

Abstract
To detect caspase activities in intact apoptotic cells at the single cell level, cell-permeable fluorogenic caspase substrates were synthesized incorporating the optimal peptide recognition motifs for caspases 1, 3/7, 6, 8, and 9. Caspase activities were then assessed at various times after in vitro treatment of mouse thymocytes with dexamethasone or anti-Fas antibody. Dexamethasone induced the following order of appearance of caspase activities as judged by flow cytometry: LEHDase, WEHDase, VEIDase, IETDase, and DEVDase. Since the relative order of caspases 3 (DEVDase) and 6 (VEIDase) in the cascade has been controversial, this caspase activation order was reexamined using confocal microscopy. The VEIDase activity appeared before DEVDase in every apoptotic cell treated with dexamethasone. In contrast, anti-Fas stimulation altered this sequence: IETDase was the first measurable caspase activity and DEVDase preceded VEIDase. In an attempt to determine the intracellular target of the potent antiapoptotic agent carbobenzoxy-valyl-alanyl-aspartyl(β-methyl ester)-fluoromethyl ketone (Z-VAD[OMe]-FMK), we examined its ability to inhibit previously activated intracellular caspases. However, no significant reductions of these activities were observed. These fluorogenic caspase substrates allow direct observation of the caspase cascade in intact apoptotic cells, showing that the order of downstream caspase activation is dependent on the apoptotic stimulus.
Keywords: apoptosis, caspase, PhiPhiLux™, thymocyte, lymphocyte
Introduction
Since the seminal finding that the Caenorhabditis elegans death gene ced-3 encodes a protein homologous to the mammalian protease IL-1β–converting enzyme (ICE) 1, a family of related proteases has been described. Termed caspases, this family is characterized by both a catalytic cysteinyl residue and a strong preference for an aspartyl residue in the P1 position of their substrate recognition sequence. Both structural and functional studies have shown that caspases also recognize the P4 amino acid, and recent studies using combinatorial chemistry have suggested a division of the caspase family into three subfamilies based on peptide substrate recognition 2. The ICE subfamily (caspases 1, 4, and 5) prefers a bulky hydrophobic amino acid such as tyrosine or tryptophan at P4, the caspase 3 subfamily (caspases 2, 3, and 7) prefers a second aspartic acid residue at this site, and the caspase 6 subfamily (caspases 6, 8, and 9) prefers a branched hydrophobic side chain such as valine.
Caspases are expressed in cells as inactive proenzymes, which must be proteolytically processed in order to acquire activity. Consistent with the finding that the prototypic cleavage sites for such processing have the distinctive aspartic acid at P1, various caspases have been found to activate other procaspases, and a cascade of activating caspases has been described in cells undergoing apoptosis. Evidence has accumulated that the caspase cascade is normally initiated by oligomerization of either procaspase 8 or procaspase 9 via Fas-associated death domain protein (FADD) or apoptotic protein activating factor 1 (apaf-1), respectively. The subsequent order of the caspase activation cascade has been analyzed by ordering caspase processing events in cytoplasmic extracts of apoptotic cells, in conjunction with specific inhibitors. However, recent studies of caspase 9 indicate that procaspase processing is necessary but not sufficient for enzymatic activity 3, and other studies attempting to order the caspase cascade have resulted in conflicting proposals regarding the relative sequence of activation of caspases 3 and 6. Two studies have suggested that caspase 6 activates procaspase 3 4 5, while two studies have suggested the reverse order 6 7. One major problem with analyzing the caspase cascade in extracts is that events controlled by the subcellular localization of regulatory components may not be accurately reproduced. The autoactivation of long prodomain caspases occurs in large complexes that are still not well understood; critical components such as cytochrome c, apoptosis inducing factor (AIF), and procaspases 2, 3, and 9 are found in the mitochondrial intramembrane space 8 9 10 11; transcriptional events clearly lie upstream of caspase activation in many examples of apoptosis; and Bcl-2 family members move from a cytosolic to membrane localization during apoptosis 12.
The above complexities point out the need for a means to monitor caspase activation in intact apoptotic cells, so that the concepts derived from study of recombinant components and extracts of apoptotic cells can be tested in a physiological setting. To this end, we have designed and synthesized cell-permeable fluorogenic caspase substrates with specificity for caspases 1, 3/7, 6, 8, and 9 13. These substrates are peptides of 18 amino acids, with caspase recognition motifs in the center, and rhodamine derivatives covalently attached near their termini. As previously shown noncovalent cyclization can occur in such modified peptides via intramolecular complexation of rhodamines with consequent quenching of the rhodamine fluorescence until proteolysis breaks the peptide linkage 13 14. The two associated rhodamine dye molecules of the intact substrate appear to form a hydrophobic surface mediating the membrane permeability of these substrates 14 15 16. We have used these intracellular caspase substrates in conjunction with flow cytometry and confocal microscopy to examine caspase activities in the classic apoptotic system of thymocytes treated with corticosteroid or anti-Fas antibody in vitro. In the former case, the caspase cascade is triggered by apaf-1–mediated aggregation of procaspase 9 17 18 while in the latter case it is via FADD-mediated procaspase 8 aggregation, largely independent of procaspase 9 and the Bid amplification loop 19 20.
We have also addressed the question of the pharmacological target of the widely used potent apoptosis blocker carbobenzoxy-valyl-alanyl-aspartyl(β-methyl ester)-fluoromethyl ketone (ZVAD[OMe]-FMK). This compound has been found to block a wide variety of apoptosis systems in vitro and has also been reported to protect mice from the lethal effects of intravenously injected anti-Fas, as well as ischemia-reperfusion in a stroke model 21 22. Since ZVAD(OMe)-FMK is widely regarded as a nonselective inhibitor of caspases, we expected that it would inhibit the intracellular caspase activities detected by our substrates. However, we found that although ZVAD(OMe)-FMK blocks intracellular caspase activation in thymocytes when added before or at the same time as dexamethasone, it does not significantly inhibit any detectable intracellular caspase activities after they have already become activated.
Materials and Methods
Materials.
Thymocytes were prepared from 4–6-wk-old C57BL/6 mice obtained from Frederick Cancer Center. RPMI 1640 and FCS were from HyClone. Dexamethasone, DMSO, Hepes, CHAPS, Triton X-100, leupeptin, E64, and iodoacetamide were from Sigma-Aldrich, and dithiothreitol (DTT) was from Pierce Chemical Co. Hamster anti–murine Fas antibody (Jo-2) was obtained from BD PharMingen, and PE-labeled goat F(ab′)2 anti–hamster IgG was from Caltag. The apoptosis inhibitor Z-VAD(OMe)-FMK was purchased from Alexis Biochemicals (some confusion exists about this compound, as not all suppliers make it clear that the normal commercial product is a methyl ester). PhiPhiLux™ and CaspaLux™ cell-permeable fluorogenic substrates were from OncoImmunin, Inc. Recombinant caspases 3, 6, 7, and 8 were obtained from BD PharMingen and Medical Biological Laboratories (Nagoya, Japan). Solvents such as HPLC grade dichloromethane, methanol, and acetonitrile were from Fisher Scientific. Propidium iodide (PI) and the fluorophores 5′,6′-carboxytetramethylrhodamine succinimidyl ester and 5′,6′-rhodamine green carboxylic acid succinimidyl ester were from Molecular Probes. Reverse phase HPLC equipment and columns were from Waters Corp. and SynChrom, Inc.
Caspase Substrates.
The reagents and methods used for peptide synthesis and derivatization have been described in detail previously 14. In brief, peptides were synthesized using both an automated peptide synthesizer and by manual solid phase methodology, and subsequently purified by reverse phase HPLC. Peptides were subjected to mass spectrometric analysis (PeptidoGenic Research) to determine the molecular mass and confirm peptide structure and composition. Each purified peptide was derivatized with the appropriate fluorophore as described previously 14. Substrates were purified into single components of homodoubly derivatized peptides by reverse phase HPLC and further characterized by absorption and fluorescence spectroscopy.
Caspase Activity Measurements in Extracts and Intact Thymocytes.
Single cell suspensions of thymocytes were cultured in RPMI 1640 with 10% FCS at a concentration of 106 cells/ml. Dexamethasone (final concentration, 0.1 μM) and Z-VAD (OMe)-FMK (final concentration, 50 μM) were added at the indicated times from stock solutions in DMSO (100 mM for dexamethasone and 200 mM for Z-VAD[OMe]-FMK). For anti-Fas stimulation, wells in a 24-well plate were coated with anti-Fas antibody (20 μg/ml; 250 μl per well) overnight at 37°C. After twice washing the wells with medium containing 10% FCS, 4 × 106 thymocytes were added per well. After culture at 37°C in a 5% CO2 atmosphere for the indicated times, cells were centrifuged and resuspended in 75 μl of each substrate (10 μM) in 1.5-ml Eppendorf tubes. The open tubes were placed in the CO2 incubator for an additional 60 min. After washing in saline, cells were analyzed by flow cytometry. Caspase activity measurements in extracts with several known inhibitors and noninhibitors were carried out using fluorometers from Photon Technology International and SLM-AMINCO. Cell lysates were prepared using a cell lysate buffer consisting of 50 mM Hepes, pH 7.5, 10% (wt/vol) sucrose, 0.1% (wt/vol) CHAPS, 0.5% (wt/vol) Triton X-100, and 10 mM DTT. The substrate (10 μM) was prepared in 50 mM Hepes, pH 7.5, with 10 mM DTT. All measurements were made at 37°C using a thermostated cell holder in a 150-μl quartz cuvette (3 × 3.45 mm; Starna Cells, Inc.). The temperature of the substrate solution was preequilibrated for 20 min, ensuring temperature equilibration by having a flat base line before cell lysate addition. A 10-μl aliquot from an apoptotic thymocyte lysate was added to 110 μl of substrate solution. The fluorescence intensity was then monitored for 30–60 min. The initial velocity was calculated from the linear portion of the fluorescence increase. Various amounts of inhibitors were added to the cell lysate for 20 min before addition of the cell lysate mixture to the substrate solution.
Flow Cytometry.
Instruments from both Becton Dickinson (FACSort™ and FACScan™) and Beckman Coulter (EPICS XL) were used in this study. 105 PI-negative cellular events were analyzed for each file using FL1 versus FL3 dot plots to establish a PI-negative gate using a polygon region. Throughout the entire time course of experiments, the determined PI-positive population of any sample was never >20%. The EPICS XL was used for the forward scatter histograms and PI-gated fluorescence histograms shown in Fig. 2. In experiments with time course activation with anti-Fas antibody and with those of dexamethasone and carbobenzoxy-valyl-alanyl-aspartyl-fluoromethyl ketone (ZVAD-FMK), the caspase activity of the cell population and the order of caspase activation were determined by obtaining the mean fluorescence channel of all PI-negative cells using CELLQuest™ (Becton Dickinson) or EXPO™ (Beckman Coulter) software.
Figure 2.


Analysis of caspase activation by flow cytometry. (A) Dexamethasone (final concentration 0.1 μM) was added to freshly isolated mouse thymocytes that were incubated at 37°C for various times before washing and adding one of five caspase substrates. Incubation with substrate was then carried out for an additional 60 min. The time of exposure of cells to dexamethasone is indicated in the left margin of each panel. Forward angle light scatter from incubation without substrate addition is shown at the left; these histograms were identical to those of cells with substrates. (B) Thymocytes cultured on anti-Fas–coated wells were analyzed as in A.
Confocal Microscopy.
Cells were incubated with various substrates at 10 μM while suspended in RPMI 1640 plus 10% FCS, 10 mM Hepes, and 0.1 μM dexamethasone. Cell suspensions were transferred to a thermostated chamber with a no. 1 coverslip bottom, allowed to settle, and viewed on a ZEISS LSM410 laser scanning confocal microscope system using a 63×, 1.4 NA objective. Substrates were present at 10 μM throughout the course of the induction and imaging. Samples were excited using a 488/518-nM krypton/argon laser, and fluorescent and Nomarski images were acquired every 5 min. Fluorescent images were acquired as single optical sections 2–3 μm in thickness, and brightness/contrast settings were adjusted so that the fluorescent signal of cells without fluorescent substrate was near background. As the substrates are cleaved in apoptotic cells, cellular fluorescence shifts from below to above the fluorescence of the bulk solution in the same plane. Changes in cell size and fluorescence were analyzed using Image-Pro® Plus (Media Cybernetics). The outlines of individual cells were manually traced from digitized RGB images, and cell area and integrated fluorescence intensities were calculated.
Results
Peptide Substrate Cleavage by Apoptotic Thymocyte Extracts.
The caspase substrates used in these studies were based on optimal peptide recognition motifs for various caspases as reported by peptide library studies 2 and are described in Table . To assess whether cleavage of these substrates in apoptotic thymocytes was due to caspases, we examined extracts from untreated fresh thymocytes, or from thymocytes treated with dexamethasone. We found that the DEVD substrate was cleaved 25-fold faster by apoptotic thymocyte extracts than by fresh thymocyte extracts (on a cell basis), as expected from studies with other DEVD substrates 23. Fig. 1 shows the effect of various protease inhibitors on the apoptotic DEVDase activity, giving an inhibition profile expected for a caspase. Thus DEVD-CHO is a potent blocker, followed by ZVAD-FMK, which together with the inhibition by iodoacetamide implicates a cysteine protease. The complete inhibition by DEVD-CHO is consistent with the absence of proteases other than caspase 3 or a very closely related protease in the apoptotic thymocyte cell lysate that might cleave the DEVDase substrate and generate fluorescence increases. Additionally, the lack of inhibition by E-64 or leupeptin is not compatible with this activity being due to lysosomal cathepsins or calpain, leaving caspases as the remaining candidate intracellular enzyme class responsible for the activity in extracts 24.
Table 1.
Amino Acid Sequence of Cell-permeable Fluorogenic Caspase Substrates Used in This Study
| Substrate | Target caspase | Activity | Sequence |
|---|---|---|---|
| PhiPhiLux | Caspase 3/7 | DEVDase | KDPC5GDEVDGIDGC5PKGY |
| CaspaLux-3PE | Procaspase 3 processing | IETDase | KDPC5GIETDSGVGC5PKGY |
| CaspaLux-6 | Caspase 6 | VEIDase | KDPC5GLVEIDNGGC5PKGY |
| CaspaLux-9 | Caspase 9 | LEHDase | KDPC5GLEHDGINGC5PKGY |
| CaspaLux-1 | Caspase 1 | WEHDase | KDPC5GWEHDGINGC5PKGY |
The amino acid residues constituting the major protease recognition motif (P4 to P1) are shown in boldface. Caspase cleavage occurs at the COOH end of the P1 amino acid. The IETD recognition sequence is preferred by caspase 8 also. The fluorophores are covalently conjugated through the ε-amino groups of lysine side chains. C5 is ε-aminocaproic acid.
Figure 1.

Inhibition of DEVDase activity in apoptotic thymocyte extract using PhiPhiLux™ substrate. An extract of mouse thymocytes exposed to 0.1 μM dexamethasone for 6 h was incubated in fluorometer cuvettes at 37°C with 10 μM PhiPhiLux™ substrate with or without the indicated protease inhibitors. Enzymatic activity was calculated from the rate of increase of fluorescence.
To confirm further that these substrates are cleaved by a caspase when loaded into apoptotic cells, we have carried out an HPLC analysis of the recovered intact substrate and the cleaved substrate fragments from the apoptotic thymocytes that have been incubated with these substrates for 15–30 min. When the fluorescent products in extracts of such apoptotic thymocytes were examined, the initial cleavage products detected were derived from a cleavage at the intended P1 aspartic acid in both cases, with subsequent secondary breakdown products formed (data not shown).
Intracellular Caspase Activities in Dexamethasone-treated Thymocytes.
Quantitation of intracellular caspase activities in apoptotic thymocytes treated with dexamethasone in vitro is shown in Fig. 2. For these experiments, the apoptotic cells were sampled after various times of incubation as indicated, the five rhodamine green–based caspase substrates were loaded into aliquots of the cell suspension, and flow cytometry was subsequently carried out. By this approach, the period of substrate exposure was equivalent for all cells analyzed, allowing a comparison between different stages in the caspase cascade.
In all cases, the 1-h time point shown in Fig. 2 A gave identical profiles to fresh thymocytes, and this single peak was assumed to represent unhydrolyzed substrate taken up uniformly by the cells. Fresh thymocytes run without exposure to substrate had a single peak with ∼10-fold less fluorescence (data not shown). Examination of all apoptotic profiles from all caspase substrates shows that a second discrete peak with an intensity ∼10-fold higher than the initial fluorescence is formed with further incubation time, and this peak increasingly dominates the profile. The VEIDase activity profiles were unique in exhibiting two discrete peaks of increased fluorescence intensity, and at 6–7 h the population was divided roughly equally among three fluorescence levels.
The sequence of caspase activation can be best seen by examination of the profiles at the 2-h time point, which shows highest activity for the LEHD substrate, followed by WEHDase (or ICE-like activity), VEIDase, and finally IETDase and DEVDase. Although about half the cells had increased their fluorescence by ∼10-fold with the LEHD substrate at 2 h after addition of dexamethasone, a comparable increase in DEVDase activity was not observed until 5 h. As expected, the onset of the IETDase activity (caspase 3 processing enzyme) preceded the appearance of the DEVDase activity. Since all five substrates showed unique patterns of increased fluorescence with time, it appears that distinct enzymes with different kinetics of activation are responsible for the proteolytic activities of these substrates in thymocytes.
Intracellular Caspase Activities in Anti-Fas–treated Thymocytes.
To compare the intracellular caspase activities in thymocytes with a distinctly different upstream activation pathway, we have carried out experiments parallel to the above with anti-Fas antibody (Fig. 2 B). Compared with dexamethasone, caspase activation by saturating amounts of anti-Fas antibody was slow, and there was more heterogeneity of caspase activity in the apoptotic cells. The order of activation of caspases was determined by plotting the mean fluorescence of the PI-negative cells between 1 and 3 h for each of the five substrates (data not shown), which increased in the order: IETDase, WEHDase, DEVDase, LEHDase, and VEIDase. Since the IETD substrate was optimal for caspase 8, this result is consistent with expectations that it initiates this cascade. Although the progression of caspase activation was less distinct for anti-Fas triggering than dexamethasone triggering, it is very clear that for the anti-Fas case DEVDase activity precedes VEIDase, in contrast to dexamethasone treatment of these cells.
Other differences between induction by the two stimuli were the distribution profiles and extent of induction. Thus, although stimulation by dexamethasone resulted in a bimodal pattern for all substrates except VEIDase (which became trimodal at later times), thymocytes stimulated with anti-Fas developed trimodal patterns with all substrates. To ascertain if anti-Fas–mediated IETDase activation of a subpopulation of thymocytes was due to differential cell surface expression of Fas, flow cytometry using the triggering anti-Fas antibody was used to compare Fas surface expression on IETDasehigh and IETDaselow subpopulations. These showed identical histograms, indicating that differences in surface Fas expression did not account for the subpopulation differences in caspase activation among thymocytes (data not shown).
Ordering of VEIDase versus DEVDase by Confocal Microscopy.
Because two recent studies with apoptotic Jurkat cell lysates showed that procaspase 6 was activated by caspase 3 6 7, the observation that VEIDase activity increased before the DEVDase activity in dexamethasone-treated thymocytes (Fig. 2 A) was unexpected. The relative order of appearance of these two activities was addressed using confocal microscopy with cells continuously exposed to a mixture of the VEID and DEVD substrates derivatized with rhodamine green and tetramethyl rhodamine fluorophores, respectively, with dexamethasone added along with the substrates at time zero. Confocal images of these thymocytes at 20-min intervals are shown in Fig. 3 A. The initial color appearing in individual cells from 90 to 190 min is invariably the green VEIDase activity. With increasing time, this VEIDase activity is replaced by the yellow color, signifying the presence of both VEIDase and DEVDase activities. Cells appearing red, indicative of DEVDase activity without accompanying VEIDase activity, are not obvious in any images. Thus, this independent approach using confocal microscopy confirms the VEIDase before DEVDase order found by flow cytometry for this apoptotic system. Fig. 3 B also shows the separate images used to construct the frames of Fig. 3 A, illustrating the distribution of fluorescent enzymatic products within these apoptotic thymocytes. It can be seen that the cells in the lower left panel with green VEIDase activity that have not yet become positive for red DEVDase display a relatively uniform distribution of fluorescence throughout the cells. However, those cells that have also become DEVDase positive and have therefore been VEIDase positive for some time appear yellow due to the accumulation of the green as well as red enzymatic products in cytoplasmic organelles. These more intensely stained cells seem likely to account for the highest intensity peak of VEIDase activity in Fig. 2 A. Fig. 3 C shows the result of quantitatively analyzing a representative individual cell from these confocal images. When examined on a frame by frame basis, the green and red fluorescent signals have distinct patterns of increase that cannot be accounted for by assuming that the red DEVDase activity is based on a less sensitive detection of the VEIDase activity.
Figure 3.

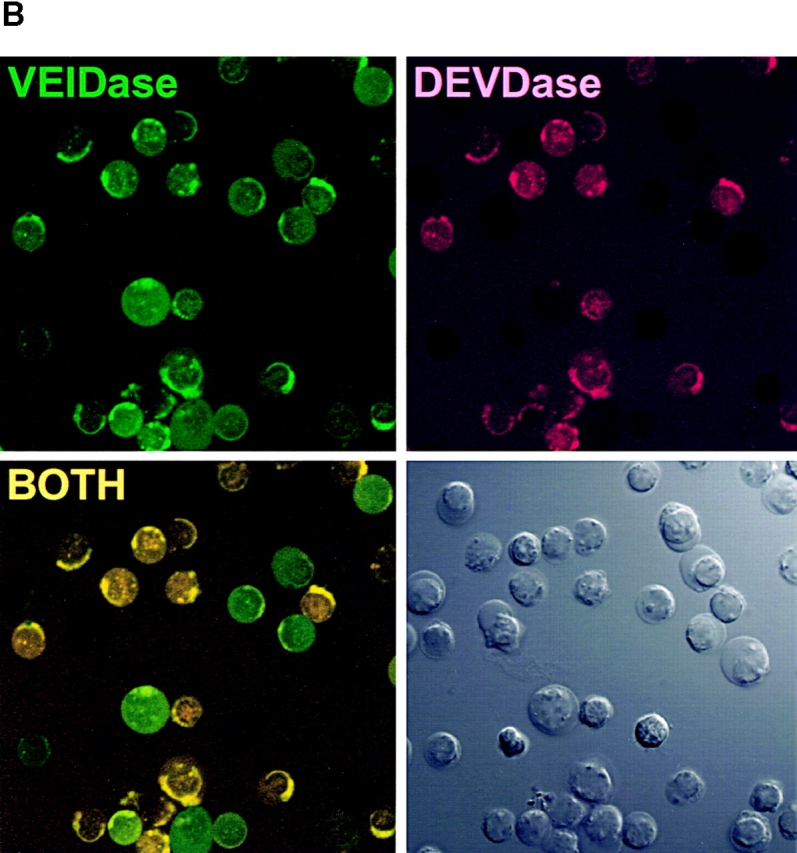

Confocal microscopy images of dexamethasone-treated thymocytes. (A) Time course of cells incubated with 10 μM DEVDase (red) and VEIDase (green) substrates. The apoptosis inducing agent, 0.1 μM dexamethasone, and two substrates were present continuously. Confocal images of cells at 20-min intervals starting at 90 min are shown here. The yellow color indicates the presence of both caspase activities. (B) Fluorescence images of caspases in apoptotic thymocytes derived from VEIDase (CaspaLux-6™) and DEVDase (PhiPhiLux™) are shown separately to assess their differential activity, as well as the additive image similar to A. These images are from the 140-min time point of the experiment shown in A. The Nomarski image is in the lower right. The dark images of cells in each fluorescence image correspond to those cells in which intracellular caspase activation is not yet detectable. (C) Time course of activation of the DEVDase (circles) and VEIDase (squares) activities compared with the cell cross-sectional area (triangles) plotted from the single cell shown. Both area and fluorescence are shown in arbitrary units, relative to the 92-min time point.
Further examination of the images in Fig. 3 reveals an apparent size increase as the cells become caspase positive. This is best seen by finding a green cell in one frame of Fig. 3 A and following it backward with time in the frames shown. In every case, the Nomarski images show that before becoming caspase positive, the thymocytes show a smaller diameter than the green (VEIDase only) cells. It appears in Fig. 3 A that the yellow cells at 190 min may also be smaller than they were when they were green in earlier frames. Nomarski images of some of these large cells are suggestive of blebbing, with smooth round membranes. When projected as a time-lapse sequence of Nomarski images without fluorescence overlay, the membranes and cytoplasmic organelles appear in vigorous motion suggestive of zeiosis (25; data not shown). Fig. 3 C quantitates the size increase of one apoptotic thymocyte in this experiment, and shows that the onset of this increase precedes the appearance of caspase cleavage products. The increase in diameter seen in confocal images appears to be reflected in the small shift to the right in the forward scatter peak at 2 h as seen in Fig. 2 A. This scatter shift was transient, as by 3 h or later the light scatter histogram returned nearly to that of the 1-h time point. It is only later, at 5–7 h, that the histograms show a peak of lower scattering particles building up that could be attributed to classical apoptotic cell shrinkage.
Pharmacological Target of ZVAD(OMe)-FMK.
The ability to detect intracellular caspase activities allowed us to study the pharmacological target of the potent apoptosis inhibitor ZVAD(OMe)-FMK. We have previously shown that ZVAD(OMe)-FMK specifically blocks the appearance of dexamethasone-induced apoptotic nuclear morphology and thymocyte death in vitro 26. In this experiment, we sought to identify which intracellular caspases were inactivated by ZVAD(OMe)-FMK treatment of thymocytes. To this end, we added ZVAD(OMe)-FMK to thymocytes at various times relative to the addition of dexamethasone and then examined caspase activity by flow cytometry at 6 h. Activity was assessed by the mean fluorescence channel number of PI-negative cells. Fig. 4 shows the results of this experiment, which confirms that the addition of ZVAD (OMe)-FMK blocks apoptosis when added before or simultaneously with dexamethasone. However, when addition of this inhibitor was delayed, the intracellular activity of all caspases detected rose, with no significant selective depletion of activity. When ZVAD(OMe)-FMK was added at 4 h, the observed activity at 6 h of all caspases was 70–80% of uninhibited levels. As seen in Fig. 4, the time course for development of ZVAD(OMe)-FMK–resistant caspase activity is strikingly parallel for the different substrates, and the constant difference between the activity ratios of the different substrates are accounted for by the method used to define caspase positivity. When thymocytes were pretreated with ZVAD(OMe)-FMK followed by washing before dexamethasone addition, minimal inhibition was seen (data not shown). As discussed below, these data provide no evidence for significant inactivation of any of the detectable intracellular caspases by ZVAD(OMe) treatment of intact cells, but do suggest that ZVAD(OMe)-FMK blocks an upstream component in the caspase activation cascade.
Figure 4.

Effect of ZVAD(OMe)-FMK treatment on in situ caspase activation. Thymocytes were treated with 0.1 μM dexamethasone in vitro and analyzed after a total of 6 h incubation. The apoptosis inhibitor ZVAD(OMe)-FMK was added at various times of incubation as indicated, and the caspase activities were assessed by flow cytometry. The results are expressed as ratios of mean fluorescence channel number comparing the activity in the presence of 50 μM Z-VAD(OMe)-FMK with the uninhibited sample for that substrate.
Discussion
Intracellular Caspase Substrates.
The cell-permeable fluorogenic substrates used this study (Table ) are based on peptides of 18 amino acids containing central optimal tetrapeptide caspase recognition/cleavage sequences, with two identical fluorophores covalently attached near their termini. Previous studies have shown that in solution such substituted peptides assume an oval-shaped structure due to the formation of intramolecular excitonic H-dimers between the two fluorophores 27 28. In such rhodamine-derivatized dimers, the fluorophore fluorescence is quenched 90–99%. When a protease cleaves the peptide backbone of this complex, the cyclic structure incorporating the fluorophores is broken and two highly fluorescent substituted peptide fragments are generated. A surprising finding has been that caspase substrates of this design are permeable to cell membranes, thus allowing reporting of a variety of intracellular environments. While normally peptides of ∼18 amino acids are impermeable to cells without a specialized means of transport, we speculate that the head-to-head complex of rhodamine fluorophores forms a hydrophobic surface capable of interacting with lipid bilayers to allow passive diffusion of these substrates across the bilayer. After intracellular cleavage, the rate of diffusion of the peptide–fluorophore reaction products across the membrane is significantly reduced since they lack the large hydrophobic surface provided by the fluorophore–fluorophore dimer of the substrate. The intracellular fluorescent signal generated by these substrates is predominantly due to the accumulation of fluorescent peptide cleavage products. Until the apoptotic loss of membrane integrity, this fluorescent signal reflects a balance between the rate of substrate hydrolysis and the rate of loss of peptide hydrolysis products from the cell. Although analyzing intracellular caspase activities using such substrates lacks many of the advantages of traditional biochemical approaches (e.g., limited numbers of purified components in dilute solutions), the ability to assess enzymatic activity in intact cells avoids some of the potential artifacts of such approaches and provides an important opportunity to test molecular models in a physiologically meaningful setting 29.
In the above description of our results, we have been careful to avoid ascribing the enzymatic activities with particular substrates to individual caspases, even though the substrates have been designed based on optimal caspase recognition motifs (Table ). An initial question arises as to whether the activities seen represent caspases, as cleavage at any peptide bond between the lysines will give a signal with these substrates. Candidate cytoplasmic proteases that could be responsible for cleavage of these substrates include lysosomal cathepsins, the proteasome, and calpain. Fig. 1 shows that activity on the DEVDase substrate in apoptotic thymocyte extracts is specifically blocked by inhibitors targeted to caspase 3/7. Moreover, HPLC analysis of fluorophore-bearing peptide substrate fragments from intracellular digestion in apoptotic thymocytes shows that the initial primary cleavage is at the expected P1 aspartate (data not shown). It is clear that all the activities detected are caspase dependent, since ZVAD(OMe)-FMK blocks their appearance (Fig. 4) as well as other detectable manifestations of dexamethasone-induced thymocyte apoptosis 26. Thus, we conclude that activity on these protease substrates in intact apoptotic cells reflects caspases as opposed to other possible intracellular proteases.
Since the demonstrated peptide cleavage selectivity with recombinant caspases is not absolute, and the in situ intracellular activities of individual caspases have not been tested, the attribution of the activities observed to particular caspases must be made cautiously. We have carried out experiments with several recombinant caspases (3 and 6) to assess the specificity of some of the substrates used here, in particular the VEIDase and DEVDase. We have found that the peptide substrates in Table demonstrate a far greater caspase selectivity than the tetrapeptide AMC substrates (Komoriya, A., B.Z. Packard, and P.A. Henkart, manuscript in preparation).
Caspase Activities in Intact Cells.
Analysis of intracellular caspase activity in dexamethasone-induced apoptotic thymocytes by flow cytometry (Fig. 2 A) strikingly shows a “quantal” distribution of caspase activity, with cells having either a background level of fluorescence due to uncleaved substrate or a 40-fold higher level of fluorescence, with few cells at an intermediate level. This quantal higher fluorescence level can most simply be explained as that achieved as a balance between maximal substrate hydrolysis and leakage of the fluorescent products out of the cell. The activity histograms for VEIDase after dexamethasone treatment as well as all five activities after anti-Fas treatment become trimodal, with an additional higher peak. This higher peak appears to correlate with the accumulation of intense fluorescence within intracellular organelles as seen in the microscope. The heterogeneity of the intracellular and intercellular caspase activities observed with these intracellular substrates is a novel observation, not readily detectable with other methods of measuring caspase activities.
Our results show that after dexamethasone treatment there is a pattern of progressive caspase activation with time, as expected for the caspase cascade proposed from traditional biochemical studies. As shown in Fig. 2 A, this progression begins with LEHDase, followed by WEHDase, VEIDase, IETDase, and DEVDase. These activities correspond to the optimal tetrapeptide recognition sequences for caspases 9, 1, 6, 8, and 3/7, respectively (Table ), and this order of caspase activation for dexamethasone-treated thymocytes is reasonable when compared with recent studies with enzyme inhibitors 26 and caspase knockout mice. This death pathway requires Apaf-1 17 and caspase 9 18 but is independent of caspases 1 and 3 30 31 and FADD 32. Our observation that the first detectable caspase signal is the LEHDase activity is consistent with caspase 9 activation as the initiator of the caspase cascade in these cells. Although WEHDase activity seen in Fig. 2 A could be due to caspase 1, it could also be due to caspases 4 or 5 2, or potentially to newly described members of the ICE subfamily. While caspase 1 is presumably not part of the apoptotic death pathway, it is possible that it is responsible for processing cytokines such as IL-1β in the dying cells 33.
The order of caspase activation for anti-Fas–treated thymocytes (Fig. 2 B) is less clear-cut but distinctly different from dexamethasone-treated thymocytes. The initial activity increases are in the order IETDase, WEHDase, DEVDase, LEHDase, and VEIDase. Since IETDase is the preferred substrate for caspase 8, which is activated by FADD-induced aggregation of its proenzyme 34, its early activation is consistent with current ideas about this pathway. The late activation of LEHDase, the preferred substrate for caspase 9, is consistent with the minor role of the Bid/mitochondrial amplification loop in the Fas death pathway in thymocytes 20 and the activation of caspase 9 by downstream caspases.
Studies in other laboratories on the relative order of activation of caspases 3 and 6 have yielded conflicting results, and our data show clearly that even in the same cells different triggering signals result in a different relative order of activation of these downstream caspases. Our observation that VEIDase activity appears before DEVDase activity in dexamethasone-treated thymocytes was unexpected given the studies of Slee et al. 7, which provided clear evidence that caspase 3 activity is upstream of caspase 6. They used Western blots to examine caspase activation/processing initiated by cytochrome c in Jurkat extracts, a model we considered relevant to thymocytes treated with dexamethasone. The most compelling experiments were those in which removal of individual caspases from the extracts blocked activation of other caspases, and it was particularly striking that depletion of caspase 3 abolished the ability of cytochrome c to trigger the processing of caspase 6, placing caspase 3 upstream of caspase 6. This same order was deduced from studies of the Fas death pathway 6 and are compatible with our observations of this pathway in thymocytes. On the other hand, an earlier study showed that addition of purified caspase 6 to a nonapoptotic extract triggered the processing of caspases 3 and 7, whereas addition of purified caspase 3 or 7 to a nonapoptotic extract failed to trigger the processing of caspase 6 4. These results suggest that caspase 6 is upstream of caspases 3 and 7. Recently, Xanthoudakis et al. assayed camptothecin-treated Jurkat extracts for an enzyme with procaspase 3 processing activity and found that this substrate formed a complex with Hsp60 which was then processed by caspase 6 5. These results also favor the activation of caspase 6 before caspase 3 in the cascade. Our results argue against a common downstream module of caspase activation, suggesting that different upstream caspases trigger distinct downstream activation pathways.
The Pharmacological Target of ZVAD(OMe)-FMK.
The finding in Fig. 4 that ZVAD(OMe)-FMK treatment of apoptotic cells fails to significantly inactivate detectable caspase activity was unexpected. This compound was synthesized as a stable substitute for the ICE inhibitor ZVAD-FMK, which has a half-life of <1 h in aqueous solution 35. It has been widely and successfully used to block apoptosis in vitro, and has shown potency in animal models for diseases involving apoptosis 21 22. ZVAD(OMe)-FMK should be membrane permeable since it lacks charges, but for it to inactivate caspases intracellular esterases are required to remove the ester, forming the active site reagent ZVAD-FMK. Since this inhibitor lacks a P4 amino acid and is a potent upstream blocker of apoptotic pathways triggered by both death domain receptors and apaf-1 36, it is widely assumed that ZVAD-FMK is a general inhibitor of caspases. A recent solution study showed that ZVAD-FMK indeed irreversibly inactivates all of the individual recombinant caspases 1–9, but with a 1,000-fold range in reaction rates 35. Caspases 1, 5, 8, and 9 show rapid inactivation, with a t 1/2 of several seconds at 1 μM ZVAD-FMK, whereas caspases 2, 4, and 6 react two to three orders of magnitude more slowly. Since caspases 8 and 9 are the initial caspases in the two major triggering pathways, these results appeared to account for the potent antiapoptotic activity of ZVAD(OMe)-FMK. Thus, we expected to find the LEHDase and WEHDase activities in apoptotic thymocytes inactivated by ZVAD(OMe)-FMK treatment. In the experiment shown in Fig. 4, we measured intracellular caspase activities at 6 h, when all substrates showed substantial activity. ZVAD(OMe)-FMK totally blocked the activation of all detectable caspases when added before or at the time of dexamethasone addition, but after these caspases were activated (e.g., at 4 h), addition of ZVAD(OMe)-FMK blocked only a minor component of the activities attributable to newly activated enzymes. There was no clear indication of a selective caspase inactivation predicted by the differing reaction rates with ZVAD-FMK 35, suggesting that ZVAD(OMe)-FMK acts upstream of the measured components of the caspase cascade. Extrapolating results from the biochemical study of ZVAD-FMK reactivity is difficult, as its cytoplasmic concentration is not known.
There are several possible explanations for our failure to observe inactivation by ZVAD(OMe)-FMK of previously activated caspases. Although we believe it unlikely for the reasons discussed, it is possible we are not measuring the caspases these substrates were designed for, and that the LEHDase activity observed is not due to caspase 9. Another possibility is that the major intracellular caspase activities are within membrane-bound organelles (as suggested by the confocal images) and thus not accessible to the active hydrophilic ZVAD-FMK inhibitor in the cytoplasm. A third possibility is that ZVAD(OMe)-FMK or ZVAD-FMK could be reacting with a critical caspase we have not measured, or perhaps a cysteine protease that is not a caspase. In this respect, it is worth noting that the cathepsin B inhibitor carbobenzoxy-phenylalanyl-alanyl-fluoromethyl ketone (ZFA-FMK) does not block dexamethasone-induced thymocyte apoptosis 26, suggesting a P1 amino acid specificity for the pharmacological target of ZVAD(OMe)-FMK. It is possible that ZVAD-FMK blocks the putative critical upstream aggregation-induced autoactivation of procaspase 9. This autoactivation activity, involving processing near the active site cysteine when the proenzymes are part of a multimeric complex, is not understood in detail but is presumably not measurable with the present fluorogenic substrates. ZVAD-FMK may selectively target this activity (and the homologous autoactivation of procaspase 8) more efficiently than it inactivates the mature caspases.
Regardless of its precise molecular target, the results in Fig. 4 clearly indicate that ZVAD(OMe)-FMK acts at an early stage in the apoptotic pathway, either by inactivating an upstream caspase or another activity. The inability of ZVAD(OMe)-FMK to significantly block caspase activation if added before dexamethasone and then washed out indicates that this activity is silent before apoptosis is induced. We thus conclude that the pharmacological target of ZVAD(OMe)-FMK has the following properties: (a) it is activated early in the apoptosis cascade; (b) it is a common element in apoptotic pathways induced by many diverse triggers; and (c) it has the reactivity of a cysteine protease with selectivity for a P1 aspartic acid.
We believe that the ability to monitor enzyme activities in intact functioning cells is an important component of our efforts to apply the knowledge of enzyme biochemistry to physiological systems. The caspase substrates described here provide the first attempt to use this approach for apoptosis, where there is ample evidence for the importance of cellular organization in regulating these critical proteolytic mediators. Given the widespread interest in selectively modulating apoptosis in vivo, we believe that these are useful tools that can be applied to a variety of other apoptotic systems.
Acknowledgments
The authors thank Dr. L. Guedez for advice in flow cytometry instrumentation and Dr. K. Allison-Ampe for thymocyte isolation pedagogy.
Footnotes
Abbreviations used in this paper: apaf-1, apoptotic protein activating factor 1; DTT, dithiothreitol; FADD, Fas-associated death domain protein; ICE, IL-1β–converting enzyme; PI, propidium iodide.
References
- Yuan J., Shaham S., Ledoux S., Ellis H.M., Horvitz H.R. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1β-converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- Thornberry N.A., Rano T.A., Peterson E.P., Rasper D.M., Timkey T., Garcia-Calvo M., Houtzager V.M., Nordstrom P.A., Roy S., Vaillancourt J.P. A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J. Biol. Chem. 1997;272:17907–17911. doi: 10.1074/jbc.272.29.17907. [DOI] [PubMed] [Google Scholar]
- Rodriguez J., Lazebnik Y. Caspase-9 and APAF-1 form an active holoenzyme. Genes Dev. 1999;13:3179–3184. doi: 10.1101/gad.13.24.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth K., O'Rourke K., Salvesen G.S., Dixit V.M. Molecular ordering of apoptotic mammalian CED-3/ICE-like proteases. J. Biol. Chem. 1996;271:20977–20980. doi: 10.1074/jbc.271.35.20977. [DOI] [PubMed] [Google Scholar]
- Xanthoudakis S., Roy S., Rasper D., Hennessey T., Aubin Y., Cassady R., Tawa P., Ruel R., Rosen A., Nicholson D.W. Hsp60 accelerates the maturation of pro-caspase-3 by upstream activator proteases during apoptosis. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:2049–2056. doi: 10.1093/emboj/18.8.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata H., Takahashi A., Kobayashi S., Yonehara S., Sawai H., Okazaki T., Yamamoto K., Sasada M. Caspases are activated in a branched protease cascade and control distinct downstream processes in fas-induced apoptosis. J. Exp. Med. 1998;187:587–600. doi: 10.1084/jem.187.4.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slee E.A., Harte M.T., Kluck R.M., Wolf B.B., Casiano C.A., Newmeyer D.D., Wang H.G., Reed J.C., Nicholson D.W., Alnemri E.S. Ordering the cytochrome c–initiated caspase cascadehierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9–dependent manner. J. Cell Biol. 1999;144:281–292. doi: 10.1083/jcb.144.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossy-Wetzel E., Newmeyer D.D., Green D.R. Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:37–49. doi: 10.1093/emboj/17.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin S.A., Lorenzo H.K., Zamzami N., Marzo I., Snow B.E., Brothers G.M., Mangion J., Jacotot E., Costantini P., Loeffler M. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Mancini M., Nicholson D.W., Roy S., Thornberry N.A., Peterson E.P., Casciola-Rosen L.A., Rosen A. The caspase-3 precursor has a cytosolic and mitochondrial distributionimplications for apoptotic signaling. J. Cell Biol. 1998;140:1485–1495. doi: 10.1083/jcb.140.6.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin S.A., Lorenzo H.K., Zamzami N., Marzo I., Brenner C., Larochette N., Prevost M.C., Alzari P.M., Kroemer G. Mitochondrial release of caspase-2 and -9 during the apoptotic process. J. Exp. Med. 1999;189:381–394. doi: 10.1084/jem.189.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechushtan A., Smith C.L., Hsu Y.T., Youle R.J. Conformation of the Bax C-terminus regulates subcellular location and cell death. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:2330–2341. doi: 10.1093/emboj/18.9.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packard B.Z., Toptygin D.D., Komoriya A., Brand L. Design of profluorescent protease substrates guided by exciton theory. Methods Enzymol. 1997;278:15–23. doi: 10.1016/s0076-6879(97)78004-2. [DOI] [PubMed] [Google Scholar]
- Packard B.Z., Toptygin D.D., Komoriya A., Brand L. Profluorescent protease substratesintramolecular dimers described by the exciton model. Proc. Natl. Acad. Sci. USA. 1996;93:11640–11645. doi: 10.1073/pnas.93.21.11640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packard B.Z., Toptygin D.D., Komoriya A., Brand L. Characterization of fluorescence quenching in bifluorophoric protease substrates. Biophys. Chem. 1997;67:167–176. doi: 10.1016/s0301-4622(97)00036-7. [DOI] [PubMed] [Google Scholar]
- Packard B.Z., Toptygin D.D., Komoriya A., Brand L. Intramolecular resonance dipole-dipole interactions in a protease substrate. J. Phys. Chem. B. 1998;102:752–758. [Google Scholar]
- Yoshida H., Kong Y.Y., Yoshida R., Elia A.J., Hakem A., Hakem R., Penninger J.M., Mak T.W. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–750. doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- Hakem R., Hakem A., Duncan G.S., Henderson J.T., Woo M., Soengas M.S., Elia A., de la Pompa J.L., Kagi D., Khoo W. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 1998;94:339–352. doi: 10.1016/s0092-8674(00)81477-4. [DOI] [PubMed] [Google Scholar]
- Zhang J., Cado D., Chen A., Kabra N.H., Winoto A. Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature. 1998;392:296–300. doi: 10.1038/32681. [DOI] [PubMed] [Google Scholar]
- Yin X.M., Wang K., Gross A., Zhao Y., Zinkel S., Klocke B., Roth K.A., Korsmeyer S.J. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature. 1999;400:886–891. doi: 10.1038/23730. [DOI] [PubMed] [Google Scholar]
- Rodriguez I., Matsuura K., Ody C., Nagata S., Vassalli P. Systemic injection of a tripeptide inhibits the intracellular activation of CPP32-like proteases in vivo and fully protects mice against Fas-mediated fulminant liver destruction and death. J. Exp. Med. 1996;184:2067–2072. doi: 10.1084/jem.184.5.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara H., Friedlander R.M., Gagliardini V., Ayata C., Fink K., Huang Z., Shimizu-Sasamata M., Yuan J., Moskowitz M.A. Inhibition of interleukin 1beta converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc. Natl. Acad. Sci. USA. 1997;94:2007–2012. doi: 10.1073/pnas.94.5.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enari M., Talanian R.V., Wong W.W., Nagata S. Sequential activation of ICE-like and CPP32-like proteases during Fas-mediated apoptosis. Nature. 1996;380:723–726. doi: 10.1038/380723a0. [DOI] [PubMed] [Google Scholar]
- Nicholson D.W., Ali A., Thornberry N.A., Vaillancourt J.P., Ding C.K., Gallant M., Gareau Y., Griffin P.R., Labelle M., Lazebnik Y.A. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- Majno G., Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am. J. Pathol. 1995;146:3–15. [PMC free article] [PubMed] [Google Scholar]
- Sarin A., Wu M.-L., Henkart P.A. Different ICE-family protease requirements for the apoptotic death of T lymphocytes triggered by diverse stimuli. J. Exp. Med. 1996;184:2445–2450. doi: 10.1084/jem.184.6.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packard B.Z., Komoriya A., Toptygin D.D., Brand L. Structural characteristics of fluorophores which form intramolecular H-type dimers ina protease substrate. J. Phys. Chem. B. 1997;101:5070–5074. [Google Scholar]
- Packard B.Z., Komoriya A., Nanda V., Brand L. Intramolecular excitonic dimers in protease substratesmodification of the backbone moiety to probe the H-dimer structure. J. Phys. Chem. B. 1998;102:1820–1827. [Google Scholar]
- Weng G., Bhalla U.S., Iyengar R. Complexity in biological signaling systems. Science. 1999;284:92–96. doi: 10.1126/science.284.5411.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida K., Lippke J.A., Ku G., Harding M.W., Livingston D.J., Su M.S.S., Flavell R.A. Altered cytokine export and apoptosis in mice deficient in interleukin-1β converting enzyme. Science. 1995;267:2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- Kuida K., Zheng T.S., Na S., Kuan C., Yang D., Karasuyama H., Rakic P., Flavell R.A. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- Zornig M., Hueber A.O., Evan G. p53-dependent impairment of T-cell proliferation in FADD dominant-negative transgenic mice. Curr. Biol. 1998;8:467–470. doi: 10.1016/s0960-9822(98)70182-4. [DOI] [PubMed] [Google Scholar]
- Miwa K., Asano M., Horai R., Iwakura Y., Nagata S., Suda T. Caspase 1-independent IL-1beta release and inflammation induced by the apoptosis inducer Fas ligand. Nat. Med. 1998;4:1287–1292. doi: 10.1038/3276. [DOI] [PubMed] [Google Scholar]
- Wallach D., Varfolomeev E.E., Malinin N.L., Goltsev Y.V., Kovalenko A.V., Boldin M.P. Tumor necrosis factor receptor and Fas signaling mechanisms. Annu. Rev. Immunol. 1999;17:331–367. doi: 10.1146/annurev.immunol.17.1.331. [DOI] [PubMed] [Google Scholar]
- Garcia-Calvo M., Peterson E.P., Leiting B., Ruel R., Nicholson D.W., Thornberry N.A. Inhibition of human caspases by peptide-based and macromolecular inhibitors. J. Biol. Chem. 1998;273:32608–32613. doi: 10.1074/jbc.273.49.32608. [DOI] [PubMed] [Google Scholar]
- Slee E.A., Zhu H.J., Chow S.C., MacFarlane M., Nicholson D.W., Cohen G.M. Benzyloxycarbonyl-Val-Ala-Asp (OMe) fluoromethylketone (Z-VAD.FMK) inhibits apoptosis by blocking the processing of CPP32. Biochem. J. 1996;315:21–24. doi: 10.1042/bj3150021. [DOI] [PMC free article] [PubMed] [Google Scholar]