

Abstract
Whereas CD40–CD40 ligand interactions are important for various dendritic cell (DC) functions in vitro, their in vivo relevance is unknown. We analyzed the DC status of CD40 ligand −/− mice using a contact hypersensitivity (CHS) model system that enables multiple functions of DCs to be assessed in vivo. Immunohistochemistry of skin sections revealed no differences in terms of numbers and morphology of dendritic epidermal Langerhans cells (LCs) in unsensitized CD40 ligand −/− mice as compared with wild-type C57BL/6 mice. However, after contact sensitization of CD40 ligand −/− mice, LCs failed to migrate out of the skin and substantially fewer DCs accumulated in draining lymph nodes (DLNs). Furthermore, very few antigen-bearing DCs could be detected in the paracortical region of lymph nodes draining sensitized skin. This defect in DC migration after hapten sensitization was associated with defective CHS responses and decreased cutaneous tumor necrosis factor (TNF)-α production and was corrected by injecting recombinant TNF-α or an agonistic anti-CD40 monoclonal antibody. Thus, CD40–CD40 ligand interactions in vivo regulate the migration of antigen-bearing DCs from the skin to DLNs via TNF-α production and play a vital role in the initiation of acquired T cell–mediated immunity.
Keywords: Langerhans cells, epidermis, TNF-α, contact hypersensitivity, acquired immunity
Introduction
A critical event in the initiation of acquired immune responses is the ferrying of Ag from peripheral nonlymphoid tissues to secondary lymphoid organs, where low frequency, Ag-specific lymphocytes congregate in discrete histological niches 1. This transport of Ag is efficiently performed by dendritic cells (DCs), a network of sentinel cells strategically located at the boundaries between the immune system and the environment that functions to initiate immune responses 1 2. Although it is known that several molecules on DCs influence their ability to activate naive T cells 1 2, pathways through which T cells might affect DCs remain unknown. One of the most dramatic influences on DCs is provided by CD40 stimulation in vitro 3 4, raising the possibility that in vivo, some DC functions might be dependent on CD40–CD40 ligand interactions. Although functional CD40 has been found on DCs, B cells, monocytes, endothelium, and keratinocytes, expression of CD40 ligand appears to be restricted to activated T cells, activated platelets, basophils, and mast cells.
The clinical relevance of CD40–CD40 ligand interactions is highlighted in the life-threatening manifestations of Hyper IgM syndrome (HIM), where accumulation of IgM and the inability of B cells to isotype switch has been attributed to mutated CD40 ligand on helper T cells 5 6. However, these defective humoral responses do not fully explain the increased opportunistic infections seen in these patients, an indication of impaired T cell responses 5 6. Indeed, cellular immunity has been shown to be impaired when CD40–CD40 ligand interactions are disrupted 5, though the precise mechanisms remain unclear. Blocking CD40–CD40 ligand interplay dramatically prolongs cardiac and skin allograft survival and inhibits T cell–proliferative responses both in vitro and in vivo 7. Likewise, development of experimental allergic encephalomyelitis (EAE) in myelin basic protein (MBP)–TCR-transgenic mice is also blocked when these animals lack CD40 ligand 5. Interestingly, Ag-specific responses are intact in MBP–TCR CD40 ligand −/− T cells in vitro, and the inability to induce EAE in these mice is corrected by reconstituting them with B7.1-transgenic APCs. This strongly suggests that the induction of costimulatory activity on APCs is defective 5. Moreover, naive T cells from CD40 ligand −/− animals display normal polyclonal as well as Ag-specific responses 8. This raises the possibility that the immunological abnormalities seen as a result of mutated CD40 or CD40 ligand are in fact due to defective upstream events in the initiation of acquired immunity, i.e., during the presentation of Ag and priming of naive T cells.
Contact hypersensitivity (CHS) is a T cell–mediated response dependent on intact DC functions. During the induction phase of CHS, these functions include Ag capture in the skin, Ag processing, transport to lymphoid tissues such as draining lymph nodes (DLNs), and Ag presentation to naive T cells 1 9 10 11 12. In this study, we investigate the in vivo role of CD40–CD40 ligand interactions on multiple DC functions using CD40 ligand −/− mice and CHS as the model system. We provide in vivo evidence that CD40–CD40 ligand interactions play a critical role in regulating the transport of Ag by epidermal DCs to DLNs and subsequently the initiation of acquired Ag-specific T cell–mediated immunity.
Materials and Methods
Mice.
Specific pathogen–free C57BL/6 mice were obtained from the National Cancer Institute Frederick Cancer Research Facility Animal Production Area (Frederick, MD), and CD40 ligand −/− mice were obtained from The Jackson Laboratory. The animals were maintained in facilities approved by the Association for Assessment and Accreditation of Laboratory Animal Care, in accordance with current regulations of the United States Department of Health and Human Services. All animal procedures were reviewed and approved by the Institutional Animal Care and Use Committee. Mice were 12–16 wk old at the start of each experiment.
Induction and Elicitation of CHS Responses.
0.4 ml (2 × 200 μl) of a 0.5% solution of FITC (Sigma-Aldrich) prepared in acetone/dibutylphthalate (1:1) was applied to the shaved abdomens of mice. 6 d later, CHS was elicited by painting the dorsal and ventral surface of each ear with 5 μl of 0.5% FITC. Ear thickness was measured using an engineers' micrometer (Mitutoyo) 24 h after challenge and compared with ear thickness before challenge. Specific ear swelling was calculated by subtracting the background swelling of mice that were not sensitized but challenged. In experiments where elicitation of CHS was not required, mice were killed 12 or 18 h after sensitization. Mice killed 18 h after sensitization were the source of abdominal skin resected to prepare epidermal sheets for immunostaining or axillary, brachial, and inguinal DLNs, which were removed and used for immunostaining and DC purification. Mice killed 12 h after topical application of FITC were the source of abdominal skin resected to prepare epidermal sheets for cytokine detection by ELISA.
Isolation of DCs from DLNs.
Mice were killed and axillary, brachial, and inguinal DLNs were excised. Single-cell suspensions were prepared from pooled DLNs taken from individual mice and DCs enriched by density gradient centrifugation as previously described 13. DCs were first assessed by direct examination using light microscopy. For each mouse in an experimental group (n = 5), five counts were made and the mean number of total DCs ± SD per DLN was calculated. Only cells showing a clear spiny appearance or well defined filiform and mobile cytoplasmic projections were considered as DCs. Cells exhibiting only pseudopod-like projections were not counted as DCs. In addition, DCs were also evaluated by May-Grünwald-Giemsa staining, immunocytochemistry of cytospin preparations, and flow cytometry.
FACS® Analysis.
DCs purified from DLNs were incubated with primary Abs or isotype-matched controls for 35 min at 4°C. DCs were then washed in cold blocking buffer (saline solution containing 0.1% BSA and 0.5% FCS), incubated with second-step reagents for 30 min at 4°C, and washed before analysis. As FITC was used as the sensitizer, gating on this fluorochrome was used concomitantly with forward and side scatter parameters to retrieve information only on FITC-bearing DCs. Primary Abs included B7.1–PE, B7.2–PE, CD8a–PE, CD11c–PE, CD14–PE, biotinylated anti-CD40, and anti–CD40 ligand (all from PharMingen) and biotinylated anti–MHC class II (Accurate Chemical). NLDC145 mAb to mouse DCs was from Serotec. PE-labeled or biotinylated isotype-matched Abs were from PharMingen. Second and third step reagents included biotinylated goat anti–rat Abs (Vector Labs.) and streptavidin Cy-5 (Southern Biotechnology), respectively. Single- and two-color analyses were performed using a Coulter Profile and ELITE V instrument (Coulter Immunology). Results are expressed as the percentage of positive DCs within the population of FITC-carrying DCs.
Epidermal Sheet Preparation and Immunostaining.
Mice were killed and the abdominal skin was shaved and resected. Fat was mechanically removed and the skin floated, dermis side down, in a 0.5 M solution of EDTA for 1 h at 37°C to achieve separation of the epidermis from the dermis. Epidermal sheets were then rinsed twice in isotonic saline solution (Baxter), fixed in cold acetone for 25 min, and washed three times in blocking buffer. Epidermal sheets from each experimental group of animals (usually five) were pooled for analysis and incubated with Abs to the DC Ag DEC205 (Rat mAb NLDC145; Serotec) at a dilution of 1:50. A rat mAb to Thy1.2 (American Type Culture Collection) was used at a dilution 1:100 to identify dendritic epidermal T cells (DETCs). Appropriate isotype-matched Abs (PharMingen) were used as controls for specificity. Secondary reagents included biotinylated anti-rat Abs (Vector Labs.) at a dilution of 1:300 and alkaline phosphatase (1:350) or peroxidase (1:250) conjugated to streptavidin (Biosource International). Red color was developed using a Fast Red™ developing kit from DAKO and blue color with True Blue–peroxidase substrate (Kirkegaard & Perry Labs.). Samples developed with peroxidase were previously blocked using a 3% solution of H202 for 15 min. A microscope with a calibrated grid was used to calculate the number of positive cells per square millimeter, evaluating at least 20 random fields per sample.
Immunostaining of Frozen Sections of DLN.
Pooled DLNs from each experimental group were fixed in cold acetone, preincubated with blocking buffer for 15 min, and then incubated with an alkaline phosphatase–conjugated mAb specific to FITC (Sigma-Aldrich) at a 1:100 dilution. An appropriate isotype-matched Ab (PharMingen) was used as a control for specificity. FITC-positive cells in the DLNs were visualized with a Fast Red™ developing kit (DAKO). Counterstaining of DLN sections was performed using methyl green.
Determination of Epidermal TNF-α Levels by ELISA.
Epidermal sheets from unsensitized and sensitized mice were prepared as described above. Each tissue sample was disrupted in a Dounce homogenizer with 0.5 ml of lysis buffer (4 M guanidium isothiocyanate, 0.5% sodium sarcosine, 25 mM sodium citrate, and 0.1 M 2-ME). The protein was desalted by ethanol precipitation and centrifuged at 10,000 g. Protein pellets were washed with ice cold 70% ethanol and then homogenized once more in 0.5 ml of 0.1 M sodium carbonate buffer, pH 8.0. The concentration of TNF-α per sample of epidermal lysate was determined using a sandwich ELISA. Capture Abs, biotinylated Abs, and rTNF-α were purchased from PharMingen and used according to the manufacturer's instructions. The test was considered positive if the absorbance value of the experimental group was at least 3 SD greater than the OD of the negative control. Generally, the limit of detection for TNF-α was no less than 1 ng/ml. The amount of TNF-α present was normalized for all the samples by comparing it with the total protein concentration of each sample. Total protein concentration was determined using the bicinchoninic acid (BCA) method (Pierce Chemical Co.).
Administration of rTNF-α and Anti-murine CD40 Ab.
100 ng of recombinant murine TNF-α (<0.1 ng endotoxin per microgram of TNF-α; Genzyme) in 50 μl of sterile isotonic, endotoxin-free saline solution (Baxter) was injected intradermally into each ear. Control animals received the same volume of saline solution only. 18 h later, mice were killed, their ears were resected, and epidermal sheets were prepared for immunostaining to detect DCs as already described. 200 μg of rat anti–murine CD40 (Clone IC10, endotoxin free; provided by Dr. M. Howard, DNAX, Palo Alto, CA) or rat control isotype (PharMingen) were injected intraperitoneally in sterile, endotoxin-free saline solution. Mice were killed 18 h later, abdominal skin was excised, and the frequency of DCs in the epidermis was determined as described above.
Statistical Analyses.
Data were expressed as the mean ± SD or SE. Statistical analyses were performed using two-tailed Student's t tests. A P value ≤ 0.05 was considered significant (*). Unless indicated otherwise, all experiments were performed at least twice and were scored by more than one investigator, each blinded to the experimental protocols employed. Each experimental group consisted of at least five mice, and the epidermal sheets of each group of mice were pooled for analysis. A microscope with a calibrated grid was used to calculate the number of epidermal DCs per square millimeter, and at least 20 random fields were examined per sample.
Results and Discussion
We first examined dendritic epidermal Langerhans cells (LCs) in the epidermis of normal C57BL/6 (Fig. 1 a) and CD40 ligand −/− (Fig. 1 b) mice by immunohistochemistry and found that these animals exhibited no striking differences in either LC counts or morphology. Another cell population of the epidermis, the Thy1.2+ DETCs 14, was also present in normal skin of control C57BL/6 (Fig. 1 c) and CD40 ligand −/− (Fig. 1 d) mice in relatively equal numbers. Likewise, DCs isolated from the lymph nodes of normal, unsensitized C57BL/6 (Fig. 1 e) and CD40 ligand −/− (Fig. 1 f) mice showed no obvious morphological differences, nor were there any differences in the number of DCs isolated from the lymph nodes of these mice (data not shown). Thus, CD40 ligand does not appear to be essential for the development and congregation of LC and DETC populations in the skin or the presence of resident DCs in lymph nodes.
Figure 1.
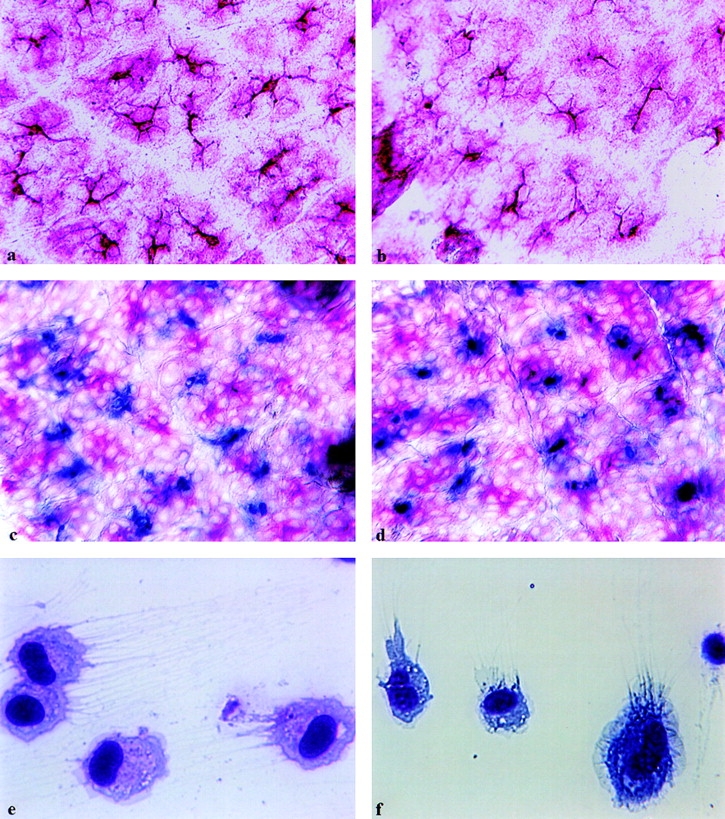
LCs, DETCs, and lymph node DCs are similar in number and morphology in C57BL/6 and CD40 ligand −/− mice. Epidermal sheets from normal C57BL/6 (a, c) or CD40 ligand −/− (b, d) mice were examined for the presence of epidermal DCs and DETCs. LCs are stained red (a–d), and Thy1.2+ DETCs are stained blue (c, d) by using the Fast Red™ and True Blue™ peroxidase substrate systems, respectively. Purified DCs from lymph nodes of normal C57BL/6 (e) or CD40 ligand −/− (f) mice were stained with May-Grünwald-Giemsa stain (e, f). The difference in color intensities between e and f could be due to the fact that these samples were stained at different times. Original magnifications: a–d, ×350; e and f, ×700.
It is well documented that cutaneous hapten sensitization induces LCs to migrate out of the skin 1 9 10 11 12. Thus, we next addressed the consequence of painting FITC on the skin of C57BL/6 and CD40 ligand −/− mice (Fig. 2). As anticipated, the number of LCs was reduced in the epidermis of sensitized C57BL/6 control (Fig. 2b and Fig. e) mice compared with unsensitized (Fig. 2, a and e) mice. In contrast, unexpectedly, we found that LC migration is dramatically reduced after hapten sensitization of CD40 ligand −/− mice (Fig. 2d and Fig. e) compared with sensitized control C57BL/6 (Fig. 2b and Fig. e) mice.
Figure 2.
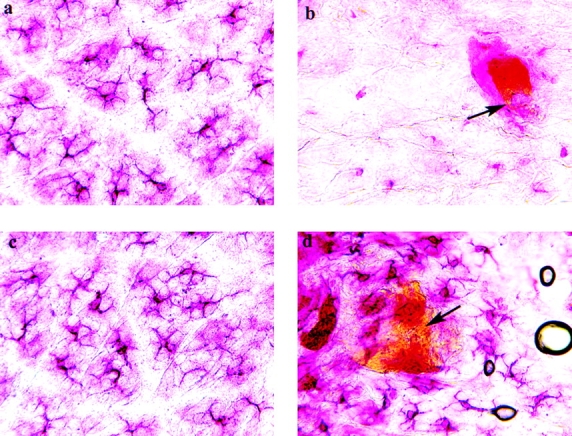

Impaired migration of epidermal DCs in CD40 ligand −/− mice upon Ag sensitization. Epidermal sheets from C57BL/6 (a, b) and CD40 ligand −/− (c, d) mice were stained for LCs (shown in red; as described). Mice in panels b and d were sensitized with FITC, and those in panels a and c were not. Yellow spots of FITC are still visible in the epidermis (b and d, arrows). DCs nearly disappear from the epidermis of control mice after FITC sensitization (b), but they are clearly present around FITC-positive regions in the epidermis of CD40 ligand −/− mice (d). To determine the frequency of epidermal DCs, we pooled epidermal sheets from at least five mice per experimental group (*P < 0.001), and at least 20 random fields were examined per sample. Results are expressed as DCs per square millimeter of epidermis (e). Original magnification, ×350.
The extent of LC migration out of the skin was surprisingly high (80%) compared with other studies reporting moderate levels of migration, ∼20–30% after FITC sensitization. This discrepancy could be accounted for by the fact that we stained for the presence of LCs on the basis of DEC205 expression rather than ATPase activity or Ia Ag expression, as carried out in other studies 15 16. In a recent study, we consistently found that more epidermal LCs stain positive for the marker DEC205 than for ATPase activity in untreated mouse skin, implying that not all LCs possess this enzyme 17. Therefore, the possibility arises that different subpopulations of LCs may exist in the epidermis, some being more susceptible to becoming mobilized upon epicutaneous FITC painting than others.
Associated with LC migration from the skin is a concomitant accumulation of DCs in lymph nodes draining FITC-sensitized skin, a large proportion of which bears FITC 18 19 20. This phenomenon is also defective in CD40 ligand −/− mice. We found that far fewer DCs accumulate in the DLNs of CD40 ligand −/− mice as a consequence of FITC sensitization compared with normal control mice (Fig. 3 a). Furthermore, immunostaining of frozen DLN sections revealed a nonrandom accumulation of FITC-bearing DCs in the paracortex of the DLNs from normal (Fig. 3b and Fig. c) animals. In contrast, considerably fewer Ag-bearing DCs were detected in DLNs from CD40 ligand −/− (Fig. 3d and Fig. e) mice. Concomitantly, and possibly as a consequence of the failure of epidermal LCs to emigrate and carry Ag to the T cell areas of DLNs, CHS reactions were substantially decreased in CD40 ligand −/− animals, compared with the response of positive control C57BL/6 animals (Fig. 4).
Figure 3.

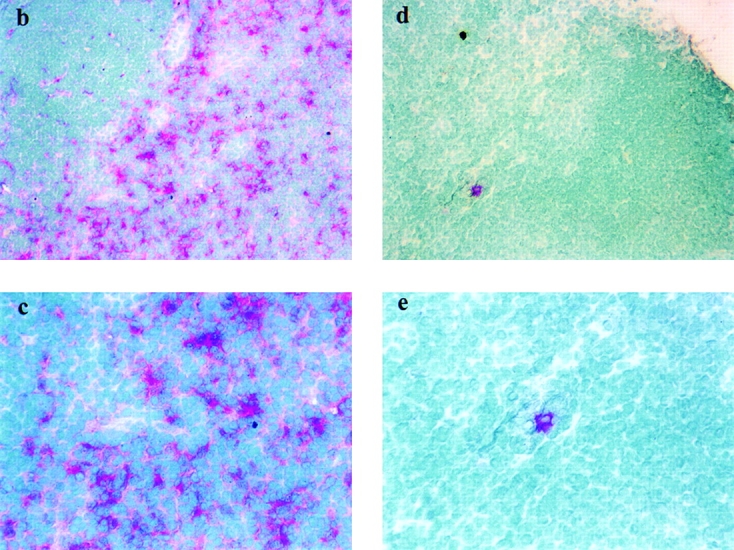
Ag-sensitized CD40 ligand −/− mice have reduced numbers of total and Ag-bearing DCs in the DLN. DLNs from FITC-sensitized C57BL/6 (a, left) and CD40 ligand −/− (a, right) animals (n = 5) were removed 18 h after sensitization, and the total number of DCs per DLN was determined for individual mice (a). To identify Ag-carrying DCs in situ, frozen sections of lymph nodes draining FITC-sensitized skin were stained with an alkaline phosphatase–labeled mAb specific for FITC and developed using the Fast Red™ substrate system. Ag-bearing DCs (stained red) are congregated in the paracortical areas of DLNs from C57BL/6 (b, c) but not CD40 ligand −/− mice (d, e). Panels b and d are low magnifications (×130), whereas panels c and e are high magnifications (×260). Sections were counterstained with methyl green.
Figure 4.

CD40 ligand −/− mice have defective CHS responses. Mice were epicutaneously sensitized on their shaved abdomens with FITC and challenged on both of their ears 6 d later. Ear thickness was measured immediately before and 24 h after challenge using an engineers' micrometer. Background swelling was obtained from control animals that were not sensitized but were challenged. Results are expressed as the mean ± SE of at least five mice per experimental group (*P < 0.001).
Since FITC was used as a sensitizer, we were able to specifically analyze the phenotype of FITC-bearing DCs from DLNs using flow cytometry (Table ). Compared with wild-type controls, fewer B7.1+ and CD40+ DCs were found in CD40 ligand −/− mice, whereas no difference in the percentage of cells expressing MHC class II or B7.2 was observed. Also, fewer DLN DCs from CD40 ligand −/− mice expressed DEC205, a prominent nonclonal receptor of the innate immune system involved in Ag presentation by DCs 21.
Table 1.
Phenotype of FITC-bearing DCs from DLNs of C57BL/6 and CD40 Ligand −/− Mice
| Percent staining | ||
|---|---|---|
| Markers | C57BL/6 | CD40 ligand −/− |
| CD80 (B7-1) | 49 | 15 |
| CD86 (B7-2) | 96 | 95 |
| CD40 | 33 | <1 |
| CD40 ligand | 0 | 0 |
| DEC205 | 60 | 23 |
| MHC class II | 99 | 98 |
| CD11c | 67 | 30 |
| CD14 | 22 | 9 |
| CD8a | 73 | 37 |
| DEC205/MHC class II | 57 | 30 |
| DEC205/CD11c | 45 | 10 |
| DEC205/CD14 | 20 | 5 |
| MHC class II/CD8a | 72 | 36 |
DCs from DLNs of FITC-sensitized mice were purified by density gradient centrifugation to enrich for DCs. They were then analyzed by flow cytometry for the markers shown. Greater than 12,000 events were acquired for each experimental group. Cells were first gated on the basis of high forward and side scatter to identify DCs. They were then gated on the basis of FITC expression; ∼5,000 gated (FITC-positive) cells were analyzed for PE and Cy-5. Results are expressed as the percentages of DCs that are positive for a given marker within the population of FITC-carrying DCs.
Though it has long been known that cutaneous Ag sensitization induces LCs to leave the skin and migrate to DLNs 1 9 10 11 12, the initial events triggering this process in the skin are not well understood. Studies using anti–TNF-α Abs 22 or rTNF-α 13 or its receptors 23 have established this cytokine as a critical mediator involved in DC migration from skin and CHS. Cutaneous Ag application has also been shown to induce mRNA for a variety of proinflammatory cytokines including TNF-α 24. Moreover, CD40 ligation induces secretion of TNF-α by CD40+ basal keratinocytes 25. We therefore speculated that the lack of LC migration observed in CD40 ligand −/− mice after epicutaneous sensitization may occur as a consequence of defective TNF-α production within the skin of these mice. Indeed, analysis by ELISA of the TNF-α content of epidermal sheets prepared from unsensitized and sensitized skin of C57BL/6 and CD40 ligand −/− mice revealed differences in TNF-α production between these mouse strains. In contrast to C57BL/6 mice, whose baseline TNF-α levels in untreated skin increased after Ag-sensitization, CD40 ligand −/− mice exhibited high levels of TNF-α in unsensitized skin equivalent to the levels seen in sensitized wild-type mice, which decreased after sensitization (Fig. 5 a). Thus, LCs may not mobilize after hapten sensitization because TNF-α production is aberrantly regulated in the epidermis of CD40 ligand −/− mice, both before and after epicutaneous sensitization.
Figure 5.
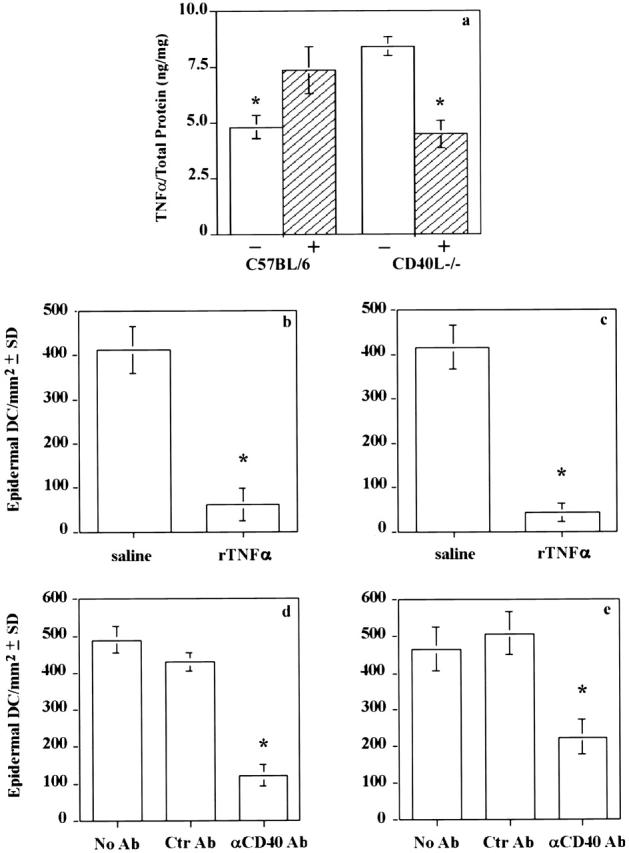
Reconstituting CD40 signaling or injecting TNF-α restores migration of epidermal DCs in CD40 ligand −/− mice. TNF-α levels in the epidermis of untreated (−) or sensitized (+) wild-type and CD40 ligand −/− mice were determined by ELISA using protein extracts of epidermal sheets (a). The data are reported as TNF-α/total protein (ng/mg) and represent the mean ± SD of each sample tested in triplicate. To assess the role of local TNF-α on DC migration, wild-type (b) and CD40 ligand −/− (c) mice were injected with saline (b and c, left bars) or rTNF-α (b and c, right bars). Mice were then killed 18 h later to remove the ears, prepare epidermal sheets, and count epidermal DCs in wild-type (b) and CD40 ligand −/− (c) mice. To reconstitute CD40 signaling in wild-type (d) and CD40 ligand −/− (e) mice, anti–murine CD40 (d and e, right bars) or control Ab (d and e, middle bars) was injected intraperitoneally and the mice were killed 18 h later. The number of epidermal DCs was then counted in wild-type (d) and CD40 ligand −/− (e) mice. Epidermal sheets from each experimental group were pooled for immunostaining. Results are expressed as the mean ± SD of DCs per square millimeter of epidermis. A minimum of five animals was used per group (*P < 0.001).
To rule out the possibility that the altered LC migration in the mutant mice was due to some intrinsic defect rather than the aberrant regulation of TNF-α production, we next had to address whether LC migration could be restored in CD40 ligand −/− mice if an exogenous source of TNF-α was provided. In fact, by administering rTNF-α intradermally into the skin of CD40 ligand −/− mice, LCs were induced to migrate out of the epidermis (Fig. 5 c) to the same extent as observed in C57BL/6 mice treated in the same manner (Fig. 5 b). Likewise, LC mobilization was also induced when the missing CD40 ligand signal was re-established by injecting agonistic anti-CD40 mAb 26 27 into CD40 ligand −/− (Fig. 5 e) and control (Fig. 5 d) mice, thus ruling out an intrinsic LC migratory defect and indicating an essential role for CD40 signaling and TNF-α in LC migration.
It is intriguing that unsensitized CD40 ligand −/− mice produced significantly higher amounts of TNF-α in their epidermis compared with wild-type age-matched control mice. It is possible that TNF production is enhanced in these mice to compensate for the deficiency in CD40 ligand production. Although unexpected, this finding is not too surprising considering the fact that TNF-α and CD40 ligand are both members of the TNF-α ligand superfamily. Thus, it would appear that TNF-α production and CD40 ligand expression are intimately linked. Indeed, in inflammatory bowel diseases such as Crohn's disease and ulcerative colitis, which are characterized by the local production of cytokines such as TNF, there is a significant increase in the number of CD40- and CD40 ligand–positive cells in the inflamed mucosa 28. Furthermore, decreased TNF production as well as impaired inflammatory responses have been described in CD40 ligand −/− mice infected with Leishmania 5. Recently, it has also been demonstrated that increased TNF-α production and induction of neuronal injury occur when activated microglia, which exhibit increased CD40 expression, are treated with CD40 ligand 29.
Although our data points to the aberrant regulation of TNF-α production in the skin of CD40 ligand −/− mice, accounting for the defect in DC migration, it is difficult to explain how FITC sensitization would decrease TNF-α levels in these mice. One possible mechanism may be via the overproduction of IL-10, an antiinflammatory cytokine recently demonstrated to play an important role in inhibiting epidermal LC migration from sensitized skin to DLNs 30.
The cellular source of CD40 ligand in the skin is unclear. Conventional T cells are an obvious candidate because they express CD40 ligand upon activation. However, they are not normally present in the skin, nor are they significantly recruited into the skin during the initiation of CHS (reference 31 and data not shown). Mast cells, on the other hand, express functionally active CD40 ligand 32 and are found in normal skin, although in low numbers. They possess an arsenal of powerful pharmacological mediators and, in addition, are the only cells known to constitutively produce and store TNF-α 33, ready for immediate release early during an inflammatory reaction. Nonlymphoid cells such as platelets may also serve this function. They can be detected localizing inside 34 and outside 35 dermal venules at sites of acute inflammation. Furthermore, human umbilical vein endothelial cells, which constitutively express CD40, can be induced to release inflammatory cytokines such as TNF-α as a result of triggering by inducible CD40 ligand on activated platelets 36.
Arguably, the finding that mast cell–deficient mice display normal CHS responses raises doubt that mast cells may play a critical role in regulating DC migration and consequently the induction of CHS. Indeed, severe depletion of platelets in mast cell–deficient mice with antiplatelet Ab has been demonstrated to strongly inhibit CHS responses 37, therefore supporting a role for platelets in this process. However, although antiplatelet Ab treatment markedly reduces CHS responses in mast cell–deficient mice, the same treatment of normal +/+ littermate control mice only minimally affects CHS responses 37. Thus, mast cell–deficient mice are unique in that they exhibit a significantly greater dependence on platelets compared with wild-type control mice for full manifestation of CHS ear swelling responses. Therefore, both mast cells and platelets likely play a role in regulating CHS responses, although to differing extents, with mast cells likely playing a more prominent role in normal mice, perhaps by influencing LC migration via CD40–CD40 ligand interactions in the skin. Interestingly, we found that mast cells fail to accumulate in the dermis of sensitized CD40 ligand −/− mice compared with wild-type controls (data not shown).
In conclusion, our findings reveal that CD40–CD40 ligand interactions in vivo play a key role in regulating the migration of Ag-bearing epidermal DCs to DLNs and subsequently the initiation of an acquired immune response. They also imply that an important outcome of these interactions is the production of cutaneous TNF-α, a known mediator of epidermal DC migration. Thus, CD40–CD40 ligand interactions might be considered as a functional bridge between innate responses and the initiation of acquired immunity.
Acknowledgments
We thank the High Resolution Microscopy facility at M.D. Anderson Cancer Center for their help. We also acknowledge with thanks the gift of anti–murine CD40 mAb from Dr. Maureen Howard, DNAX, Palo Alto, CA.
This work was supported by grants CA 52457-08 and CA 75575 from the National Cancer Institute and ES 07327 from the National Institute of Environmental Health Sciences, National Institutes of Health. The animal facilities at the M.D. Anderson Cancer Center are supported in part by a core grant (CA 16672) from the National Cancer Institute. V. Shreedhar was supported by the Rosalie B. Hite Fellowship.
Footnotes
A.M. Moodycliffe and V. Shreedhar contributed equally to this work.
Abbreviations used in this paper: CHS, contact hypersensitivity; DCs, dendritic cells; DETCs, dendritic epidermal T cells; DLNs, draining lymph nodes; LCs, dendritic epidermal Langerhans cells.
References
- Banchereau J., Steinman R.M. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Steinman R.M. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 1991;9:271–296. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- Caux C., Massacrier C., Vanbervliet B., Dubois B., Kooten C.V., Durand I., Banchereau J. Activation of human dendritic cells through CD40 cross-linking. J. Exp. Med. 1994;180:1263–1272. doi: 10.1084/jem.180.4.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorck P., Banchereau J., Flores-Romo L. CD40 ligation counteracts Fas-induced apoptosis of human dendritic cells. Int. Immunol. 1997;9:365–372. doi: 10.1093/intimm/9.3.365. [DOI] [PubMed] [Google Scholar]
- Grewal I.S., Flavell R.A. A central role of CD40 ligand in the regulation of CD4+ T-cell responses. Immunol. Today. 1996;17:736–738. doi: 10.1016/0167-5699(96)10030-x. [DOI] [PubMed] [Google Scholar]
- Banchereau J., Bazan F., Blanchard D., Briere F., Galizzi J.P., Kooten C.V., Liu Y.J., Rousset F., Saeland S. The CD40 molecule and its ligand. Annu. Rev. Immunol. 1994;12:881–992. doi: 10.1146/annurev.iy.12.040194.004313. [DOI] [PubMed] [Google Scholar]
- Larsen C.P., Elwood E.T., Alexander D.Z., Ritchie S.C., Hendrix R., Tucker-Burden C., Cho H.R., Aruffo A., Hollenbaugh D., Linsley P.S. Long-term acceptance of skin and cardiac allografts after blocking CD40 and CD28 pathways. Nature. 1996;381:434–438. doi: 10.1038/381434a0. [DOI] [PubMed] [Google Scholar]
- Grewal I.S., Xu J., Flavell R.A. Impairment of antigen-specific T-cell priming in mice lacking CD40 ligand. Nature. 1995;378:617–620. doi: 10.1038/378617a0. [DOI] [PubMed] [Google Scholar]
- Kripke M.L., Munn C.G., Jeevan A., Tang J.-M., Bucana C. Evidence that cutaneous antigen-presenting cells migrate to regional lymph nodes during contact sensitization. J. Immunol. 1990;145:2833–2838. [PubMed] [Google Scholar]
- Steinman R.M., Hoffman L., Pope M. Maturation and migration of cutaneous dendritic cells. J. Invest. Dermatol. 1995;105:2S–7S. doi: 10.1111/1523-1747.ep12315162. [DOI] [PubMed] [Google Scholar]
- Austyn J.M. New insights into the mobilization and phagocytic activity of dendritic cells. J. Exp. Med. 1996;183:1287–1292. doi: 10.1084/jem.183.4.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moodycliffe A.M., Ullrich S.E. Role of cytokines in the regulation of hypersensitivity responses. . In: Aggarwal B., Puri R., editors. Human CytokinesTheir Role in Disease and Therapy. Blackwell Science, Inc; Cambridge, MA: 1995. pp. 131–152. [Google Scholar]
- Cumberbatch M., Kimber I. Dermal tumour necrosis factor-α induces dendritic cell migration to draining lymph nodes, and possibly provides one stimulus for Langerhans' cell migration. Immunology. 1992;75:257–263. [PMC free article] [PubMed] [Google Scholar]
- Tigelaar R.E., Lewis J.M. Immunobiology of mouse dendritic epidermal T cellsa decade later, some answers, but still more questions. J. Invest. Dermatol. 1995;105:43S–49S. doi: 10.1111/1523-1747.ep12315280. [DOI] [PubMed] [Google Scholar]
- Doi S., Kobayashi M., Sugiura Y., Sakamoto T., Torii S. Heterogeneous reactivity of murine epidermal Langerhans cells after application of FITCa histochemical evaluation. Arch. Histol. Cytol. 1999;62:363–373. doi: 10.1679/aohc.62.363. [DOI] [PubMed] [Google Scholar]
- Woods G.M., Qu M., Ragg S.J., Muller H.K. Chemical carcinogens and antigens induce immune suppression via Langerhans' cell depletion. Immunology. 1996;88:134–139. doi: 10.1046/j.1365-2567.1996.d01-645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shreedhar V., Moodycliffe A.M., Ullrich S.E., Bucana C., Kripke M.L., Flores-Romo L. Dendritic cells require T cells for functional maturation in vivo. Immunity. 1999;11:625–636. doi: 10.1016/s1074-7613(00)80137-5. [DOI] [PubMed] [Google Scholar]
- Macatonia S.E., Edwards A.J., Knight S.C. Dendritic cells and the initiation of contact sensitivity to fluorescein isothiocyanate. Immunology. 1986;59:509–514. [PMC free article] [PubMed] [Google Scholar]
- Macatonia S.E., Knight S.C., Edwards A.J., Griffiths S., Fryer P. Localization of antigen on lymph node dendritic cells after exposure to the contact sensitizer fluorescein isothiocyanate. Functional and morphological studies. J. Exp. Med. 1987;166:1654–1657. doi: 10.1084/jem.166.6.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wilsem E.J.G., Breve J., Kleijmeer M., Kraal G. Antigen-bearing Langerhans cells in skin draining lymph nodesphenotype and kinetics of migration. J. Invest. Dermatol. 1994;103:217–220. doi: 10.1111/1523-1747.ep12393088. [DOI] [PubMed] [Google Scholar]
- Jiang W., Swiggard W.J., Heufler C., Peng M., Mirza A., Steinman R.M., Nussenzweig M.C. The receptor DEC-205 expressed by dendritic cells and thymic epithelial cells is involved in antigen processing. Nature. 1995;375:151–155. doi: 10.1038/375151a0. [DOI] [PubMed] [Google Scholar]
- Piguet P.F., Grau G.E., Hauser C., Vassalli P. Tumor necrosis factor is a critical mediator in hapten-induced irritant and contact hypersensitivity reactions. J. Exp. Med. 1991;173:673–679. doi: 10.1084/jem.173.3.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B., Fujisawa H., Zhuang L., Kondo S., Shivji G.M., Kim C.S., Mak T.W., Sauder D.N. Depressed Langerhans cell migration and reduced contact hypersensitivity response in mice lacking TNF receptor p75. J. Immunol. 1997;159:6148–6155. [PubMed] [Google Scholar]
- Enk A.H., Katz S.I. Early molecular events in the induction phase of contact sensitivity. Proc. Natl. Acad. Sci. USA. 1992;89:1398–1402. doi: 10.1073/pnas.89.4.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peguet-Navarro J., Dalbiez-Gauthier C., Moulon C., Berthier O., Reano A., Gaucherand M., Banchereau J., Rousset F., Schmitt D. CD40 ligation of human keratinocytes inhibits their proliferation and induces their differentiation. J. Immunol. 1997;158:144–152. [PubMed] [Google Scholar]
- Heath A.W., Wu W.W., Howard M.C. Monoclonal antibodies to murine CD40 define two distinct functional epitopes. Eur. J. Immunol. 1994;24:1828–1834. doi: 10.1002/eji.1830240816. [DOI] [PubMed] [Google Scholar]
- Ferlin W.G., von der Weid T., Cottrez F., Ferrick D.A., Coffman R.L., Howard M.C. The induction of a protective response in Leishmania major-infected BALB/c mice with anti-CD40 mAb. Eur. J. Immunol. 1998;28:525–531. doi: 10.1002/(SICI)1521-4141(199802)28:02<525::AID-IMMU525>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Liu Z., Colpaert S., D'Haens G.R., Kasran A., de Boer M., Rutgeerts P., Geboes K., Ceuppens J.L. Hyper-expression of CD40 ligand (CD154) in inflammatory bowel disease and its contribution to pathogenic cytokine production. J. Immunol. 1999;163:4049–4057. [PubMed] [Google Scholar]
- Tan J., Town T., Paris D., Mori T., Suo Z., Crawford F., Mattson M.P., Flavell R.A., Mullan M. Microglial activation resulting from CD40-CD40L interaction after β-amyloid stimulation. Science. 1999;286:2352–2355. doi: 10.1126/science.286.5448.2352. [DOI] [PubMed] [Google Scholar]
- Wang B., Zhuang L., Fujisawa H., Shinder G.A., Feliciani C., Shivji G.M., Suzuki H., Amerio P., Toto P., Sauder D.N. Enhanced epidermal Langerhans cell migration in IL-10 knockout mice. J. Immunol. 1999;162:277–283. [PubMed] [Google Scholar]
- Grabbe S., Schwarz T. Immunoregulatory mechanisms involved in elicitation of allergic contact hypersensitivity. Immunol. Today. 1998;19:38–45. doi: 10.1016/s0167-5699(97)01186-9. [DOI] [PubMed] [Google Scholar]
- Gauchat J.F., Henchoz S., Mazzei G., Aubry J.-P., Brunner T., Blasey H., Life P., Talabot D., Flores-Romo L., Thompson J. Induction of human IgE synthesis in B cells by mast cells and basophils. Nature. 1993;365:340–343. doi: 10.1038/365340a0. [DOI] [PubMed] [Google Scholar]
- Galli S.J., Wershil B.K. The two faces of the mast cell. Nature. 1996;381:21–22. doi: 10.1038/381021a0. [DOI] [PubMed] [Google Scholar]
- Senaldi G., Piguet P.F. Platelets play a role in the pathogenesis of the irritant reaction in mice. J. Invest. Dermatol. 1997;108:248–252. doi: 10.1111/1523-1747.ep12286444. [DOI] [PubMed] [Google Scholar]
- Feng D., Nagy J.A., Pyne K., Dvorak H.F., Dvorak A.M. Platelets exit venules by a transcellular pathway at sites of F-met peptide-induced acute inflammation in guinea pigs. Int. Arch. Allergy. Immunol. 1998;116:188–195. doi: 10.1159/000023944. [DOI] [PubMed] [Google Scholar]
- Henn V., Slupsky J.R., Grafe M., Anagnostopoulos I., Forster R., Muller-Berghaus G., Kroczek R.A. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. 1998;391:591–594. doi: 10.1038/35393. [DOI] [PubMed] [Google Scholar]
- Geba G.P., Ptak W., Anderson G.M., Paliwal V., Ratzlaff R.E., Levin J., Askenase P.W. Delayed-type hypersensitivity in mast cell-deficient micedependence on platelets for expression of contact sensitivity. J. Immunol. 1996;157:557–565. [PubMed] [Google Scholar]