

Abstract
Bone marrow–derived antigen-presenting cells (APCs) take up cell-associated antigens and present them in the context of major histocompatibility complex (MHC) class I molecules to CD8+ T cells in a process referred to as cross-priming. Cross-priming is essential for the induction of CD8+ T cell responses directed towards antigens not expressed in professional APCs. Although in vitro experiments have shown that dendritic cells (DCs) and macrophages are capable of presenting exogenous antigens in association with MHC class I, the cross-presenting cell in vivo has not been identified. We have isolated splenic DCs after in vivo priming with ovalbumin-loaded β2-microglobulin–deficient splenocytes and show that they indeed present cell-associated antigens in the context of MHC class I molecules. This process is transporter associated with antigen presentation (TAP) dependent, suggesting an endosome to cytosol transport. To determine whether a specific subset of splenic DCs is involved in this cross-presentation, we negatively and positively selected for CD8− and CD8+ DCs. Only the CD8+, and not the CD8−, DC subset demonstrates cross-priming ability. FACS® studies after injection of splenocytes loaded with fluorescent beads showed that 1 and 0.6% of the CD8+ and the CD8− DC subsets, respectively, had one or more associated beads. These results indicate that CD8+ DCs play an important role in the generation of cytotoxic T lymphocyte responses specific for cell-associated antigens.
Keywords: major histocompatibility complex class I, antigen presentation, antigen-presenting cell, cytotoxic T lymphocyte, cross-priming
Introduction
Naive CD8+ T cells are stimulated to proliferate and to develop into cytotoxic effector T cells after recognition of short peptides associated with MHC class I molecules on professional APCs. “Endogenous” cytosolic proteins are generally the source of MHC class I–restricted antigens. These proteins are degraded by the proteasome, and peptides are transported to the endoplasmic reticulum by the transporter associated with antigen presentation (TAP) transporter, where they can bind to newly synthesized MHC class I molecules. In contrast, membrane-associated proteins and endocytosed “exogenous” proteins are mainly presented by MHC class II molecules for recognition by CD4+ T cells. However, the division of the endogenous and exogenous pathway is not absolute. 25 years ago, it was recognized that immunization with cells lacking host MHC alleles, but bearing foreign minor histocompatibility antigens, leads to minor specific CD8+ T cell activation that is restricted to the host MHC allele 1 2. This suggested that host APCs can process exogenous cell-associated antigens and present them in the context of MHC class I molecules, a process termed cross-priming or cross-presentation. Since then, cross-priming has been shown to be important in initiating MHC class I–restricted responses to tumors, peripheral self, viral, and bacterial antigens 3 4 5 6. This indicates that cross-presentation is a general mechanism for the induction of T cells specific for antigens not expressed by APCs themselves and is involved in a variety of T cell responses 7. In addition to T cell activation, cross-presentation has been shown to induce T cell tolerance 8 9.
Bone marrow chimera studies demonstrated that the ability to cross-present cell-associated antigens is restricted to bone marrow–derived APCs 4 10. Several groups have investigated the role of macrophages and dendritic cells (DCs) in presenting exogenously derived antigens in MHC class I in vitro (for a review, see reference 7). Exogenous antigens in the form of soluble proteins, particulate antigens, and cell-associated antigens derived from apoptotic or necrotic cells have been shown to be taken up, processed, and presented to CD8+ T cells by both cell types 11 12 13 14 15 16 17. Other reports have indicated that cross-presentation is a specific property of DCs 18 19 20. However, which bone marrow–derived APC is involved in MHC class I–restricted presentation of exogenous antigens in vivo has not been examined.
DCs comprise populations of cells with heterogeneous phenotypes 21 22. Murine splenic DCs can be divided into two subsets, both of which express high levels of CD11c. One DC population is further characterized by high expression of the myeloid marker CD11b and by absence of expression of CD8α and DEC-205, whereas the other is CD11blowCD8α+DEC-205+ 23 24 25 26 27. The CD8−CD11bhigh DCs can be further divided into a CD4+ and CD4− population 28. Some evidence suggests that the CD8+ and CD8− DC subsets belong to different lineages. Several transcription factor knockout and mutant mice exhibited differential effects on the development of each subset in vivo 29 30 31 32. In addition, CD8+ DCs were shown to be derived from a thymic progenitor that could not generate myeloid cells, whereas CD8− DCs can be grown from bone marrow using GM-CSF 33 34 35. Therefore, CD8+ DCs are thought to relate to the lymphoid lineage, whereas CD8− DCs are considered myeloid related. For convenience, in this paper we will use the terms lymphoid DC and myeloid DC to describe the CD8+ and CD8− DC subsets.
In addition to phenotypic differences, the lymphoid and myeloid DCs reside in different areas of the spleen. Whereas lymphoid DCs are localized in the T cell–rich areas of the periarteriolar lymphatic sheaths (PALS), myeloid DCs can be found in the marginal zone 21 36. Functional distinctions between lymphoid and myeloid DCs have also been reported. Initial in vitro studies indicated that lymphoid DCs could suppress T cell responses by causing apoptosis of CD4+ T cells and by limiting IL-2 production by CD8+ T cells, whereas myeloid DCs were shown to be strong stimulators of primary T cell responses 37 38. These findings led to the suggestion that myeloid and lymphoid DCs function as T cell stimulators and tolerizers, respectively. However, in a recent study, peptide-coated DCs of both subsets were shown to induce strong CD8+ T cell responses, indicating no inherent tolerizing function for lymphoid DCs 39. Both DC subsets do appear to differ in their ability to activate CD4+ cells, as myeloid DCs preferentially stimulate Th2 responses, whereas lymphoid DCs preferentially induce Th1 responses 40 41 42. The role of both DC subsets in cross-priming of CD8+ T cells in vivo has not been investigated.
To determine the cell types responsible for cross-priming in vivo, we primed C57BL/6 (B6) mice with irradiated, β2-microglobulin−/− (β2m−/−) splenocytes loaded with OVA in their cytoplasm. Such β2m−/− splenocytes are unable to present the OVA epitope in association with MHC class I molecules, but they elicit, when injected in B6 mice, OVA-specific, MHC class I–restricted CTL responses. We show that host splenic DCs take up and present cell-associated OVA to CD8+ T cells. Analysis of the OVA-presenting DCs indicated that only lymphoid DCs, and not myeloid DCs, cross-present antigens in vivo. This result points to an important and distinct role for lymphoid DCs in the generation of CD8 T cell responses directed towards cell-associated antigens.
Materials and Methods
Mice.
β2m−/− and B6 mice were purchased from Taconic Farms. OT-I recombination-activating gene (RAG)2−/− and Thy1.1+ OT-I mice were bred in our specific pathogen-free facility and have a transgenic Vα2Vβ5 TCR specific for the OVA257–264 epitope in the context of H2-Kb 43.
Antibodies.
CD11c-, CD11b-, CD8α-, CD8β-, Kb/Db-, and I-Ab–specific antibodies were purchased from BD PharMingen. Biotinylated DEC-205–specific antibodies were a gift from A. Rudensky (University of Washington, Seattle, WA). Flow cytometry was conducted on a FACSCalibur™ and analyzed using CELLQuest™ software (Becton Dickinson). Cell sorting was performed in HBSS with 25 mM Hepes using a FACS Vantage™ (Becton Dickinson).
Cross-priming with OVA-loaded β2m−/− Cells.
Single cell suspensions were prepared in serum-free medium from spleen and cervical, axillary, brachial, inguinal, and mesenteric lymph nodes from female β2m−/− mice. Cells were loaded with OVA by osmotic shock as described previously 44. In short, ∼15 × 107 cells were incubated in 1 ml of hypertonic medium (0.5 M sucrose, 10% wt/vol polyethylene glycol 1000, and 10 mM Hepes in RPMI 1640, pH 7.2) containing 10 mg/ml OVA (Calbiochem) for 10 min at 37°C. 13 ml of prewarmed hypotonic medium (40% H2O, 60% RPMI 1640) was added and the cells were incubated for an additional 2 min at 37°C. The cells were centrifuged, washed twice with cold PBS, and irradiated (1,350 rads). 20–35 × 106 OVA-loaded cells were injected in 200 μl of PBS into the tail vein. In one experiment, β2m−/− cells were shocked with both OVA and yellow/green fluorescent 0.2 μm beads (Molecular Probes). The beads were washed two times in PBS by centrifugation for 10 min at 1,400 rpm and sonication for 10 min. The beads were resuspended to 0.004% (wt/vol) in hypertonic solution containing OVA before shocking. After osmotic shocking of the cells, free beads were removed by three washes in PBS.
CFSE Labeling of OT-I Cells.
OT-I cells from spleen and lymph nodes were washed twice in PBS containing 0.1% BSA. To label cells with the intracellular fluorescent dye carboxyfluorescein diacetate succinimidyl ester (CFSE; Molecular Probes) the cells were resuspended at 5 × 107 cells/ml in PBS/0.1% BSA with 10 μM CFSE for 10 min at 37°C. Cells were washed twice with cold RPMI 1640/10% FCS (RP10) followed by two washes in PBS. 106 CD8+Vα2+CFSE+ OT-I cells in 200 μl of PBS were injected into the tail vein.
Low Density Cell Preparation.
14 h after injection with irradiated OVA-loaded β2m−/− cells, spleens of 5–35 mice were cut into grain size pieces and incubated in 1 ml per spleen of 1 mg/ml collagenase/dispase (Sigma-Aldrich) and 50 μg/ml DNase I (Boehringer) in PBS with continuous stirring at 37°C for 30 min or until digested. EDTA was added to a 10 mM final concentration, and the cell suspension was incubated for an additional 5 min at room temperature. RP10/10 mM EDTA/20 mM Hepes (RP10/HE) was added and the cells were pelleted. Red blood cells were lysed with ACK lysis buffer. Cells were washed once with RP10 and the cells were resuspended in 10 ml/5 spleens of RP10/HE. Undigested material was removed by filtration through a wire mesh screen and the cell suspension was loaded on 14.5% Accudenz (Accurate Chemical & Scientific Corporation) gradients in RP10/HE and centrifuged at 530 g for 20 min at room temperature 25. The low density fraction was recovered and washed once in RP10/HE and resuspended in RP10.
Magnetic Bead Depletion.
5 × 106 low density cells were incubated with 300 μl 2.4G2 supernatant (anti-Fc receptor) for 15 min on ice. Cells were then incubated with 200–250 μl of biotinylated antibody specific for CD11c, CD11b, CD8α, or CD8β in PBS/0.1% BSA for 30 min at 4°C under slow rotation. Depletion of antibody-bound cells with streptavidin-coated magnetic beads (Dynal) was performed in PBS/0.1% BSA according to the manufacturer's instructions.
Proliferation Assay.
To detect OVA cross-priming, different DC preparations from injected mice were used as stimulators for naive OT-I cells in a [3H]thymidine incorporation assay. Indicated numbers of irradiated (2,250 rads) DCs were incubated with 105 OT-I RAG2−/− cells in flat-bottomed plates in 200 μl RP10. As a positive control, stimulator cells were coated with 1 μM OVA257–264 peptide for 1 h and washed three times. After 48 h, the plates were pulsed for 16 h with 1 μCi/well of [3H]thymidine and harvested.
Results
Cross-presentation to CD8+ T Cells In Vivo.
To study the MHC class I–restricted cross-presentation of cell-associated antigens, we injected B6 mice intravenously with β2m−/− cells that had been osmotically loaded with OVA. As expected, the injected cells are efficiently taken up and processed for MHC class I presentation by host APCs. This is demonstrated by the proliferative response of adoptively transferred, CFSE-labeled H2-Kb–restricted, OVA-specific TCR transgenic OT-I cells that serve as an indicator for efficient presentation in vivo. 3 d after priming, the transferred OT-I cells had divided up to seven times, as revealed by the sequential loss of CFSE intensity (Fig. 1 A). In contrast, OT-I cells did not proliferate in mice that did not receive OVA-loaded cells (Fig. 1 B).
Figure 1.

CD8+ T cell proliferation in vivo after priming with OVA-loaded β2m−/− (β2m0/0) cells. Thy1.1+ CFSE-labeled OT-I cells were transferred into B6 mice. 3 d later, mice were primed by injection with irradiated OVA-loaded β2m−/− cells. Spleen cells were isolated 3 d after priming and stained for Thy1.1, Vα2, and CD8. (A) CFSE profile of Thy1.1+Vα2+CD8+ OT-I cells from mice primed with OVA-loaded β2m−/− cells. (B) The same profile from a mouse that received no OVA-loaded β2m−/− cells.
Low Density Cells Cross-present Cell-associated Antigens in a TAP-dependent Manner.
Because we immunized mice via the intravenous route, we focussed our attention on the spleen as the source of the cross-presenting APC. To identify the cross-presenting cell, we isolated low density spleen cells from mice 14 h after injection of β2m−/− cells loaded with BSA or OVA. These cells were then used to stimulate naive OT-I cells in an in vitro proliferation assay in the absence of additional antigen. The low density cell preparation was enriched for DCs and generally contained <1% Db/Kb-negative cells, indicating the paucity of injected OVA-loaded β2m−/− cells (data not shown). Low density spleen cells from mice injected with OVA/β2m−/− cells, but not from mice injected with BSA/β2m−/− cells, were able to stimulate naive OT-I cells in vitro (Fig. 2 A). Low density cells from both types of mice were able to stimulate proliferation after peptide pulsing in vitro (Fig. 2 A, inset).
Figure 2.

Low density spleen cells cross-present OVA in a TAP-dependent fashion. B6 (A) and TAP−/− (TAP0/0) mice (B) were primed with OVA- or BSA-loaded β2m−/− cells. Low density spleen cells were isolated 14 h after injection and analyzed for their ability to stimulate OT-I cells in vitro. As indicated, the different bars represent titered numbers of low density cells added per well. Error bars indicate SEM of triplicate wells. The stimulatory capacity of low density cells after pulsing with OVA257–264 peptide in vitro is shown in the insets.
Processing of exogenous antigens can involve an endosome to cytosol pathway in which the endocytosed antigen is transported to the cytosol and further processed and presented via the “classical” endogenous pathway 20. The TAP transporter is essential in this pathway, in that it transports peptides that are generated by the proteasome from the cytosol to the endoplasmic reticulum where they bind nascent MHC class I molecules. TAP has been shown to be essential for cross-priming MHC class I–restricted responses in vivo 3 45. Alternatively, a TAP-independent pathway of class I MHC cross-presentation has also been described 7 46 47. To determine which pathway is used for the cross-presentation of cell-associated antigens in vivo, BSA- or OVA-loaded β2m−/− cells were injected into TAP−/− mice and low density spleen cells from these mice were evaluated for their capacity to stimulate OT-I in vitro. Low density cells from TAP−/− mice could not stimulate OT-I cells, indicating that the endosome to cytosol pathway is employed for cross-presentation of cell-associated antigens in this system (Fig. 2 B). This result also shows that contaminating APCs in the OT-I cell preparation did not contribute to the proliferation in vitro. The various low density spleen cell preparations did not differ in their presentation capacity, as exogenous OVA peptide pulsing of all preparations showed similarly strong stimulatory capacity for OT-I T cells.
Only Lymphoid DCs Cross-present OVA Antigen to CD8+ T Cells.
To investigate further which cell population within the low density cell preparation was responsible for cross-presentation, we first evaluated which cell types were present. Flow cytometric analysis of the low density cells revealed four cell populations as defined by CD11c and CD11b staining (Fig. 3). A population of CD11c−CD11b− cells that consisted mainly of T and B cells made up 10–20% of the preparation. Besides these residual T and B cells, a population of CD11c− and CD11b− cells were detected that were autofluorescent in the FL-1, FL-2, and FL-3 channels, which could be misinterpreted as low level expression of CD11c and CD11b. The autofluorescent cells expressed MHC class I, heterogeneous levels of MHC class II, and low levels of the macrophage marker F4/80, but no CD4, CD8α, or DEC205 (data not shown). This phenotype is consistent with recently described autofluorescent cells present in DC cell preparations that were identified as macrophages 28. Our low density cell preparation contained ∼30–40% myeloid and 10–30% lymphoid DCs, both expressing high levels of CD11c but different levels of CD11b (Fig. 3). Analysis of the myeloid and lymphoid DC subsets revealed high MHC class II expression by both subsets, high CD8α and DEC205 expression by the lymphoid DCs, and heterogenous CD4 and low DEC-205 expression by the myeloid DC subset, consistent with previously published studies 25 26 28.
Figure 3.

Phenotypic analysis of low density cell preparation. Low density spleen cell preparations were isolated from B6 mice 14 h after priming with OVA-loaded β2m−/− cells. The cells were stained for CD11c, CD11b, and the indicated third reagent, and analyzed by FACS®. Gated myeloid DCs (CD11bhighCD11chigh) and lymphoid DCs (CD11blow CD11chigh) were further analyzed for CD8α, DEC-205, CD4, and MHC class II expression. MyDC, myeloid DCs; LyDC, lymphoid DCs; AFC, autofluorescent cells, mostly macrophages.
We asked whether cross-presentation is a specific characteristic of DCs and, if so, whether it is a feature of a particular DC subset. Therefore, we depleted the low density cell preparation from OVA/β2m−/−-primed mice of specific cell subsets using antibodies and magnetic beads and the remaining cells were evaluated for their capacity to stimulate OT-I cells in vitro (Fig. 4). Depletion of CD11c+ cells in the DC preparation resulted in the loss of both the myeloid and lymphoid DC subsets (66% CD11c+ cells were present before depletion, and 8% after depletion, data not shown). The remaining cells consisted of autofluorescent and CD11c−CD11b− cells and showed almost no OVA presentation. These results demonstrate that cross-presentation of cell-associated OVA is mediated entirely by CD11c+ DCs. Depletion of F4/80+ cells resulted in no loss of activity (data not shown). Therefore, CD11c− macrophages do not seem to play an important in vivo role in cross-presenting cell-associated antigens.
Figure 4.
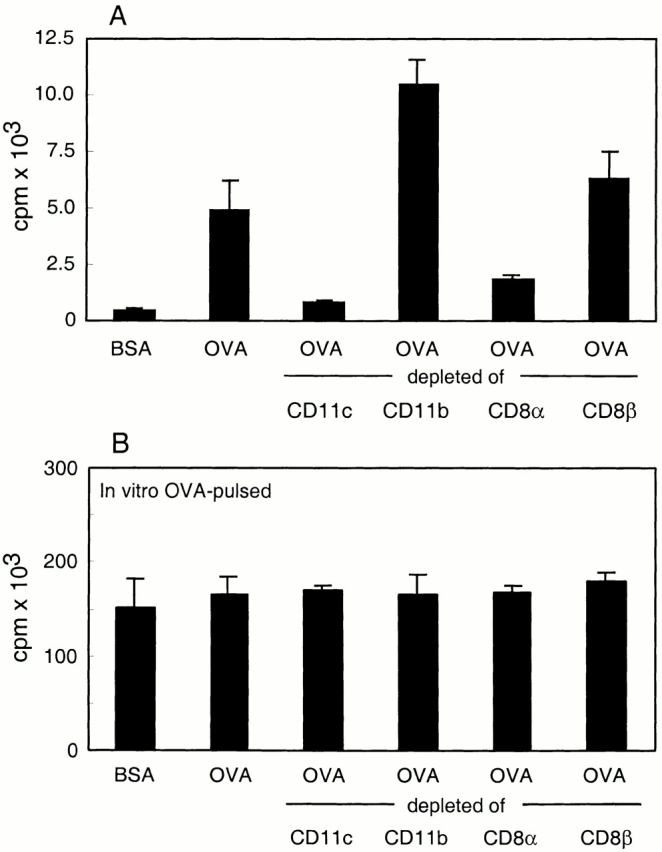
OVA cross-presentation by low density cells depleted of different cell subsets. Low density spleen cells were isolated from B6 mice 14 h after priming with BSA/β2m−/− or OVA/β2m−/− cells. The low density cells from OVA/β2m−/−-injected mice were depleted of CD11c+, CD11bhigh, CD8αhigh, and CD8β+ cells by magnetic beads and the depleted populations were tested (A) directly for their presenting activity in an in vitro OT-I proliferation assay (2 × 105 stimulator cells/well) or (B) after pulsing with OVA peptide in vitro (2 × 104 cells/well). Error bars indicate SEM of triplicate wells.
Depletion of CD11b+ cells specifically removed myeloid DC (38% predepletion to 9% postdepletion) and concomitantly increased the percentage of lymphoid DCs (26% predepletion to 48% postdepletion). This resulted in a higher stimulatory activity of the DC preparation (Fig. 4). To specifically deplete for lymphoid DCs, we used anti-CD8α antibodies. However, this depletion of the lymphoid DC subset was only ∼50% complete (22% predepletion to 13% postdepletion) with the preferential loss of the CD8αhigh-expressing lymphoid DCs (data not shown). The specific depletion of CD8αhigh lymphoid DCs resulted in significant loss of stimulatory activity (Fig. 4). Finally, depletion for CD8β+ cells did not lead to a significant change in numbers of the different subsets or in OVA cross-presenting activity. These results suggest that the lymphoid DC was the main cross-presenting APC.
To confirm our findings, we isolated low density cells from mice injected previously with OVA-loaded β2m−/− cells and sorted myeloid and lymphoid DCs on the basis of their CD11b and CD11c expression. Sorting for myeloid and lymphoid DC populations resulted in 87 and 84% pure populations, respectively (Fig. 5 A). The sorted DC subsets were used as APCs for OT-I in an in vitro proliferation assay. Whereas sorted myeloid DCs lacked stimulatory capacity, sorted lymphoid DCs had stimulatory activity that was stronger than that of the starting DC preparation (Fig. 5 B). This result clearly demonstrates that only lymphoid DCs have the ability to cross-present antigen acquired in vivo from injected β2m−/− cells.
Figure 5.

Lymphoid DCs, but not myeloid DCs, cross-present OVA antigen. Low density cells were isolated from mice primed previously with OVA- or BSA-loaded β2m−/− cells. Myeloid and lymphoid DCs from OVA/β2m−/−-injected mice were (A) FACS® sorted based on their differential CD11b/CD11c expression and (B) tested for their ability to stimulate naive OT-I cells in vitro. The different bars represent titered numbers of DCs in the well. The proliferation of OT-I induced by peptide-pulsed cells is depicted in the inset. One of three experiments with similar results is shown. Error bars indicate SEM of triplicate wells. MyDC, myeloid DCs; LyDC, lymphoid DCs; AFC, autofluorescent cells, mostly macrophages; n.t., not tested.
Lymphoid DCs Rapidly Acquire Antigen In Vivo.
It has been suggested that antigens are transferred from migratory DCs to DCs in the T cell areas 41 48 49. Other studies clearly indicate a migration of DCs from the marginal zone where the myeloid DCs are localized into the central T cell areas of the PALS where lymphoid DCs can be found 50 51 52. This could indicate that marginal zone DCs can transfer antigens to the lymphoid DCs or that marginal zone DCs may mature into lymphoid DCs. Both alternatives would predict that early after injection of OVA/β2m−/− cells, the OVA stimulatory capacity would not be restricted to the lymphoid DC subset. We therefore isolated myeloid and lymphoid DCs soon after injection of OVA/β2m−/− cells and evaluated their OT-I stimulatory capacity (Fig. 6). As early as 2 h after injection, all the OVA stimulatory activity was found in the lymphoid DC subset. Unless transfer of antigens between DCs or the maturation of myeloid DCs into lymphoid DCs is an extremely rapid process, this result suggests that the lymphoid DCs themselves take up and present the cell-associated antigens in vivo.
Figure 6.

MHC class I–restricted cross-presentation is restricted to lymphoid DCs as early as 2 h after priming. Low density cells were isolated from mice 2 or 14 h after priming with OVA-loaded β2m−/− cells. Myeloid and lymphoid DCs were FACS® sorted based on their differential CD11b/CD11c expression and tested for their ability to stimulate naive OT-I cells in vitro. The different bars represent titered numbers of DCs in the well. Error bars indicate SEM of triplicate wells. MyDC, myeloid DCs; LyDC, lymphoid DCs; n.t., not tested.
Myeloid DCs Take Up Cell-associated Antigens.
The lack of presentation by myeloid DCs could be explained if they do not have access to the injected, antigen-bearing cells. This could either be due to the failure of the injected cells to home to the marginal zone areas where myeloid DCs reside, or the myeloid DCs may lack specific surface receptors for the internalization of cell-associated antigen. To determine whether myeloid DCs take up cell-associated antigens, β2m−/− cells were shocked with both OVA and yellow/green fluorescent beads and injected into B6 mice. Splenic low density cells were isolated 14 h after injection, stained for CD11c and CD11b, and analyzed by flow cytometry (Fig. 7 A). Cells containing one or more fluorescent beads were detected in the FITC channel. The CD11c and CD11b FACS® profile of bead-positive cells clearly shows that in addition to lymphoid DCs, both myeloid DCs and autofluorescent macrophages have taken up beads derived from β2m−/− cells (Fig. 7 B). This result indicates that the restricted presentation by lymphoid DCs is not due to restricted uptake of cell-associated antigens.
Figure 7.

Uptake of cell-associated beads by both DC subsets and autofluorescent macrophages. Low density cells were isolated from mice 14 h after priming with OVA/bead-loaded β2m−/− cells. (A) The cells were stained for CD11c and CD11b and analyzed by FACS®. MyDC, myeloid DCs; LyDC, lymphoid DCs; AFC, autofluorescent cells, mostly macrophages. (B) CD11c and CD11b profile of the gated low density cells containing beads.
Only a Small Fraction of Lymphoid DCs Present OVA.
We made two estimates of the fraction of cells within the isolated low density preparation that could present OVA to OT-1 after in vivo uptake of antigen. At 14 h after injection of cells loaded with OVA and fluorescent beads, 0.8% of the total low density cell preparation was bead-positive, as determined by FITC fluorescence (Table ). Within the lymphoid DC subset, which made up 27% of the cells, 1% were bead-positive. Therefore, only 0.27% of the total low density cell preparation was estimated to contain and present OVA to OT-1 cells.
Table 1.
In Vivo Uptake of Cell-associated Beads
| Percentage of total | Percentage of bead+ cells in each subset | |
|---|---|---|
| Low density cell preparation | 100 | 0.8 |
| Myeloid DCs | 41 | 0.6 |
| Lymphoid DCs | 27 | 1.0 |
| Autofluorescent cells | 13 | 1.5 |
The percentage of different subsets in low density cell preparation and the percentage of each that contain beads.
Second, the amount of OT-1 proliferation stimulated by in vivo–derived low density cells was compared with that stimulated by a titration of in vitro peptide-pulsed low density cells. This comparison suggested that 0.32% of lymphoid DCs, or 0.06% of low density cells, presented OVA (average of three experiments, data not shown). This is presumably an underestimate of the number of OVA-presenting cells, as the amount of OVA acquired in vivo is probably lower than the amount of OVA used for exogenous peptide pulsing. Together, these experiments suggest that only ∼1% of the lymphoid DC subset present OVA in vitro.
Discussion
Cross-presentation of exogenous antigens in association with MHC class I molecules has been studied extensively in vitro. Both macrophages and DCs have been shown to present peptides derived from soluble and particulate antigens in the context of MHC class I molecules (for a review, see reference 7). In addition, antigens from apoptotic or necrotic cells can be processed and presented in MHC class I by both cell types in vitro 15 19 53 54. In contrast, there is little information concerning the identity of cross-presenting cells in vivo. DCs have been shown to present soluble protein-derived and cell-associated antigens in MHC class II molecules 48 51 55, but the cell that cross-presents antigens in association with MHC class I molecules in vivo has remained elusive. In our study, we have isolated low density spleen cells from in vivo–primed mice and shown that they present exogenously derived antigens in association with MHC class I in a TAP-dependent manner. Further analysis of the cross-presenting cells identified them as CD11c+ DCs. This is the first isolation and characterization of DCs in the central lymphoid organs that have taken up cell-associated antigens to present them to CD8+ T cells in vivo.
Further experiments showed that only the lymphoid DC subset cross-presents cell-associated antigens in MHC class I molecules in the spleen. The number of actual OVA-presenting cells is very low. We estimated that only 1% of lymphoid DCs present OVA to T cells. The lymphoid DC subset–restricted presentation was evident as early as 2 h after immunization, which strongly suggests that lymphoid DCs themselves take up the exogenous cell-associated antigen. Transfer of antigen between different DC subsets, as had been suggested, is therefore unlikely 41 48 49. In support of the notion of direct uptake of antigen by lymphoid DCs, rat DCs containing apoptotic cell remnants have been found to migrate to the T cell areas of lymph nodes. These DCs were CD4− and may be the rat equivalent of the mouse lymphoid DCs 56. Our finding that only lymphoid DCs cross-present cell-associated antigens in the spleen does not preclude the presence of other types of APCs with cross-priming function in other tissues. APCs have been isolated from tumors and islets of Langerhans that could stimulate CD8+ T cells specific for tumor and islet antigens, respectively 57 58 59. One of these studies defined the phenotype of the APCs as macrophage like 57.
Why do only lymphoid and not myeloid DCs cross-present cell-associated antigens to CD8+ T cells? In principle, it could be the result of differences in the uptake of cell debris, processing ability, or their T cell stimulatory capacity. We show that, in addition to lymphoid DCs, both autofluorescent macrophages and myeloid DCs contain cell-associated beads. This indicates that uptake of cell-associated antigen is not restricted to lymphoid DCs. This contrasts with the study by Fossum and Rolstad, in which allogeneic lymphocyte cell debris was found mainly in lymphoid DCs 60. However, differences in the in vivo model systems used may explain this discrepancy. Second, myeloid and lymphoid DCs do not seem to differ in their capacity to stimulate T cells, as both DC subsets have been shown to stimulate CD8+ T cells efficiently in vitro and in vivo 39. This leaves differences in antigen processing pathways between myeloid and lymphoid DCs as the most likely explanation. A selective transport of internalized antigens to the cytosol has been shown to occur in DCs and to be absent in macrophages 20. Both bone marrow–derived DCs and a splenic-derived DC line exhibited this ability, but ex vivo DCs were not evaluated. Our results suggest that this endosome to cytosol transport will be limited to lymphoid DCs.
Cross-presentation of cell-associated antigens to CD8+ T cells has been shown to result in either T cell activation or tolerance 1 2 8 9 10 61 62. In both cases, bone marrow–derived cells are necessary 8 9 10. Lymphoid DCs have been speculated to mediate peripheral tolerance, whereas myeloid DCs have been suggested to be essential for T cell stimulation 49 63. Our studies do not specifically address the outcome of the DC–T cell interaction, as T cell proliferation is involved in both processes. However, our method of intravenous immunization with OVA-loaded cells has repeatedly been shown to lead to T cell activation and memory 10 61 64. Therefore, we are convinced that we have isolated the APC required for CD8+ T cell cross-priming.
An attractive alternative explanation for the different outcomes of DC–T cell interaction is offered by several studies indicating that the activation state of the DCs is an important factor. DCs involved in cross-priming are activated via CD40–CD40L interaction provided by CD4+ T cells 64 65 66. CD8+ T cell cross-priming does not occur in the absence of CD4+ T cells, illustrating the crucial role of CD4+ T cells in this process 10. Likewise, CD4+ T cells play an important role in preventing the induction of CD8+ T cell tolerance to self-antigens, as the addition of antigen-specific CD4+ T cells converts CD8+ T cell tolerance induction into CD8+ T cell activation 67. Furthermore, in several other tolerance induction systems, in vivo CD40 activation resulted in abrogation of tolerance and in T cell activation 68 69 70 71. Taken together, these studies strongly suggest that the activation state of the DCs is an important factor in determining the outcome of the DC–T cell interaction and that lymphoid DCs may be implicated in both activation and tolerization of antigen-specific CD8+ T cells.
Recent studies suggest different functions for lymphoid and myeloid DCs in the activation of CD4+ T cells, preferentially inducing Th1 and Th2 responses, respectively 40 41 42. The capacity of lymphoid DCs to drive Th1 responses is explained by their production of IL-12 and IFN-γ 27 52 72. Furthermore, the localization of the different DC and Th subsets appears to be similar. Th1 cells primarily reside in proximity to the lymphoid DCs in the central T cell zone of the PALS, whereas Th2 cells are found in the outer PALS near the B cell follicles and the marginal zone where myeloid DCs are located 73. Differential chemokine receptor expression patterns in both Th and DC subsets are essential in establishing this homing pattern 73 74 75 76. Our data add a new role for the lymphoid DCs as the main stimulator of CD8+ T cell responses specific for cell-associated antigens. This points to an interaction between lymphoid DCs, CD8+ T cells, and Th1 cells. It remains to be investigated whether CD8+ T cell cross-priming is specifically associated with and dependent on Th1 responses.
In summary, our data demonstrate that lymphoid DCs play a key role in cross-presentation of cell-associated antigens to CD8+ T cells in vivo. Because cross-presentation enables the activation of naive T cells specific for antigens that are not expressed by the APCs themselves, we envisage that lymphoid DCs will be essential for the induction of a large spectrum of CD8+ T cell responses.
Acknowledgments
We thank Drs. A. Norment, S. Clarke, K. Urdahl, and P. Fink for critical reading of the manuscript.
This work was supported by the Howard Hughes Medical Institute and by postdoctoral fellowships from the European Molecular Biology Organization (ALTF 115-1998) and from the Dutch Cancer Society to J.M.M. den Haan.
Footnotes
Abbreviations used in this paper: B6, C57BL/6; β2m, β2-microglobulin; CFSE, carboxyfluorescein diacetate succinimidyl ester; DC, dendritic cell; PALS, periarteriolar lymphatic sheaths; TAP, transporter associated with antigen presentation.
References
- Bevan M.J. Minor H antigens introduced on H-2 different stimulating cells cross-react at the cytotoxic T cell level during in vivo priming. J. Immunol. 1976;117:2233–2238. [PubMed] [Google Scholar]
- Bevan M.J. Cross-priming for a secondary cytotoxic response to minor H antigens with H-2 congenic cells which do not cross-react in the cytotoxic assay. J. Exp. Med. 1976;143:1283–1288. doi: 10.1084/jem.143.5.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigal L.J., Crotty S., Andino R., Rock K.L. Cytotoxic T-cell immunity to virus-infected non-haematopoietic cells requires presentation of exogenous antigen. Nature. 1999;398:77–80. doi: 10.1038/18038. [DOI] [PubMed] [Google Scholar]
- Huang A.Y., Golumbek P., Ahmadzadeh M., Jaffee E., Pardoll D., Levitsky H. Role of bone marrow-derived cells in presenting MHC class I-restricted tumor antigens. Science. 1994;264:961–965. doi: 10.1126/science.7513904. [DOI] [PubMed] [Google Scholar]
- Kurts C., Heath W.R., Carbone F.R., Allison J., Miller J.F., Kosaka H. Constitutive class I–restricted exogenous presentation of self antigens in vivo. J. Exp. Med. 1996;184:923–930. doi: 10.1084/jem.184.3.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz L.L., Butz E.A., Bevan M.J. Requirements for bone marrow–derived antigen-presenting cells in priming cytotoxic T cell responses to intracellular pathogens. J. Exp. Med. 2000;192:1135–1142. doi: 10.1084/jem.192.8.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yewdell J.W., Norbury C.C., Bennink J.R. Mechanisms of exogenous antigen presentation by MHC class I molecules in vitro and in vivoimplications for generating CD8+ T cell responses to infectious agents, tumors, transplants, and vaccines. Adv. Immunol. 1999;73:1–77. doi: 10.1016/s0065-2776(08)60785-3. [DOI] [PubMed] [Google Scholar]
- Kurts C., Kosaka H., Carbone F.R., Miller J.F., Heath W.R. Class I–restricted cross-presentation of exogenous self-antigens leads to deletion of autoreactive CD8+ T cells. J. Exp. Med. 1997;186:239–245. doi: 10.1084/jem.186.2.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler A.J., Marsh D.W., Yochum G.S., Guzzo J.L., Nigam A., Nelson W.G., Pardoll D.M. CD4+ T cell tolerance to parenchymal self-antigens requires presentation by bone marrow–derived antigen-presenting cells. J. Exp. Med. 1998;187:1555–1564. doi: 10.1084/jem.187.10.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett S.R., Carbone F.R., Karamalis F., Miller J.F., Heath W.R. Induction of a CD8+ cytotoxic T lymphocyte response by cross-priming requires cognate CD4+ T cell help. J. Exp. Med. 1997;186:65–70. doi: 10.1084/jem.186.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock K.L., Rothstein L., Gamble S., Fleischacker C. Characterization of antigen-presenting cells that present exogenous antigens in association with class I MHC molecules. J. Immunol. 1993;150:438–446. [PubMed] [Google Scholar]
- Kovacsovics-Bankowski M., Clark K., Benacerraf B., Rock K.L. Efficient major histocompatibility complex class I presentation of exogenous antigen upon phagocytosis by macrophages. Proc. Natl. Acad. Sci. USA. 1993;90:4942–4946. doi: 10.1073/pnas.90.11.4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norbury C.C., Hewlett L.J., Prescott A.R., Shastri N., Watts C. Class I MHC presentation of exogenous soluble antigen via macropinocytosis in bone marrow macrophages. Immunity. 1995;3:783–791. doi: 10.1016/1074-7613(95)90067-5. [DOI] [PubMed] [Google Scholar]
- Norbury C.C., Chambers B.J., Prescott A.R., Ljunggren H.G., Watts C. Constitutive macropinocytosis allows TAP-dependent major histocompatibility complex class I presentation of exogenous soluble antigen by bone marrow-derived dendritic cells. Eur. J. Immunol. 1997;27:280–288. doi: 10.1002/eji.1830270141. [DOI] [PubMed] [Google Scholar]
- Bellone M., Iezzi G., Rovere P., Galati G., Ronchetti A., Protti M.P., Davoust J., Rugarli C., Manfredi A.A. Processing of engulfed apoptotic bodies yields T cell epitopes. J. Immunol. 1997;159:5391–5399. [PubMed] [Google Scholar]
- Brossart P., Bevan M.J. Presentation of exogenous protein antigens on major histocompatibility complex class I molecules by dendritic cellspathway of presentation and regulation by cytokines. Blood. 1997;90:1594–1599. [PMC free article] [PubMed] [Google Scholar]
- Shen Z., Reznikoff G., Dranoff G., Rock K.L. Cloned dendritic cells can present exogenous antigens on both MHC class I and class II molecules. J. Immunol. 1997;158:2723–2730. [PubMed] [Google Scholar]
- Mitchell D.A., Nair S.K., Gilboa E. Dendritic cell/macrophage precursors capture exogenous antigen for MHC class I presentation by dendritic cells. Eur. J. Immunol. 1998;28:1923–1933. doi: 10.1002/(SICI)1521-4141(199806)28:06<1923::AID-IMMU1923>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Albert M.L., Sauter B., Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature. 1998;392:86–89. doi: 10.1038/32183. [DOI] [PubMed] [Google Scholar]
- Rodriguez A., Regnault A., Kleijmeer M., Ricciardi-Castagnoli P., Amigorena S. Selective transport of internalized antigens to the cytosol for MHC class I presentation in dendritic cells. Nat. Cell Biol. 1999;1:362–368. doi: 10.1038/14058. [DOI] [PubMed] [Google Scholar]
- Steinman R.M., Pack M., Inaba K. Dendritic cells in the T-cell areas of lymphoid organs. Immunol. Rev. 1997;156:25–37. doi: 10.1111/j.1600-065x.1997.tb00956.x. [DOI] [PubMed] [Google Scholar]
- Banchereau J., Steinman R.M. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Vremec D., Zorbas M., Scollay R., Saunders D.J., Ardavin C.F., Wu L., Shortman K. The surface phenotype of dendritic cells purified from mouse thymus and spleeninvestigation of the CD8 expression by a subpopulation of dendritic cells. J. Exp. Med. 1992;176:47–58. doi: 10.1084/jem.176.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vremec D., Shortman K. Dendritic cell subtypes in mouse lymphoid organscross-correlation of surface markers, changes with incubation, and differences among thymus, spleen, and lymph nodes. J. Immunol. 1997;159:565–573. [PubMed] [Google Scholar]
- Leenen P.J., Radosevic K., Voerman J.S., Salomon B., van Rooijen N., Klatzmann D., van Ewijk W. Heterogeneity of mouse spleen dendritic cellsin vivo phagocytic activity, expression of macrophage markers, and subpopulation turnover. J. Immunol. 1998;160:2166–2173. [PubMed] [Google Scholar]
- Anjuere F., Martin P., Ferrero I., Fraga M.L., del Hoyo G.M., Wright N., Ardavin C. Definition of dendritic cell subpopulations present in the spleen, Peyer's patches, lymph nodes, and skin of the mouse. Blood. 1999;93:590–598. [PubMed] [Google Scholar]
- Pulendran B., Lingappa J., Kennedy M.K., Smith J., Teepe M., Rudensky A., Maliszewski C.R., Maraskovsky E. Developmental pathways of dendritic cells in vivodistinct function, phenotype, and localization of dendritic cell subsets in FLT3 ligand-treated mice. J. Immunol. 1997;159:2222–2231. [PubMed] [Google Scholar]
- Vremec D., Pooley J., Hochrein H., Wu L., Shortman K. CD4 and CD8 expression by dendritic cell subtypes in mouse thymus and spleen. J. Immunol. 2000;164:2978–2986. doi: 10.4049/jimmunol.164.6.2978. [DOI] [PubMed] [Google Scholar]
- Burkly L., Hession C., Ogata L., Reilly C., Marconi L.A., Olson D., Tizard R., Cate R., Lo D. Expression of relB is required for the development of thymic medulla and dendritic cells. Nature. 1995;373:531–536. doi: 10.1038/373531a0. [DOI] [PubMed] [Google Scholar]
- Wu L., Nichogiannopoulou A., Shortman K., Georgopoulos K. Cell-autonomous defects in dendritic cell populations of Ikaros mutant mice point to a developmental relationship with the lymphoid lineage. Immunity. 1997;7:483–492. doi: 10.1016/s1074-7613(00)80370-2. [DOI] [PubMed] [Google Scholar]
- Wu L., D'Amico A., Winkel K.D., Suter M., Lo D., Shortman K. RelB is essential for the development of myeloid-related CD8α− dendritic cells but not of lymphoid-related CD8α1 dendritic cells. Immunity. 1998;9:839–847. doi: 10.1016/s1074-7613(00)80649-4. [DOI] [PubMed] [Google Scholar]
- Galy A., Christopherson I., Ferlazzo G., Liu G., Spits H., Georgopoulos K. Distinct signals control the hematopoiesis of lymphoid-related dendritic cells. Blood. 2000;95:128–137. [PubMed] [Google Scholar]
- Wu L., Li C.L., Shortman K. Thymic dendritic cell precursorsrelationship to the T lymphocyte lineage and phenotype of the dendritic cell progeny. J. Exp. Med. 1996;184:903–911. doi: 10.1084/jem.184.3.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba K., Inaba M., Romani N., Aya H., Deguchi M., Ikehara S., Muramatsu S., Steinman R.M. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman R.M., Inaba K. Myeloid dendritic cells. J. Leukoc. Biol. 1999;66:205–208. doi: 10.1002/jlb.66.2.205. [DOI] [PubMed] [Google Scholar]
- Banchereau J., Briere F., Caux C., Davoust J., Lebecque S., Liu Y.J., Pulendran B., Palucka K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- Kronin V., Winkel K., Suss G., Kelso A., Heath W., Kirberg J., von Boehmer H., Shortman K. A subclass of dendritic cells regulates the response of naive CD8 T cells by limiting their IL-2 production. J. Immunol. 1996;157:3819–3827. [PubMed] [Google Scholar]
- Suss G., Shortman K. A subclass of dendritic cells kills CD4 T cells via Fas/Fas-ligand–induced apoptosis. J. Exp. Med. 1996;183:1789–1796. doi: 10.1084/jem.183.4.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruedl C., Bachmann M.F. CTL priming by CD8+ and CD8− dendritic cells in vivo. Eur. J. Immunol. 1999;29:3762–3767. doi: 10.1002/(SICI)1521-4141(199911)29:11<3762::AID-IMMU3762>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Pulendran B., Smith J.L., Caspary G., Brasel K., Pettit D., Maraskovsky E., Maliszewski C.R. Distinct dendritic cell subsets differentially regulate the class of immune response in vivo. Proc. Natl. Acad. Sci. USA. 1999;96:1036–1041. doi: 10.1073/pnas.96.3.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A.L., de St Groth B.F. Antigen-pulsed CD8α1 dendritic cells generate an immune response after subcutaneous injection without homing to the draining lymph node. J. Exp. Med. 1999;189:593–598. doi: 10.1084/jem.189.3.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado-Lopez R., De Smedt T., Michel P., Godfroid J., Pajak B., Heirman C., Thielemans K., Leo O., Urbain J., Moser M. CD8α1 and CD8α2 subclasses of dendritic cells direct the development of distinct T helper cells in vivo. J. Exp. Med. 1999;189:587–592. doi: 10.1084/jem.189.3.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogquist K.A., Jameson S.C., Heath W.R., Howard J.L., Bevan M.J., Carbone F.R. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- Moore M.W., Carbone F.R., Bevan M.J. Introduction of soluble protein into the class I pathway of antigen processing and presentation. Cell. 1988;54:777–785. doi: 10.1016/s0092-8674(88)91043-4. [DOI] [PubMed] [Google Scholar]
- Huang A.Y., Bruce A.T., Pardoll D.M., Levitsky H.I. In vivo cross-priming of MHC class I-restricted antigens requires the TAP transporter. Immunity. 1996;4:349–355. doi: 10.1016/s1074-7613(00)80248-4. [DOI] [PubMed] [Google Scholar]
- Rock K.L., Goldberg A.L. Degradation of cell proteins and the generation of MHC class I-presented peptides. Annu. Rev. Immunol. 1999;17:739–779. doi: 10.1146/annurev.immunol.17.1.739. [DOI] [PubMed] [Google Scholar]
- Castellino F., Boucher P.E., Eichelberg K., Mayhew M., Rothman J.E., Houghton A.N., Germain R.N. Receptor-mediated uptake of antigen/heat shock protein complexes results in major histocompatibility complex class I antigen presentation via two distinct processing pathways. J. Exp. Med. 2000;191:1957–1964. doi: 10.1084/jem.191.11.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba K., Turley S., Yamaide F., Iyoda T., Mahnke K., Inaba M., Pack M., Subklewe M., Sauter B., Sheff D. Efficient presentation of phagocytosed cellular fragments on the major histocompatibility complex class II products of dendritic cells. J. Exp. Med. 1998;188:2163–2173. doi: 10.1084/jem.188.11.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman R.M., Turley S., Mellman I., Inaba K. The induction of tolerance by dendritic cells that have captured apoptotic cells. J. Exp. Med. 2000;191:411–416. doi: 10.1084/jem.191.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Smedt T., Pajak B., Muraille E., Lespagnard L., Heinen E., De Baetselier P., Urbain J., Leo O., Moser M. Regulation of dendritic cell numbers and maturation by lipopolysaccharide in vivo. J. Exp. Med. 1996;184:1413–1424. doi: 10.1084/jem.184.4.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis e Sousa C., Germain R.N. Analysis of adjuvant function by direct visualization of antigen presentation in vivoendotoxin promotes accumulation of antigen-bearing dendritic cells in the T cell areas of lymphoid tissue. J. Immunol. 1999;162:6552–6561. [PubMed] [Google Scholar]
- Reis e Sousa C.R., Hieny S., Scharton-Kersten T., Jankovic D., Charest H., Germain R.N., Sher A. In vivo microbial stimulation induces rapid CD40 ligand–independent production of interleukin 12 by dendritic cells and their redistribution to T cell areas. J. Exp. Med. 1997;186:1819–1829. doi: 10.1084/jem.186.11.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovere P., Vallinoto C., Bondanza A., Crosti M.C., Rescigno M., Ricciardi-Castagnoli P., Rugarli C., Manfredi A.A. Bystander apoptosis triggers dendritic cell maturation and antigen-presenting function. J. Immunol. 1998;161:4467–4471. [PubMed] [Google Scholar]
- Sauter B., Albert M.L., Francisco L., Larsson M., Somersan S., Bhardwaj N. Consequences of cell deathexposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J. Exp. Med. 2000;191:423–434. doi: 10.1084/jem.191.3.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley M., Inaba K., Steinman R.M. Dendritic cells are the principal cells in mouse spleen bearing immunogenic fragments of foreign proteins. J. Exp. Med. 1990;172:383–386. doi: 10.1084/jem.172.1.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F.P., Platt N., Wykes M., Major J.R., Powell T.J., Jenkins C.D., MacPherson G.G. A discrete subpopulation of dendritic cells transports apoptotic intestinal epithelial cells to T cell areas of mesenteric lymph nodes. J. Exp. Med. 2000;191:435–444. doi: 10.1084/jem.191.3.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulaski B.A., Yeh K.Y., Shastri N., Maltby K.M., Penney D.P., Lord E.M., Frelinger J.G. Interleukin 3 enhances cytotoxic T lymphocyte development and class I major histocompatibility complex “re-presentation” of exogenous antigen by tumor-infiltrating antigen-presenting cells. Proc. Natl. Acad. Sci. USA. 1996;93:3669–3674. doi: 10.1073/pnas.93.8.3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green E.A., Wong F.S., Eshima K., Mora C., Flavell R.A. Neonatal tumor necrosis factor alpha promotes diabetes in nonobese diabetic mice by CD154-independent antigen presentation to CD8+ T cells. J. Exp. Med. 2000;191:225–238. doi: 10.1084/jem.191.2.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiodoni C., Paglia P., Stoppacciaro A., Rodolfo M., Parenza M., Colombo M.P. Dendritic cells infiltrating tumors cotransduced with granulocyte/macrophage colony-stimulating factor (GM-CSF) and CD40 ligand genes take up and present endogenous tumor-associated antigens, and prime naive mice for a cytotoxic T lymphocyte response. J. Exp. Med. 1999;190:125–133. doi: 10.1084/jem.190.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fossum S., Rolstad B. The roles of interdigitating cells and natural killer cells in the rapid rejection of allogeneic lymphocytes. Eur. J. Immunol. 1986;16:440–450. doi: 10.1002/eji.1830160422. [DOI] [PubMed] [Google Scholar]
- Carbone F.R., Bevan M.J. Class I–restricted processing and presentation of exogenous cell-associated antigen in vivo. J. Exp. Med. 1990;171:377–387. doi: 10.1084/jem.171.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toes R.E., Blom R.J., van der Voort E., Offringa R., Melief C.J., Kast W.M. Protective antitumor immunity induced by immunization with completely allogeneic tumor cells. Cancer Res. 1996;56:3782–3787. [PubMed] [Google Scholar]
- de St Groth B.F. The evolution of self-tolerancea new cell arises to meet the challenge of self-reactivity. Immunol. Today. 1998;19:448–454. doi: 10.1016/s0167-5699(98)01328-0. [DOI] [PubMed] [Google Scholar]
- Bennett S.R., Carbone F.R., Karamalis F., Flavell R.A., Miller J.F., Heath W.R. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 1998;393:478–480. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- Ridge J.P., Di Rosa F., Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 1998;393:474–478. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- Schoenberger S.P., Toes R.E., van der Voort E.I., Offringa R., Melief C.J. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- Kurts C., Carbone F.R., Barnden M., Blanas E., Allison J., Heath W.R., Miller J.F. CD4+ T cell help impairs CD8+ T cell deletion induced by cross-presentation of self-antigens and favors autoimmunity. J. Exp. Med. 1997;186:2057–2062. doi: 10.1084/jem.186.12.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl L., den Boer A.T., Schoenberger S.P., van der Voort E.I., Schumacher T.N., Melief C.J., Offringa R., Toes R.E. CD40 activation in vivo overcomes peptide-induced peripheral cytotoxic T-lymphocyte tolerance and augments anti-tumor vaccine efficacy. Nat. Med. 1999;5:774–779. doi: 10.1038/10495. [DOI] [PubMed] [Google Scholar]
- Sotomayor E.M., Borrello I., Tubb E., Rattis F.M., Bien H., Lu Z., Fein S., Schoenberger S., Levitsky H.I. Conversion of tumor-specific CD4+ T-cell tolerance to T-cell priming through in vivo ligation of CD40. Nat. Med. 1999;5:780–787. doi: 10.1038/10503. [DOI] [PubMed] [Google Scholar]
- Garza K.M., Chan S.M., Suri R., Nguyen L.T., Odermatt B., Schoenberger S.P., Ohashi P.S. Role of antigen-presenting cells in mediating tolerance and autoimmunity. J. Exp. Med. 2000;191:2021–2028. doi: 10.1084/jem.191.11.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefrancois L., Altman J.D., Williams K., Olson S. Soluble antigen and CD40 triggering are sufficient to induce primary and memory cytotoxic T cells. J. Immunol. 2000;164:725–732. doi: 10.4049/jimmunol.164.2.725. [DOI] [PubMed] [Google Scholar]
- Ohteki T., Fukao T., Suzue K., Maki C., Ito M., Nakamura M., Koyasu S. Interleukin 12–dependent interferon γ production by CD8α1 lymphoid dendritic cells. J. Exp. Med. 1999;189:1981–1986. doi: 10.1084/jem.189.12.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randolph D.A., Huang G., Carruthers C.J., Bromley L.E., Chaplin D.D. The role of CCR7 in TH1 and TH2 cell localization and delivery of B cell help in vivo. Science. 1999;286:2159–2162. doi: 10.1126/science.286.5447.2159. [DOI] [PubMed] [Google Scholar]
- Cyster J.G. Leukocyte migrationscent of the T zone. Curr. Biol. 2000;10:R30–R33. doi: 10.1016/s0960-9822(99)00253-5. [DOI] [PubMed] [Google Scholar]
- Aliberti J., Reis e Sousa C., Schito M., Hieny S., Wells T., Huffnagle G.B., Sher A. CCR5 provides a signal for microbial induced production of IL-12 by CD8α1 dendritic cells. Nat. Immunol. 2000;1:83–87. doi: 10.1038/76957. [DOI] [PubMed] [Google Scholar]
- Sato N., Ahuja S.K., Quinones M., Kostecki V., Reddick R.L., Melby P.C., Kuziel W.A., Ahuja S.S. CC chemokine receptor (CCR)2 is required for langerhans cell migration and localization of T helper cell type 1 (Th1)-inducing dendritic cells. Absence of CCR2 shifts the Leishmania major–resistant phenotype to a susceptible state dominated by Th2 cytokines, B cell outgrowth, and sustained neutrophilic inflammation. J. Exp. Med. 2000;192:205–218. doi: 10.1084/jem.192.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]