

Abstract
The pathogenesis of immunoglobulin A (IgA) nephropathy (IgAN), the most prevalent form of glomerulonephritis worldwide, involves circulating macromolecular IgA1 complexes. However, the molecular mechanism(s) of the disease remain poorly understood. We report here the presence of circulating soluble FcαR (CD89)-IgA complexes in patients with IgAN. Soluble CD89 was identified as a glycoprotein with a 24-kD backbone that corresponds to the expected size of CD89 extracellular domains. To demonstrate their pathogenic role, we generated transgenic (Tg) mice expressing human CD89 on macrophage/monocytes, as no CD89 homologue is found in mice. These mice spontaneously developed massive mesangial IgA deposition, glomerular and interstitial macrophage infiltration, mesangial matrix expansion, hematuria, and mild proteinuria. The molecular mechanism was shown to involve soluble CD89 released after interaction with IgA. This release was independent of CD89 association with the FcRγ chain. The disease was induced in recombination activating gene (RAG)2−/− mice by injection of serum from Tg mice, and in severe combined immunodeficiency (SCID)-Tg mice by injection of patients' IgA. Depletion of soluble CD89 from serum abolished this effect. These results reveal the key role of soluble CD89 in the pathogenesis of IgAN and provide an in vivo model that will be useful for developing new treatments.
Keywords: IgA nephropathy, IgA, Fc receptor, monocytes, transgenic mice
Introduction
IgA nephropathy (IgAN), or Berger's disease, is characterized by hematuria and IgA deposits in the mesangium 1 2. The disease is associated with mononuclear cell infiltration in the kidney, which correlates with glomerular damage and interstitial tissue injury 3 4 5. Within 10–20 yr of onset, about one third of patients develop renal failure. The pathogenic mechanisms underlying IgAN are poorly understood. They include a galactose deficiency of the IgA1 hinge region 6 7 and formation of immune complexes (ICs) containing polymeric IgA1 molecules 8 9 10. The role of IgA-ICs in the pathogenesis of IgAN is supported by the recurrence of IgA deposits in renal grafts 11. Some of the components of IgA-containing ICs have been identified, such as IgG, fibronectin, and collagen 2 7 12. Studies in mice have shown that mesangial IgA deposition depends on multiple elements generating the macromolecular IgA-IC, such as the polymeric nature of IgA 13 14 and levels of uteroglobin, which control IgA–fibronectin IC formation 15. However, it is noteworthy that not all patients with IgAN have fibronectin–IgA complexes 16, and most components responsible for IgA-IC deposition remain poorly identified 17.
Based on observations of patients with IgAN, we postulated that the IgA Fc receptor, CD89, was strongly involved in the pathogenic mechanism of the disease 18 19. In healthy individuals, CD89 is constitutively expressed on human myeloid cells as 55–75-kD glycoproteins that bind IgA polymers more than monomers 20 21 22. In IgAN, we observed a marked decrease in monocyte CD89 expression 18. Despite low receptor numbers, increased CD89 occupancy by IgA1 is found on monocytes of IgAN patients and is linked to the severity of renal lesions such as glomerulosclerosis and mesangial proliferation.
In this study, we first detected soluble CD89 complexed with IgA in the serum of patients with IgAN, contrary to patients with other IgA-related and -unrelated diseases. To determine the role of soluble CD89 in the pathogenesis of IgAN, we then generated CD89 transgenic (Tg) mice with high level protein expression on monocyte/macrophages, driven by the mouse CD11b promoter. We obtained direct evidence that soluble CD89 is involved in IgA-IC formation, as CD89 Tg mice developed IgAN with macrophage infiltration in damaged glomeruli. The heavy hematuria in mice that received Tg serum or patients' IgA suggests that soluble CD89–IgA complexes are pathogenic and that the nature of the IgA controls the formation of these complexes.
Materials and Methods
Subjects.
Serum was obtained from 30 patients with biopsy-proven IgAN (23 males and 7 females), 30 healthy laboratory volunteers (17 males and 13 females), 11 patients with minimal change disease (MCD), 14 patients with rheumatoid arthritis, and 20 patients with alcoholic liver cirrhosis (ALC). Patients were selected on the basis of no severe renal failure and no steroid treatment. Some experiments used heparinized blood. The study was approved by the Hôpital Necker Ethics Committee.
Constructs, Tg Mice, and Cell Lines.
A construct encoding human CD89 was obtained by inserting a 896-bp BamHI fragment containing the human CD89 a.1 isoform gene 23 into the BamHI site of the pB202–D736 construct containing the 1.7-kb mouse CD11b promoter (provided by D. Tenen, Harvard Medical School, Boston, MA) 24. An electroeluted 4.8-kb DNA XhoI-NotI fragment was microinjected into fertilized nonobese diabetic (NOD) eggs and reimplanted into (NOD × C57BL/6)F1 foster mothers as described elsewhere 25. Three offspring were found to contain the human FcαR cDNA by Southern blot or by PCR of tail DNA using either a cDNA probe (provided by C. Maliszewski, Immunex Corp., Seattle, WA; reference 22) or transgene-specific primers 5′-GGGGAATTCGACGCAAACAAGGCAGGGCGC-3′ and 5′-GGGGTCGACCTTGCAGACACTTGGTGTTCG-3′. The C57BL/6 background was introduced into CD89 Tg mice of all three lines by more than four consecutive crosses with C57BL/6 mice. The SCID mutations were introduced into CD89-CD11b Tg mice (line 83) by two consecutive crosses with SCID-NOD mice. Tg mice homozygous for the SCID defect were first selected by immunofluorescence of blood lymphocytes and then confirmed by CD89 PCR typing of their progeny. We used heterozygous mice because they provide littermate controls. All mouse strains used in this study were raised and housed in strictly controlled specific pathogen-free conditions. Recombination activating gene (RAG)-2−/− mice, backcrossed nine times to C57BL/6, were obtained from the Centre National de la Recherche Scientifique Central Animal Facility. Rat basophilic cell lines (RBL-2H3) expressing CD89 (wild-type [WT]), and a mutated version with Leu instead of Arg at position 209 of the transmembrane region (R209L) were used as described previously 26. CD89 R209L expression was stable on the cell surface and was not associated with the FcR γ chain, as expected 26. Recombinant soluble CD89 was produced as described previously 27.
Antibodies.
The murine IgG1 anti-FcαR mAbs A3, A62, and A77, recognizing epitopes on the extracellular domains of FcαR 28, were purified in our laboratory. F(ab′)2 fragments of A3 and A77 mAbs were prepared by pepsin digestion (Sigma-Aldrich) as described elsewhere 29. The murine IgM anti-CD89 mAb My43, which blocks IgA binding, was provided by L. Shen (Dartmouth Medical School, Lebanon, NH; reference 30). To generate anti-CD89 intracytoplasmic (Cy-α) region antibodies, Cy-α was PCR amplified and introduced into a glutathione S-transferase (GST) gene fusion vector (pGEX; Amersham Pharmacia Biotech). GST-Cy-α was produced and purified to immunize rabbits (with help from U. Blank, Institut Pasteur). Purified anti–Cy-α antibodies were able to immunoprecipitate CD89 molecules from lysates of U937 cells (not shown). mAb A62, mAb A77, and a polyclonal anti–Cy-α antibody were coupled to biotin. PE-conjugated A59 was purchased from BD PharMingen. Goat anti–human IgA, and anti–human IgG coupled to alkaline phosphatase (AP) were purchased from Southern Biotechnology Associates, Inc. Biotinylated mouse anti–rat antibodies were purchased from Jackson ImmunoResearch Laboratories. Mac-1 and F4/80 hybridomas were provided by F. Lepault (Centre National de la Recherche Scientifique, URA 1461, Paris, France). Human dimeric monoclonal IgA1κ was obtained from a patient with myeloma 26. Mouse monoclonal dimeric IgA purified from ascites of the IgA-C5 hybridoma was provided by A. Phalipon (Institut Pasteur) 31. MOPC315 IgA was purchased from American Type Culture Collection. Purified IgA was biotinylated as described elsewhere 26.
Flow Cytometry.
Cell suspensions (106) from the peritoneal cavity and heparinized blood were incubated with 2.4G2 and human IgG to block FcγR. For two-color staining, cells were stained with PE-conjugated A59 and with FITC-labeled mac-1 for 30 min at 4°C before red cell lysis with Becton Dickinson lysis buffer. In some experiments cells were stained with biotinylated mAb A77 (0.1 mg/ml) followed by streptavidin-PE as developing reagent. Stained cells were analyzed on a FACSCalibur™ (Becton Dickinson) using CELLQuest™ software.
Cell Radiolabeling and Immunoprecipitation.
Cell surface iodination with Na125I (1 mCi; Amersham Pharmacia Biotech) used the lactoperoxidase method. For immunoprecipitation, cells (107/ml) were lysed for 30 min at 4°C in PBS containing 0.5% NP-40 (Sigma-Aldrich), 0.02% sodium azide, 1% aprotinin, 1 mM diisopropylfluorophosphate, 5 mM iodoacetamide, and 1 mM PMSF. After centrifugation at 14,000 g for 30 min to remove insoluble material, cleared lysates were immunodepleted of FcγR by using human IgG and mAb 2.4G2 precipitated with A77 F(ab′)2 fragments coupled to Sepharose 4B as described previously 26. Bound materials were treated or untreated with peptide N-glycosidase F (Oxford GlycoSystems) and samples were subsequently prepared for SDS-PAGE 32. For metabolic labeling, blood monocytes (5 × 106) were first starved for 1 h at 37°C in Met−/Cys− RPMI medium, and then pulsed for 18 h in 2 ml of Met−/Cys− RPMI containing 1 mCi of 35S-Met/Cys Translabel (specific activity 1,175 Ci/mmol; ICN Biomedicals), 10% Met−/Cys− FCS (Myoclone; GIBCO BRL), and 2 mM glutamine. After washes, cells were chased in RPMI containing 10% FCS for 48 h. Cells were microfuged and 1.5 ml of supernatant was collected. The cell pellet was lysed in 0.5% NP-40 lysis buffer as described above. Lysates and supernatants were immunoprecipitated as described above, and proteins were separated by SDS-12% PAGE and revealed by autoradiography.
Solid-Phase ELISA for Soluble CD89 and IgA.
To detect soluble CD89, we used two mAbs directed against epitopes expressed on the extracellular domain and a polyclonal antibody directed against CD89-Cy-α. 96-well plates (Dynex Technologies, Inc.) were coated with F(ab′)2 fragments of mAb A3 (5 μg/ml), blocked with PBS containing 1% BSA, and incubated overnight at 4°C with test samples (1:2 to 1:20 dilutions in PBS containing 0.05% Tween 20). Recombinant soluble CD89 27 and a U937 cell lysate were used as standard positive controls. After washes, biotin-labeled anti-FcαR antibodies (5 μg/ml) recognizing either the extracellular domains (clone A62 or A77) or the cytoplasmic tail (Cy-α) were added. In some experiments, biotin-labeled anti-IgA or anti-IgG was used to detect sCD89–Ig complexes. After 2 h of incubation at room temperature, the plates were washed and incubated with streptavidin-AP (1:2,000 dilution). The reaction was developed by adding the AP substrate, and absorbance was read at 405 nm. To determine circulating ICs containing soluble CD89, serum samples were precipitated with polyethylene glycol (PEG) 6000 (Merck-Clevenot) as described elsewhere 10. Before analysis, each PEG precipitate was redissolved in 500 μl of 0.01 M phosphate buffer, pH 7.4, containing 0.5 M NaCl and 0.05% Tween 20. Levels of IC-containing FcαR were then estimated as described above. Patients' results are expressed in SD units (SDU) according to the formula: SDU = (X t – X n)/SDn, where X t is the mean value (absorbance) of each test sample, X n is the mean value of samples from 12 healthy blood donors assayed simultaneously, and SDn is the SD of the mean value obtained for the panel of healthy donors as described elsewhere 10. Mouse IgA was measured by coating plates with a rat anti–mouse IgA mAb (clone R5-140; BD PharMingen) at 10 μg/ml and developed by adding biotinylated goat anti–mouse IgA (Southern Biotechnology Associates, Inc.) plus streptavidin-AP. The standard curve was constructed with purified mouse IgA (Sigma-Aldrich).
Morphologic Analyses.
Kidney tissues were fixed in 3.6% paraformaldehyde, dehydrated in graded alcohols, and embedded in paraffin. Sections 2–3 μm thick were deparaffinized and rehydrated before staining with hematoxylin and eosin and periodic acid-Schiff.
Immunohistochemistry.
Frozen tissue sections 3-4 μm thick were adhered to microscope slides (Superfrost Plus; VWR Scientific), fixed in acetone (HPLC grade; Sigma-Aldrich) for 10 min at 4°C, and then allowed to air dry for at least 1 h. Slides were washed in PBS for 5–10 min, followed by the avidin/biotin blocking steps. Slides were loaded with normal horse serum (Vector Labs) for 30 min to block nonspecific sites. The primary antibody (rat mAb) was incubated for 60 min in a humid chamber (Vector Labs), and the biotinylated mouse anti–rat Ig (Jackson ImmunoResearch Laboratories) was added and supplemented with 2% normal mouse serum. The avidin/biotin-horseradish peroxidase complex (ABC-HRP reagent; Vector Labs) was then added, followed by 3,3′-diaminobenzidine tetrahydrochloride (DAB). Hematoxylin (1 min) and blueing steps were completed. Slides were then water washed, dehydrated with ethanol, cleared in xylene, and coverslipped.
Albuminuria and Blood Urea Nitrogen.
96-well plates (Dynex) were coated with mouse serum albumin (MSA, 3 μg/ml; Sigma-Aldrich), blocked with 1% casein (Sigma-Aldrich) in PBS, and incubated for 1 h at 37°C with urine samples (pure and 1:2 dilutions) in PBS containing 1:5,000-diluted rabbit anti-MSA antibodies (Cappel Laboratories). MSA was used as the standard control. After washes, HRP-labeled anti–rabbit antibodies (5 μg/ml; Jackson ImmunoResearch Laboratories) were added followed by 2,2′-azino-bis(3-ethylbenz-thiazoline-6-sulfonic acid) (ABTS). Blood urea nitrogen (BUN) levels were determined using an autoanalyzer (Hitachi 917; Boehringer) with commercially available kits.
Statistical Analyses.
Differences between groups were determined by Student's t test and the nonparametric Mann-Whitney U test, as indicated. Results are presented as means ± SEM.
Results
Identification of Soluble CD89–IgA Complexes in Circulating ICs from Patients with IgAN.
High levels of soluble CD89 were detected in 40% (12/30) of PEG precipitates from IgAN patients' sera, whereas only one patient had increased soluble CD89 in serum as free form using a sandwich ELISA with two mAbs specific for CD89 extracellular domains (Fig. 1 A). No significant increase in soluble CD89 was detected in PEG precipitates of serum from patients with other IC diseases (e.g., rheumatoid arthritis), elevated serum IgA (e.g., ALC), or another renal disease (e.g., MCD) (Fig. 1 A). To investigate whether soluble CD89 was complexed with IgA within the IC, we developed an ELISA based on anti–CD89 F(ab′)2 mAb (clone A3) as capture antibody and anti–human IgA as developing antibody. High levels of IgA were detected in ICs containing soluble CD89 from IgAN patients (Fig. 1 B). In contrast, no IgG was detected, indicating that the interaction of soluble CD89 with IgA is isotype specific. To characterize the soluble CD89, we cultured metabolically labeled blood monocytes from two IgAN patients. After 48 h of culture, heavily glycosylated CD89 molecules (50–70 kD) were specifically immunoprecipitated from supernatants of both patients' samples by an anti-CD89 F(ab′)2 mAb (Fig. 1 C). N-glycosidase F treatment of A77 F(ab′)2-reactive molecules precipitated from supernatants gave rise to a 24-kD protein core and a 33-kD band, whereas treatment of the corresponding molecules from cell lysates resulted in the expected 32-kD backbone only (Fig. 1 C, lanes 4 and 8). To determine if the soluble CD89 found in PEG precipitates from IgAN patients contained an intracytoplasmic tail (Cy-α), we used an ELISA method based on mAb A3 and anti–CD89-Cy-α polyclonal antibody as capture and/or developing antibodies. No reactivity was observed, indicating that soluble CD89 lacks the cytoplasmic tail. In contrast, high reactivity (>1.2 OD) was obtained with the control CD89 (membrane form from U937 cell lysates; data not shown).
Figure 1.
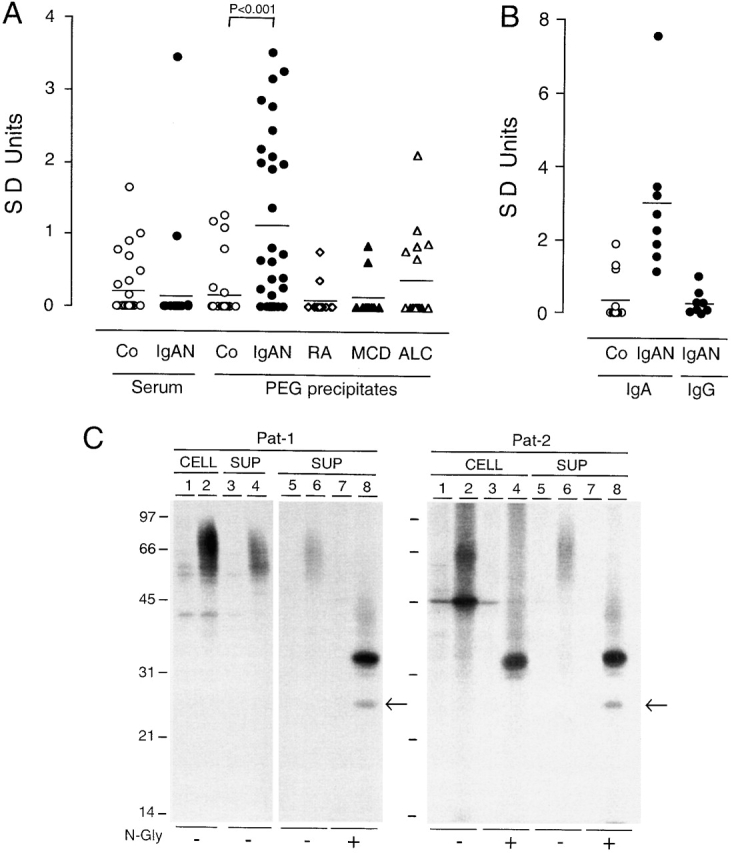
Characterization of soluble CD89 in serum ICs of patients with IgAN. (A) Soluble CD89 was detected in PEG precipitates from patients with IgAN (n = 30), but not in their serum (except for one patient), by using a sandwich ELISA with two anti-CD89 mAbs (A3 F[ab′]2/A77 as capture/developing antibodies). In contrast, no significant increase in soluble CD89 was found in PEG precipitates from normal, healthy controls (Co) and patients with rheumatoid arthritis (RA, n = 14), MCD (n = 11), and ALC (n = 20). Results are expressed in SD units as described in Materials and Methods. (B) Detection of IgA within CD89+ PEG precipitates by ELISA using A3 F(ab′)2 and anti–human IgA antibodies. No IgG was found with anti-IgG antibodies. (C) Biochemical characterization of soluble FcαR (CD89). Blood monocytes (107) from two IgAN patients (Pat-1 and Pat-2) were pulsed for 18 h in 35S-Met/Cys and chased for 48 h. Cells were spun and supernatants (SUP) were collected. Cell pellets were lysed and immunoprecipitated with either an irrelevant mouse IgG1κ mAb (lanes 1, 3, 5, and 7) or anti-CD89 A77 F(ab′)2 coupled to beads (lanes 2, 4, 6, and 8). After immunoprecipitation, materials were treated or untreated with N-glycosidase F (N-gly) as described elsewhere (reference 21). Materials were then separated by SDS-12% PAGE. The arrows indicate the 24-kD band of deglycosylated soluble CD89.
Generation of Human FcαR (CD89) Tg Mice.
As no CD89 homologue has been found in mice 33 34, we examined whether expression of human CD89 on mouse monocyte/macrophages would induce alterations in the mouse immune system. We generated three Tg mouse lines in the C57BL/6 and SCID backgrounds, designated 73, 83, and 96, using a construct containing human full-length CD89 cDNA under the control of the mouse CD11b promoter 24 that confers high level protein expression on monocyte/macrophages (Fig. 2 A). Macrophages isolated from the peritoneal cavity of C57BL/6-Tg and SCID-Tg mice expressed CD89 (Fig. 2 B). Macrophage CD89 expression was stable in these mice from 6–24 wk of age (data not shown). CD89 was heavily glycosylated, yielding 32- and 36-kD bands after N-glycosydase F treatment, as described on human myeloid cells 21 (Fig. 2 C). The level of transgene expression was ∼10-fold higher on macrophages from line 83 than from the other two lines (Fig. 2 D). We next analyzed the ability of CD89 to bind purified human and mouse IgA, using macrophages from SCID-Tg mice (line 83). Binding of dimeric human and mouse IgA was specifically inhibited by the anti–CD89 mAb My43 (84 and 80%, respectively; Fig. 2 E). No specific binding of monomeric IgA was observed. These experiments indicated that CD89 could bind dimeric but not monomeric mouse IgA. This was confirmed by increased endogenous IgA binding to blood monocytes of C57BL/6-Tg mice, as shown with anti-IgA antibodies (data not shown).
Figure 2.

Generation and characterization of three lines of Tg mice expressing human CD89. (A) Structure of the transgene consisting of a 4.8-kb insert carrying the cDNA encoding the human CD89 (huCD89) and pB202/D736 construct containing the 1.7-kb mouse CD11b promoter. (B) Expression of CD89 on peritoneal macrophages of Tg mice, detected by two-color FACS® staining with anti-CD89 (A59-PE) and anti-CD11b (mac-1–FITC) mAbs. (C) Biochemical nature of CD89 on macrophages of Tg mice. Equal numbers of cells (107) from CD89-Tg (lines 83 and 96) and littermate controls (Co) were iodinated, solubilized and immunoprecipitated by anti-CD89 A77 F(ab′)2 coupled to beads. Half the resulting material was treated with N-glycosidase F as described elsewhere (reference 21) and separated on SDS-10% PAGE. (D) Comparative analysis of CD89 expression on peritoneal macrophages from the three Tg lines and line 83 SCID mice using overlay histograms of anti-CD89 mAb (A59-PE) staining. (E) IgA binding capacity of peritoneal macrophages from SCID-Tg mice (line 83, black bars). Biotinylated human dimeric IgA1 (dIgA1), mouse dIgA (IgA-C5) (0.2 mg/ml), and monomeric sIgA (mIgA; MOPC315) were used. Hatched bars represent inhibition of IgA binding by the anti-CD89 mAb My43 (percentage of inhibition is indicated at the top of the columns), calculated as described elsewhere (reference 26), after correction for background obtained with non-Tg–SCID littermate controls (SCID, white bars).
Spontaneous Mesangial IgA Deposition and Hematuria in Mice Expressing CD89.
Kidneys from C57BL/6-Tg mice showed massive mesangial IgA deposition at 24 wk of age (Fig. 3). This was more intense in line 83 than in the two other lines, and correlated with the level of transgene expression on peritoneal macrophages (Fig. 2 and Fig. 3). Marked infiltration by mac-1+ mononuclear cells was observed in the intra- and periglomerular regions surrounding the Bowman space, indicating the presence of macrophages in and around damaged glomeruli (Fig. 3, bottom). Interstitial macrophage infiltration was also present in these Tg mice (Fig. 3, and data not shown). Mac-1+ cells positively stained with F4/80 mAb, a macrophage-specific marker, ruling out the presence of neutrophils (not shown). Time sequence experiments (in line 83) revealed that IgA deposits and infiltration by mac-1+ cells were visible in CD89-Tg mice older than 12 wk (Fig. 4 A). CD89-Tg mice had significant spontaneous microscopic hematuria after 18 wk (Fig. 4 B) and significant albuminuria at 24 wk (Fig. 4 C). Histological analyses showed focal and segmental lesions with glomerular hypertrophy, mesangial matrix expansion without cellular proliferation, congested glomerular capillary, and moderate interstitial mononuclear cell infiltration focally condensed around glomeruli in 24-wk-old Tg mice (Fig. 4 D). BUN levels were slightly, but not significantly, increased in 24-wk-old Tg mice (Fig. 4 E). However, these mice had no increased mortality over a 1 yr period. No CD89 staining was detected on kidney sections of CD89-Tg mice using biotinylated anti-CD89 mAbs plus biotin/avidin-HRP as a developing reagent (data not shown).
Figure 3.
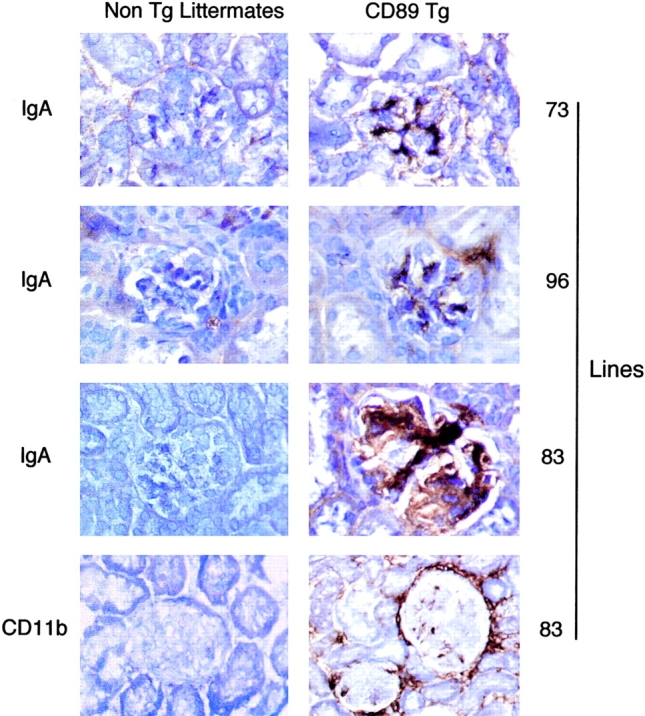
Mesangial IgA deposits and macrophage infiltration in the kidneys of CD89-Tg mice (C57BL/6) at 24 wk of age. IgA and CD11b immunohistochemical staining was performed with rat anti–mouse IgA mAb and mac-1 mAb, respectively, as indicated on the left. The CD89-Tg lines (73, 96, and 83) are indicated on the right. Tg and their non-Tg littermate controls are indicated at the top.
Figure 4.

Time sequence of pathologic features in CD89 Tg mice of line 83. (A) IgA and CD11b stainings (indicated on the left) of three Tg mice at different ages. Concomitant appearance of mesangial IgA deposits and periglomerular CD11b+ cells was seen in kidneys after 12 wk of age. (B) Significant hematuria appeared in Tg mice after 18 wk of age (groups of 25 Tg mice, •, and 23 non-Tg littermates, ○). (C) Mild albuminuria detected in urine samples of 24-wk-old Tg mice. (D) Histologic kidney sections after periodic acid-Schiff (PAS) and hematoxylin and eosin (HE) staining from representative Tg mice and a non-Tg littermates (Lt) at different ages. Focal and segmental mesangial matrix expansion (white arrowhead) associated with congested glomerular capillary (black arrowhead) and with glomerular hypertrophy was seen in 24-wk-old Tg mice but not in their littermate controls. (E) BUN levels in Tg and non-Tg mice. *P < 0.001, and **P < 0.01.
IgA-dependent Release of Soluble CD89 in Tg Mice.
To determine the molecular mechanism involved in the pathogenesis of IgAN in CD89-Tg mice, we first compared CD89 expression on blood cells of C57BL/6-Tg and SCID-Tg mice of line 83. CD89 expression was decreased ∼10-fold on blood monocytes of CD89-Tg mice in the C57BL/6 background compared with the same cell population of SCID-Tg mice of line 83 (Fig. 5A and Fig. B). Similar results were obtained with two other lines (73 and 96). Dim CD89 expression was detected on neutrophils, except for line 96, which has a subpopulation of neutrophils expressing CD89, and no expression was observed on lymphocytes of any mice of the three lines (data not shown). Therefore, we analyzed CD89 expression as a function of age. Blood cells from mice of line 83 were analyzed from 6 to 24 wk of age. As shown in Fig. 5 C, CD89 expression fell after 12 wk of age as serum IgA levels increased. In contrast, stable CD89 expression level was observed on SCID-Tg mice (line 83) in which serum IgA was undetectable. As a control, CD11b antigen expression on monocytes was similar in C57BL/6- and SCID-Tg mice (Fig. 5 D). To explain the decrease in CD89 expression on monocytes, we sought for soluble CD89 in serum of Tg mice by using a sandwich ELISA with either human IgA and an anti-CD89 mAb or two anti-CD89 mAbs, as capture and developing reagents. Significant reactivity of human IgA/anti-CD89 mAb (clone A3) and of two mAbs (A3/A62) towards soluble CD89 was obtained in serum from 24-wk-old Tg mice (line 83) compared with littermate controls (Fig. 5 E). Similar results were obtained with serum from Tg mice of lines 73 and 96 (data not shown). Soluble CD89 was only detected after 12 wk of age. No soluble CD89 was detected in serum of SCID-Tg mice (line 83; data not shown). To determine whether IgA could indeed induce the release of soluble CD89, we used rat mast-cell line (RBL-2H3) transfectants expressing either the full-length CD89 (WT), or a receptor with a point mutation (R to L at position 209) that is unable to associate with the γ chain 26. Culture of transfectants for 36 h with IgA induced the release of soluble CD89 into supernatants of both WT and R209L transfectants; this release was stronger with dimeric IgA1 than monomeric IgA1 (Fig. 5 F). It was specific for IgA, as it was not observed after culture with IgG (Fig. 5 F). As R209L transfectants express only γ-less CD89, and as WT transfectants express both forms of CD89 26, our results indicate that soluble CD89 release from cells is not dependent on the association of CD89 with the γ chain.
Figure 5.

Decreased expression of CD89 on blood monocytes of CD89 Tg mice (line 83) is associated with release of soluble CD89. (A) Decreased CD89 expression on blood monocytes from 6-wk-old CD89 C57BL/6-Tg mice compared with SCID-Tg mice. Double staining was performed with anti-CD89 (A59) and anti-CD11b (mac-1). (B) Comparative analysis of CD89 expression on blood monocytes determined by overlay histograms. (C) Analysis of CD89 expression and IgA levels as a function of the age of C57BL/6-Tg (•) and SCID-Tg mice (○). Every 3–6 wk, blood cells were stained as stated above and IgA was measured in serum (triangles). Intensity of expression was evaluated by measuring fluorescence intensity (FI) as described elsewhere (reference 21). (D) No differences in CD11b expression were observed between C57BL/6-Tg (▪) and SCID-Tg mice (□). (E) Detection of soluble CD89 in serum of Tg mice. Serum from 24-wk-old CD89 Tg mice and their littermate controls (Co) was tested with a sandwich ELISA using human IgA and/or anti-CD89 mAbs as indicated. Results are expressed in arbitrary units as described elsewhere (reference 25). (F) Release of soluble CD89 was induced by IgA and was independent of γ chain association. RBL transfectants expressing either WT or mutant (R209L) CD89 were cultured with 0.25 mg/ml biotinylated IgG or 0.25 mg/ml biotinylated monomeric or dimeric human IgA1κ. After 36 h, cells were spun and supernatants were collected. Soluble CD89 was measured with an ELISA using A3 F(ab′)2-coated plates and streptavidin-HRP as developing reagent. Results are expressed in OD.
Transfer of IgAN Disease into RAG-2−/− Mice by Serum Soluble CD89 from CD89 Tg Mice.
To test the pathogenicity of soluble CD89 complexed to mouse IgA, we injected serum from C57BL/6-Tg mice or littermate controls (24-wk-old mice from line 83) into 8-wk-old RAG-2−/− recipients. A single injection of serum (300 μl) of Tg mice induced heavy, macroscopic hematuria for 24 h (Fig. 6 A) accompanied by mesangial IgA deposits and mac-1+ cell infiltration in intra- and periglomerular regions (Fig. 6 B). No IgA deposition or cellular infiltration in glomerular regions of RAG-2−/− mice receiving serum from littermate controls was seen (Fig. 6 B). To demonstrate that soluble CD89 mediates the transfer of IgAN disease, we preadsorbed serum from Tg mice with either an irrelevant control mAb (IgG1) or four anti-CD89 mAbs before injection into RAG-2−/− recipients. A major decrease in hematuria, IgA deposits, and mac-1+ cell infiltration was observed when serum was preadsorbed with anti-CD89 mAbs, indicating the presence of nephritogenic soluble CD89 complexes in the serum of Tg mice (Fig. 6 C).
Figure 6.

Features of IgAN induced by cotransfer of serum from CD89 Tg donors into RAG-2−/− recipients. 12 RAG-2−/− males (6 wk old) were injected intravenously with 300 μl of pooled serum from 6 Tg donors or 6 littermate controls (line 83). (A) Hematuria in RAG-2−/− mice receiving serum from Tg (•, n = 6) or littermate mice (○, n = 6). (B) RAG-2−/− kidneys were stained with rat anti–mouse IgA mAb (IgA) or with mac-1 mAb (CD11b), as indicated. (C) Decreased hematuria in RAG-2−/− mice receiving serum from C57BL/6-Tg donors after adsorption (four times) with anti-CD89 mAb (A3, A59, A62, A77) coupled to Sepharose beads (three in each group) as indicated. As control, Tg serum was adsorbed with an irrelevant IgG1 mAb coupled to Sepharose beads.
Induction of IgAN Features in SCID–CD89 Tg Mice by Injection of Patient IgA.
In previous studies, we found that increased interaction between patient IgA and CD89 results in CD89 downregulation on the monocyte surface 18. To determine whether patient IgA can downregulate CD89 expression in vivo and induce mesangial lesions and clinical manifestations, we injected purified patient IgA and normal IgA (100 μg) into 6-wk-old SCID-Tg mice (line 83) and monitored CD89 expression on blood monocytes. As shown in Fig. 7 A, injection of patient IgA induced a decrease of CD89 expression, with a maximal effect 2 h after injection. Patient IgA had a significantly stronger negative effect on CD89 expression than did normal serum IgA (Fig. 7 B). Injection of patient IgA was followed by macrophage infiltration in periglomerular areas 48 h later (Fig. 7 E), whereas no such cells were found in the kidney of non-Tg–SCID mice injected with patient IgA (Fig. 7 D). Weak mesangial human IgA deposition was also observed (not shown). Interestingly, patient IgA induced heavy and sustained macroscopic hematuria for a 72-h period, whereas normal IgA only induced transient microscopic hematuria that disappeared after 12 h (Fig. 7 C). Only transient hematuria was obtained after injection of patient IgA into non-Tg–SCID mice (not shown). No hematuria was seen with normal IgA in non-Tg–SCID mice (not shown).
Figure 7.

Induction of gross hematuria and macrophage infiltration by injection of patient IgA into 6-wk-old SCID-Tg mice (line 83). IgA, purified from a normal donor and from a patient with IgAN, was injected intravenously into SCID-Tg mice (n = 12). Blood was collected at the times indicated and double stained for CD89 (A59-PE) and CD11b. (A) Overlay histogram profiles of CD89 staining at the monocyte gate after injection of patient IgA. (B) Comparative analyses of percentage variation in CD89 fluorescence intensity after injection of patient and normal IgA, estimated as described elsewhere (reference 21). Open and filled symbols represent injections of normal and patient IgA, respectively; *P < 0.03. (C) Erythrocyte counts at the times indicated. (D and E) Mac-1 and anti-CD11b kidney staining 48 h after injection of patient IgA into a SCID mouse and a SCID-Tg mouse, respectively.
Discussion
Our data point to the existence of soluble CD89 in serum of IgAN patients being most frequently found in this disease in a complexed form with their ligand, the IgA. Soluble FcR is a common feature of the FcR family 35. This is one of the first identifications of a self component in circulating macromolecular IgA complexes 2, and would explain the marked decrease in CD89 expression on patients' monocytes 18. Monocyte soluble CD89 was characterized as 50–70-kD glycoproteins with a protein core of 24 kD, which differs from the 55–75-kD CD89 with a 32-kD CD89 backbone isolated from cell lysates. This is consistent with the molecular weight of the previously reported recombinant soluble CD89 (23,700), which corresponds to receptor extracellular domains 27. Thus, the 24-kD backbone of soluble CD89 is probably a product of proteolytic shedding. The loss of 60 amino acids (19 of transmembrane, and 41 of intracytoplasmic domains) would only decrease the M r by 8 kD, and might not affect SDS-PAGE mobility due to the heavy glycosylation of the extracellular domain. Shedding of soluble CD89 is also supported by experiments in which IgA, but not IgG, induces its release from mast cell transfectants expressing either full-length CD89 (that may or may not be associated with the γ chain) or a mutated version that cannot associate with the γ chain 26. This led us to the conclusion that soluble CD89 release is independent of the γ chain association. The additional 33-kD band observed in supernatants could be explained by partial digestion by N-glycosidase F, as described for the recombinant soluble CD89 produced by COS cells, which gives rise to a major broad 25–30-kD band and a smaller 23-kD protein core 27. However, we cannot rule out from our experiments that the 33-kD band in the supernatant corresponds to the product of an additional soluble form of CD89 lacking only the transmembrane region that would be secreted by monocytes. Transcripts encoding for a soluble CD89 have been described in neutrophils and eosinophils 36.
The results in CD89-Tg mice demonstrate that soluble CD89 plays an essential role in the pathogenesis of IgAN. Expression of CD89 on mouse monocyte/macrophages was detrimental, mediating the development of IgAN through the release of soluble CD89 after interaction with mouse IgA. The mechanism underlying spontaneous IgAN disease onset in CD89-Tg mice is probably linked to the nature of mouse serum IgA, which is predominantly polymeric (70–80% of total serum IgA), contrary to the situation in humans 37, and also to the possible absence of a CD89 homologue in mice 33 34. We deduce that the interaction between mouse polymeric IgA and the CD89 transgene product results in a release of pathogenic soluble CD89–IgA complexes. This is supported by two observations: polymeric IgA induces a stronger release of soluble CD89 than monomeric IgA in vitro, and the absence of CD89 shedding from blood monocytes in SCID-Tg mice in association with undetectable serum IgA levels is due to their inherent immunodeficient status (Fig. 5). It is interesting to note that increased levels of polymeric IgA observed in the ddY mice induce IgA deposits without hematuria or proteinuria 14. This reinforces the role of CD89 in IgAN disease onset. Although CD89 was not directly detectable in the mesangium, possibly owing to the poor sensitivity of anti-CD89 mAbs in immunohistochemical staining, our CD89 Tg mice are a valid model of spontaneous IgAN, as they develop physical manifestations including microscopic hematuria and mild albuminuria. Indeed, clinical IgAN onset in humans is usually defined by the presence of microscopic hematuria associated with mild proteinuria 1 17. Regarding renal histology, the characteristic alterations in CD89 Tg mice were progressive focal and segmental mesangial matrix expansion with congested capillary and glomerular hypertrophy. However, BUN levels were not significantly altered. The absence of severe renal failure in CD89-Tg mice suggests that progression of IgAN towards renal failure requires other factors, such as inappropriate protein expression by mesangial cells (e.g., megsin 38) and/or T cell infiltration 5.
The pathogenic role of soluble CD89–IgA complexes was demonstrated by transfer of the disease to RAG-2−/− mice injected with serum from Tg mice, but not with serum from control mice, or serum from Tg mice adsorbed with anti-CD89 mAbs (see Fig. 6). As IgA induces soluble CD89 release in vitro (Fig. 5 F), and to determine the role of circulating soluble CD89 complexed with IgA, we performed in vivo experiments in SCID-Tg mice using both normal and patient IgA. It is important to note that CD89 downregulation was stronger with patient IgA than with normal IgA, suggesting that the previously reported IgA galactosylation deficiency and increased proportion of polymeric IgA in IgAN patients play a role in CD89 shedding 2. This is consistent with our previous data showing that patient IgA binds more than normal IgA to monocytes 18. The gross hematuria observed after injection of patient IgA in SCID-Tg but not SCID–non-Tg mice further supports the pathogenic role of soluble CD89–IgA complexes and provides a possible explanation for the linkage between CD89 alterations and renal lesions in patients with IgAN 18. The transient hematuria observed after injection of normal human IgA into SCID-Tg mice might be explained by binding of monomeric IgA to CD89, which can induce soluble CD89 release (Fig. 5 F). Indeed, transient monomeric IgA binding to CD89 has been demonstrated previously 20 39. Together, our data point to a pathogenic role of patient IgA in the soluble CD89 release and IgA complex formation in IgAN.
This study demonstrates that soluble CD89–IgA complex deposition induces an influx of mononuclear cells, mainly macrophages (mac-1+F4-80+), into the glomerular and interstitial regions. This is consistent with observations in IgAN patient kidney specimens 3 4. Transfer experiments demonstrated that macrophage influx was also dependent on soluble CD89 complexed with IgA, as serum from Tg mice adsorbed with anti-CD89 mAbs were not able to transfer the disease. Mesangial deposition of soluble CD89–IgA, which might depend on expression of other IgA receptors by mesangial cells 40 41, could lead to cell activation and release of chemokines or other factors resulting in macrophage recruitment 42. Indeed, enhanced kidney expression of monocyte chemotactic peptide 1 has been observed in patients with IgAN 43. Moreover, macrophage activation could trigger the release of proinflammatory cytokines (e.g., IL-6 and TNF-α) and/or chemokines that might induce mesangial matrix expansion, hematuria, and proteinuria 44 45 46 47. Taken together, our data point to a central role of soluble CD89–IgA complexes in the pathogenesis of IgAN and provide evidence that the nature (monomeric/polymeric and/or glycosylation status) of the IgA controls the formation of these complexes.
Acknowledgments
We thank J.F. Bach, P. Ronco, C. Carnaud, M. Benhamou, and K. Lassoued for suggestions or critical review of the manuscript; P. Jungers, A.T. Nguyen, the staff of the Nephrology Department, C. Silvain, and V. Montenegro for providing patient sera; I. Cisse for mouse care; M. Leborgne, O. Babin, and N. Brousse for assistance in immunohistochemistry; A. Mason and T. Nguyen-Khoa for help with techniques in renal function analyses; M. Netter for preparing prints; and D. Young for critical reading.
R.C. Monteiro was supported by ARC grant 5349, and Assistance Publique-Hôpitaux de Paris grant AOA94047. M. Arcos-Fajardo was the recipient of FAPESP (Brazil) grant 98/06422-2.
Note added in proof. CD89 Tg mice backcrossed five times into BALB/c background also developed the pathologic features of IgA nephropathy.
Footnotes
Pierre Launay and Béatrice Grossetête contributed equally to this work.
Abbreviations used in this paper: ALC, alcoholic liver cirrhosis; AP, alkaline phosphatase; BUN, blood urea nitrogen; FcR, receptor for Ig Fc fragment; HRP, horseradish peroxidase; IgAN, IgA nephropathy; IC, immune complex; MCD, minimal change disease; MSA, mouse serum albumin; NOD, nonobese diabetic; PEG, polyethylene glycol; RAG, recombination activating gene; Tg, transgenic; WT, wild-type.
References
- Berger J. IgA glomerular deposits in renal disease. Transplant. Proc. 1969;1:939–944. [PubMed] [Google Scholar]
- D'Amico G. Pathogenesis of immunoglobulin A nephropathy. Curr. Opin. Nephrol. Hypertens. 1998;7:247–250. doi: 10.1097/00041552-199805000-00001. [DOI] [PubMed] [Google Scholar]
- Arima S., Nakayama M., Naito M., Sato T., Takahashi K. Significance of mononuclear phagocytes in IgA nephropathy. Kidney Int. 1991;39:684–692. doi: 10.1038/ki.1991.82. [DOI] [PubMed] [Google Scholar]
- Nagata M., Akioka Y., Tsunoda Y., Komatsu Y., Kawaguchi H., Yamaguchi Y., Ito K. Macrophages in childhood IgA nephropathy. Kidney Int. 1995;48:527–535. doi: 10.1038/ki.1995.323. [DOI] [PubMed] [Google Scholar]
- Falk M.C., Ng G., Zhang G.Y., Fanning G.C., Roy L.P., Bannister K.M., Thomas A.C., Clarkson A.R., Woodroffe A.J., Knight J.F. Infiltration of the kidney by αβ and γδ T cellseffect on progression in IgA nephropathy. Kidney Int. 1995;47:177–185. doi: 10.1038/ki.1995.21. [DOI] [PubMed] [Google Scholar]
- Allen A.C., Harper S.J., Feehally J. Galactosylation of N- and O-linked carbohydrate moieties of IgA1 and IgG in IgA nephropathy. Clin. Exp. Immunol. 1995;100:470–474. doi: 10.1111/j.1365-2249.1995.tb03724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomana M., Novak J., Julian B.A., Matousovic K., Konecny K., Mestecky J. Circulating immune complexes in IgA nephropathy consist of IgA1 with galactose-deficient hinge region and antiglycan antibodies. J. Clin. Invest. 1999;104:73–81. doi: 10.1172/JCI5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trascasa M.L., Egido J., Sancho L., Hernando L. IgA glomerulonephritis (Berger's disease)evidence of high serum levels of polymeric IgA. Clin. Exp. Immunol. 1980;42:247–254. [PMC free article] [PubMed] [Google Scholar]
- Monteiro R.C., Halbwachs-Mecarelli L., Roque-Barreira M.C., Noel L.H., Berger J., Lesavre P. Charge and size of mesangial IgA in IgA nephropathy. Kidney Int. 1985;28:666–671. doi: 10.1038/ki.1985.181. [DOI] [PubMed] [Google Scholar]
- Czerkinsky C., Koopman W.J., Jackson S., Collins J.E., Crago S.S., Schrohenloher R.E., Julian B.A., Galla J.H., Mestecky J. Circulating immune complexes and immunoglobulin A rheumatoid factor in patients with mesangial immunoglobulin A nephropathies. J. Clin. Invest. 1986;77:1931–1938. doi: 10.1172/JCI112522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmacht C., Kliem V., Burg M., Nashan B., Schlitt H.J., Brunkhorst R., Koch K.M., Floege J. Recurrent immunoglobulin A nephropathy after renal transplantationa significant contributor to graft loss. Transplantation. 1997;64:1493–1496. doi: 10.1097/00007890-199711270-00024. [DOI] [PubMed] [Google Scholar]
- Cederholm B., Wieslander J., Bygren P., Heinegard D. Circulating complexes containing IgA and fibronectin in patients with primary IgA nephropathy. Proc. Natl. Acad. Sci. USA. 1988;85:4865–4868. doi: 10.1073/pnas.85.13.4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifai A., Small P.A., Jr., Teague P.O., Ayoub E.M. Experimental IgA nephropathy. J. Exp. Med. 1979;150:1161–1173. doi: 10.1084/jem.150.5.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muso E., Yoshida H., Takeuchi E., Yashiro M., Matsushima H., Oyama A., Suyama K., Kawamura T., Kamata T. Enhanced production of glomerular extracellular matrix in a new mouse strain of high serum IgA ddY mice. Kidney Int. 1996;50:1946–1957. doi: 10.1038/ki.1996.517. [DOI] [PubMed] [Google Scholar]
- Zheng F., Kundu G.C., Zhang Z., Ward J., DeMayo F., Mukherjee A.B. Uteroglobin is essential in preventing immunoglobulin A nephropathy in mice. Nat. Med. 1999;5:1018–1025. doi: 10.1038/12458. [DOI] [PubMed] [Google Scholar]
- Baldree L.A., Wyatt R.J., Julian B.A., Falk R.J., Jennette J.C. Immunoglobulin A-fibronectin aggregate levels in children and adults with immunoglobulin A nephropathy. Am. J. Kidney Dis. 1993;22:1–4. doi: 10.1016/s0272-6386(12)70159-2. [DOI] [PubMed] [Google Scholar]
- Galla J.H. IgA nephropathy. Kidney Int. 1995;47:377–387. doi: 10.1038/ki.1995.50. [DOI] [PubMed] [Google Scholar]
- Grossetete B., Launay P., Lehuen A., Jungers P., Bach J.F., Monteiro R.C. Down-regulation of Fcα receptors on blood cells of IgA nephropathy patientsevidence for a negative regulatory role of serum IgA. Kidney Int. 1998;53:1321–1335. doi: 10.1046/j.1523-1755.1998.00885.x. [DOI] [PubMed] [Google Scholar]
- Montenegro V., Monteiro R.C. Elevation of serum IgA in spondyloarthropathies and IgA nephropathy and its pathogenic role. Curr. Opin. Rheumatol. 1999;11:265–272. doi: 10.1097/00002281-199907000-00007. [DOI] [PubMed] [Google Scholar]
- Chevailler A., Monteiro R.C., Kubagawa H., Cooper M.D. Immunofluorescence analysis of IgA binding by human mononuclear cells in blood and lymphoid tissue. J. Immunol. 1989;142:2244–2249. [PubMed] [Google Scholar]
- Monteiro R.C., Kubagawa H., Cooper M.D. Cellular distribution, regulation, and biochemical nature of an Fcα receptor in humans. J. Exp. Med. 1990;171:597–613. doi: 10.1084/jem.171.3.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maliszewski C.R., March C.J., Schoenborn M.A., Gimpel S., Shen L. Expression cloning of a human Fc receptor for IgA. J. Exp. Med. 1990;172:1665–1672. doi: 10.1084/jem.172.6.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patry C., Sibille Y., Lehuen A., Monteiro R.C. Identification of Fcα receptor (CD89) isoforms generated by alternative splicing that are differentially expressed between blood monocytes and alveolar macrophages. J. Immunol. 1996;156:4442–4448. [PubMed] [Google Scholar]
- Dziennis S., Van Etten R.A., Pahl H.L., Morris D.L., Rothstein T.L., Blosch C.M., Perlmutter R.M., Tenen D.G. The CD11b promoter directs high-level expression of reporter genes in macrophages in transgenic mice. Blood. 1995;85:319–329. [PubMed] [Google Scholar]
- Lehuen A., Lantz O., Beaudoin L., Laloux V., Carnaud C., Bendelac A., Bach J.F., Monteiro R.C. Overexpression of natural killer T cells protects Vα14-Jα281 transgenic nonobese diabetic mice against diabetes. J. Exp. Med. 1998;188:1831–1839. doi: 10.1084/jem.188.10.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Launay P., Patry C., Lehuen A., Pasquier B., Blank U., Monteiro R.C. Alternative endocytic pathway for immunoglobulin A Fc receptors (CD89) depends on the lack of FcRγ association and protects against degradation of bound ligand. J. Biol. Chem. 1999;274:7216–7225. doi: 10.1074/jbc.274.11.7216. [DOI] [PubMed] [Google Scholar]
- Maliszewski C.R., VandenBos T., Shen L., Schoenborn M.A., Kubagawa H., Beckmann M.P., Monteiro R.C. Recombinant soluble IgA Fc receptorgeneration, biochemical characterization, and functional analysis of the recombinant protein. J. Leukoc. Biol. 1993;53:223–232. doi: 10.1002/jlb.53.3.223. [DOI] [PubMed] [Google Scholar]
- Monteiro R.C., Cooper M.D., Kubagawa H. Molecular heterogeneity of Fcα receptors detected by receptor-specific monoclonal antibodies. J. Immunol. 1992;148:1764–1770. [PubMed] [Google Scholar]
- Silvain C., Patry C., Launay P., Lehuen A., Monteiro R.C. Altered expression of monocyte IgA Fc receptors is associated with defective endocytosis in patients with alcoholic cirrhosis. Potential role for IFN-γ. J. Immunol. 1995;155:1606–1618. [PubMed] [Google Scholar]
- Shen L., Lasser R., Fanger M.W. My 43, a monoclonal antibody that reacts with human myeloid cells inhibits monocyte IgA binding and triggers function. J. Immunol. 1989;143:4117–4122. [PubMed] [Google Scholar]
- Phalipon A., Kaufmann M., Michetti P., Cavaillon J.M., Huerre M., Sansonetti P., Kraehenbuhl J.P. Monoclonal immunoglobulin A antibody directed against serotype-specific epitope of Shigella flexneri lipopolysaccharide protects against murine experimental shigellosis. J. Exp. Med. 1995;182:769–778. doi: 10.1084/jem.182.3.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli U.K. Cleavage of structural proteins during the assembly of the head of the bacteriophage T4. Nature. 1970;277:680–684. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Kubagawa H., Burrows P., Cooper M.D. A novel pair of immunoglobulin-like receptors expressed by B cells and myeloid cells. Proc. Natl. Acad. Sci. USA. 1997;94:5261–5266. doi: 10.1073/pnas.94.10.5261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayami K., Fukuta D., Nishikawa Y., Yamashita Y., Inui M., Ohyama Y., Hikida M., Ohmori H., Takai T. Molecular cloning of a novel murine cell-surface glycoprotein homologous to killer cell inhibitory receptors. J. Biol. Chem. 1997;272:7320–7327. doi: 10.1074/jbc.272.11.7320. [DOI] [PubMed] [Google Scholar]
- Galon J., Paulet P., Galinha A., Lores P., Bonnerot C., Jami J., Fridman W.H., Sautes C. Soluble Fcγ receptorsinteraction with ligands and biological consequences. Int. Rev. Immunol. 1997;16:87–111. doi: 10.3109/08830189709045704. [DOI] [PubMed] [Google Scholar]
- van Dijk T.B., Bracke M., Caldenhoven E., Raaijmakers J.A., Lammers J.W., Koenderman L., de Groot R.P. Cloning and characterization of FcαRb, a novel Fcα receptor (CD89) isoform expressed in eosinophils and neutrophils. Blood. 1996;88:4229–4238. [PubMed] [Google Scholar]
- Kerr M.A. The structure and function of human IgA. Biochem. J. 1990;271:285–296. doi: 10.1042/bj2710285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata T., Nangaku M., Suzuki D., Inagi R., Uragami K., Sakai H., Okubo K., Kurokawa K. A mesangium-predominant gene, megsin, is a new serpin upregulated in IgA nephropathy. J. Clin. Invest. 1998;102:828–836. doi: 10.1172/JCI2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wines B.D., Hulet M.D., Jamieson G.P., Trist H.M., Spratt J.M., Hogarth P.M. Identification of residues in the first domain of human Fcα receptor essential for interaction with IgA. J. Immunol. 1999;162:2146–2153. [PubMed] [Google Scholar]
- Diven S.C., Caflish C.R., Hammond D.K., Weigel P.H., Oka J.A., Goldblum R.M. IgA induced activation of human mesangial cellsindependent of FcαRI (CD89) Kidney Int. 1998;54:837–847. doi: 10.1046/j.1523-1755.1998.00054.x. [DOI] [PubMed] [Google Scholar]
- Gomez-Guerrero C., Duque N., Egido J. Mesangial cells possess an asiologlycoprotein receptor with affinity for human immunoglobulin A. J. Am. Soc. Nephrol. 1998;9:568–576. doi: 10.1681/ASN.V94568. [DOI] [PubMed] [Google Scholar]
- Zlotnik A., Morales J., Hedrick J.A. Recent advances in chemokines and chemokine receptors. Crit. Rev. Immunol. 1999;19:1–47. [PubMed] [Google Scholar]
- Grandaliano G., Gesualdo L., Ranieri E., Monno R., Montinaro V., Marra F., Schena F.P. Monocyte chemotactic peptide-1 expression in acute and chronic human nephritidesa pathogenetic role in interstitial monocytes recruitment. J. Am. Soc. Nephrol. 1996;7:906–913. doi: 10.1681/ASN.V76906. [DOI] [PubMed] [Google Scholar]
- Horii Y., Muraguchi A., Iwano M., Matsuda T., Hirayama T., Yamada H., Fujii Y., Dohi K., Ishikawa H., Ohmoto Y. Involvement of IL-6 in mesangial proliferative glomerulonephritis. J. Immunol. 1989;143:3949–3955. [PubMed] [Google Scholar]
- Liu Z.H., Striker G.E., Steller-Stevenson M., Fukushima P., Patel A., Striker L.J. TNF-α and IL-1α induce mannose receptors and apoptosis in glomerular mesangial but not endothelial cells. Am. J. Physiol. 1996;270:595–601. doi: 10.1152/ajpcell.1996.270.6.C1595. [DOI] [PubMed] [Google Scholar]
- Saitoh A., Suzuki Y., Takeda M., Kubota K., Itoh K., Tomino Y. Urinary levels of monocyte chemoattractant protein (MCP)-1 and disease activity in patients with IgA nephropathy. J. Clin. Lab. Anal. 1998;12:1–5. doi: 10.1002/(SICI)1098-2825(1998)12:1<1::AID-JCLA1>3.0.CO;2-F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama H., Wada T., Furuichi K., Segawa C., Shimizu M., Kobayashi K., Su S., Mukaida N., Matsushima K. Urinary levels of chemokines (MCAF/MCP-1, IL-8) reflect distinct disease activities and phases of human IgA nephropathy. J. Leukoc. Biol. 1998;63:493–499. doi: 10.1002/jlb.63.4.493. [DOI] [PubMed] [Google Scholar]