed consensus domain which takes the form of a hydrophobic region flanked by positively charged residues adjacent to a positively charged tail 36. Interestingly, COOH-terminal targeting motifs responsible for directing other proteins involved in apoptosis to the outer mitochondrial membrane are also found to conform to this consensus (Fig. 5). Included in this category are Bcl-2 31 39, Bcl-XL 40, BHRF-1 41, Nip3 41 42, and Nix 43. This motif is also present in the Bcl-2 family members Boo/Diva 44 45, CED-9 46, and KSbcl-2 from human herpesvirus 8 47, although the intracellular localization of these proteins and/or the precise role of this motif in targeting are unknown at present.
We have shown that M11L prevents mitochondria from undergoing a permeability transition after initiation of an apoptotic signal by staurosporine. We demonstrated this both in Rat2 cells stably expressing M11L and in HeLa cells transiently transfected with a GFP-M11L construct. Although there are pitfalls associated with the use of fluorescent dyes for monitoring mitochondrial function 48 49, we obtained the same result using two different fluorescent probes in the two systems, supporting the conclusion that M11L protects mitochondria from apoptotic changes. In addition, the correct localization of M11L to mitochondria appears to be essential for this function. It is therefore likely that M11L acts as a viral survival effector by preventing amplification of apoptotic cascades that proceed via the mitochondrial pathway. Two other viral antiapoptotic proteins, herpesvirus saimiri Bcl-2 and human CMV vMIA, both expressed by herpesviruses, are similarly important for preservation of mitochondrial function after exposure to apoptosis-inducing agents 14 15. In the case of myxoma virus and CMV, this function is also required to sustain viral replication 15 23.
To investigate the apparently contradictory observation that the loss of M11L function results in the induction of both apoptosis and a massive inflammatory response in infected rabbits, we tested the ability of the M11L knockout virus, as well as various other deletion mutant viruses, to induce apoptosis during infection of primary rabbit monocytes. Whereas a number of these myxoma virus deletion mutants induce apoptosis in the RL-5 lymphocyte cell line, only the M11L knockout virus elicited an apoptotic response in primary rabbit monocytes. This indicates that expression of M11L might be particularly important in allowing myxoma virus to productively infect cells of the monocyte/macrophage lineage, and is interesting in view of the long-recognized importance of macrophages of the reticulo-endothelial system in the development of myxomatosis 50. Elevated levels of apoptosis in tissues infected by the knockout virus would not be anticipated in association with signs of an increased inflammatory response except in the unusual situation of apoptosis in cells of the monocyte/macrophage lineage. This proinflammatory effect could be attributed to a variety of causes. Monocyte apoptosis has been shown to result in the processing and release of the inflammatory cytokine IL-1β 38. In addition, monocyte-derived macrophages are responsible for limiting the inflammatory effects of activated neutrophils and other granulocytes. Hence, depletion of the monocyte population could impair the normal regulatory processes of the immune system, which are designed to contain the potentially dangerous effects of uncontrolled inflammation 37.
In conclusion, we show that M11L is an antiapoptotic protein that localizes to the exterior of mitochondria by means of a 25–amino acid COOH-terminal targeting motif and protects this organelle from changes associated with apoptosis induction. M11L may be particularly important for circumventing an apoptotic response in monocytes/macrophages that infiltrate into lesions during myxoma virus infection of rabbits. It will now be of interest to investigate the role of M11L in protecting cells from additional apoptotic inducers and to ascertain in more detail the role of this protein in modulating mitochondrial function.
Acknowledgments
We wish to thank Dr. D. Nicholson for providing the anti–caspase-3 antibody, Nigel Waterhouse and Luc Berthiaume for helpful discussions, and Robert Maranchuk and Wei Zeng for preparation of virus stocks.
This work was funded by the National Cancer Institute of Canada and the Medical Research Council of Canada. H. Everett gratefully acknowledges past support from the Alberta Heritage Foundation for Medical Research and the 75th Anniversary Award of the Faculty of Medicine, University of Alberta, and is currently the recipient of a Province of Alberta Graduate Fellowship. M. Barry was supported by an Alberta Heritage Foundation for Medical Research Fellowship. R.C. Bleackley is an Alberta Heritage Foundation for Medical Research Medical Scientist, Medical Research Council Distinguished Scientist, and Howard Hughes Medical Institute International Research Scholar. G. McFadden is a Medical Research Council Senior Scientist.
Footnotes
Abbreviations used in this paper: CCCP, carbonyl cyanide m-chlorophenyl hydrazone; COX IV, cytochrome c oxidase subunit IV; DiOC6, 3,3′-dihexyloxacarbocyanine iodide; FBS, fetal bovine serum; GFP, green fluorescent protein; MOI, multiplicity of infection; TMRE, ethyl ester of tetramethylrhodamine; TUNEL, terminal deoxynucleotidyl transferase–mediated dUTP-fluorescein nick end labeling; vFLIP, viral FLICE inhibitory protein.
References
- O'Brien V. Viruses and apoptosis. J. Gen. Virol. 1998;79:1833–1845. doi: 10.1099/0022-1317-79-8-1833. [DOI] [PubMed] [Google Scholar]
- Tschopp J., Thome M., Hofmann K., Meinl E. The fight of viruses against apoptosis. Curr. Opin. Genet. Dev. 1998;8:82–87. doi: 10.1016/s0959-437x(98)80066-x. [DOI] [PubMed] [Google Scholar]
- Everett H., McFadden G. Apoptosisan innate immune response to virus infection. Trends Microbiol. 1999;7:160–165. doi: 10.1016/s0966-842x(99)01487-0. [DOI] [PubMed] [Google Scholar]
- Barry M., McFadden G. Apoptosis regulators from DNA viruses. Curr. Opin. Immunol. 1998;10:422–430. doi: 10.1016/s0952-7915(98)80116-7. [DOI] [PubMed] [Google Scholar]
- Hardwick J.M. Viral interference with apoptosis. Semin. Cell Dev. Biol. 1998;9:339–349. doi: 10.1006/scdb.1998.0243. [DOI] [PubMed] [Google Scholar]
- Roulston A., Marcellus R.C., Branton P.E. Viruses and apoptosis. Annu. Rev. Microbiol. 1999;53:577–628. doi: 10.1146/annurev.micro.53.1.577. [DOI] [PubMed] [Google Scholar]
- Meinl E., Fickenscher H., Thome M., Tschopp J., Fleckenstein B. Anti-apoptotic strategies of lymphotropic viruses. Immunol. Today. 1998;19:474–479. doi: 10.1016/s0167-5699(98)01309-7. [DOI] [PubMed] [Google Scholar]
- Shisler J.L., Gooding L.R. Adenoviral inhibitors of the apoptotic cascade. Trends Microbiol. 1998;6:337–339. doi: 10.1016/s0966-842x(98)01342-0. [DOI] [PubMed] [Google Scholar]
- Deveraux Q.L., Reed J.C. IAP family proteins—suppressors of apoptosis. Genes Dev. 1999;13:239–252. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- Green D.R., Reed J.C. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Gross A., McDonnell J.M., Korsmeyer S.J. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- Kroemer G., Dallaporta B., Resche-Rigon M. The mitochondrial death/life regulators in apoptosis and necrosis. Annu. Rev. Physiol. 1998;60:619–642. doi: 10.1146/annurev.physiol.60.1.619. [DOI] [PubMed] [Google Scholar]
- Vander Heiden M.G., Thompson C.B. Bcl-2 proteinsregulators of apoptosis or of mitochondrial homeostasis? Nat. Cell Biol. 1999;1:209–214. doi: 10.1038/70237. [DOI] [PubMed] [Google Scholar]
- Derfuss T., Fickenscher H., Kraft M.S., Henning G., Lengenfelder D., Fleckenstein B., Meinl E. Antiapoptotic activity of the herpesvirus saimiri-encoded Bcl-2 homologstabilization of mitochondria and inhibition of caspase-3-like activity. J. Virol. 1998;72:5897–5904. doi: 10.1128/jvi.72.7.5897-5904.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmacher V.S., Bartle L.M., Skaletskaya A., Dionne C.A., Kedersha N.L., Vater C.A., Han J.-W., Lutz R.J., Watanabe S., McFarland E.D. A cytomegalovirus-encoded mitochondria-localized inhibitor of apoptosis structurally unrelated to Bcl-2. Proc. Natl. Acad. Sci. USA. 1999;96:12536–12541. doi: 10.1073/pnas.96.22.12536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden G., Barry M. How poxviruses oppose apoptosis. Semin. Virol. 1998;8:429–442. [Google Scholar]
- Nash P., Barrett J., Cao J.-X., Hota-Mitchell S., Lalani A., Everett H., Xu X.-M., Robichaud J., Hnatiuk S., Ainslie C. Immunomodulation by virusesthe myxoma virus story. Immunol. Rev. 1999;168:103–120. doi: 10.1111/j.1600-065x.1999.tb01286.x. [DOI] [PubMed] [Google Scholar]
- DiGiacomo R.F., Maré C.J. Viral diseases. In: Manning P.J., Ringler D.H., Newcomer C.E., editors. The Biology of the Laboratory Rabbit. 2nd ed. Academic Press; New York/London: 1994. pp. 171–204. [Google Scholar]
- Barry M., Hnatiuk S., Mossman K., Lee S.-F., Boshkov L., McFadden G. The myxoma virus M-T4 gene encodes a novel RDEL-containing protein that is retained within the endoplasmic reticulum and is important for the productive infection of lymphocytes. Virology. 1997;239:360–377. doi: 10.1006/viro.1997.8894. [DOI] [PubMed] [Google Scholar]
- Macen J.L., Graham K.A., Lee S.F., Schreiber M., Boshkov L.K., McFadden G. Expression of the myxoma virus tumor necrosis factor receptor homologue (T2) and M11L genes is required to prevent virus-induced apoptosis in infected rabbit T lymphocytes. Virology. 1996;218:232–237. doi: 10.1006/viro.1996.0183. [DOI] [PubMed] [Google Scholar]
- Mossman K., Lee S.F., Barry M., Boshkov L., McFadden G. Disruption of M-T5, a novel myxoma virus gene member of the poxvirus host range superfamily, results in dramatic attenuation of myxomatosis in infected European rabbits. J. Virol. 1996;70:4394–4410. doi: 10.1128/jvi.70.7.4394-4410.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham K.A., Opgenorth A., Upton C., McFadden G. Myxoma virus M11L ORF encodes a protein for which cell surface localization is critical for manifestation of viral virulence. Virology. 1992;191:112–124. doi: 10.1016/0042-6822(92)90172-l. [DOI] [PubMed] [Google Scholar]
- Opgenorth A., Graham K., Nation N., Strayer D., McFadden G. Deletion analysis of two tandemly arranged virulence genes in myxoma virus, M11L and myxoma growth factor. J. Virol. 1992;66:4720–4731. doi: 10.1128/jvi.66.8.4720-4731.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton C., Macen J.L., Schreiber M., McFadden G. Myxoma virus expresses a secreted protein with homology to the tumor necrosis factor receptor gene family that contributes to viral virulence. Virology. 1991;184:370–382. doi: 10.1016/0042-6822(91)90853-4. [DOI] [PubMed] [Google Scholar]
- Macen J.L., Upton C., Nation N., McFadden G. SERP-1, a serine proteinase inhibitor encoded by myxoma virus, is a secreted glycoprotein that interferes with inflammation. Virology. 1993;195:348–363. doi: 10.1006/viro.1993.1385. [DOI] [PubMed] [Google Scholar]
- Morgenstern J.P., Land H. Advanced mammalian gene transferhigh titer retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990;18:3587–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pear W.S., Nolan G.P., Scott M.L., Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. USA. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans K., Schifferli K., Hawle-Nelson P. High-efficiency transfection of HeLa cells. Focus. 1999;21:15. [Google Scholar]
- Sgonc R., Boeck G., Dietrich H., Gruber J., Recheis H., Wick G. Simultaneous determination of cell surface antigens and apoptosis. Trends Genet. 1994;10:41–42. doi: 10.1016/0168-9525(94)90140-6. [DOI] [PubMed] [Google Scholar]
- Atkinson E.A., Barry M., Darmon A.J., Shostak I., Turner P.C., Moyer R.W., Bleackley R.C. Cytotoxic T lymphocyte-assisted suicide. J. Biol. Chem. 1998;273:21261–21266. doi: 10.1074/jbc.273.33.21261. [DOI] [PubMed] [Google Scholar]
- Nguyen M., Millar D.G., Youg V.W., Korsmeyer S.J., Shore G.C. Targeting of Bcl-2 to the mitochondrial outer membrane by a COOH-terminal signal anchor sequence. J. Biol. Chem. 1993;268:25265–25268. [PubMed] [Google Scholar]
- Metivier D., Dallaporta B., Zamzami N., Larochette N., Susin S.A., Marzo I., Kroemer G. Cytofluorometric detection of mitochondrial alterations in early CD95/Fas/Apo-1-triggered apoptosis of Jurkat T lymphoma cells. Comparison of seven mitochondrion-specific fluorochromes. Immunol. Lett. 1998;61:157–163. doi: 10.1016/s0165-2478(98)00013-3. [DOI] [PubMed] [Google Scholar]
- Ehrenberg B., Montana V., Wei M.-D., Wuskell J.P., Loew L.J. Membrane potential can be determined in individual cells from the Nernstian distribution of cationic dyes. Biophys. J. 1988;53:785–794. doi: 10.1016/S0006-3495(88)83158-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas D.L., Wei M., Febbroriello P., Carson J.H., Loew L.M. Simultaneous imaging of cell and mitochondrial membrane potential. Biophys. J. 1989;56:1053–1069. doi: 10.1016/S0006-3495(89)82754-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazebnik Y.A., Kaufmann S.H., Desnoyers S., Poirier G.G., Earnshaw W.C. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- Isenmann S., Khew-Goodall Y., Gamble J., Vadas M., Wattenberg B.W. A splice-isoform of vesicle-associated membrane protein-1 (VAMP-1) contains a mitochondrial targeting signal. Mol. Biol. Cell. 1998;9:1649–1660. doi: 10.1091/mbc.9.7.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savill J. Apoptosis in the resolution of inflammation. J. Leukoc. Biol. 1997;61:375–380. doi: 10.1002/jlb.61.4.375. [DOI] [PubMed] [Google Scholar]
- Hogquist K., Nett M.A., Unanue E.R., Chaplin D.D. Interleukin 1 is processed and released during apoptosis. Proc. Natl. Acad. Sci. USA. 1991;88:8485–8489. doi: 10.1073/pnas.88.19.8485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janiak F., Leber B., Andrews D.W. Assembly of Bcl-2 into microsomal and outer mitochondrial membranes. J. Biol. Chem. 1994;269:9842–9849. [PubMed] [Google Scholar]
- Wolter K.G., Hsu Y.-T., Smith C.L., Nechushtan A., Xi X.-G., Youle R.J. Movement of Bax from the cytosol to mitochondria during apoptosis. J. Cell Biol. 1997;139:1281–1292. doi: 10.1083/jcb.139.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda M., Theodorakis P., Subramanian T., Chinnadurai G. Adenovirus E1B-19K/Bcl-2 interacting protein BNIP3 contains a BH3 domain and a mitochondrial targeting sequence. J. Biol. Chem. 1998;273:12415–12421. doi: 10.1074/jbc.273.20.12415. [DOI] [PubMed] [Google Scholar]
- Chen B.G., Ray R., Dubik D., Shi L., Cizeau J., Bleackley R.C., Saxena S., Geitz R.D., Greenberg A.H. The E1B 19k/bcl-2 binding protein Nip 3 is a dimeric mitochondrial protein that activates apoptosis. J. Exp. Med. 1997;186:1975–1983. doi: 10.1084/jem.186.12.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G., Cizeau J., Vande Velde C., Park J.H., Bozek G., Bolton J., Shi L., Dubik D., Greenberg A. Nix and Nip3 form a subfamily of pro-apoptotic mitochondrial proteins. J. Biol. Chem. 1999;274:7–10. doi: 10.1074/jbc.274.1.7. [DOI] [PubMed] [Google Scholar]
- Song Q., Kuang Y., Dixit V., Vincenz C. Boo, a novel negative regulator of cell death, interacts with Apaf-1. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:167–178. doi: 10.1093/emboj/18.1.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inohara N., Gourley T.S., Carrio R., Muniz M., Merino J., Garcia I., Koseki T., Hu T., Chen S., Nunez G. Diva, a Bcl-2 homolog that binds directly to Apaf-1 and induces BH3-independent cell death. J. Biol. Chem. 1998;273:32479–32486. doi: 10.1074/jbc.273.49.32479. [DOI] [PubMed] [Google Scholar]
- Wu D., Wallen H.D., Nunez G. Interaction and regulation of subcellular localization of CED-4 by CED-9. Science. 1997;275:1126–1129. doi: 10.1126/science.275.5303.1126. [DOI] [PubMed] [Google Scholar]
- Cheng E.H.-Y., Nicholas J., Bellows D.S., Hayward G.S., Guo H.-G., Reitz M.S., Hardwick J.M. A bcl-2 homolog encoded by Kaposi sarcoma-associated virus, human herpesvirus 8, inhibits apoptosis but does not heterodimerize with Bax or Bak. Proc. Natl. Acad. Sci. USA. 1997;94:690–694. doi: 10.1073/pnas.94.2.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P., Scorrano L., Colonna R., Petronilli V., Di Lisa F. Mitochondria and cell death. Eur. J. Biochem. 1999;264:687–701. doi: 10.1046/j.1432-1327.1999.00725.x. [DOI] [PubMed] [Google Scholar]
- Poot M., Pierce R.C. Detection of apoptosis and changes in mitochondrial membrane potential with chloromethyl-x-rosamine. Cytometry. 1999;36:359–360. doi: 10.1002/(sici)1097-0320(19990801)36:4<359::aid-cyto12>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Ahlstrom C.G. On the anatomical character of the infectious myxoma of rabbits. Acta Pathol. 1940;18:377–393. [Google Scholar]