

Abstract
Shigella, the causative agent of bacillary dysentery, is capable of directing its movement within host cells by exploiting actin dynamics. The VirG protein expressed at one pole of the bacterium can recruit neural Wiskott-Aldrich syndrome protein (N-WASP), a downstream effector of Cdc42. Here, we show that Cdc42 is required for the actin-based motility of Shigella. Microinjection of a dominant active mutant Cdc42, but not Rac1 or RhoA, into Swiss 3T3 cells accelerated Shigella motility. In add-back experiments in Xenopus egg extracts, addition of a guanine nucleotide dissociation inhibitor for the Rho family, RhoGDI, greatly diminished the bacterial motility or actin assembly, which was restored by adding activated Cdc42. In N-WASP–depleted extracts, the bacterial movement almost arrested was restored by adding exogenous N-WASP but not H208D, an N-WASP mutant defective in binding to Cdc42. In pyrene actin assay, Cdc42 enhanced VirG-stimulating actin polymerization by N-WASP–actin-related protein (Arp)2/3 complex. Actually, Cdc42 stimulated actin cloud formation on the surface of bacteria expressing VirG in a solution containing N-WASP, Arp2/3 complex, and G-actin. Immunohistological study of Shigella-infected cells expressing green fluorescent protein–tagged Cdc42 revealed that Cdc42 accumulated by being colocalized with actin cloud at one pole of intracellular bacterium. Furthermore, overexpression of H208D mutant in cells interfered with the actin assembly of infected Shigella and diminished the intra- and intercellular spreading. These results suggest that Cdc42 activity is involved in initiating actin nucleation mediated by VirG–N-WASP–Arp2/3 complex formed on intracellular Shigella.
Keywords: bacterial infections, bacterial protein, microfilament proteins, actins, Wiskott-Aldrich syndrome
Introduction
The ability of Shigella to move within the cytoplasm of infected epithelial cells and the subsequent intercellular spreading are essential for bacillary dysentery. Intracellular Shigella is capable of exploiting the host cellular functions to direct actin polymerization at one pole of the bacterial surface, through which the bacterium gains a propulsive force in the cytoplasm 1 2. Upon making contact with the inner surface of the host cell plasma membrane, a motile Shigella develops a long membranous protrusion (filopodium) with the F-actin tail behind it that is endocytosed by the adjacent cells, resulting in the bacterium being surrounded by a double membrane 3 4 5 6 7. Shigella then disrupts the membranes and disseminates into the new host cytoplasm and multiplies again 3 4 7. Thus, the bacterial ability to spread intra- and intercellularly is a predominant feature of the pathogenesis of Shigella.
VirG (IcsA) protein of S. flexneri plays a crucial role in intra- and intercellular spreading 1 8, and VirG can directly interact with some of the host proteins involved in the modulation of actin dynamics 9 10. The VirG polypeptide is composed of 1,102 amino acids and contains 3 distinctive domains 6 11 12: the NH2-terminal signal peptide (residues 1–52); the surface-exposed VirG α domain (residues 53–758), which possesses 6 glycine-rich repeats and is essential for mediating actin assembly from motile Shigella in mammalian cells 9, whereas the remaining portion is required for the asymmetric distribution of VirG on the bacterial body 9; and the COOH-terminal portion, named VirG β core (residues 759–1,102), embedded in the outer membrane of S. flexneri and required for mediating the surface presentation of the α domain (residues 53–758) 12.
The motility of S. flexneri in mammalian cells requires many host factors. Using immunofluorescence of fixed infected cells with antibodies, several actin-associated proteins such as vinculin 3 9 13 14, plastin (fimbrin) 5, filamin 5, α-actinin 15 16, vasodilator-simulated phosphoprotein (VASP) 17, neural Wiskott-Aldrich syndrome protein (N-WASP) 10, zyxin 14, actin-related protein (Arp)2/3 complex 14 16 18, ezrin, cofilin, and CapZ 16 have been identified as being localized to the actin tail or at the posterior end of intracellular bacteria. N-WASP, ∼50% identical to WASP 19 20, has been reported to be functionally involved in generating the actin tail from motile S. flexneri 10 18. Arp2/3 complex is also required for actin-based motility of Shigella, in which N-WASP stimulates actin nucleation activity of Arp2/3 complex in vitro 18. Recently, actin depolymerizing factor (ADF)/cofilin, capping protein, α-actinin, and profilin have been reported to be involved in the regulation of actin turnover and stabilization of the actin tail in vitro 21. Besides Shigella, Listeria monocytogenes (for reviews, see references 22 and 23), spotted fever group Rickettsia 24 25, and the vaccinia virus 26 also induce polarized actin assembly at their surface. Listeria surface protein ActA is crucial for actin-based motility 27 28. ActA has a multidomain for interacting with two essential host factors, Arp2/3 complex 29 and Drosophila Enabled (Ena)/VASP family proteins 17 30. The NH2-terminal domain of ActA can not only interact with Arp2/3 complex, but also stimulate its actin nucleation activity 31. The central proline-rich domain of ActA interacts with Ena/VASP proteins that are proposed to mediate insertional actin polymerization on the surface of Listeria 32. The mechanism of Rickettsia movement, including the bacterial factor mediating actin assembly in mammalian cells, is still unknown. Vaccinia surface protein A36R can link to N-WASP through binding to adaptor protein Nck, which is implicated in the actin-based motility of virus particles, although the signal transduction required for the actin assembly appears to be different from that for Shigella motility 33. Therefore, although the precise mechanisms underlying the actin polymerization involved in the motility of the pathogens still remain to be elucidated, each strategy for modulating the host functions required for the reorganization of actin cytoskeletons will somehow differ among pathogens.
N-WASP is a member of the WASP family of proteins that includes human WASP 19 20, Saccharomyces cerevisiae WASP-like protein Las17p/Bee1p 34 35, and more distantly related Scar/WAVE proteins 36 37 38. N-WASP (and WASP) possesses several distinctive domains: a pleckstrin homology (PH) domain that binds phosphatidylinositol 4,5-bisphosphate (PtdIns[4,5]P2), a GTPase binding domain (GBD) that binds Cdc42, a proline-rich (P) region, a G-actin–binding verprolin homology (V) domain, a domain (C) with homology to the actin-depolymerizing protein cofilin, and finally a COOH-terminal acidic (A) segment 20. The COOH-terminal VCA domain interacts with the Arp2/3 complex and stimulates actin polymerization by activating the complex 39. Recently, it has been proposed that N-WASP exists in two conformations: an inactive form in which the VCA domain is masked, and an active form in which it is exposed. Both states are dependent on whether Cdc42 is bound to the GBD, thus somehow affecting the intramolecular interaction between the A segment and the basic amino acids near the GBD 40. Therefore, unmasked N-WASP, or the VCA domain itself, is thought to be able to promote rapid actin polymerization in filopodium formation in locomoting mammalian cells 40. Indeed, N-WASP has been shown to be required for Cdc42-mediated microspike formation in cells or Cdc42-mediated actin assembly by stimulating Arp2/3 complex in vitro 39 40. Although the precise mechanisms of N-WASP activation remain to be investigated, Cdc42 activity is considered to be the key player for regulating the state of N-WASP in vivo (for a review, see reference 41).
Rho family GTPases are involved in the reorganization of F-actin structures in response to extracellular stimuli, acting as modulators between membrane receptor signaling and the downstream effectors involved in regulating actin dynamics (for reviews, see references 42 43 44). Rho induces the assembly of contractile actomyosin filaments involved in the formation of actin stress fibers and focal adhesions, whereas Rac and Cdc42 control actin polymerization involved in the formation of lamellipodial and filopodial membrane protrusions, respectively 45 46 47. As described above, since Cdc42-dependent N-WASP activation is required for filopodial protrusions 40 and capable of interacting with VirG expressed on Shigella surface 10, we investigated whether or not Rho family GTPases including Cdc42 could be involved in the actin-based motility of Shigella.
Materials and Methods
Bacterial Strains, Cell Culture, and Media.
The S. flexneri 2a YSH6000 and L. monocytogenes serotype 1/2a EGD strains have been described previously 27 48. Escherichia coli MC1061 ompT::Km carrying pD10-1 (a pBR322-derived plasmid encoding virG gene) was prepared as described previously 10. L. monocytogenes serotype 1/2a EGD carrying pERL3::prfA 7973 was a gift of T. Chakraborty (Universität Giessen, Giessen, Germany). All bacteria were routinely grown in brain heart infusion broth (BHI; Difco) at 37°C. Swiss 3T3 cells, COS-7 cells, and Madin-Darby canine kidney (MDCK) cells were grown in DMEM (Sigma-Aldrich) containing 10% FCS (Nichirei). Sf9 insect cells for the baculovirus expression system were cultured in Sf-900 II SFM (Life Technologies) containing 10% FCS at 27°C under vigorous shaking.
Antibodies.
The rabbit anti-VirG (VRG-N2) antibody and the rabbit anti–N-WASP specific antibody have been described previously 10. The rat anti–ZO-1 mAb and mouse antiactin mAb were obtained from Chemicon International. The rabbit anti-Cdc42 polyclonal antibody and mouse anti–E-cadherin mAb were obtained from Calbiochem and Transduction Laboratories, respectively.
Protein Purification.
Glutathione S-transferase (GST)-VirG α domain fusion protein (GST-α1 and GST-α3) was constructed as described previously 9. E. coli expressing human Rho family GTPases, Cdc42-G12V (valine substituted for glycine at residue 12), Rac1-G12V, and RhoA-G14V (valine substituted for glycine at residue 14) were prepared as GST fusion proteins 49. The biological activities of these purified GTPases were confirmed by microinjection into serum-starved Swiss 3T3 cells 45 46 47. For the preparation of baculovirus expressing GST-fused Rho family GTPases, the DNA fragments encoding these fusion proteins were generated by PCR and cloned into pFASTBAC1 (Life Technologies). The membrane fractions of Sf9 cells including membrane-bound GST fusion proteins were solubilized in a buffer containing 50 mM Tris-HCl, 50 mM NaCl, 5 mM MgCl2, 1 mM dithiothreitol, 1 mM PMSF, and 1% CHAPS (Dojinkagaku), pH 7.6. The solubilized GST fusion proteins were purified using glutathione-Sepharose beads (Amersham Pharmacia Biotech). Recombinant rat N-WASP and its mutant H208D (aspartic acid substituted for histidine at residue 208) expressed by baculovirus 40 were prepared by using HiTrap Heparin and HiTrap Q columns (Amersham Pharmacia Biotech). Histidine-tagged rat N-WASP expressed by baculovirus was purified using Ni-NTA-Agarose (Qiagen). E. coli expressing RhoGDI were purified as a GST fusion protein. The GST portion of the fusion proteins was removed by digesting with thrombin in thrombin buffer (50 mM Tris-HCl, 150 mM NaCl, 2.5 mM CaCl2, 5 mM MgCl2, 1 mM dithiothreitol, pH 8.0). To activate Rho family GTPases in an actin tail assay, fusion proteins bound to beads were loaded with 5 mM GTPγS in loading buffer (100 mM Tris-HCl, 5 mM EDTA, pH 7.5) for 30 min at 30°C before thrombin digestion. The Arp2/3 complex was purified from bovine brain by the method described previously 39.
Infection of Cultured Cells and Immunofluorescence Microscopy.
All cells were grown on glass coverslips to ∼60% confluency in the absence of antibiotics. Cells were infected with Shigella at a multiplicity of infection of ∼100 per cell. The plates were centrifuged at 700 g for 10 min after adding bacteria. After 20 min, the plates were extensively washed with HBSS, and fresh medium supplemented with gentamicin (100 μg/ml) and kanamycin (60 μg/ml) was added. The infected cells were incubated for an additional 40 min to 1 h at 37°C before microinjection experiments or fixation in 4% paraformaldehyde in PBS for 20 min. For immunofluorescence studies, the coverslips were incubated in 50 mM NH4Cl in PBS for 10 min and the permeabilization of cells was carried out in 0.2% Triton X-100 in PBS for 20 min. After blocking for 30 min in 2% BSA in Tris-buffered saline (TBS: 50 mM Tris-HCl, 150 mM NaCl, pH 7.4), the coverslips were incubated with primary antibodies in TBS for 1 h. After washing, the samples were incubated with FITC-conjugated secondary antibodies (Amersham Pharmacia Biotech) for 1 h. Rhodamine-phalloidin (Molecular Probes) was used to visualize actin filaments. The coverslips were mounted in Vectashield (Vector Laboratories) and observed with a confocal laser-scanning microscope (MicroRadiance Plus; Bio-Rad). An accumulation of N-WASP or F-actin around the intracellular bacteria was defined as an area where the intensity was higher than an arbitrary threshold of 80 estimated in noninfected cells by using image processing software (LaserSharp Radianceplus v3.2; Bio-Rad).
Microinjection.
Purified Rho family GTPases were microinjected into the cytoplasm of Swiss 3T3 cells infected with S. flexneri. The microscope stage was maintained at 37°C and 5% CO2 during the time of injection and data acquisition.
Actin Tail Assay in Xenopus Egg Extracts.
Meiotically arrested cytoplasmic extracts of Xenopus laevis were prepared as described previously 50 51. For immunodepletion, preimmune rabbit IgG or anti–N-WASP antibody was bound to 30 μl of protein A–conjugated Sepharose 4B (Sigma-Aldrich) in TBS. The pellets were washed twice with 1 ml of Xenopus buffer (XB: 100 mM KCl, 50 mM sucrose, 5 mM EGTA, 2 mM MgCl2, 0.1 mM CaCl2, 10 mM Hepes, pH 7.7) and incubated with 30 μl of extracts for 1 h at 4°C. Purified G-actin from rabbit skeletal muscle was covalently labeled with tetramethyl rhodamine (TMR)-iodoacetamide (Molecular Probes) as an actin tail tracer. E. coli MC1061 ompT::Km carrying pD10-1 (expressing VirG protein) and L. monocytogenes serotype 1/2a EGD carrying pERL3::prfA 7973 were labeled with 4,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich). Bacterial motility was assayed by mixing 5 μl of depleted extracts with 0.5 μl of DAPI-labeled bacteria, 0.05 mg/ml of TMR-labeled G-actin, and other components. In experiments where various proteins were added, XB or proteins were added in a volume of 0.5 μl. Static or moving images of actin tails were collected using a cooled CCD camera with a time-lapse imaging system (Roper Scientific). The speeds of the moving bacteria, the lengths of actin tail, and the total tail fluorescence were quantitated at intervals of 5 min, and the average value of each set of data was calculated using IPLab Spectrum software (Signal Analytics Corporation). The length in pixels was converted to microns using a micrometer standard. Total fluorescence in the actin tails was measured by multiplying the pixel area by the average pixel intensity of a selected area minus the average pixel intensity of a selected background area. The CCD camera responds linearly to fluorescence intensity in the range of 10–2,000 counts/pixel, and the illumination levels were used to avoid saturating the signal.
Pyrene Actin Assay.
Pyrene actin was labeled according to an established protocol 52 using rabbit skeletal muscle actin and N-(1-pyrene)iodoacetamide (Molecular Probes). Pyrene labeled G-actin and unlabeled G-actin were prepared by centrifugation at 400,000 g for 1 h at 4°C after incubating freshly thawed proteins in G buffer (5 mM Tris-HCl, pH 7.8, 0.1 mM CaCl2, 0.2 mM ATP, and 1 mM dithiothreitol). For pyrene actin assays using Xenopus egg extracts, the extracts were ultracentrifuged at 400,000 g for 1 h at 4°C, diluted threefold in XB to prevent the high background fluorescence in the extracts, and mixed with pyrene actin (final 1 μM) and the other proteins as described previously 53. The actin concentration is ∼1 mg in high speed supernatants as estimated by Western blotting. For assays using purified components, 2.2 μM G-actin (10% pyrenyl-labeled) and various proteins were incubated in F buffer (5 mM Tris-HCl, pH 7.8, 0.1 mM CaCl2, 0.2 mM ATP, 1 mM dithiothreitol, 1 mM MgCl2, and 0.1 M KCl). Actin polymerization was measured as the change in pyrenyl actin fluorescence by using a spectrofluorometer (FP-777; JASCO Corporation) thermostated at 20°C, with excitation at 365 nm and emission at 407 nm.
In Vitro Protein Binding Assay.
Various amounts of recombinant VirG-α1, GTPγS-charged Cdc42-G12V expressed by baculovirus, and 0.1 μM histidine-tagged N-WASP were mixed in 100 μl of binding buffer (10 mM Tris-HCl, pH 7.5, 1 mM MgCl2, 0.2 mM ATP, 1 mM dithiothreitol, 0.1% Tween 20, 0.5 mg/ml BSA, and 0.1 M KCl) and incubated for 1 h at 4°C. The protein solutions were added to 10 μl of Ni-NTA-Agarose (Qiagen) and incubated for 1 h at 4°C on a rotating wheel. The beads were washed twice with washing buffer (binding buffer plus 60 mM imidazole without BSA), and the solubilized proteins were subjected to Western blotting. For immunoprecipitation, VirG-α1, nontagged N-WASP, and Cdc42 were incubated in binding buffer as described above. VRG-N2 antibody was added to the mixture (final 10 μg/ml) and further incubated for 1 h at 4°C. Immunocomplexes were precipitated by adding 10 μl of protein A–Sepharose (Sigma-Aldrich) and incubated for 1 h at 4°C on a rotating wheel. The beads were washed with washing buffer without 60 mM imidazole.
Transient Expression of Green Fluorescent Protein–tagged Cdc42, N-WASP, and Its Mutant in COS-7 Cells.
A DNA fragment encoding human Cdc42 was cloned into pEGFP-C1 mammalian expression vector (CLONTECH Laboratories, Inc.). Expression plasmid encoding wild-type N-WASP or H208D mutant was constructed as described previously 20 40. Cells (0.5 × 107) were mixed with purified plasmid (10 μg) and transfected by electroporation. The cells were replated and cultured for 24–48 h before bacterial infection.
Stable Expression of N-WASP and Its Mutant in MDCK Cells and Plaque-forming Assay.
For construction of MDCK cell lines stably expressing wild-type N-WASP (or its mutant), MDCK cells were transfected by electroporation and independent cell clones were isolated by resistance to 1 mg/ml of G418 (geneticin; Life Technologies). The expression levels of N-WASP in each clone were examined by Western blotting of total cell lysates and immunostaining using anti–N-WASP antibody. The plaque-forming assay was performed as described previously 54.
Results
Stimulation of Shigella Motility in Mammalian Cells by Microinjection of Cdc42.
To explore whether or not Rho family GTPases are involved in the motility of intracellular S. flexneri, we attempted to microinject a recombinant dominant active mutant of Cdc42, Rac1, or RhoA into Swiss 3T3 cells at 1 h after infection with S. flexneri YSH6000. The concentrations of Cdc42-G12V, Rac1-G12V, and RhoA-G14V in needles for the microinjections were 1.5, 0.6, and 0.3 mg/ml, respectively, as in previous reports 45 46 47. To monitor the effect of each mutant Rho family GTPase on the bacterial motility, we measured bacterial motility at 4 min before the injection through to 5 min after injection using a phase–contrast microscope equipped with a cooled CCD camera with a time-lapse imaging system. Rac1-G12V and RhoA-G14V injection had no substantial effect on Shigella motility (Fig. 1B and Fig. C), whereas Cdc42-G12V injection stimulated it (Fig. 1 A), though the levels of increased motility varied among bacteria. When these proteins were microinjected into the cells infected with L. monocytogenes 1/2a EGD, significant changes of bacterial motility were not observed (data not shown), in agreement with recent studies 55 56. The speed of Shigella originally moving at <5 μm/min increased 4.5-fold at 5 min after injection with Cdc42-G12V, whereas that of the bacterium originally moving at >5 μm/min was only increased 1.6-fold, indicating that the response of a low speed bacterium to Cdc42-G12V injection is much greater than that of a high speed bacterium (Fig. 1 A). Although the reason for the different responses is unclear, the reduced response to Cdc42-G12V may reflect an activated state of assembly of the actin tail on the bacterial surface; the increase in bacterial motility was not observed, even though the needle concentration of either Rac1 or RhoA increased 1.5 mg/ml, the same as that of Cdc42 (data not shown).
Figure 1.

Effects of the dominant-active Rho family GTPases on the speed of Shigella motility in Swiss 3T3 cells. Relative velocity values before and after microinjection with (A) Cdc42-G12V; (B) Rac1-G12V; and (C) RhoA-G14V. Arrows indicate the time of microinjection. •, bacteria moving at low speeds (<5 μm/min) before microinjection; ○, bacteria moving at high speeds (>5 μm/min) before microinjection. The numbers of intracellular bacteria analyzed for microinjection were: Cdc42-G12V, 32; Rac1-G12V, 22; and RhoA-G12V, 28; in over five experiments. Error bars represent SEM.
Inactivation of Rho Family GTPases from Xenopus Egg Extract Inhibits VirG-directed Bacterial Movement.
To verify the involvement of Cdc42 in Shigella motility, we carried out add-back experiments on Rho family GTPases in Xenopus egg extracts using E. coli K-12 MC1061 ompT::Km carrying pD10-1 10. To suppress the endogenous activity of Rho family GTPases, RhoGDI prepared as a GST fusion protein was added to the extracts and assayed for the effect on bacterial motility or assembly of the actin tail as described in Materials and Methods. In untreated extracts, the formation of actin clouds around the bacteria was observed within 5 min, and then bacteria started to move with a long actin tail (Fig. 2 C, panel a). The movement of bacteria continued steadily within 1 h. The average speed of moving bacteria was 5.32 ± 0.79 μm/min (n = 34). When RhoGDI at 100 nM was added to the extracts, although initial actin clouds were formed around ∼50% of bacteria within 10 min, the bacterial movements decreased (47 ± 18%, n = 35) and the amounts of assembled actin filament decreased to <5% of original levels (Fig. 2A and Fig. B). When a further amount of RhoGDI (400 nM) was added to the extracts, initial actin cloud formation was abolished completely (Fig. 2A–C, panel b) and then some clouds began to appear after 20 min of incubation. Although the tail-like actin assembly was observed on ∼6% of bacteria, its length was extremely short. Similar effects of RhoGDI on the actin assembly of endogenous vesicles stimulated by GTPγS to that reported by Ma et al. 53 and Moreau and Way 56 were reproducibly observed within 1 h (data not shown). Importantly, actin assembly induced by L. monocytogenes 1/2a EGD harboring pERL3::prfA 7973 in the extract was not affected by the addition of RhoGDI at 400 nM (Fig. 2A–C, panels c and d), implying that the inhibitory effect by RhoGDI is Shigella specific.
Figure 2.
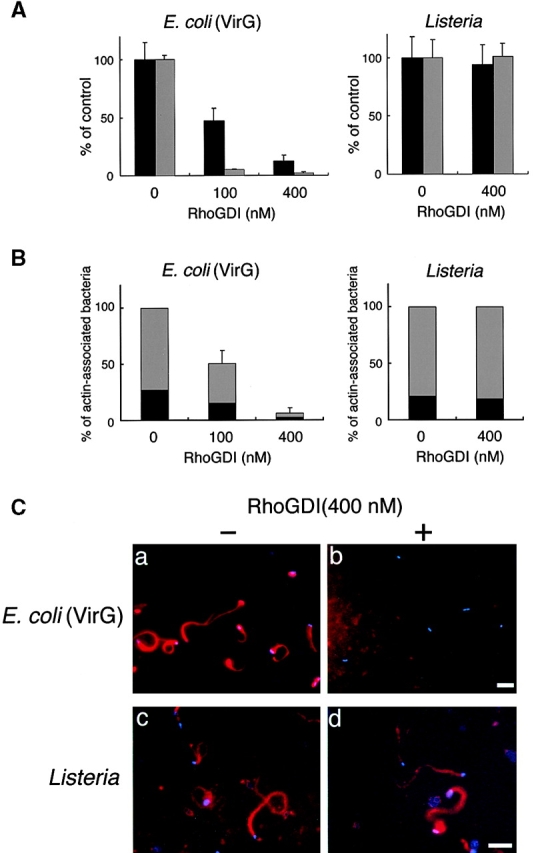
The effect of RhoGDI on the actin assembly from E. coli (VirG) and Listeria in Xenopus egg extracts. (A) RhoGDI was added to the egg extracts and assayed for the effect on the bacterial motility and assembly of actin (see text for details). Black bars, bacterial speeds; gray bars, F-actin intensities. All activities are normalized to untreated extracts. (B) The percentage of bacteria associating actin clouds (black bars) or actin tails (gray bars). Error bars in A and B represent SEM from three separate experiments. (C) Inhibition of the actin assembly by RhoGDI in the extracts as viewed with DAPI-bacteria (blue) and TMR-actin (red): a and b, actin assembly from E. coli (VirG); c and d, actin assembly from Listeria; a and c, untreated extracts; b and d, RhoGDI (at 400 nM). All images were observed 10 min after mixing bacteria with extracts. Bars, 10 μm.
To examine whether or not the inhibitory effect of RhoGDI on the bacterial motility resulted from the inactivation of Cdc42, Cdc42-G12V charged with GTPγS expressed by baculovirus, an activated form of Cdc42, was supplied to the extracts. The addition of Cdc42-G12V (GTPγS) at 200 and at 400 nM induced actin cloud formation around the bacteria within 5 min, and restored the bacterial motility by 46% (46 ± 17%, n = 29) and 70% (70 ± 19%, n = 38) of the original level, respectively (Fig. 3 A). Similar results were also obtained in the actin assembly (Fig. 3a Fig. b Fig. c, panel a). In contrast, addition of Rac1-G12V (GTPγS) expressed by baculovirus at 200 or 400 nM had no effect at all on the restoration of the bacterial motility and actin assembly (Fig. 3a Fig. b Fig. c, panel b). Consistent with this, when Cdc42-T17N (asparagine substituted for threonine at residue 17), a Cdc42 dominant negative mutant expressed by baculovirus, was added to the untreated extract (without addition of RhoGDI) at 400 and at 800 nM, the bacterial motility declined by 30% (30 ± 3.0%, n = 35) and 22% (22 ± 2.1%, n = 41) of the original level (100%), respectively, and the extent of assembly of actin tail at 400 and 800 nM was <5% of the original (Fig. 3a Fig. b Fig. c, panel c). The results thus further indicate that motility and actin assembly of Shigella depends on Cdc42 activity.
Figure 3.



The effect of Rho family GTPases on the VirG-directed actin assembly in Xenopus egg extracts. (A) GTPγS-charged Cdc42-G12V or Rac1-G12V was added to egg extracts along with RhoGDI (at 400 nM). Cdc42-T17N, a dominant negative mutant of Cdc42, was added in the absence of RhoGDI. Black bars, bacterial speeds; gray bars, F-actin intensities. All activities are normalized to untreated extracts. (B) The percentage of bacteria associating actin clouds (black bars) or actin tails (gray bars). Error bars in A and B represent SEM from three separate experiments. (C) Actin assembly as viewed with DAPI-bacteria (blue) and TMR-actin (red) in the presence of: a, RhoGDI and Cdc42-G12V (GTPγS); b, RhoGDI and Rac1-G12V (GTPγS); and c, Cdc42-T17N. The images were observed 10 min after mixing bacteria with extracts. Bar, 10 μm.
N-WASP Activated by Cdc42 Is Involved in Shigella Motility.
N-WASP has been shown to be able to mediate the formation of an extremely long filopodium from mammalian cells when coexpressed with activated Cdc42 40 or to directly interact with VirG in vitro and in vivo 10. Hence, we reasoned that the stimulation of Shigella motility in mammalian cells by activated Cdc42 could be due to the activation of N-WASP. First, we examined whether or not the interaction of N-WASP with VirG is affected by the addition of RhoGDI to Xenopus egg extracts. Addition of RhoGDI at 400 nM to the extract greatly inhibited the actin assembly of MC1061 ompT::Km carrying pD10-1 (Fig. 4 A, panels b, d, f, and h, and B), whereas using Cy2-labeled N-WASP at 75 nM, N-WASP accumulation on the bacterial surface was only ∼10% decreased as judged by immunofluorescence microscopy (Fig. 4 A, panels c and d), indicating that RhoGDI does not markedly inhibit the interaction between N-WASP and VirG in this assay. Next, to investigate the role of Cdc42 interaction with N-WASP in actin assembly induced by Shigella, extracts immunodepleted using anti–N-WASP antibody with or without RhoGDI were examined for the activity to support actin assembly. When exogenous N-WASP was added back at a physiological concentration (45 nM) without RhoGDI, the motility of bacteria and the assembly of the actin tail were restored (Fig. 5A Fig. c, panel b). When N-WASP (at 45 nM) was added to the depleted extract with RhoGDI (at 400 nM), the initial actin assembly around the bacteria was abolished similarly to RhoGDI treatment in Fig. 2 (Fig. 5A Fig. c, panel c), suggesting that addition of N-WASP alone was not sufficient to support actin assembly in the extract-inactivated Rho family GTPases. Furthermore, when N-WASP (at 45 nM) was added together with Cdc42-G12V (GTPγS) at 400 nM to the N-WASP–depleted extract with RhoGDI, the bacterial motility and actin assembly were increased up to 90% (Fig. 5A Fig. c, panel d), suggesting that Cdc42-G12V (GTPγS) addition could circumvent the inhibitory effect of RhoGDI. To further clarify the roles of N-WASP and Cdc42 in VirG-directed bacterial movement in Xenopus egg extracts, we added H208D mutant to N-WASP–depleted extracts to examine the effect on the bacterial motility. H208D was defective in the ability to interact with Cdc42 due to a replacement of histidine (H) 208 with aspartic acid (D) in the GBD 40. When H208D mutant was added instead of wild-type N-WASP to the N-WASP–depleted extract, the bacterial motility and actin assembly were significantly decreased, but not completely abolished, suggesting that H208D is slightly functional for actin assembly in the extracts (see the Discussion). The motility and actin assembly were not restored even by Cdc42-G12V (GTPγS) at 400 nM (Fig. 5A Fig. c, panel e). Since H208D was still capable of interacting with VirG in vivo (see below), these results indicate that the capacity of N-WASP to interact with Cdc42 is critical for promoting the actin assembly of Shigella.
Figure 4.
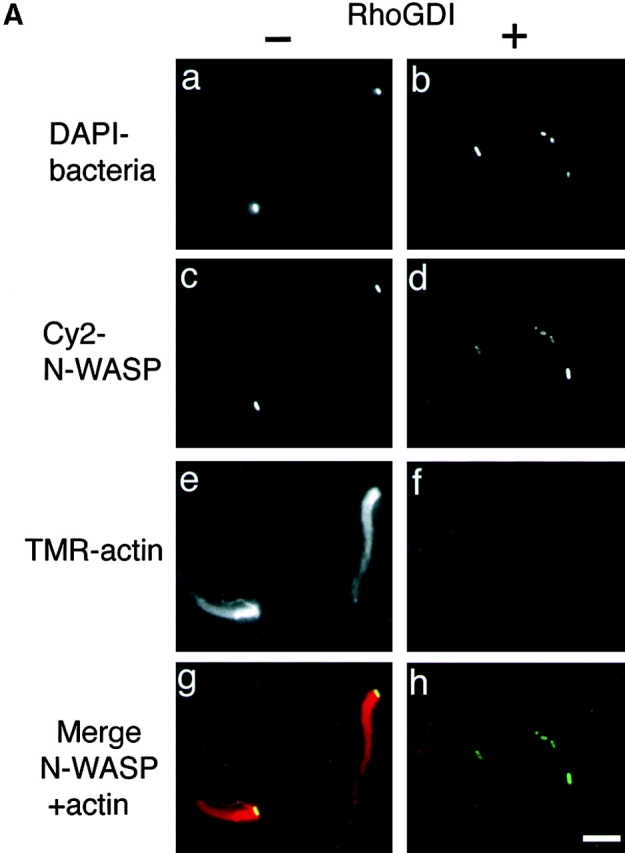
The effect of RhoGDI on the interaction between N-WASP and VirG in Xenopus egg extracts. (A) Cy2-labeled recombinant N-WASP (at 75 nM) was added to the egg extracts without (a, c, e, and g) or with (b, d, f, and h) RhoGDI (at 400 nM). a and b, DAPI-labeled bacteria; c and d, Cy2-labeled N-WASP; e and f, TMR-actin. The yellow color in the combined image shown in g and h indicates the colocalization between N-WASP (green) and actin (red). The images were observed 10 min after mixing bacteria with extracts. (B) Quantitation of N-WASP intensity, bacterial motility, and F-actin intensity without or with RhoGDI (at 400 nM). White bars, N-WASP intensities; black bars, bacterial speeds; gray bars, F-actin intensities. All activities are normalized to untreated extracts. Error bars represent SEM from three separate experiments. Bar, 10 μm.
Figure 5.
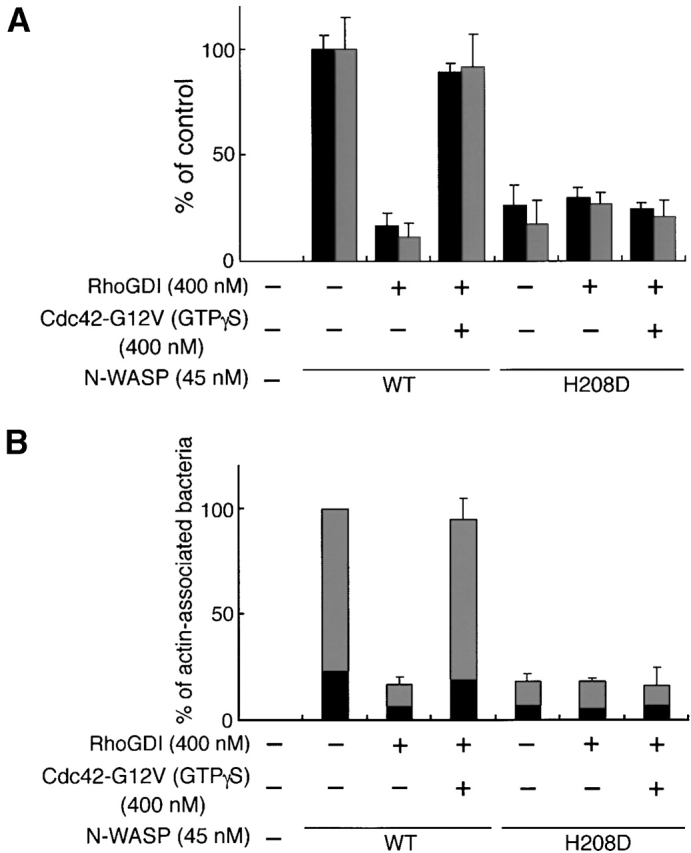

The capacity of N-WASP to interact with Cdc42 is critical for promoting actin assembly in Xenopus egg extracts. (A) Recombinant wild-type (WT) N-WASP (or H208D mutant) (at 45 nM) was added back to the N-WASP–depleted extracts in the absence or presence of RhoGDI (at 400 nM) or Cdc42-G12V (GTPγS) (at 400 nM). Black bars, bacterial speeds; gray bars, F-actin intensities. All activities are normalized to depleted extracts plus wild-type N-WASP. (B) The percentage of bacteria associating actin clouds (black bars) or actin tails (gray bars). Error bars in A and B represent SEM from three separate experiments. (C) Actin assembly as viewed with DAPI-bacteria (blue) and TMR-actin (red) in the N-WASP–depleted extracts in the presence of: a, no addition; b, wild-type (WT) N-WASP; c, wild-type N-WASP and RhoGDI; d, wild-type N-WASP, RhoGDI, and Cdc42-G12V (GTPγS); e, H208D mutant, RhoGDI, and Cdc42-G12V (GTPγS). The images were observed 10 min after mixing bacteria with extracts. Bar, 10 μm.
RhoGDI Inhibits Actin Nucleation Induced by VirG In Vitro.
In the visual actin tail assay, the data clearly indicated that Cdc42 was required for the restoration of the actin assembly inhibited by RhoGDI; however, it was not clarified which of the steps of the actin-based process, actin nucleation, tail assembly, or bacterial propulsion, was affected. To examine whether or not RhoGDI treatment affects the actin nucleation in the extracts, we used the well-established pyrene actin assay 57. To eliminate the effects of endogenous lipid vesicles, high speed supernatants were prepared by ultracentrifugation as described previously 53. Recombinant VirG-α1 protein encompassing Thr53–Arg759 of VirG polypeptide, which we previously showed to be exposed on the Shigella surface 12, was prepared as a GST fusion protein after removal of the GST portion by thrombin treatment, and the VirG-α1 protein was then added to the pyrene actin–labeled high speed supernatant. The addition of VirG-α1 at 100 or 250 nM to the supernatant caused a rapid increase in fluorescence within the first 100 s in a dose-dependent manner (Fig. 6 A). When RhoGDI (400 nM) was added to the supernatant in the presence of VirG-α1 at 250 nM, the actin nucleation was almost completely abolished, indicating that RhoGDI blocks the VirG-α1–mediated actin nucleation step.
Figure 6.

Cdc42 stimulates the VirG-induced actin polymerization by N-WASP–Arp2/3 complex in vitro. (A) Inhibition of VirG-induced actin polymerization by RhoGDI. High speed supernatants containing pyrene actin (∼10% labeled) were incubated with buffer (×), 100 nM VirG-α1 (○), 250 nM VirG-α1 (•), 400 nM RhoGDI (▴), or 250 nM VirG-α1 and 400 nM RhoGDI (▵). (B–D) G-actin (2.2 μM, 10% pyrenyl labeled) was induced to polymerize by addition of 0.1 M KCl and 1 mM MgCl2 in the presence or absence of Arp2/3 complex (20 nM) and other components as indicated. (B) VirG-α1 activates N-WASP–Arp2/3 complex. Actin was polymerized alone (×); in the presence of 20 nM Arp2/3 (○); in the presence of Arp2/3 and 0.1 μM VirG-α1 (•); Arp2/3 and 0.25 μM N-WASP (□); Arp2/3, 0.25 μM N-WASP, and 0.1 μM (▪) or 0.5 μM (▵) VirG-α1. (C) Activation of N-WASP–Arp2/3 complex by VirG-α1 is more enhanced in the presence of Cdc42. Actin was polymerized in the presence of Arp2/3 and 0.25 μM N-WASP (×); Arp2/3, N-WASP, and 0.25 μM Cdc42-G12V (GTPγS) expressed by baculovirus (○); Arp2/3, N-WASP, and 0.1 μM VirG-α1 (•); Arp2/3, N-WASP, 0.25 μM Cdc42-G12V (GTPγS), and 0.1 μM (▵) or 0.5 μM (▴) VirG-α1; Arp2/3, 0.25 μM H208D mutant of N-WASP, 0.25 μM Cdc42-G12V (GTPγS), and 0.1 μM VirG-α1 (□). (D) Effect of VirG-α3 on actin polymerization mediated by N-WASP–Arp2/3 complex. Actin was polymerized in the presence of Arp2/3 and 0.25 μM N-WASP (×); Arp2/3, N-WASP, and 0.1 μM VirG-α3 (○); Arp2/3, N-WASP, 0.25 μM Cdc42-G12V (GTPγS), and 0.1 μM (▵) or 0.5 μM (▴) VirG-α3.
Cdc42 Stimulates Actin Nucleation Induced by VirG In Vitro.
Recently, Rohatgi et al. have indicated that Cdc42-activated N-WASP stimulates actin polymerization through the interaction of N-WASP with Arp2/3 complex 39, whereas Egile et al. have shown that VirG can activate N-WASP, thus stimulating actin polymerization activity of Arp2/3 complex 18. Therefore, to investigate whether or not Cdc42 is involved in the actin nucleation induced by VirG, the individual proteins VirG-α1, N-WASP, and Cdc42, and Arp2/3 complexes, were examined for their role in VirG-mediated actin polymerization using an actin pyrene assay with purified proteins. When VirG-α1 was added to the mixture in the presence of 0.25 μM N-WASP, 20 nM Arp2/3, and 2.2 μM actin, the actin polymerization activity was slightly stimulated (1.6-fold at 0.5 μM VirG-α1 at the maximum) in a concentration-dependent manner (Fig. 6 B). VirG-α1 itself did not affect the polymerization of Arp2/3-actin or actin alone. The activity of N-WASP–Arp2/3 complex was slightly enhanced (1.2-fold) in the presence of 0.25 μM GTPγS-bound Cdc42-G12V expressed by baculovirus (Fig. 6 C), whereas in the presence of both VirG-α1 and Cdc42, the activity of N-WASP–Arp2/3 was further stimulated (about fourfold), concentration-dependent on VirG-α1 and in a saturable fashion (Fig. 6 C). As the activation was greater than that of VirG-α1 or Cdc42 alone, our results suggested a synergistic effect of VirG-α1 and Cdc42 on the actin polymerization by N-WASP–Arp2/3 complex. Consistent with the data in Fig. 5 A on replacement of wild-type N-WASP with H208D mutant, the activity was unable to be stimulated by Cdc42. Since Egile et al. 18 have indicated that the truncated portion encompassing Thr53–Thr508 of VirG polypeptide, which we previously named VirG-α3 9, markedly stimulates actin polymerization mediated by N-WASP–Arp2/3 complex, we examined the activity of VirG-α3 on actin polymerization. When VirG-α3 (0.1 μM) was added to the mixture in the presence of 0.25 μM N-WASP, 20 nM Arp2/3, and 2.2 μM actin, the stimulation by VirG-α3 was obviously higher (3.5-fold) than that of VirG-α1 under the same conditions. Furthermore, in the presence of both VirG-α3 and Cdc42, the activity of N-WASP–Arp2/3 was dramatically stimulated (6.3-fold), concentration-dependent on VirG-α3 and in a saturable fashion (Fig. 6 D). Although the molecular mechanism of the activation of N-WASP by Cdc42 and VirG is still to be investigated, these results indicated that Cdc42 enhances actin nucleation induced by VirG.
Cdc42 Enhances Interaction of VirG with N-WASP.
We expected that the interaction of N-WASP with VirG would also be promoted by the binding of Cdc42 to N-WASP. To test this, we carried out pull-down assays using recombinant histidine-tagged N-WASP (His-N-WASP). His-N-WASP was incubated with various concentrations of VirG-α1 or GTPγS-charged Cdc42-G12V after incubation with nickel affinity resin. The amounts of VirG-α1 or Cdc42 coprecipitated with His-N-WASP were then compared in Western blotting with anti-VirG antibody or anti–N-WASP antibody. The same amounts of His-N-WASP were confirmed to be precipitated in the presence or absence of either VirG-α1 or Cdc42. As shown in Fig. 7 A, VirG-α1 was precipitated by His-N-WASP, and the amounts increased as the concentration of Cdc42 increased. Although the amounts of the coprecipitated VirG-α1 did not increase markedly (up to 1.2-fold), the results were reproducible. In contrast, VirG-α1 had no effect on Cdc42 binding to N-WASP. To further confirm this, equal molar amounts of VirG and N-WASP (nontagged) were incubated with increasing amounts of Cdc42-G12V (GTPγS). Proteins were then immunoprecipitated by using anti-VirG antibody and analyzed in Western blotting with anti–N-WASP antibody. Similarly, the amounts of precipitated N-WASP increased up to 2.2-fold in the presence of Cdc42 in a concentration-dependent manner (Fig. 7 B). In contrast, when H208D was incubated with VirG-α1 and Cdc42-G12V (GTPγS) instead of wild-type N-WASP, the amount of H208D coprecipitated with VirG-α1 did not change (data not shown). These results indicated that the interaction of N-WASP with VirG is more enhanced in the presence than absence of Cdc42 in vitro.
Figure 7.
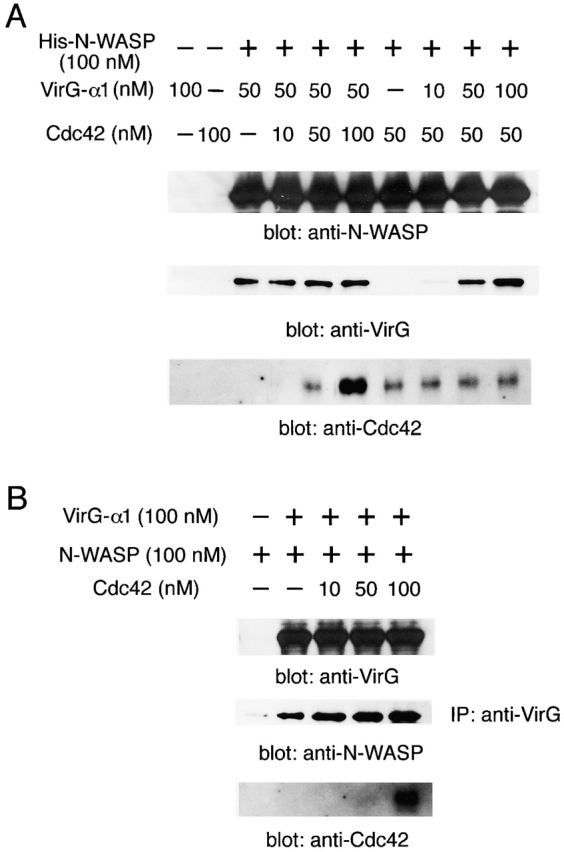
The effect of Cdc42 on the interaction between N-WASP and VirG in vitro. (A) Recombinant histidine-tagged N-WASP (His-N-WASP) was incubated with various concentrations of VirG-α1 or GTPγS-charged Cdc42-G12V. Proteins precipitated with nickel affinity resin were subjected to Western blotting. (B) Recombinant VirG-α1 and N-WASP were incubated with increasing amounts of GTPγS-charged Cdc42-G12V. Immunoprecipitated proteins (IP) with anti-VirG antibody were analyzed by Western blotting.
Cdc42 Is Required for In Vitro Reconstitution of Actin Assembly Induced by VirG.
To further confirm the role of Cdc42 in VirG-induced actin nucleation, we performed in vitro reconstitution for actin assembly by using E. coli expressing VirG and purified proteins. First, bacteria (MC1061 ompT::Km carrying pD10-1) were incubated in F buffer containing N-WASP, Arp2/3 complex, and TMR-actin. Formation of actin clouds around the bacteria was not observed immediately, but after 20 min of incubation, the actin clouds were barely detectable in ∼20% of bacteria with indistinguishable levels from background (Fig. 8 A, panel b). Preincubation of bacteria with N-WASP for 5 min caused an increase in the number of bacteria associated with weak actin clouds (∼80%). In contrast, when GTPγS-bound Cdc42-G12V was added to the solution, actin clouds with strong fluorescence were immediately observed in ∼80% of bacteria within a few minutes (Fig. 8 A, panel e), indicating that Cdc42 enhances the initiation of actin cloud formation around the bacteria in vitro. No actin assembly was observed at all under the same conditions with control E. coli MC1061 ompT::Km that did not express VirG (data not shown). Since a recent report showed that PtdIns(4,5)P2 and Cdc42 synergistically enhanced the capacity of N-WASP to stimulate Arp2/3 complex–induced actin nucleation in vitro 39, we examined the effect of PtdIns(4,5)P2 on the actin assembly induced by VirG. The addition of lipid vesicles containing phosphatidylcholine, phosphatidylinositol, and PtdIns(4,5)P2 in a 48:48:4 ratio into the solution in the presence of all components did not result in further enhancement of actin assembly (data not shown). On the other hand, in agreement with Welch et al. 31, actin assembly around the bacterial surface of Listeria was observed in the presence of Arp2/3 complex and actin (Fig. 8 B).
Figure 8.

In vitro reconstitution of actin assembly on E. coli (VirG) and Listeria surface. (A) E. coli expressing VirG were incubated in F buffer containing 0.4 μM N-WASP, 70 nM Arp2/3 complex, and 1 μM TMR-actin, with or without 0.4 μM Cdc42-G12V (GTPγS) expressed by baculovirus. (B) Listeria were incubated in F buffer containing 70 nM Arp2/3 complex and 1 μM TMR-actin: a, d, and g, DAPI-labeled bacteria; b, e, and h, TMR-actin; c, f, and i, merged images. Insets show magnified images. Bar, 10 μm.
Cdc42 Is Involved in Initial Actin Cloud Formation on Shigella in Infected Cells.
The data presented above strongly suggested the functional involvement of Cdc42 in initial actin cloud formation on Shigella in infected cells. Therefore, to gain more insight into the role of Cdc42 in Shigella-infected mammalian cells, the localization of Cdc42 in infected cells was examined by using COS-7 cells expressing GFP-Cdc42 fusion protein. The cells were infected with S. flexneri YSH6000, and the immunolocalization of GFP-Cdc42 and actin filaments on the intracellular bacterium was examined by confocal microscopy. After 40 min of infection, accumulation of GFP-Cdc42 at one pole of the bacterium was observed in ∼80% of bacteria associating actin clouds (Fig. 9). In contrast, GFP-Cdc42 accumulation was hardly detected on the bacterium assembled long actin tail, which was considered to be moving fast. Under the same conditions, GFP alone did not accumulate at all on the surface of bacteria (data not shown). These results indicated that Cdc42 is involved in the initial actin cloud formation on the Shigella surface in infected cells in vivo.
Figure 9.

Localization of Cdc42 in COS-7 cells expressing GFP-Cdc42 infected with Shigella. (A) Phase–contrast image; (B) localization of GFP-Cdc42; (C) localization of F-actin visualized by staining with rhodamine-phalloidin. The yellow color in the combined image (D) indicates colocalization between GFP-Cdc42 (green) and F-actin (red). Arrows and arrowheads show Shigella-associated actin clouds and actin tails, respectively. Bar, 10 μm.
Effect of Overexpression of H208D in COS-7 Cells on Actin Assembly of Shigella.
To pursue the importance of the Cdc42 interaction with N-WASP for promoting the actin-based motility of Shigella in infected epithelial cells, COS-7 cells overexpressing wild-type N-WASP or H208D mutant were infected with wild-type S. flexneri YSH6000 and examined for the effect on actin assembly of intracellular Shigella using double immunostaining with FITC-labeled rabbit anti–N-WASP and rhodamine-phalloidin. After 40 min to 1 h of infection, N-WASP was highly concentrated at one pole of the intracellular bacterium from which a long actin tail appeared (Fig. 10 A, panels a and b). In contrast, although H208D mutant was still confined to the site of VirG expression at one pole of the bacterium in cells, the number of bacteria associating actin assembly was significantly decreased (Fig. 10 A, panels d and e). The percentage of bacteria associating actin assembly in COS-7 cells overexpressing H208D was decreased by ∼30% (33.7 ± 8.5%, n = 66) compared with wild-type N-WASP–expressing cells (88.7 ± 10.4%, n = 46) (P < 0.01) (Fig. 10 B).
Figure 10.

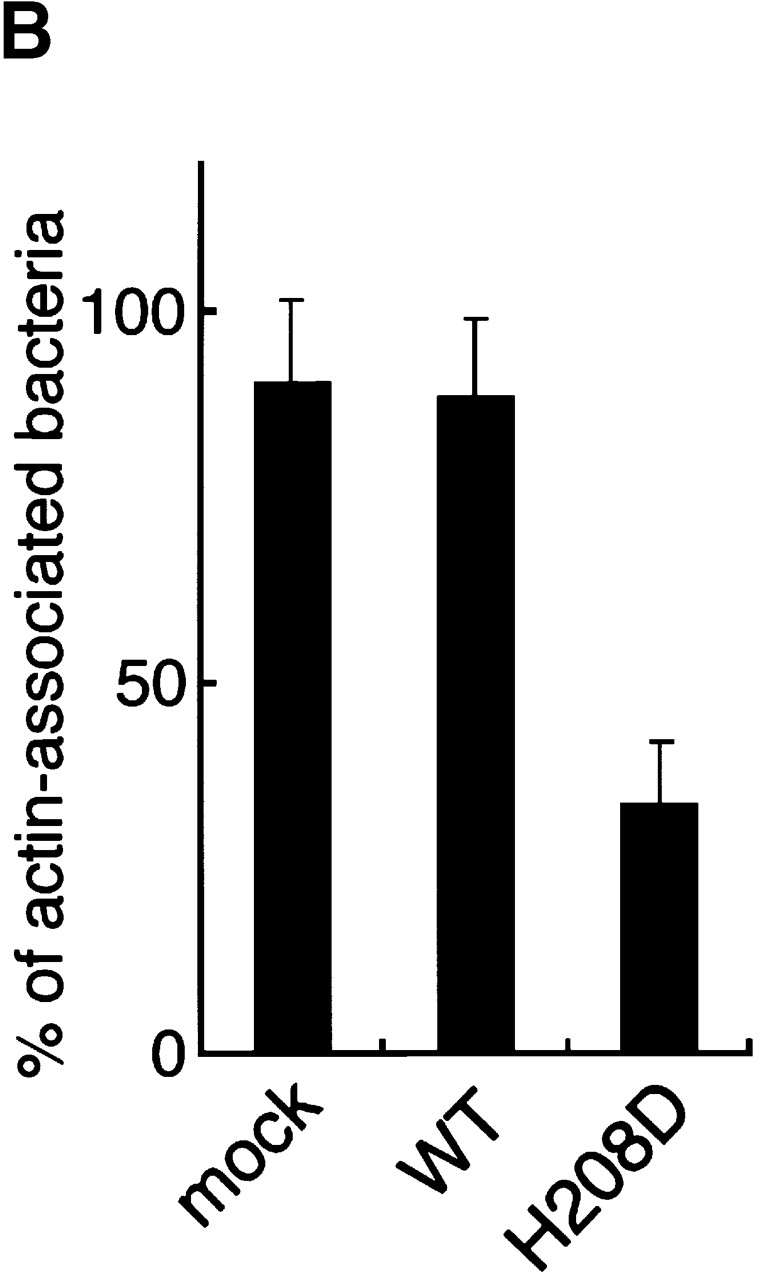
Actin assembly from Shigella in infected COS-7 cells expressing the H208D mutant. (A) COS-7 cells overexpressing wild-type (WT) N-WASP (a–c) or H208D mutant (d–f) were infected with Shigella and immunostained using FITC-labeled anti–N-WASP antibody (a and d) and rhodamine-phalloidin (b and e). The yellow color in the combined image shown in c and f indicates colocalization between N-WASP (green) and actin (red). The arrowheads indicate intracellular Shigella. (B) The percentage of intracellular bacteria associating actin assembly. Error bars represent SEM from three separate experiments. Bar, 10 μm.
Effect of Overexpression of H208D in MDCK Cells on Cell to Cell Spreading of Shigella.
Since the ability of Shigella to direct actin-based motility within the host cytoplasm has been shown to seriously reflect the dissemination of bacteria into the adjacent epithelial cells 1 8, we wished to elucidate the important role of Cdc42 in Shigella intercellular spreading. 12 independent MDCK cell lines stably expressing wild-type N-WASP or H208D mutant were constructed. The expression level of wild-type N-WASP and H208D mutant in each cell line was approximately threefold that of the mock transfectant as determined by Western blotting with anti–N-WASP antibody, whereas there was no significant effect on the levels of E-cadherin or actin (Fig. 11 A). The MDCK cell monolayers infected by wild-type S. flexneri YSH6000 were examined for the ability to form plaques. As represented in Fig. 11 B, the average size of plaques formed by YSH6000 in MDCK cell monolayers expressing H208D at 3 d after infection was significantly smaller (∼17%, 0.28 ± 0.05 mm2; n = 18) than that in cells expressing wild-type N-WASP (∼120%, 1.9 ± 0.26 mm2; n = 25) or mock transfectant (100% as a control, 1.6 ± 0.14 mm2; n = 22). Since ectopic expression of wild-type N-WASP or H208D mutant in MDCK cells had no significant effect on cell to cell junctions as judged by immunostainings with anti–E-cadherin and ZO-1 antibodies (Fig. 11 C), these results thus further indicated that the interaction between Cdc42 and N-WASP is critical for promoting the actin-based intercellular spreading of Shigella.
Figure 11.
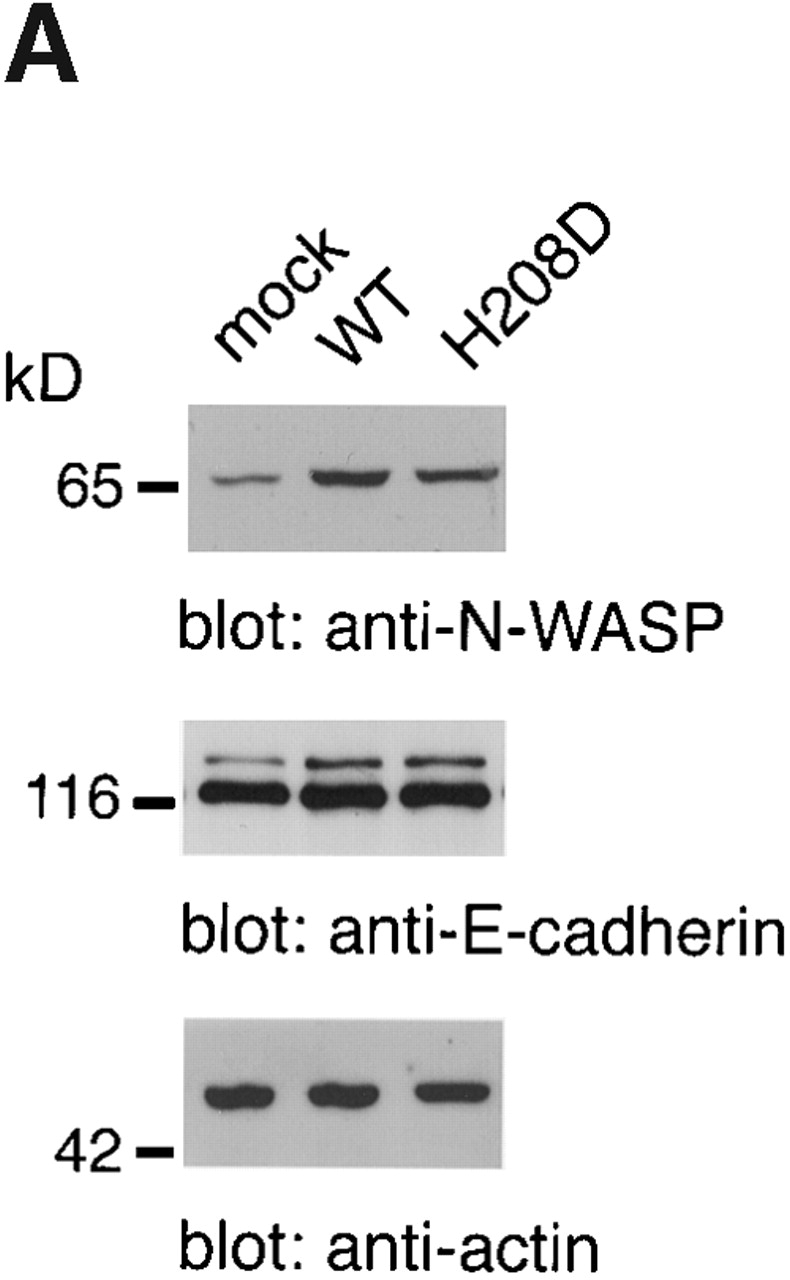
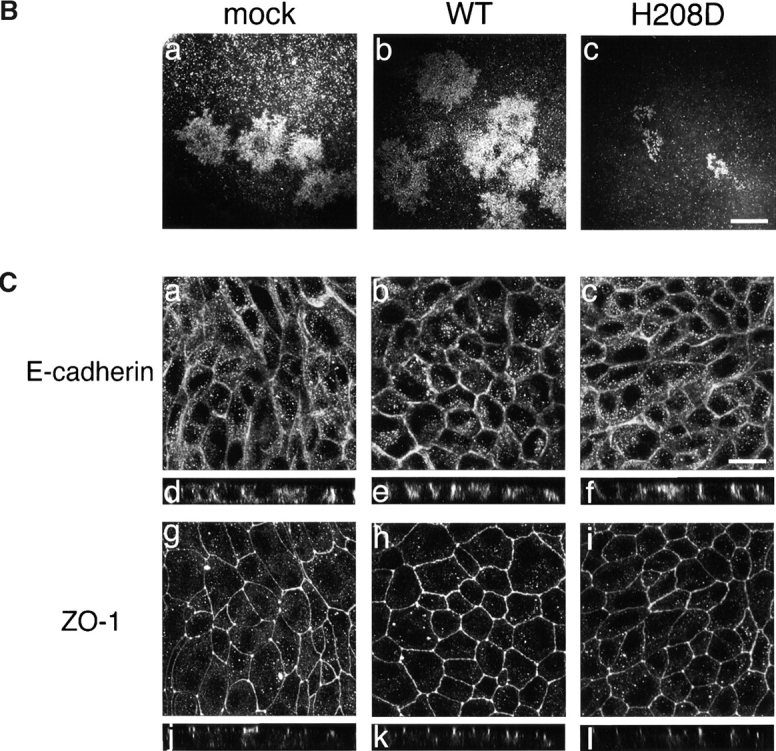
Cell to cell spreading of Shigella in infected MDCK cells stably expressing wild-type (WT) N-WASP or H208D mutant. (A) Expression of N-WASP variants in stable transfectants of MDCK cell lines. Total cell lysates were subjected to Western blotting with anti–E-cadherin, anti–N-WASP, and antiactin antibodies. Representative data are shown. (B) Dark field photomicrographs of plaque formation in MDCK cells stably expressing N-WASP variants 3 d after infection with Shigella. The results shown are representative of three independent experiments: a, mock-transfected cells; b, wild-type N-WASP; c, H208D mutant. (C) Localization of E-cadherin and ZO-1 in MDCK cells stably expressing N-WASP variants. Mock-transfected cells (a, d, g, and j), wild-type expressing cells (b, e, h, and k), and H208D mutant expressing cells (c, f, i, and l) were double-stained with anti–E-cadherin antibody (a–f) and anti–ZO-1 antibody (g–l). a–c and g–i, junctional levels; d–f and j–l, vertical sections. Bars, (B) 1 mm; (C) 10 μm.
Discussion
Rho family GTPases Cdc42, Rac and Rho have been implicated in regulating the cytoskeletal reorganization, cell adhesion, phagocytosis, and gene transcription 42 43 44 45 46 47. Their activity is also involved in the early stage of bacterial infection of epithelial cells; Cdc42/Rac1/RhoA and Cdc42/Rac1 activities are required for the invasion of epithelial cells by Shigella 58 59 60 61 and Salmonella 62 63 64, respectively, whereas Rac1 activity is involved in the signaling cascade in Opa52-dependent internalization of Neisseria gonorrhoeae 65. Although the precise mechanisms underlying bacteria-directed cellular signaling or the downstream effectors involved in inducing actin reorganization remain to be elucidated, studies have clearly indicated that pathogens are capable of variously activating cellular signal transduction pathways regulated by Rho family GTPases and exploiting their downstream cellular responses such as formations of focal adhesions, lamellipodia or filopodia, for their own internalization into the target host cells. In this study, we showed that Cdc42, but not Rac1 or RhoA, plays a crucial role in initiating the actin-based motility of S. flexneri in mammalian cells, in which Cdc42 appears to be required for triggering the activation of N-WASP, a critical host factor mediating actin polymerization on the surface of intracellular Shigella 10.
We observed that when activated Cdc42 was injected into Shigella-infected Swiss 3T3 fibroblasts, intracellular bacterial motility was stimulated. Importantly, upon injection of Cdc42, low speed bacteria moving at <5 μm/min in cells showed a remarkable increase in movement, whereas high speed bacteria did not (Fig. 1 A). Since the different responses of intracellular Shigella to activated Cdc42 injection were reproducible and the stimulation was Cdc42 specific, we presumed that Cdc42 activity is required at an initial stage of Shigella movement. As microinjection of the activated Cdc42 into serum-starved Swiss 3T3 cells can provoke a rapid filopodial formation 47, we therefore assumed that similar cellular events downstream of the Cdc42-activated signal would be involved in initiating Shigella movement in infected epithelial cells. The requirement of Cdc42 for Shigella motility in mammalian cells was also revealed in the actin tail assay using Xenopus oocyte extracts. When Rho GTPases activities in the extracts were blocked by adding RhoGDI, bacterial motility including the ability to form actin tail of an E. coli K-12 strain expressing VirG was inhibited in a RhoGDI dose–dependent manner. A similar inhibitory effect by RhoGDI on actin assembly from PtdIns(4,5)P2-containing lipid vesicles was observed previously 53, but RhoGDI addition had no effect on the motility of L. monocytogenes in Xenopus extracts 56. As addition of the activated Cdc42, but not activated Rac1 or RhoA, into the RhoGDI-containing Xenopus egg extracts restored VirG-mediated bacterial motility, we concluded that Shigella actin–based intracellular motility requires Cdc42 activity.
N-WASP binds Shigella VirG, which is required for bacterial movement in mammalian cells 10. N-WASP is one of the effector molecules regulated by Cdc42 involved in formation of filopodium. In mammalian cells, N-WASP exists in two states, either resting or activating, and transits stochastically between the two states 40. In the resting state, the N-WASP molecule is folded by intramolecular interaction between the basic amino acid region near the GBD and the COOH-terminal acidic residues, whereas when Cdc42 is activated such as on receiving an extracellular signal, the activated Cdc42 binds to GBD, which leads to unmasking of the VCA domain of N-WASP and a change to the active state 40. In the activated state, the VCA domain can directly interact with and stimulate actin nucleation activity of Arp2/3 complex (39, and for a review, see reference 66), whereas the VCA domain of N-WASP, like profilin, possesses actin sequestering activity that facilitates efficient actin assembly at the barbed end of F-actin 18. Therefore, it has been proposed that N-WASP recruited by VirG on Shigella can mediate actin polymerization by Arp2/3 complex, although the way in which N-WASP is activated, such as by binding by Cdc42 and/or VirG, is controversial 18 60. In the present study, the involvement of Cdc42 activity in Shigella motility, which is required for activation of N-WASP, was clearly demonstrated in add-back experiments of N-WASP or H208D mutant defective in binding to Cdc42 in RhoGDI-treated Xenopus egg extracts. Indeed, the bacterial motility was restored by adding activated Cdc42 together with N-WASP but not H208D. Furthermore, the importance of Cdc42 interaction with N-WASP was revealed in vivo, since actin assembly by Shigella in COS-7 cells overexpressing H208D was greatly decreased compared with that of cells expressing wild-type N-WASP.
To investigate which of the steps in actin-based Shigella motility, actin nucleation, elongation of F-actin tail, or stability of the actin tail, requires Cdc42 activity, we used the pyrene actin assay. In the presence of RhoGDI at 400 nM, actin nucleation mediated by VirG-α1 in a high speed centrifuged supernatant of Xenopus egg extracts was greatly inhibited (Fig. 6), suggesting that Cdc42 activity is involved in the actin nucleation. Furthermore, using purified components required for VirG-mediated actin nucleation that included VirG-α1, G-actin, N-WASP, and Arp2/3 complex in vitro 18 39, we tested whether Cdc42 can stimulate actin nucleation mediated by VirG–N-WASP–Arp2/3 complex in the pyrene actin assay. As revealed by Fig. 6, under optimum actin nucleation conditions the rate of actin polymerization was increased 1.6-fold by adding VirG-α1 at 0.5 μM, and 1.2-fold by adding activated Cdc42 at 0.25 μM. The ability of VirG-α1 to stimulate N-WASP activity as determined by actin nucleation was consistent with the results obtained by Egile et al. 18, albeit to a lesser extent compared with the level by IcsA (VirG) protein (see below). When N-WASP was replaced with H208D mutant, the actin nucleation in the presence of activated Cdc42 was significantly decreased to the level in the absence of Cdc42. The partial activation of H208D mutant was also observed in add-back experiments using H208D mutant in N-WASP–depleted extracts (Fig. 5). In this context, some bacteria in RhoGDI-treated extracts were observed to induce weak actin assembly at a later stage (Fig. 2). We speculate that these residual activities are caused by VirG–N-WASP interaction, as indicated by Egile et al. 18 and by our assay, that can somewhat affect the state of N-WASP. Importantly, the rate of actin nucleation was greatly (fourfold) enhanced by adding Cdc42 and VirG-α1 together into the pyrene actin assay system. In this regard, our results on the activation by VirG-α1 were apparently different from those of Egile et al.: they observed a dramatic activation of N-WASP on adding IcsA (VirG)53–508 18. Indeed, as revealed by Fig. 6, the extent of N-WASP activation by VirG-α1 (VirG53–759) as judged by actin nucleation was low compared with that by IcsA (VirG)53–508 used by Egile et al. 18. Since VirG-α1 used in our study encompasses the full length of 706 amino acids of VirG α domain, whereas VirG53–508 encompasses the NH2-terminal 446 amino acids, it is thus possible that the small VirG segment (VirG53–508) is more accessible to N-WASP than VirG-α1 in the resting state, and that the binding of the small VirG segment may stably sustain the unmasked form of N-WASP more than VirG-α1 in vitro. Since VirG-α1 encompasses the entire surface-exposed VirG portion on Shigella 12, and in infected mammalian cells VirG cannot be processed to generate the VirG53–508 portion 67, the marked stimulation of N-WASP–Arp2/3 complex–mediated actin nucleation by VirG53–508 in the absence of activated Cdc42 reported by Egile et al. 18 may be due to the effect of the aberrant small VirG version under in vitro conditions.
Enhancement of actin nucleation by Cdc42 on bacterial surface expressing VirG was also confirmed by an in vitro actin assembly reconstitution assay 18 31. When the E. coli K-12 strain expressing VirG was incubated in F buffer containing G-actin, N-WASP, and Arp2/3 complex, ∼20% of bacteria were surrounded by actin clouds but very weakly. When the bacteria were preincubated with N-WASP, the percentage of bacterial particles surrounded by actin clouds was significantly (∼80%) increased, as reported by Egile et al. 18, though the extent of actin condensation on the bacterial surface as judged by TMR signal was still significantly weak compared with that induced in the presence of activated Cdc42. Thus, as pointed out above, although VirG binding to N-WASP partly contributes to stimulating N-WASP activity, Cdc42 binding to N-WASP would be critical for initiating efficient actin nucleation. As reported by Loisel et al. 21, additional host factors such as ADF/cofilin, capping protein, α-actinin, and profilin are involved in the subsequent elongation and stabilization of the actin tail generated by the activated N-WASP–Arp2/3 complex. Thus, the elongation of the actin tail mediated by activated N-WASP–Arp2/3 complex linked to VirG may be less dependent on Cdc42 activity.
As indicated above, the involvement of Cdc42 activity in initiating movement of the E. coli K-12 strain expressing VirG revealed in this study is apparently controversial when compared with the results of Mounier et al. 60. They revealed that addition of GTPγS or Clostridium difficile toxin TcdB-10463, an inhibitor for Rho GTPases, into Xenopus oocyte extracts had no effect on VirG (IcsA)-mediated bacterial motility, including the formation of the actin tail 60. Furthermore, Egile et al. reported that when the E. coli K-12 strain expressing VirG was preincubated with N-WASP in the absence of Cdc42, the bacteria induced actin cloud formation in the in vitro reconstitution assay 18. The exact reasons for the different results are unclear; however, it is worth mentioning that the level of VirG expression from the E. coli K-12 strain used in other studies is very high compared with this study, since Mounier et al. or Egile et al. used a pUC8-derived vector for VirG expression 18 60 while we used a pBR322-derived vector 10. Therefore, it is likely that at a high concentration of VirG on the bacterial surface, the activation of N-WASP is easily achieved through binding by VirG. Because wild-type S. flexneri possesses only one copy of the virG gene in the large plasmid 8 11, we assume that a limited number of VirG molecules at one pole of the bacterium would still be insufficient to trigger activation of N-WASP in infected epithelial cells.
The above notion was supported by the results for the in vivo Shigella-infected cell model. Immunohistochemical study of intracellular Shigella at an early stage of infection (at 40 min after infection) using COS-7 cells expressing GFP-Cdc42 revealed that Cdc42 was mostly colocalized with the actin cloud at one pole of the bacterium, which is thought to be just before movement although a trace amount of Cdc42 was detected at the front of the actin tail generated by rapidly moving bacteria (Fig. 9). Importantly, at a later stage of Shigella infection, such as at 90 min after infection, Cdc42 was poorly detected at the front of a long actin tail or along the actin tail generated by rapidly moving bacteria, strongly suggesting that the role of Cdc42 in the actin-based Shigella motility in infected epithelial cells lies at the initial stage of bacterial movement. The results of the unipolar accumulation of Cdc42 together with the actin cloud on the intracellular bacterium agreed with the results in Fig. 1, where bacteria originally moving slowly were stimulated by the microinjection of activated Cdc42. If this notion is true, our results would still be compatible with the results of Mounier et al. 60, that addition of toxin TcdB-10463 or overexpression of dominant negative Cdc42 in a Shigella-infected cell had no effect on the motile Shigella: their results may reflect the steady state bacterial motility in mammalian cells. In the later stage of infection, the activity of N-WASP–Arp2/3 complex may still be sustained in the host cells, where the endogenous activity of Cdc42 may be downregulated. However, once the stable complex consisting of VirG, N-WASP, Arp2/3 complex, and actin filaments was formed on the bacterial surface, the actin polymerization activity mediated by VirG–N-WASP–Arp2/3 complex would be maintained as an active state. Although we cannot rule out other possibilities such as differences in strains (S. flexneri 2a in this study versus S. flexneri 5 in the other studies) or experimental conditions, we believe that the different stages of bacterial motility resulted in the different conclusions regarding the dependency of Cdc42 activity.
The essential role of Cdc42 activity in activating N-WASP in intracellular as well as intercellular movement of Shigella was conclusively demonstrated in the cell to cell spreading assay using mammalian cells stably expressing H208D mutant of N-WASP, where Shigella motility including intercellular movement was greatly inhibited as judged by the size of plaques formed in MDCK monolayers (Fig. 11). Our result is especially important because the pathogen must reinitiate actin assembly through its spreading process in the new host's cytoplasm in order to move within the cells and into adjacent host cells 8, where the interference by H208D of the actin nucleation mediated by N-WASP–Arp2/3 complex has a serious effect on bacterial spreading ability, a prerequisite for bacillary dysentery.
Acknowledgments
We are grateful to Alan Hall for GST-Rho family GTPase plasmids, Marie-France Carlier for the protocol, and Trinad Chakraborty for Listeria strains.
This work was supported by the Research for the Future Program of the Japan Society for the Promotion of Science, and Grant-in-aid for Scientific Research from the Japanese Ministry of Education, Science, Sports and Culture.
Footnotes
Abbreviations used in this paper: Arp, actin-related protein; GBD, GTPase binding domain; GFP, green fluorescent protein; GST, glutathione S-transferase; MDCK, Madin-Darby canine kidney; N-WASP, neural WASP; TBS, Tris-buffered saline; TMR, tetramethylrhodamine; VASP, vasodilator-stimulated phosphoprotein; WASP, Wiskott-Aldrich syndrome protein.
References
- Bernardini M.L., Mounier J., D'Hauteville H., Coquis-Rondon M., Sansonetti P.J. Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra and intercellular spread through interaction with F-actin. Proc. Natl. Acad. Sci. USA. 1989;86:3867–3871. doi: 10.1073/pnas.86.10.3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriot J.A., Mitchison T.J., Tilney L.G., Portnoy D.A. The rate of actin-based motility of intracellular Listeria monocytogenes equals the rate of actin polymerization. Nature. 1992;357:257–260. doi: 10.1038/357257a0. [DOI] [PubMed] [Google Scholar]
- Kadurugamuwa J.L., Rohde M., Wehland J., Timmis K.N. Intercellular spread of Shigella flexneri through a monolayer mediated by membranous protrusions and associated with reorganization of the cytoskeletal protein vinculin. Infect. Immun. 1991;59:3463–3471. doi: 10.1128/iai.59.10.3463-3471.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allaoui A., Mounier J., Prévost C., Sansonetti P.J., Parsot C. icsBa Shigella flexneri virulence gene necessary for the lysis of protrusions during intercellular spread. Mol. Microbiol. 1992;6:1605–1616. doi: 10.1111/j.1365-2958.1992.tb00885.x. [DOI] [PubMed] [Google Scholar]
- Prévost M.-C., Lesourd M., Arpin F., Vernel F., Mounier J., Hellio R., Sansonetti P.J. Unipolar reorganization of F-actin layer at bacterial division and bundling of actin filaments by plastin correlate with movement of Shigella flexneri within HeLa cells. Infect. Immun. 1992;60:4088–4099. doi: 10.1128/iai.60.10.4088-4099.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg M.B., Barzu O., Parsot C., Sansonetti P.J. Unipolar localization and ATPase activity of IcsA, a Shigella flexneri protein involved in intracellular movement. J. Bacteriol. 1993;175:2189–2196. doi: 10.1128/jb.175.8.2189-2196.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T., Murai T., Fukuda I., Tobe T., Yoshikawa M., Sasakawa C. Identification and characterization of a chromosomal virulence gene, vacJ, required for intercellular spreading of Shigella flexneri . Mol. Microbiol. 1994;11:31–41. doi: 10.1111/j.1365-2958.1994.tb00287.x. [DOI] [PubMed] [Google Scholar]
- Makino S., Sasakawa C., Kamata T., Yoshikawa M. A genetic determinant required for continuous reinfection of adjacent cells on a large plasmid in Shigella flexneri 2a. Cell. 1986;46:551–555. doi: 10.1016/0092-8674(86)90880-9. [DOI] [PubMed] [Google Scholar]
- Suzuki T., Saga S., Sasakawa C. Functional analysis of Shigella VirG domains essential for interaction with vinculin and actin-based motility. J. Biol. Chem. 1996;271:21878–21885. doi: 10.1074/jbc.271.36.21878. [DOI] [PubMed] [Google Scholar]
- Suzuki T., Miki H., Takenawa T., Sasakawa C. Neural Wiskott-Aldrich syndrome protein is implicated in actin-based motility of Shigella flexneri . EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:2767–2776. doi: 10.1093/emboj/17.10.2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lett M.-C., Sasakawa C., Okada N., Sakai T., Makino S., Yamada M., Komatsu K., Yoshikawa M. virG, a plasmid-coded virulence gene of Shigella flexneriidentification of the virG protein and determination of the complete coding sequence. J. Bacteriol. 1989;171:353–359. doi: 10.1128/jb.171.1.353-359.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T., Lett M.-C., Sasakawa C. Extracellular transport of VirG protein in Shigella . J. Biol. Chem. 1995;270:30874–30880. doi: 10.1074/jbc.270.52.30874. [DOI] [PubMed] [Google Scholar]
- Laine R.O., Zeile W., Kang F., Purich D.L., Southwick F.S. Vinculin proteolysis unmasks an ActA homolog for actin-based Shigella motility. J. Cell Biol. 1997;138:1255–1264. doi: 10.1083/jcb.138.6.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischknecht F., Cudmore S., Moreau V., Reckmann I., Rottger S., Way M. Tyrosine phosphorylation is required for actin-based motility of vaccinia but not Listeria or Shigella . Curr. Biol. 1999;9:89–92. doi: 10.1016/s0960-9822(99)80020-7. [DOI] [PubMed] [Google Scholar]
- Zeile W.L., Purich D.L., Southwich F.S. Recognition of two classes of oligoproline sequences in profilin-mediated acceleration of actin-based Shigella motility. J. Cell Biol. 1996;133:49–59. doi: 10.1083/jcb.133.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouin E., Gantelet H., Egile C., Lasa I., Ohayon H., Villiers V., Gounon P., Sansonetti P.J., Cossart P. A comparative study of the actin-based motilities of the pathogenic bacteria Listeria monocytogenes, Shigella flexneri and Rickettsia conorii . J. Cell Sci. 1999;112:1697–1708. doi: 10.1242/jcs.112.11.1697. [DOI] [PubMed] [Google Scholar]
- Chakraborty T., Ebel F., Domann E., Niebuhr K., Gerstel B., Pistor S., Temm-Grove C.J., Jockusch B.M., Reinhard M., Walter U., Wehland J. A focal adhesion factor directly linking intracellularly motile Listeria monocytogenes and Listeria ivanovii to the actin-based cytoskeleton of mammalian cells. EMBO (Eur. Mol. Biol. Organ.) J. 1995;14:1314–1321. doi: 10.1002/j.1460-2075.1995.tb07117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egile C., Loisel T.P., Laurent V., Li R., Pantaloni D., Sansonetti P.J., Carlier M.-F. Activation of the CDC42 effector N-WASP by the Shigella flexneri IcsA protein promotes actin nucleation by Arp2/3 complex and bacterial actin-based motility. J. Cell Biol. 1999;146:1319–1332. doi: 10.1083/jcb.146.6.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derry J.M.J., Ochs H.D., Francke U. Isolation of a novel gene mutated in Wiskott-Aldrich syndrome. Cell. 1994;78:635–644. doi: 10.1016/0092-8674(94)90528-2. [DOI] [PubMed] [Google Scholar]
- Miki H., Miura K., Takenawa T. N-WASP, a novel actin-depolymerizing protein, regulates the cortical cytoskeletal rearrangement in a PIP2-dependent manner downstream of tyrosine kinases. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:5326–5335. [PMC free article] [PubMed] [Google Scholar]
- Loisel T.P., Boujemaa R., Pantaloni D., Carlier M.-F. Reconstitution of actin-based motility of Listeria and Shigella using pure proteins. Nature. 1999;401:613–616. doi: 10.1038/44183. [DOI] [PubMed] [Google Scholar]
- Cossart P., Lecuit M. Interactions of Listeria monocytogenes with mammalian cells during entry and actin-based movementbacterial factors, cellular ligands and signaling. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:3797–3806. doi: 10.1093/emboj/17.14.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dramsi S., Cossart P. Intracellular pathogens and the actin cytoskeleton. Annu. Rev. Cell Dev. Biol. 1998;14:137–166. doi: 10.1146/annurev.cellbio.14.1.137. [DOI] [PubMed] [Google Scholar]
- Heinzen R.A., Hayes S.F., Peacock M.G., Hackstadt T. Directional actin polymerization associated with spotted fever group Rickettsia infection of Vero cells. Infect. Immun. 1993;61:1926–1935. doi: 10.1128/iai.61.5.1926-1935.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teysseire N., Chichi-Portiche C., Raoult D. Intracellular movements of Rickettsia conorii and R. typhi based on actin polymerization. Res. Microbiol. 1992;143:821–829. doi: 10.1016/0923-2508(92)90069-z. [DOI] [PubMed] [Google Scholar]
- Cudmore S., Cossart P., Griffiths G., Way M. Actin-based motility of vaccinia virus. Nature. 1995;378:636–638. doi: 10.1038/378636a0. [DOI] [PubMed] [Google Scholar]
- Domann E., Wehland J., Rohde M., Pistor S., Hartl M., Goebel W., Leimeister-Wachter M., Wuenscher M., Chakraborty T. A novel bacterial gene in Listeria monocytogenes required for host cell microfilament interaction with homology to the proline-rich region of vinculin. EMBO (Eur. Mol. Biol. Organ.) J. 1992;11:1981–1990. doi: 10.1002/j.1460-2075.1992.tb05252.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocks C., Gouin E., Tabouret M., Berche P., Ohayon H., Cossart P. Listeria monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell. 1992;68:521–531. doi: 10.1016/0092-8674(92)90188-i. [DOI] [PubMed] [Google Scholar]
- Welch M.D., Iwamatsu A., Mitchison T.J. Actin polymerization is induced by Arp2/3 protein complex at the surface of Listeria monocytogenes . Nature. 1997;385:265–269. doi: 10.1038/385265a0. [DOI] [PubMed] [Google Scholar]
- Niebuhr K., Ebel F., Frank R., Reinhard M., Domann E., Carl U.D., Walter U., Gertler F.B., Wehland J., Chakraborty T. A novel proline-rich motif present in ActA of Listeria monocytogenes and cytoskeletal proteins is the ligand for the EVH1 domain, a protein module present in the Ena/VASP family. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:5433–5444. doi: 10.1093/emboj/16.17.5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch M.D., Rosenblatt J., Skoble J., Portnoy D.A., Mitchison T.J. Interaction of human Arp2/3 and the Listeria monocytogenes ActA protein in actin filament nucleation. Science. 1998;281:105–108. doi: 10.1126/science.281.5373.105. [DOI] [PubMed] [Google Scholar]
- Laurent V., Loisel T.P., Harbeck B., Wehman A., Gröbe L., Jockusch B.M., Wehland J., Gertler F.B., Carlier M.-F. Role of proteins of the Ena/VASP family in actin-based motility of Listeria monocytogenes . J. Cell Biol. 1999;144:1245–1258. doi: 10.1083/jcb.144.6.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischknecht F., Moreau V., Rottger S., Gonfloni S., Reckmann I., Superti-Furga G., Way M. Actin-based motility of vaccinia virus mimics receptor tyrosine kinase signalling. Nature. 1999;401:926–929. doi: 10.1038/44860. [DOI] [PubMed] [Google Scholar]
- Lechler T., Li R. In vitro reconstitution of cortical actin assembly sites in budding yeast. J. Cell Biol. 1997;138:95–103. doi: 10.1083/jcb.138.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R. Bee1, a yeast protein with homology to Wiskott-Aldrich syndrome protein, is critical for the assembly of cortical actin cytoskeleton. J. Cell Biol. 1997;136:649–658. doi: 10.1083/jcb.136.3.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear J.E., Rawis J.F., Saxe C.L., III. SCAR, a WASP-related protein, isolated as a suppressor of receptor defects in late Dictyostelium development. J. Cell Biol. 1998;142:1325–1335. doi: 10.1083/jcb.142.5.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki H., Suetsugu S., Takenawa T. WAVE, a novel-WASP family protein involved in actin reorganization induced by Rac. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:6932–6941. doi: 10.1093/emboj/17.23.6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suetsugu S., Miki H., Takenawa T. Identification of two human WAVE/SCAR homologues as general actin regulatory molecules which associate with the Arp2/3 complex. Biochem. Biophys. Res. Commun. 1999;260:296–302. doi: 10.1006/bbrc.1999.0894. [DOI] [PubMed] [Google Scholar]
- Rohatgi R., Ma L., Miki H., Lopez M., Kirchhausen T., Takenawa T., Kirschner M.W. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell. 1999;97:221–231. doi: 10.1016/s0092-8674(00)80732-1. [DOI] [PubMed] [Google Scholar]
- Miki H., Sasaki T., Takai Y., Takenawa T. Induction of filopodium formation by a WASP-related actin-depolymerizing protein N-WASP. Nature. 1998;391:93–96. doi: 10.1038/34208. [DOI] [PubMed] [Google Scholar]
- Carlier M.-F., Ducruix A., Pantaloni D. Signalling to actinthe Cdc42–N-WASP–Arp2/3 connection. Chem. Biol. 1999;6:R235–R240. doi: 10.1016/s1074-5521(99)80107-0. [DOI] [PubMed] [Google Scholar]
- Aelst L.V., D'Souza-Schorey C. Rho GTPases and signaling networks. Genes Dev. 1997;11:2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Tanaka K., Takai Y. Control of reorganization of the actin cytoskeleton by Rho family small GTP-binding proteins in yeast. Curr. Opin. Cell Biol. 1998;10:112–116. doi: 10.1016/s0955-0674(98)80093-8. [DOI] [PubMed] [Google Scholar]
- Ridley A.J., Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- Ridley A.J., Paterson H.F., Johnston C.L., Diekmann D., Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- Nobes C.D., Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- Sasakawa C., Kamata K., Sakai T., Murayama Y., Makino S., Yoshikawa M. Molecular alteration of the 140-megadalton plasmid associated with loss of virulence and Congo red binding activity in Shigella flexneri . Infect. Immun. 1986;51:470–475. doi: 10.1128/iai.51.2.470-475.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Self A.J., Hall A. Purification of recombinant Rho/Rac/G25K from Escherichia coli . Methods Enzymol. 1995;256:3–10. doi: 10.1016/0076-6879(95)56003-3. [DOI] [PubMed] [Google Scholar]
- Rosenblatt J., Agnew B.J., Abe H., Bamburg J.R., Mitchison T.J. Xenopus actin depolymerizing factor/cofilin (XAC) is responsible for the turnover of actin filaments in Listeria monocytogenes tails. J. Cell Biol. 1997;136:1323–1332. doi: 10.1083/jcb.136.6.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriot J.A., Fung D.C. Listeria monocytogenes-based assays for actin assembly factors. Methods Enzymol. 1998;298:114–122. doi: 10.1016/s0076-6879(98)98013-2. [DOI] [PubMed] [Google Scholar]
- Kouyama T., Mihashi K. Fluorimetry study of N-(1-pyrenyl)iodoacetamide-labelled F-actin. Eur. J. Biochem. 1981;114:33–38. [PubMed] [Google Scholar]
- Ma L., Cantley L.C., Janmey P.A., Kirschner M.W. Corequirement of specific phosphoinositides and small GTP-binding protein cdc42 in inducing actin assembly in Xenopus egg extracts. J. Cell Biol. 1998;140:1125–1136. doi: 10.1083/jcb.140.5.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasakawa C., Kamata K., Sakai T., Makino S., Yamada M., Okada N., Yoshikawa M. Virulence-associated genetic regions comprising 31 kilobases of the 230-kilobase plasmid in Shigella flexneri 2a. J. Bacteriol. 1988;170:2480–2484. doi: 10.1128/jb.170.6.2480-2484.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchand J.-B., Moreau P., Paoletti A., Cossart P., Carlier M.-F., Pantaloni D. Actin-based movement of Listeria Monocytogenesactin assembly results from the local maintenance of uncapped filament barbed ends at the bacterium surface. J. Cell Biol. 1995;130:331–343. doi: 10.1083/jcb.130.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau V., Way M. Cdc42 is required for membrane dependent actin polymerization in vitro. FEBS Lett. 1998;427:353–356. doi: 10.1016/s0014-5793(98)00443-8. [DOI] [PubMed] [Google Scholar]
- Cooper J.A., Walker S.B., Pollard T.D. Pyrene actindocumentation of the validity of a sensitive assay for actin polymerization. J. Muscle Res. Cell Motil. 1983;4:253–262. doi: 10.1007/BF00712034. [DOI] [PubMed] [Google Scholar]
- Adam T., Giry M., Boquet P., Sansonetti P.J. Rho-dependent membrane folding causes Shigella entry into epithelial cells. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:3315–3321. [PMC free article] [PubMed] [Google Scholar]
- Watarai M., Kamata Y., Kozaki S., Sasakawa C. rho, a small GTP-binding protein, is essential for Shigella invasion of epithelial cells. J. Exp. Med. 1997;185:281–292. doi: 10.1084/jem.185.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mounier J., Laurent V., Hall A., Fort P., Carlier M.-F., Sansonetti P.J., Egile C. Rho family GTPases control entry of Shigella flexneri into epithelial cells but not intracellular motility. J. Cell Sci. 1999;112:2069–2080. doi: 10.1242/jcs.112.13.2069. [DOI] [PubMed] [Google Scholar]
- Tran Van Nhieu G., Caron E., Hall A., Sansonetti P.J. IpaC induces actin polymerization and filopodia formation during Shigella entry into epithelial cells. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:3249–3262. doi: 10.1093/emboj/18.12.3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.M., Hobbie S., Galán J.E. Requirement of CDC42 for Salmonella typhimurium-induced cytoskeletal reorganization and nuclear responses. Science. 1996;274:2115–2118. doi: 10.1126/science.274.5295.2115. [DOI] [PubMed] [Google Scholar]
- Hardt W.-D., Chen L.-M., Schuebel K.E., Bustelo X.R., Galán J.E. S. typhimurium encodes an activator of Rho GTPases that induces membrane ruffling and nuclear responses in host cells. Cell. 1998;93:815–826. doi: 10.1016/s0092-8674(00)81442-7. [DOI] [PubMed] [Google Scholar]
- Fu Y., Galán J.E. A Salmonella protein antagonizes Rac-1 and Cdc42 to mediate host-cell recovery after bacterial invasion. Nature. 1999;401:293–297. doi: 10.1038/45829. [DOI] [PubMed] [Google Scholar]
- Hauck C.R., Meyer T.F., Lang F., Gulbins E. CD66-mediated phagocytosis of Opa52 Neisseria gonorrhoeae requires a Src-like tyrosine kinase- and Rac1-dependent signalling pathway. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:443–454. doi: 10.1093/emboj/17.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch M.D. The world according to Arpregulation of actin nucleation by the Arp2/3 complex. Trends Cell Biol. 1999;9:423–427. doi: 10.1016/s0962-8924(99)01651-7. [DOI] [PubMed] [Google Scholar]
- Fukuda I., Suzuki T., Munakata H., Hayashi N., Katayama E., Yoshikawa M., Sasakawa C. Cleavage of Shigella surface protein VirG occurs at a specific site, but the secretion is not essential for intracellular spreading. J. Bacteriol. 1995;177:1719–1726. doi: 10.1128/jb.177.7.1719-1726.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]