

Abstract
DM facilitates formation of high affinity complexes of peptide–major histocompatibility complex (MHC) by release of class II MHC–associated invariant chain peptide (CLIP). This has been proposed to occur through discrimination of complex stability. By probing kinetic and conformational intermediates of the wild-type and mutant human histocompatibility leukocyte antigen (HLA)-DR1–peptide complexes, and examining their reactivities with DM, we propose that DM interacts with the flexible hydrophobic pocket 1 of DR1 and converts the molecule into a conformation that is highly peptide receptive. A more rigid conformation, generated upon filling of pocket 1, is less susceptible to DM effects. Thus, DM edits peptide–MHC by recognition of the flexibility rather than stability of the complex.
Keywords: antigen processing, molecular conformation, HLA-DR antigens, surface plasmon resonance, fluorometry
Introduction
HLA-DM or H2-M in mice, a nonclassical HLA molecule, has been shown to play a critical role in loading antigenic peptides to newly synthesized MHC class II molecules. The significance of DM function is exemplified by the finding that MHC class II molecules isolated from DM-negative cells were largely occupied with class II MHC–associated invariant chain peptide (CLIP) 1. Consequently, DR molecules isolated from these mutant cells were prone to SDS denaturation at room temperature 2. Since its discovery, several other functions have been attributed to DM, including stabilization of empty class II molecules 3 4 and peptide editing for selection of peptides that form stable complexes with MHC class II 5 by an enzyme-like mechanism 6 7. Nevertheless, although DM accelerates dissociation of some peptide–MHC complexes, other complexes remain resistant to DM-induced dissociation 5 7 8 9 10 11. Furthermore, a chaperone-like function for DM has also been proposed 5. The crystal structures of HLA-DM 12 and H2-M 13 have been resolved and they do not exhibit a groove to accommodate a peptide. Thus, the mechanism of DM reactivity remains unsolved.
It is now established that conformational changes in the structure of MHC molecules are associated with peptide binding 14 15 16 17 18 19 20 21. Class II molecules that are empty or have lost their peptide are less rigid (floppy conformation) and migrate with a larger apparent molecular mass or fall apart on SDS-PAGE. Stable peptide–class II complexes migrate faster than the floppy conformation, indicating their more compact and rigid conformation (compact dimer [14–17, 22]). Recently, we demonstrated that the SDS stability of DR1 complexes correlates with the burial of hydrophobic residues in pocket 1. DR1 molecules in complex with peptides that lack anchor residue in corresponding pocket 1 form floppy conformations and fall apart in SDS gels 23. When a peptide like influenza virus hemagglutinin (HA)306–318, which has a tyrosine anchor fitting in pocket 1, binds DR1, the complex becomes compact and resists SDS-induced denaturation 17 24.
The rate-limiting step in formation of stable peptide–MHC complexes occurs in generation of a receptive class II conformation 19 20. A receptive conformation is created when a resident peptide dissociates. At this time, a second peptide can bind class II almost spontaneously and stochiometrically 19 20. This receptive molecule has a flexible conformation with a very short half-life. In the absence of any free peptide, this flexible molecule rapidly reverts to a “closed” conformation that binds slowly to the subsequently offered peptide 20. A mutant DR1, DR1βG86Y, that remains permanently in receptive or “open” form was designed by introducing a single site-specific mutation. β86G was changed to tyrosine presumably to fill pocket 1 and rescue the flexible pocket from collapsing 23 24. Thus, the conformation of mutant DR1 partially resembles wild-type (wt)DR1 in complex with a peptide that has tyrosine as the main anchor. The molecule is rigid and open, and thus can bind and dissociate peptides efficiently.
In accord with these in vitro findings, the process of synthesis, maturation, and export of class II molecules through the endocytic pathway involves conformational changes in the structure of MHC 25 26. A class II invariant chain peptide, CLIP, acts as a surrogate short-lived peptide for shaping of MHC class II 27. In peptide-loading compartments, CLIP dissociates, leaving a receptive groove “open” for efficient peptide binding. Similar to empty soluble DR1 molecules that aggregate in the absence of peptides 22, class II molecules tend to aggregate in the absence of invariant chains 28.
As DM seems to be involved in class II peptide–loading processes, we considered different conformations of MHC as potential ligands for DM. By probing conformational changes that exist between wt and DR1βG86Y and their reactivities with DM, we propose the following mechanism for DM function: through hydrophobic interactions around pocket 1, DM recognizes a flexible conformation of pocket 1 of DR1 and accelerates dissociation of CLIP or a peptide that lacks the pocket 1 anchor. This interaction converts the molecule into a highly receptive conformation for peptide binding. A compact class II that is generated upon binding of a peptide with a large hydrophobic side chain at corresponding pocket 1 yields a rigid conformation that is less responsive to DM.
Materials and Methods
Production of Recombinant Soluble DR1 and DM Proteins.
Soluble DR1 proteins were expressed and purified as described 23. DM, expressed and isolated as described previously 8, was further purified by gel filtration (Superdex 200; Amersham Pharmacia Biotech) and ion-exchange chromatography (Mono-Q Sepharose; Amersham Pharmacia Biotech [12]).
Peptide Synthesis and Labeling.
HA peptides, CPKYVKQNTLKLAT, HA305–318, and several variants, HAY308A (CPKAVKQNTLKLAT), and Cys-HAAnchorless (CPKAVKANGAKAAT) were purified to apparent homogeneity of >95% by reverse-phase preparative HPLC and their identities were confirmed by mass spectrometry. Then, 0.15 mM HA305–318 or HAAnchorless, containing a single cysteine in 10 ml PBS, was incubated with 25 μl of 75 mM fluorescein-5-maleimide (Molecular Probes) in N,N-dimethylformamide (DMF) for 1 h at room temperature. The samples were concentrated to 1 ml by SpeedVac (Savant Instruments, Inc.). The excess free fluorescence label was removed by passing through a Sephadex G-10 column (Amersham Pharmacia Biotech). The concentration was determined by the extinction coefficient of fluorescein-5-maleimide (83 mM−1cm−1).
Peptide Association and Dissociation Assays.
Purified wtDR1 or DR1βG86Y (2.4 μM) was incubated in the absence or presence of 1 μM DM with 100 μM fluorescence-labeled HAAnchorless peptide for various times in 0.15 M citric phosphate buffer, pH 6.0, at 37°C. After removal of free peptides by a Sephadex G-50 spin column equilibrated with PBS, fluorescence emission of the DR–FITC–HAAnchorless complex was measured at 514 nm with an excitation at 490 nm on an LS-50B spectrofluorimeter (PerkinElmer) at room temperature. The fluorescence emission of the DR–7-amino-4-methyl-coumarin-3-acetic acid (AMCA)–HAAnchorless complex was measured at 445 nm with excitation at 350 nm. Dissociation experiments with wtDR1 and DR1βG86Y complexes were performed essentially as described previously 20 23.
Data Analysis.
All the raw association data in Fig. 2 were fitted into either single or double exponential association equations as follows:
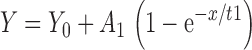 |
1 |
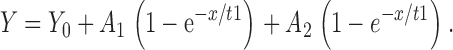 |
2 |
Figure 2.



The effects of DM on the generation of receptive DR1. (a and b) The effects of DM on the binding of HAAnchorless peptide to wt and DR1βG86Y. 2.4 μM DR1βG86Y (a) and wt (b) were incubated with 100 μM FITC-HAAnchorless peptide for various times in the absence (○) and presence (▪) of DM (1 μM, 0.15 M citrate phosphate buffer, pH 6.0, at 37°C). The calculated rates for peptide binding to DR1βG86Y, in the absence and presence of DM, to wtDR1 in the presence of DM, and to the receptive wtDR1, were all very similar and ranged between 0.47 and 0.61 s−1μM−1. (c) The kinetics of HA peptide binding to dissociating DR1–HAAnchorless complex. 50 μM FITC-HA was incubated with 2.4 μM DR1–AMCA–HAAnchorless complex (isolated by size-exclusion chromatography, citrate buffer, pH 6.0) for various times at 37°C. The unbound peptides were then removed by Sephadex G-50 spin columns in PBS. Samples were read at two wavelengths of 345 (AMCA) and 514 nm (fluorescein). The dissociation data (▪) were fit to a single exponential decay curve. The HA association (○) data were fit to a single exponential binding curve.
All the raw dissociation data were fitted into either single or double exponential dissociation equations as follows:
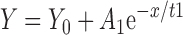 |
3 |
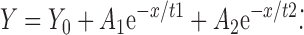 |
4 |
Intrinsic Fluorescence Measurement.
Steady-state tryptophan fluorescence measurements were done in 0.15 M citric phosphate buffer, pH 6.0, at 37°C. For this experiment, 1 μM DM and 2 μM DR1 were used. An SLM 48000 spectrofluorimeter configured in the T format was used. Emission was monitored from 310 to 500 nm with excitation at 295 nm. The excitation slit width was 4 nm. The temperature was controlled by a circulating temperature bath (Neslab), and samples were continuously stirred by a magnetic stirrer.
Real Time Binding Experiments.
For peptide immobilization to the biosensor CM5 chip (BIAcore), the surface was activated by injecting a mixture containing equal amounts of 0.05 M N-hydroxy-succinimide (NHS) and 0.2 M N-ethyl-N′ (dimethylaminopropyl) carbodiimide (EDC) at a low rate of 10 μl/min for 7 min. Amino groups were generated by injection of ethylenediamine hydrochloride (1 M, pH 6.0) for 8 min at a flow rate of 10 μl/min. To introduce maleimido groups, a solution of the heterobifunctional reagent, Sulfo-succinimidyl 4-(p-maleimidophenyl)-butyrate (SMPB; Pierce Chemical Co.), was pumped over the surface for 10 min. Cysteine peptides (100–200 μM) were injected to desired levels.
All binding experiments with use of biosensor were performed in 0.15 M citrate/phosphate buffer at pH 6.0. The pH 6.0 was chosen because the optimal activity of DM is shown to be at the pH range of 4.5–6. However, at pH levels <6.0, the BIAcore dextran chip undergoes some changes that lead to a false increase in the resonance unit (RU) levels. Thus, we tested the above pH range for our BIAcore experiments, and selected pH 6.0 because the observed bindings were real, and complexes formed dissociated within the expected dissociation rates for different pairs of peptide–MHC. On the contrary, at pH ≤ 5.8 the nonspecific effects were apparent. Soluble DR1 was incubated with 200 μM HAY308A peptide at 37°C for 36 h. Size-exclusion HPLC (Superdex 200 gel filtration column; Amersham Pharmacia Biotech) was then used to isolate the sDR1–HAY308A complexes. The complexes were further concentrated by centrifugal filter devices (Millipore). The concentration of the complex was determined by absorbance at 280 nm with the extinction coefficient of 77,000 M−1cm−1. The complex was incubated at 37°C for 20 min with or without 1 μM DM and injected to the peptide surfaces. For dissociation experiments, 9 μM DM in citrate buffer, pH 6.0, was injected over the preformed complexes, wtDR1–HA306–318, wtDR1–HAAnchorless, and DR1βG86Y–HAAnchorless, and dissociation sensograms were collected. DR1βG86Y–HA305–318 surface was used as a control surface for establishing the background signal levels due to injection of DM.
For biosensor experiments with extended contact time, a BIAcore X instrument was used in the external injection mode. Samples were delivered by an externally computer-controlled syringe pump (model 402; Gilson) in a configuration similar to that described previously 29, modified to allow the use of smaller sample volumes. Because of the temperature, 37°C, and long duration (3 h), microscopic bubbles formed that caused the unusual appearance of the sensogram. For other experiments, we used a BIAcore 2000.
Results
DR1βG86Y Molecules That Have a Shallow Pocket 1.
HAAnchorless and HAY308A are short-lived peptides that dissociate from DR1 with t 1/2 of 30–120 min and thus leave DR1 that is receptive to peptide binding. Experimental evidence has shown that alanine substitution for threonine at pocket 6 of HA306–318 sequence improved binding by threefold 30. To design a true HAAnchorless peptide, we made a threonine to glycine substitution at position 313 20 23.
To generate DR1 molecules that would not bind peptides stably and would mimic kinetic and structural intermediates, we designed a mutant DR1 molecule with a shallow pocket 1 that would not allow fitting of a large anchor residue. A single site-specific mutation for glycine at position β86 to tyrosine was introduced. Residue β86 was chosen because DR alleles exhibit glycine-valine dimorphism at that position, suggesting that a bigger sidechain would not cause gross structural changes in the protein. In support of this, I-Ek has a phenylalanine for β86 31. βG86 was changed to a tyrosine because HA306–318, which is both the best binding and best characterized peptide of DR1, has a tyrosine at that position as seen from the DR1–HA306–318 crystal structure 32.
As predicted, DR1βG86Y–HAAnchorless complexes dissociated with rates similar to that of wtDR1–HAAnchorless (Fig. 1) and exhibited a more rigid and compact structure resistant to denaturation by SDS and heat, even in the absence of peptides (23 24; Table ). DR1βG86Y binds effectively to peptides that contain alanine at the corresponding pocket 1 position via a kinetic on rate that fits a single exponential equation (Fig. 2 a). This is in contrast to empty sDR1 that binds the same peptide slowly via biphasic kinetics, and maximal binding was not obtained even after 24 h of incubation in the presence of 100 μM peptide (Fig. 2 b). The binding of wtDR1 that has just lost tenant peptide, and is in receptive form, follows a single exponential rate (Fig. 2 c). The biphasic kinetic relation in Fig. 2 b might be due to heterogeneity of DR1 molecules, some binding peptide rapidly and others that bind slowly. The fast phase might be because of DR1 molecules in complex with unknown short-lived peptides that dissociate rapidly.
Figure 1.

Peptide dissociation from wt and DR1βG86Y. Dissociation kinetics of wt (○) and sDR1βG86Y (•) complexes. DR1 in complex with FITC-labeled HAAnchorless was produced and separated. The labeled complexes were dissociated in the presence of 100 times molar excess of relevant unlabeled peptides at 37°C in PBS for various times. The fluorescence of the labeled complex before dissociation (f 0) is arbitrarily assigned a value of 1.0. The fluorescence of the labeled complex after dissociation for various times is expressed as a percentage of f 0. The dissociation data are fitted to a single exponential curve that yields t 1/2 of 1.8 ± 0.25 h. The y axis represents arbitrary fluorescence units.
Table 1.
DR1 βG86Y Has a More Rigid Conformation than WtDR1
| MHC protein | Pocket 1 | SDS stability | Tm |
|---|---|---|---|
| °C | |||
| DR1 (−P) | Deep | Unstable | 67 |
| DR1βG86Y (−P) | Shallow | Stable | 72 |
| DR1–HA306–318 | − | Stable | 84 |
Effect of DM on Peptide Release from Wt and DR1βG86Y.
Previous work has shown that DM enhances dissociation of short-lived peptide–class II complexes 5 8 33 34 35. To determine if DM affects the wt and DR1βG86Y in different ways, we examined the molar ratio of DM necessary to facilitate dissociation of wtDR1 in complex with FITC-HAAnchorless. At the lowest tested DM to wtDR1 ratio of 1:5, >40% of the complexes dissociated within 20 min incubation (Fig. 3 a). However, after 40 min incubation, >90% of complexes had dissociated (Fig. 3 b). In contrast, DM did not affect dissociation of DRβG86Y–HAAnchorless at this molar ratio. Furthermore, upon a 20-min incubation (Fig. 3 a), only 10–15% of complexes dissociated at the highest molar ratio. However, at the maximum ratio of ∼1:1 of DM to DRβG86Y–HAAnchorless, only 50% of complexes dissociated within 40 min (Fig. 3 b). For maximum difference in dissociation of complexes of the wt and DRβG86Y upon DM effects, we chose the DM/DR ratio of 1:2.5, and an incubation time of 20 min for future experiments.
Figure 3.


The effects of DM on dissociation of DR1–peptide complex in pH 6.0. 2.4 μM wtDR1 (black bars) and DR1βG86Y (white bars) in complex with FITC–HAAnchorless were incubated with DM (citrate buffer, pH 6.0) for 20 min (a) or 40 min (b), and dissociation of the complexes was measured. At the physiological ratio of 1:5 DM/DR (reference 45), DM did not affect dissociation of the DR1βG86Y–HAAnchorless complexes, whereas at higher molar ratios, up to 50% of the complexes dissociated after 40 min of incubation.
We compared the formation of FITC-peptide–MHC complexes for the DR1βG86Y and wtDR1 in the presence of 1 μM DM. Fig. 2, a and b, show comparison of association rates of the DR1βG86Y and wtDR1 interacting with HAAnchorless in the absence and after addition of DM. In the absence of DM, a clear difference in the shape and the rates of wt and DR1βG86Y binding to HAAnchorless established the receptive conformation of the DR1βG86Y. Remarkably, DM converted the biphasic peptide-binding pattern of wtDR1 to the monophasic-binding pattern of DR1βG86Y. A small (10–15%) increase in the plateau level might be due to further stabilization of the DR1βG86Y by DM.
Peptide Binding of DR1 Assisted by DM in Real Time.
To determine the effects of DM on binding of receptive DR1 to peptides and dissociation of complexes formed in real time, we developed a peptide-binding assay by BIAcore surface plasmon resonance (SPR), as the fluorescence assay does not allow accurate measurement of very rapid interactions. We took advantage of the fact that prior association with low affinity peptides induces a receptive conformation in class II 19 20. First, DR1βG86Y was used to establish the assay because it binds peptide more efficiently and stays in receptive form once the peptide is dissociated (20; Fig. 2 a). The peptide surfaces were generated first by Cys coupling to the CM5 chip. Fig. 4 a shows a sensogram of DR1βG86Y binding to the HAAnchorless surface. As the steady-state binding can be reached 9,000 s after protein injection, in the following experiments only the early binding curves were generated. The estimated dissociation rate was 5.6 × 10−5 s−1, and the t 1/2 ∼ 3.4 h. This is in agreement with the rates measured by the use of conventional fluorescence assays (23; Fig. 1).
Figure 4.



Peptide binding of DR1 by BIAcore SPR in real time. (a) The binding of DR1βG86Y to immobilized peptide in real time. Cys-HAAnchorless peptide was immobilized in a biosensor flow cell and sensograms were obtained by injection of DR1βG86Y–HAY308A complexes (citrate buffer, pH 6.0). The long-term experiments were performed in a BIAcore X instrument (for details, see Materials and Methods). The binding curve is expressed as RU as a function of time. The DR1βG86Y–HAY308A complexes were prepared by preincubation of DR1βG86Y and HAY308A peptide followed by size-exclusion purification. Running buffer was citrate buffer plus 0.01% Tween 20, pH 6.0. One out of two experiments is shown. (b and c) The effects of DM on binding of the wtDR1 (b) and DR1βG86Y (c) to the peptide surfaces; 2.4 μM wtDR1 or DR1βG86Y, in complex with HAY308A peptide, was isolated by the procedure described in the legend to Fig. 1, and then was incubated at 37°C with or without 1 μM DM for 20 min. Samples (citrate buffer, pH 6.0) were injected over three different peptide surfaces (cys-HA, cys-HAAnchorless, and the control peptide) of BIAcore 2000 at a flow rate of 4 μl/min for 7 min followed by washout (citrate buffer plus 0.01% Tween 20, pH 6.0) at a flow rate of 5 μl/min. Sensograms of DR1 binding are shown in different colors: in the absence of DM (blue), presence of DM (red), and DM plus soluble peptide for blocking (green). One out of three experiments is shown.
To examine formation of DR1–peptide complexes in real time, HA305–318, HAAnchorless, and a non–DR1-binding peptide, CLSPFPFDL, were conjugated to different surfaces of a CM5 chip. Wt or DR1βG86Y was shaped to be receptive by incubating with short-lived HAY308A peptide (see details in Materials and Methods) before it was injected over the surface and binding was monitored. Fig. 4 b shows wtDR1 binding to HAAnchorless and HA305–318 surfaces in the presence and the absence of DM. Binding was significantly higher if DM was preincubated with wtDR1 and was present in the sample mix during the injection. DM enhanced binding of wtDR1 to the peptide surface, perhaps by increasing the dissociation of HAY308A from DR1 and by maintaining the receptive conformation of the empty protein once the bound peptide was dissociated. The specificity of DR1 binding to the peptide surface was tested by mixing soluble HA306–318 peptide (5 μM) with wtDR1 1 min before injection over the peptide surfaces. As DR1 is in receptive form, stable DR1–HA306–318 complexes form in solution and therefore, when the sample is injected over the peptide surface, no binding should occur 20. Indeed, as shown, soluble HA306–318 blocked wtDR1 from binding to HAAnchorless and HA305–318 surfaces.
In contrast to wtDR1 that bound to peptide surface poorly in the absence of DM, DR1βG86Y bound to HAAnchorless surface efficiently. Preincubation with DM increased peptide binding by only 10–15%. Again, the resultant DR1βG86Y when in complex with soluble peptide failed to bind to the immobilized peptide surface. Accessibility of immobilized peptides for efficient binding might be a limitation in this assay. Also, the competition between the dissociated peptides (from DR1–HAY308A under the DM influence) present in the sample mixture together with DM that enhances their binding as well may generate binding profiles that represent slower rates than those that might be obtained by solution studies. Nonetheless, the strength of this assay is in measurement of fast dissociation rates such as those shown in Fig. 5.
Figure 5.

Dissociation of wt and DR1βG86Y complexes by DM in real time. Peptide–DR1 surfaces were generated as in the legend to Fig. 4, and 9 μM DM was passed through four different peptide–DR1 complex surfaces. For better comparison, all surfaces indicating different RU values were given zero values as the initiation point. DR1βG86Y–HA surface was used as a negative control, as binding was negligible (Fig. 4). The negative sensograms are due to lower salt concentration in DM-containing buffer, and perhaps lack of Tween 20 that was present in the running buffer. Shown are wtDR1–HAAnchorless (red), wtDR1–HA (blue), DR1βG86Y–HAAnchorless (red), and the control mock DR1βG86Y–HA (blue). In the presence of DM, dissociation rates for the wtDR1–HAAnchorless were estimated to be 3.8 × 10−3 s−1 (t 1/2 ∼ 3.0 min); for wtDR1–HA, 5.6 × 10−7 s−1 (t 1/2 ∼ 344 h); and for the DR1βG86Y–HAAnchorless, 8.5 × 10−5 s−1 (t 1/2 ∼ 2.25 h). Inset, a replot of data after subtraction of the mock DR1βG86Y–HA control surface to provide an easy evaluation of the dissociation rates of the complexes in the presence of DM. One out of two experiments is shown.
Dissociation of the WtDR1 and DR1βG86Y from HAAnchorless Surface by DM.
The observation that DM did not act on DR1βG86Y efficiently suggested that DM recognized conformations of empty pocket 1 of wtDR1. In the previous experiment in Fig. 4, wtDR1 formed complexes with HA306-318 and HAAnchorless peptide surfaces, and DR1βG86Y formed complexes with HAAnchorless peptide on the biosensor chip. Fig. 5 depicts the effects of injected DM on the dissociation of these complexes. Only the wtDR1–HAAnchorless surface showed a significant decrease (40 RU) after DM injection (t 1/2 ∼ 3 min). The half-time for dissociation of DR1βG86Y–HAAnchorless complexes in the presence of DM was 135 min; thus, the presence of DM accelerated dissociation of the wtDR1–HAAnchorless compared with the DR1βG86Y–HAAnchorless complexes by 45-fold. The HA306–318 complexes dissociated within a t 1/2 ∼ 344 h that is consistent with long-term solution studies, and was not accelerated upon DM effects. The data clearly demonstrate that DM effectively enhanced dissociation of wtDR1 in complex with a peptide with alanine at pocket 1 position but that the wtDR1–HA306–318 and DR1βG86Y–HAAnchorless surfaces were resistant to DM dissociation.
Measurements of Tryptophan Fluorescence upon DR1 and DM Interaction.
Wiley's group 12 has suggested that two tryptophan residues, α62 and β120, located on the lateral surface of DM, are partially exposed and are candidates for binding to DR. Moreover, a glycosylation mutant of DR that interfered with DM effects was suggestive of the NH2-terminal side of DR1 as a target for DM 36. We examined this possibility by detecting intrinsic tryptophan fluorescence of wtDR1 and DR1βG86Y and DM. The intrinsic tryptophan of each sample was excited at 295 nm to minimize the interference of tyrosine fluorescence, and the emission spectra were obtained from 310 to 500 nm. The fluorescence spectra of DM and empty wtDR1 alone have intensity maxima ∼340 nm (Fig. 6 a). These two spectra can be additive if the DM and DR1 do not interact with each other. However, if the DM and DR interact, causing the exposed tryptophans to be buried at their interface, tryptophan fluorescence should increase. In accord with this, when DM and empty wtDR1 were mixed, the intensity maxima of the emission fluorescence spectra increased by 10–12% (Fig. 6 a). Consistent with the rapid rate of interactions between DM and DR, there was no further significant changes detected for wtDR1 and DM monitored for 1 h after mixing (Fig. 6 a). Interestingly, mixing of DM with wtDR1–HAAnchorless produced 7% increase in fluorescence (Fig. 6 b). Moreover, in agreement with the above data that DR1βG86Y is not an optimal ligand for DM, empty DR1βG86Y and DM did not yield increased fluorescence (Fig. 6 c). In addition, no fluorescence increase was observed when DM was mixed with the wtDR1–HA306–318 complex, suggesting lack of interactions that would bury exposed tryptophans (Fig. 6 d). Overall, these observations support the notion that DM interacts with empty and flexible pocket 1 of DR1 and that the interaction causes burial of exposed tryptophans present in DR1 and DM.
Figure 6.
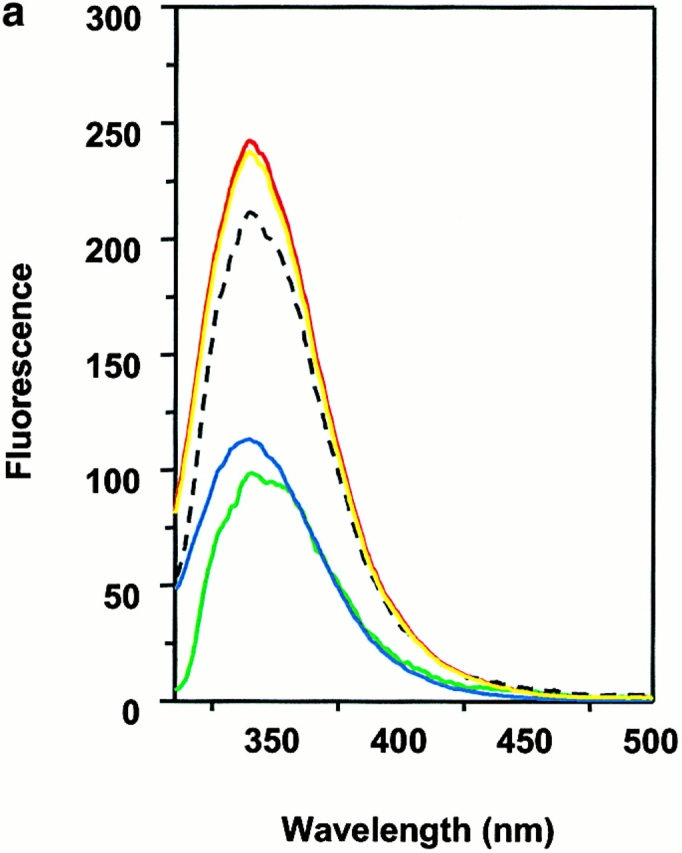
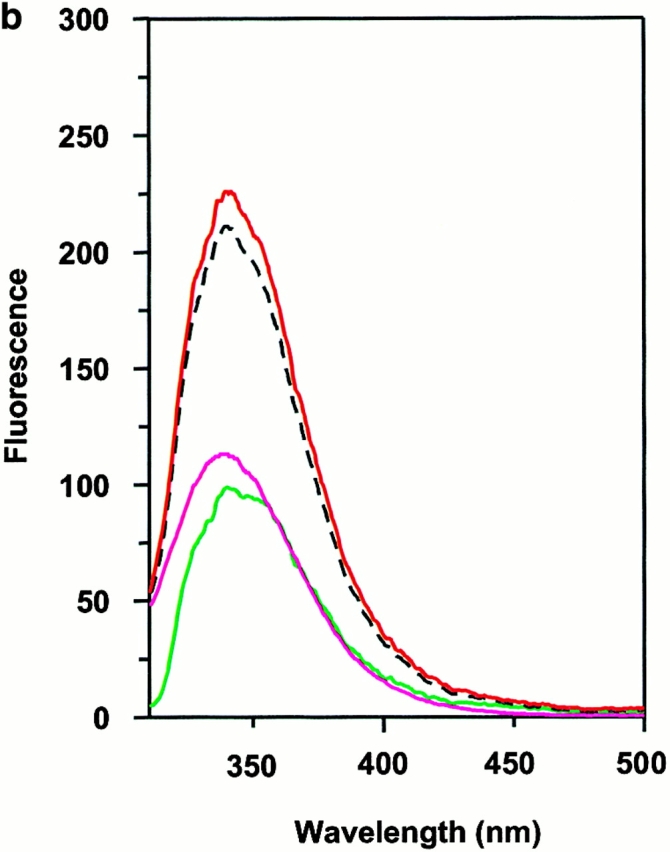
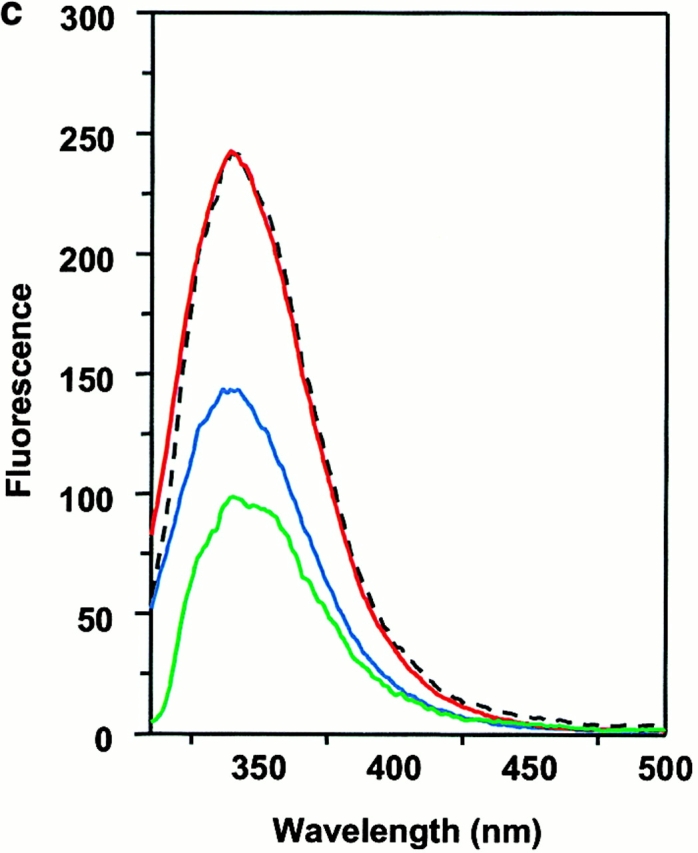
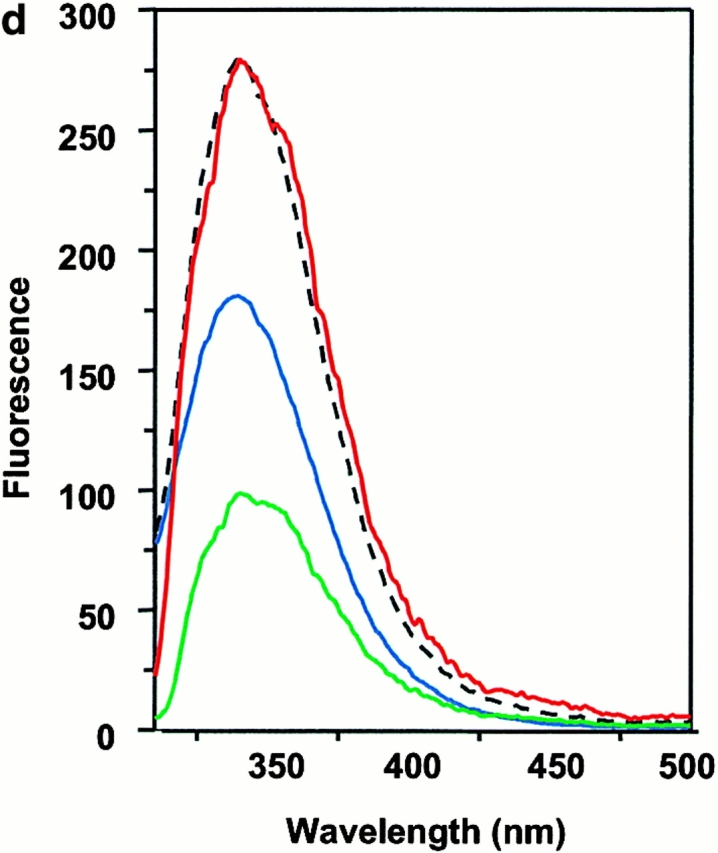
Interaction between DM and DR1 involves intrinsic tryptophan fluorescence changes. Intrinsic tryptophan fluorescence of DM interacting with empty wtDR1 (a), wtDR1–HAAnchorless (b), empty DR1βG86Y (c), and wtDR1–HA (d) complexes. DM and DR1 molecules (in citrate buffer, pH 6.0) were mixed and tryptophan fluorescence was measured immediately (red) and 1 h after mixing (yellow) for wtDR1 plus DM sample. The intrinsic tryptophans of each sample were excited at 295 nm to minimize the interference of tyrosine fluorescence and the emission spectra were obtained from 310 to 500 nm. Measurements for DM alone (green), DR alone (blue), and the mathematical sum (dashed line) are shown. The average fluorescence intensities from two separate measurements are plotted.
Discussion
In this paper, we show that DM may function through conformational recognition. Several observations support this model. Previously published results have proposed that the stability of peptide–MHC complexes is the determinant of DM reactivity. Our results here oppose this view by providing evidence that the conformational differences in an empty/flexible hydrophobic pocket 1 of DR1 is what is recognized by DM.
Formation of peptide–MHC kinetic and structural intermediate complexes leads to shaping of the peptide-binding site of class II to a conformation that is highly receptive to stable peptide binding. The rate-limiting step in the formation of stable peptide binding is therefore the generation of this peptide-receptive conformation. In this study, we have used several experimental approaches and generated a mutant DR1 with a single amino acid substitution inside its pocket 1 to reduce the depth and hydrophobicity of this pocket. The peptide-binding characteristics of DR1βG86Y resemble that of wtDR1 in a peptide-receptive conformation (Fig. 2). The receptive characteristics of this mutant might be due to the presence of tyrosine that partially fills the pocket and prohibits the protein from stable peptide binding. In addition, the presence of tyrosine induces rigidity of pocket 1 as shown by resistance to SDS and an increase in melting temperature (T m) by 5°C (Table ). This rigidity may render empty DR1βG86Y less susceptible to collapsing. We show that DM recognizes an intermediate flexible conformation of wtDR1 that is characteristic of empty DR1, or DR1 that is occupied with peptides lacking the main anchor. This interaction generates and maintains a peptide-receptive form of the protein.
DR1βG86Y forms only short-lived complexes with peptides yet serves as a poor substrate for DM function. As the conformations of DR1βG86Y and wtDR1 in complex with short-lived peptides are different, and DM can distinguish these complexes, we propose that DM recognizes its substrate via structural differences. In the absence of DM, a clear difference in the shape and the rates of wt and DR1βG86Y binding to HAAnchorless documents the receptive conformation of the DR1βG86Y (Fig. 2).
It is remarkable that DM converts the biphasic peptide-binding pattern of wtDR1 to the monophasic-binding pattern of DR1βG86Y. A small (10–15%) increase in the plateau level from the DM interaction might be the result of further stabilization of the DR1βG86Y by DM. The earlier phase in binding of wtDR1 in the presence of DM may represent binding of a cohort of the wtDR1 that is in receptive form and binds peptide rapidly, whereas the second phase represents HLA-DR1 that binds peptide slowly 20. Similar biphasic binding patterns have previously been observed for DR1 molecules binding peptides in acidic, but not neutral pH 19. Rabinowitz et al. also demonstrated that at acidic pH, preshaped wtDR1 binding to HA306–318 peptide is monophasic 19. Interestingly, adding DM to the empty wtDR1 and peptide samples converts biphasic binding to monophasic binding. The monophasic peptide-binding rate is very similar to the binding rate of the DR1βG86Y and preshaped wtDR1. This dramatic change in peptide-binding pattern marks an important and novel function of DM: that is, generation of a homogeneous population of MHC proteins capable of binding peptides rapidly.
To further elucidate the mechanism catalyzed by DM, we developed a real time peptide-binding assay. Although the BIAcore SPR assay for binding of the MHC class I to immobilized peptides has been successful 37 38, this approach has failed for class II peptide binding, presumably because the peptide-binding grooves of class II molecules are either occupied, or collapse after peptide release 17. As we and others now have established that class II molecules can be shaped to a peptide-receptive conformation that exhibits accelerated peptide association, it was likely that direct MHC class II binding to peptides could be measured in real time. Using BIAcore SPR, we show that DM assists peptide exchange more efficiently for wtDR1 than for DR1βG86Y. A three- to fourfold increase in binding of the DR1βG86Y (rate ∼119 × 10−3 s−1) versus wtDR1 (rate ∼35 × 10−3 s−1) in binding to HAAnchorless surface in the absence of DM was observed. A reason for a difference in efficiency of DR1βG86Y in binding to the peptide surface relative to the wt protein is the extreme susceptibility of the wtDR1 in receptive form to partial denaturation unless stabilized by loose binding to another peptide or DM. In contrast, DR1βG86Y is mostly in receptive conformation with little need for stabilization by either peptide or DM. As preparations of wtDR1 had been incubated in the absence of DM for a minimum of 20 min before injection over immobilized peptide surfaces, up to 50% of them 20 have already converted to the low binding conformation during this time. We also demonstrate, for the first time, using SPR, that acceleration of peptide release with addition of DM only occurs with the wtDR1 in complex with peptides that contain alanine instead of tyrosine at the corresponding pocket 1 position. Our SPR assay establishes that analysis of the behavior of class II molecules can be monitored in real time. Because of great sensitivity, interactions of MHC II with peptides and other accessory molecules, such as DO with DM, can be studied in a more controlled environment.
Can DM select its substrate by distinguishing the class II–peptide complex stability? Our results show that the wt and DR1βG86Y molecules that follow the same dissociation rates for HAAnchorless or HAY308A are differentially recognized by DM. This strongly suggests that DM recognizes a specific and flexible conformation of class II rather than the previously proposed unstable MHC–peptide. The interaction may resemble a chaperone function for DM. Just like a chaperone that is required for proper folding and/or assembly of another protein or protein complex, DM binds to a partially folded flexible class II 18 that is either empty or loosely bound to peptides. Because class II without a bound peptide is prone to denaturation and peptide binding prevents this process, DM facilitates binding and release of loosely binding peptides until a peptide that can complete the folding of the nascent protein is encountered 18. Once a stable peptide–MHC complex is formed, the conformation of the complex is no longer suitable for DM binding. At this point, class II is free to move to the cell surface. This mechanism can also explain the suitability of the DR1–CLIP complex as a DM substrate. Methionine, which fits in pocket 1 of DR, has a distinctive flexible side chain 39 that would not induce rigidity around pocket 1. This pattern of recognition for DM is also consistent with lack of recognition of I-Ek, which has a shallow pocket 1 31 very similar to that of the DR1βG86Y 11 40. Moreover, effects of H2-M on selection of immunodominant epitopes of hen egg white lysozyme may reflect its preference for more rigid ligand structures 41.
Intrinsic fluorescence assay showed that DM–DR interaction may involve the pocket 1 of DR1. An increase in intrinsic fluorescence was observed when DM was mixed with the wtDR1, whereas addition of DR1βG86Y or empty wtDR1 in complex with HA306–318 peptide did not enhance fluorescence. The observed 10–12% change in tryptophan fluorescence is significant because of minimal changes in the control groups, i.e., complexes of wtDR1–HA306–318 or DR1βG86Y–HAAnchorless, and the reproducibility of these results. Thus, such a response pattern suggests that the tryptophan-rich lateral surface of DM may be buried by interactions with empty wtDR1. This is consistent with predictions that Trp 62 on the lateral face of DM makes contact with Phe 51 of DR around pocket 1 12. Although it has been shown that when DM and DR were mixed, the nonpolar dye, anilinonaphthalene sulfonic acid (ANS), which binds to hydrophobic patches of proteins, emitted reduced fluorescence 42, the specific interaction sites were not defined. Our findings substantiate that the hydrophobic pocket 1 of DR1 is the main target for the DM-induced conformational changes in this system.
The observations in this paper point to a central role for conformational changes that take place upon peptide binding to MHC class II. These conformational adjustments regulate class II peptide loading, trafficking to the proper vesicular compartments, and cell surface expression. They monitor interactions with DM and selection of peptides that ultimately exit to the cell membrane and stimulate T cells. It is likely that in class I presentation, a chaperone/editor protein, tapasin, also plays similar roles 43 44. It will be fascinating to find out if tapasin also functions through mechanisms similar to those of DM.
Acknowledgments
We thank Drs. Peter Schuck for 4 a, Lydia Mosyak, Efstratios Stratikos, and Don Wiley for DM, Dennis Zaller for the DM cell line, and Don Wiley and Gladys Tan for discussions and reading of the manuscript.
Supported by National Institutes of Health grant R01 GM53549 (S. Sadegh-Nasseri).
Footnotes
Abbreviations used in this paper: AMCA, 7-amino-4-methyl-coumarin-3-acetic acid; CLIP, class II MHC–associated invariant chain peptide; HA, hemagglutinin; RU, resonance unit; SPR, surface plasmon resonance; wt, wild-type.
References
- Riberdy J.M., Newcomb J.R., Surman M.J., Barbosa J.A., Cresswell P. HLA-DR molecules from an antigen-processing mutant cell line are associated with invariant chain peptides. Nature. 1992;360:474–477. doi: 10.1038/360474a0. [DOI] [PubMed] [Google Scholar]
- Mellins E., Smith L., Arp B., Cotner T., Celis E., Pious D. Defective processing and presentation of exogenous antigens in mutants with normal HLA class II genes. Nature. 1990;343:71–74. doi: 10.1038/343071a0. [DOI] [PubMed] [Google Scholar]
- Denzin L.K., Hammond C., Cresswell P. HLA-DM interactions with intermediates in HLA-DR maturation and a role for HLA-DM in stabilizing empty HLA-DR molecules. J. Exp. Med. 1996;184:2153–2165. doi: 10.1084/jem.184.6.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kropshofer H., Arndt S.O., Moldenhauer G., Hammerling G.J., Vogt A.B. HLA-DM acts as a molecular chaperone and rescues empty HLA-DR molecules at lysosomal pH. Immunity. 1997;6:293–302. doi: 10.1016/s1074-7613(00)80332-5. [DOI] [PubMed] [Google Scholar]
- Kropshofer H., Vogt A.B., Moldenhauer G., Hammer J., Blum J.S., Hammerling G.J. Editing of the HLA-DR-peptide repertoire by HLA-DM. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:6144–6154. [PMC free article] [PubMed] [Google Scholar]
- Vogt A.B., Kropshofer H., Moldenhauer G., Hammerling G.J. Kinetic analysis of peptide loading onto HLA-DR molecules mediated by HLA-DM. Proc. Natl. Acad. Sci. USA. 1996;93:9724–9729. doi: 10.1073/pnas.93.18.9724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber D.A., Evavold B.D., Jensen P.E. Enhanced dissociation of HLA-DR-bound peptides in the presence of HLA-DM. Science. 1996;274:618–620. doi: 10.1126/science.274.5287.618. [DOI] [PubMed] [Google Scholar]
- Sloan V.S., Cameron P., Porter G., Gammon M., Amaya M., Mellins E., Zaller D.M. Mediation by HLA-DM of dissociation of peptides from HLA-DR. Nature. 1995;375:802–806. doi: 10.1038/375802a0. [DOI] [PubMed] [Google Scholar]
- Liljedahl M., Winqvist O., Surh C.D., Wong P., Ngo K., Teyton L., Peterson P.A., Brunmark A., Rudensky A.Y., Fung-Leung W.P., Karlsson L. Altered antigen presentation in mice lacking H2-O. Immunity. 1998;8:233–243. doi: 10.1016/s1074-7613(00)80475-6. [DOI] [PubMed] [Google Scholar]
- Stebbins C.C., Loss G.E., Jr., Elias C.G., Chervonsky A., Sant A.J. The requirement for DM in class II–restricted antigen presentation and SDS-stable dimer formation is allele and species dependent. J. Exp. Med. 1995;181:223–234. doi: 10.1084/jem.181.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks A.G., Campbell P.L., Reynolds P., Gautam A.M., McCluskey J. Antigen presentation and assembly by mouse I-Ak class II molecules in human APC containing deleted or mutated HLA DM genes. J. Immunol. 1994;153:5382–5392. [PubMed] [Google Scholar]
- Mosyak L., Zaller D.M., Wiley D.C. The structure of HLA-DM, the peptide exchange catalyst that loads antigen onto class II MHC molecules during antigen presentation. Immunity. 1998;9:377–383. doi: 10.1016/s1074-7613(00)80620-2. [DOI] [PubMed] [Google Scholar]
- Fremont D.H., Crawford F., Marrack P., Hendrickson W.A., Kappler J. Crystal structure of mouse H2-M. Immunity. 1998;9:385–393. doi: 10.1016/s1074-7613(00)80621-4. [DOI] [PubMed] [Google Scholar]
- Dornmair K., Rothenhausler B., McConnell H.M. Structural intermediates in the reactions of antigenic peptides with MHC molecules. Cold Spring Harbor Symp. Quant. Biol. 1989;1:409–416. doi: 10.1101/sqb.1989.054.01.050. [DOI] [PubMed] [Google Scholar]
- Sadegh-Nasseri S., McConnell H.M. A kinetic intermediate in the reaction of an antigenic peptide and I-Ek. Nature. 1989;337:274–276. doi: 10.1038/337274a0. [DOI] [PubMed] [Google Scholar]
- Sadegh-Nasseri S., Germain R.N. A role for peptide in determining MHC class II structure. Nature. 1991;353:167–170. doi: 10.1038/353167a0. [DOI] [PubMed] [Google Scholar]
- Sadegh-Nasseri S., Stern L.J., Wiley D.C., Germain R.N. MHC class II function preserved by low-affinity peptide interactions preceding stable binding. Nature. 1994;370:647–650. doi: 10.1038/370647a0. [DOI] [PubMed] [Google Scholar]
- Sadegh-Nasseri S., Germain R.N. How MHC class II molecules workpeptide-dependent completion of protein folding. Immunol. Today. 1992;13:43–46. doi: 10.1016/0167-5699(92)90131-P. [DOI] [PubMed] [Google Scholar]
- Rabinowitz J.D., Vrljic M., Kasson P.M., Liang M.N., Busch R., Boniface J.J., Davis M.M., McConnell H.M. Formation of a highly peptide-receptive state of class II MHC. Immunity. 1998;9:699–709. doi: 10.1016/s1074-7613(00)80667-6. [DOI] [PubMed] [Google Scholar]
- Natarajan S.K., Assadi M., Sadegh-Nasseri S. Stable peptide binding to MHC class II molecule is rapid and is determined by a receptive conformation shaped by prior association with low affinity peptides. J. Immunol. 1999;162:4030–4036. [PubMed] [Google Scholar]
- Tampe R., Clark B.R., McConnell H.M. Energy transfer between two peptides bound to one MHC class II molecule. Science. 1991;254:87–89. doi: 10.1126/science.1656526. [DOI] [PubMed] [Google Scholar]
- Stern L.J., Wiley D.C. The human class II MHC protein HLA-DR1 assembles as empty alpha beta heterodimers in the absence of antigenic peptide. Cell. 1992;68:465–477. doi: 10.1016/0092-8674(92)90184-e. [DOI] [PubMed] [Google Scholar]
- Natarajan S.K., Stern L.J., Sadegh-Nasseri S. Sodium dodecyl sulfate stability of HLA-DR1 complexes correlates with burial of hydrophobic residues in pocket 1. J. Immunol. 1999;162:3463–3470. [PubMed] [Google Scholar]
- Sato A.K., Zarutskie J.A., Rushe M.M., Lomakin A., Natarajan S.K., Sadegh-Nasseri S., Benedek G.B., Stern L.J. Determinants of the peptide-induced conformational change in the human class II major histocompatibility complex protein HLA-DR1. J. Biol. Chem. 2000;275:2165–2173. doi: 10.1074/jbc.275.3.2165. [DOI] [PubMed] [Google Scholar]
- Germain R.N., Hendrix L.R. MHC class II structure, occupancy and surface expression determined by post-endoplasmic reticulum antigen binding. Nature. 1991;353:134–139. doi: 10.1038/353134a0. [DOI] [PubMed] [Google Scholar]
- Germain R.N., Castellino F., Han R., Reis e Sousa C., Romagnoli P., Sadegh-Nasseri S., Zhong G.M. Processing and presentation of endocytically acquired protein antigens by MHC class II and class I molecules. Immunol. Rev. 1996;151:5–30. doi: 10.1111/j.1600-065x.1996.tb00701.x. [DOI] [PubMed] [Google Scholar]
- Romagnoli P., Germain R.N. The CLIP region of invariant chain plays a critical role in regulating major histocompatibility complex class II folding, transport, and peptide occupancy. J. Exp. Med. 1994;180:1107–1113. doi: 10.1084/jem.180.3.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain R.N., Rinker A., Jr. Peptide binding inhibits protein aggregation of invariant-chain free class II dimers and promotes surface expression of occupied molecules. Nature. 1993;363:725–728. doi: 10.1038/363725a0. [DOI] [PubMed] [Google Scholar]
- Schuck P., Millar D.B., Kortt A.A. Determination of binding constants by equilibrium titration with circulating sample in a surface plasmon resonance biosensor. Anal. Biochem. 1998;265:79–91. doi: 10.1006/abio.1998.2872. [DOI] [PubMed] [Google Scholar]
- Hammer J., Belunis C., Bolin D., Papadopoulos J., Walsky R., Higelin J., Danho W., Sinigaglia F., Nagy Z.A. High-affinity binding of short peptides to major histocompatibility complex class II molecules by anchor combinations. Proc. Natl. Acad. Sci. USA. 1994;91:4456–4460. doi: 10.1073/pnas.91.10.4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremont D.H., Hendrickson W.A., Marrack P., Kappler J. Structures of an MHC class II molecule with covalently bound single peptides. Science. 1996;272:1001–1004. doi: 10.1126/science.272.5264.1001. [DOI] [PubMed] [Google Scholar]
- Stern L.J., Brown J.H., Jardetzky T.S., Gorga J.C., Urban R.G., Strominger J.L., Wiley D.C. Crystal structure of the human class II MHC protein HLA-DR1 complexed with an influenza virus peptide. Nature. 1994;368:215–221. doi: 10.1038/368215a0. [DOI] [PubMed] [Google Scholar]
- Sherman M.A., Weber D.A., Jensen P.E. DM enhances peptide binding to class II MHC by release of invariant chain-derived peptide. Immunity. 1995;3:197–205. doi: 10.1016/1074-7613(95)90089-6. [DOI] [PubMed] [Google Scholar]
- Denzin L.K., Cresswell P. HLA-DM induces CLIP dissociation from MHC class II alpha beta dimers and facilitates peptide loading. Cell. 1995;82:155–165. doi: 10.1016/0092-8674(95)90061-6. [DOI] [PubMed] [Google Scholar]
- van Ham S.M., Gruneberg U., Malcherek G., Broker I., Melms A., Trowsdale J. Human histocompatibility leukocyte antigen (HLA)-DM edits peptides presented by HLA-DR according to their ligand binding motifs. J. Exp. Med. 1996;184:2019–2024. doi: 10.1084/jem.184.5.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra C.B., Busch R., Doebele R.C., Liu W., Sawada T., Kwok W.W., Chang M.D., Mellins E.D. Novel glycosylation of HLA-DRalpha disrupts antigen presentation without altering endosomal localization. J. Immunol. 1998;160:4289–4297. [PubMed] [Google Scholar]
- Khilko S.N., Corr M., Boyd L.F., Lees A., Inman J.K., Margulies D.H. Direct detection of major histocompatibility complex class I binding to antigenic peptides using surface plasmon resonance. Peptide immobilization and characterization of binding specificity. J. Biol. Chem. 1993;268:15425–15434. [PubMed] [Google Scholar]
- Khilko S.N., Jelonek M.T., Corr M., Boyd L.F., Bothwell A.L., Margulies D.H. Measuring interactions of MHC class I molecules using surface plasmon resonance. J. Immunol. Methods. 1995;183:77–94. doi: 10.1016/0022-1759(95)00033-7. [DOI] [PubMed] [Google Scholar]
- Lee C., McConnell H.M. A general model of invariant chain association with class II major histocompatibility complex proteins. Proc. Natl. Acad. Sci. USA. 1995;92:8269–8273. doi: 10.1073/pnas.92.18.8269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf P.R., Tourne S., Miyazaki T., Benoist C., Mathis D., Ploegh H.L. The phenotype of H-2M-deficient mice is dependent on the MHC class II molecules expressed. Eur. J. Immunol. 1998;28:2605–2618. doi: 10.1002/(SICI)1521-4141(199809)28:09<2605::AID-IMMU2605>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Nanda N.K., Sant A.J. DM determines the cryptic and immunodominant fate of T cell epitopes. J. Exp. Med. 2000;192:781–788. doi: 10.1084/jem.192.6.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich H.J., Doring K., Gruneberg U., Jahnig F., Trowsdale J., van Ham S.M. Interaction between HLA-DM and HLA-DR involves regions that undergo conformational changes at lysosomal pH. Proc. Natl. Acad. Sci. USA. 1997;94:13163–13168. doi: 10.1073/pnas.94.24.13163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortmann B., Copeman J., Lehner P.J., Sadasivan B., Herberg J.A., Grandea A.G., Riddell S.R., Tampe R., Spies T., Trowsdale J., Cresswell P. A critical role for tapasin in the assembly and function of multimeric MHC class I-TAP complexes. Science. 1997;277:1306–1309. doi: 10.1126/science.277.5330.1306. [DOI] [PubMed] [Google Scholar]
- Garbi N., Tan P., Diehl A.D., Chambers B.J., Ljunggren H.-G., Momburg F., Hämmerling G.J. Impaired immune responses and altered peptide repertoire in tapasin-deficient mice. Nat. Immunol. 2000;1:234–238. doi: 10.1038/79775. [DOI] [PubMed] [Google Scholar]
- Schafer P.H., Green J.M., Malapati S., Gu L., Pierce S.K. HLA-DM is present in one-fifth the amount of HLA-DR in the class II peptide-loading compartment where it associates with leupeptin-induced peptide (LIP)-HLA-DR complexes. J. Immunol. 1996;157:5487–5495. [PubMed] [Google Scholar]