

Abstract
The mechanisms that determine whether receptor stimulation leads to lymphocyte tolerance versus activation remain poorly understood. We have used rat insulin promoter (RIP)-gp/P14 double-transgenic mice expressing the lymphocytic choriomeningitis virus (LCMV) glycoprotein (gp) on pancreatic β-islet cells together with T cells expressing an LCMV-gp–specific T cell receptor to assess the requirements for the induction of autoimmunity. Our studies have shown that administration of the gp peptide gp33 leads to the activation of P14-transgenic T cells, as measured by the upregulation of activation markers and the induction of effector cytotoxic activity. This treatment also leads to expansion and deletion of P14 T cells. Despite the induction of cytotoxic T lymphocyte activity, peptide administration is not sufficient to induce diabetes. However, the administration of gp peptide together with an activating anti-CD40 antibody rapidly induces diabetes. These findings suggest that the induction of tolerance versus autoimmunity is determined by resting versus activated antigen-presenting cells.
Keywords: activation, CTL, diabetes, CD40, costimulation
Introduction
Specific T lymphocyte interactions that lead to the induction of peripheral tolerance versus autoimmunity remain poorly understood. Models have proposed that costimulation and the presence of second signals are essential for appropriate T cell activation and the induction of immunity 1 2. These models suggest that in the absence of costimulatory molecular interactions, such as that between B7-1 or B7-2 on the APC and CD28 receptor on T cells, TCR stimulation alone leads to tolerance induction. Alternate models suggest that different subpopulations of APCs define the outcome of T cell interaction with ligand, where one population induces tolerance and a second induces T cell activation 3 4 5.
Studies have demonstrated, however, that events associated with mature T cell tolerance induction, particularly the induction of T cell deletion, share common features with T cell activation: activation markers are upregulated, proliferation occurs, and effector functions are induced 6 7 8. However, the ultimate outcome of tolerance versus immunity or autoimmunity has a dramatically different consequence in vivo.
We have used a transgenic model based on lymphocytic choriomeningitis virus (LCMV) to examine events that lead to tolerance versus activation. Rat insulin promoter (RIP)-glycoprotein (gp)–transgenic mice express LCMV-gp on pancreatic β-islet cells under the control of RIP, and the P14-transgenic mice express a LCMV-gp–specific TCR 9. In the double-transgenic model (RIP-gp/P14), LCMV-gp–specific CD8+ T cells are not tolerized but remain immunologically unaware of the presence of their antigen and are fully capable of responding to antigen under the appropriate conditions. Using P14 TCR single-transgenic mice, previous studies have shown that peptide administration leads to tolerance 10. In addition, previous studies using RIP-gp single-transgenic mice have shown that LCMV infection induced diabetes 9. We now combine the models to examine the requirements of how gp33-induced tolerance can be switched to autoimmunity. Here, we report that specific TCR–peptide–MHC interactions between T cells and APCs that normally lead to tolerance can be biased toward autoimmunity when the APCs are activated using an agonistic anti-CD40 antibody in vivo. Therefore, resting versus activated APCs may be the key to determining whether peripheral tolerance or immunity is induced.
Materials and Methods
Mice and Glucose Measurements.
The P14, RIP-gp, and RIP-gp/P14 mouse lines have been previously described 9 11. Blood glucose measurements were determined with the Chemstrip bG test kit (Boehringer Mannheim) and quantitated with Accu-Chek III (Boehringer Mannheim). Mice with blood glucose levels >14 mM/liter were considered diabetic.
Peptides.
Peptides were generated at the Amgen Institute by a solid-phase method using the Fmoc/tBu-based protocol on an ABI-431 instrument. The crude product was purified by HPLC and exceeded 95% purity. Peptide gp33 (KAVYNFATM) defines the major CTL epitope on the LCMV-gp and is H-2Db restricted. AV (SGPSNTPPEI) is an H-2Db–restricted adenovirus-derived peptide.
Peptide and Antibody Treatment.
Mice received either one or three intravenous administrations of 5 μg of gp33 in HBSS. When administered three times, peptide was given every third day (beginning on day 0). The mice received an additional intravenous administration of 100 μg of purified rat anti–mouse CD40-activating antibody FGK45 12 or 100 μg of a rat polyclonal serum (Bio/Can Scientific) 2 d after each peptide treatment.
Flow Cytometry.
Single-cell suspensions of spleens were stained at 4°C in PBS, containing FCS and 0.2% NaN3, with FITC-conjugated rat anti–mouse CD8 or CD4 and PE-conjugated rat anti–mouse Vα2 (PharMingen) to assess numbers of transgenic T cells. Additional staining with biotin-conjugated rat anti–mouse CD25, CD44, and CD69 (PharMingen) and streptavidin-conjugated red-670 (Life Technologies) was performed to assess T cell activation. Viable cells (10,000 per sample), gated for lymphocytes by a combination of forward and side scatter, were analyzed on a FACScan™ flow cytometer (Becton Dickinson) using LYSIS II™ software.
Cytotoxicity Assay.
Ex vivo cytolytic activities of spleen cells were determined in a 51Cr-release assay. Single spleen cell suspensions from mice treated 2 d before with gp33 were prepared in IMDM supplemented with 10% FCS, glutamine, and 2-β-ME. EL4 (H-2b) target cells were coated with gp33 or AV at a concentration of 1 μM and labeled with 51Cr for 1 h. After washing, 104 target cells were mixed with spleen effector cells at ratios of 90:1, 30:1, 10:1, and 3:1 in 96-well round-bottomed plates. Cells were incubated for 5 h at 37°C, and 70 μl of supernatant was removed and assayed.
Lymphocyte Proliferation Assay.
Spleens were collected and dispersed into single-cell suspensions in IMEM complete media (10% FCS). Cells were cultured in flat-bottomed 96-well plates (105 cells per well) in a total volume of 200 μl in the presence of 105 cells per well of normal C57BL/6 splenocytes prepulsed with increasing peptide concentrations. Cells were incubated for 48 h at 37°C in 5% CO2, at which time 0.5 μCi of [3H]thymidine was added. The cells were harvested 18 h later, and cell-associated radioactivity was determined.
IFN-γ Detection.
IFN-γ was detected by sandwich ELISA using the PharMingen cytokine detection protocol. Microtiter ELISA plates were coated overnight with capture anti–IFN-γ antibody (R4-6A2; PharMingen) at 2 μg/ml at 4°C. Plates were blocked at room temperature with 3% BSA in PBS. After washing, serum samples were added neat, in duplicate, for 2 h at room temperature. The plates were later incubated with 1 μg/ml of biotin-conjugated anti–IFN-γ (XMG1.2; PharMingen) and were subsequently incubated with horseradish peroxidase–labeled avidin (Vector Labs.). The enzyme substrate O-phenylenediamine (Sigma-Aldrich) was used for color development. IFN-γ concentrations were calculated against murine recombinant IFN-γ (PharMingen).
Immunohistochemistry.
Freshly removed pancreata were immersed in PBS and snap frozen in liquid nitrogen. 5-μm-thick tissue sections of were cut and fixed in acetone for 10 min. Sections were then incubated with primary antibody for 30 min at room temperature. Antibodies used included mAb 169 (anti-CD8) and mAb M114 (anti–class I MHC). Primary antibodies were followed by a two-step indirect immunoenzymatic staining procedure. First, alkaline phosphatase–labeled goat antibodies to rat Igs were applied for 30 min. Alkaline phosphatase was then detected by a red color reaction using naphtho-AS-BI phosphate and New Fuchsin as substrate. Endogenous alkaline phosphatase was blocked by Levamisol. Sections were counterstained with Mayer's hemalum for 2 min. The severity of pancreatic islet infiltration by CD8+ cells (insulitis) was measured on a scale of 0–3, where a severity of 0 = no insulitis, 1 = periinsulitis/mild insulitis, 2 = partial insulitis, and 3 = complete insulitis.
Results and Discussion
In the P14 TCR single-transgenic model, tolerance can be induced with repeated intravenous administration of the LCMV-gp peptide epitope gp33. Peptide administration caused the upregulation of T cell activation markers such as CD69 (Fig. 1 a), CD25, and CD44 (data not shown). In addition, whereas transgenic T cells from untreated mice were incapable of lysing peptide pulsed targets ex vivo, in vivo peptide treatment induced T cell cytolytic activity (Fig. 1 b). Moreover, peptide administration also induced the expansion of transgenic T cells, followed by deletion, as determined by the shift in the number of transgenic T cells above and below normal baseline numbers (Fig. 1 c). The remaining P14-transgenic T cells were anergic and no longer proliferated in response to the gp peptide gp33 (Fig. 1 d; reference 10). Using the RIP-gp/P14 double-transgenic model, we examined whether intravenous administration of gp33 induced diabetes. Although LCMV-gp–specific cytotoxic activity was induced, this surprisingly did not lead to the induction of diabetes (Fig. 2 a).
Figure 1.
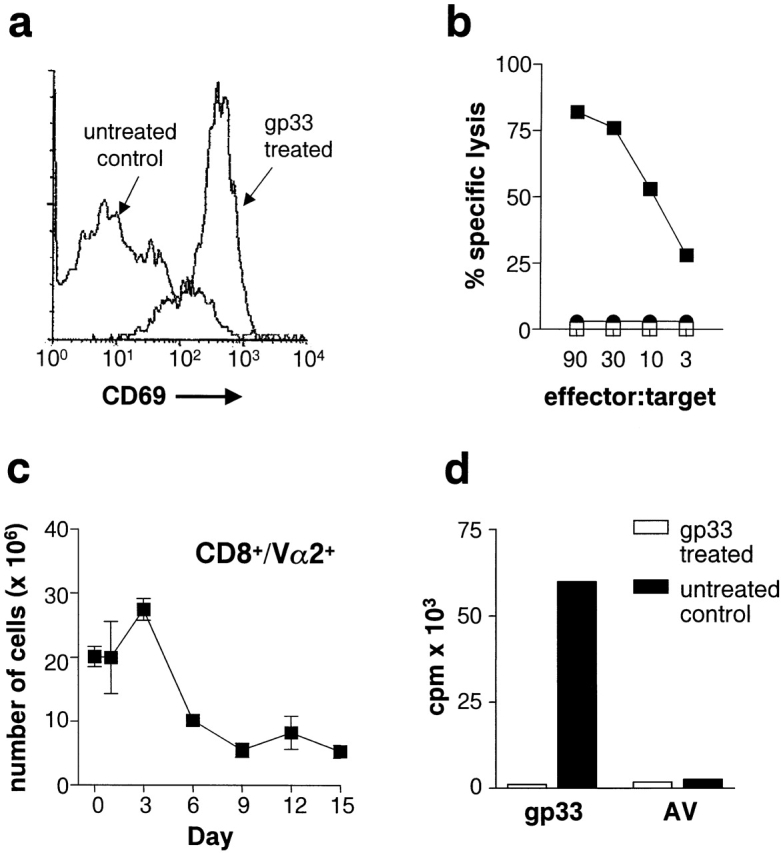
In vivo peptide treatment of P14-transgenic mice leads to T cell activation followed by tolerance. P14-transgenic mice were treated three times, 3 d apart, starting on day 0, with 5 μg of gp33 intravenously. 2 d after the first peptide treatment, spleens were obtained and transgenic T cells were assessed for cell surface expression of CD69 (a) and for CTL effector function against gp33-pulsed targets (▪) or against control peptide–pulsed targets (□) (b). As negative controls, transgenic T cells from spleens of untreated P14 mice were assessed for CD69 expression (a) and for CTL activity against gp33-pulsed targets (•) (b). The number of splenic transgenic T cells in peptide-treated P14 mice was followed over time in different cohorts of animals by determining the percentage of CD8+Vα2+ T cells (c) (n = 3 mice per time point). In vitro splenic proliferative responses against gp33 and control peptide (AV) were determined on day 9 for peptide-treated mice (open bars) and nontreated controls (closed bars) (n = 2 mice per group; variance <10%) (d). Data is presented as mean ± SEM and represents one of two to three experiments.
Figure 2.




Activation of APCs together with peptide treatment leads to insulitis and diabetes. RIP-gp/P14 double-transgenic mice were treated with the indicated peptide on day 0 and antibody on days 0 and 2. Blood glucose levels were monitored over time (a) (n > 20). Pancreatic islet infiltration by CD8+ cells was determined on day 3. Three nonserial sections per mouse were assessed for islet number and severity of infiltration, where a severity of 0 = no insulitis, 1 = periinsulitis/mild insulitis, 2 = partial insulitis, and 3 = complete insulitis (b) (n = 5 animals per group, 10–20 islets per mouse). Immunohistochemical analysis for CD8+ T cells is shown for gp33 and control antibody–treated mice (c) or gp33- and anti-CD40–treated mice (d).
Previous studies using the RIP-gp model have shown that diabetes can be induced upon viral infection 9. Viral infections generally lead to the induction of immunity, which encompasses the events of APC activation and the induction of inflammatory responses. Therefore, to examine the mechanism that leads to autoimmunity rather than tolerance, we focused on the role of APCs. Recent reports have demonstrated that the maturation and activation of APCs can be induced with the ligation of CD40, resulting in increased capacity to present antigen 13 14 15 and the induction of CD8+ immunity 16 17 18. Therefore, RIP-gp/P14 double-transgenic mice were immunized intravenously with gp33 and a rat anti–mouse CD40 activating antibody FGK45 12 or rat polyclonal antiserum as an isotype-matched control. Studies have shown that administration of 100 μg of FGK45 in vivo led to the activation of APCs and the induction of T cell function (reference 17, 18; data not shown). All of the mice that received gp33 plus anti-CD40 antibody were diabetic (Fig. 2 a), in contrast to control transgenic mice receiving gp33 and the control antibody. Thus, the in vivo activation of APCs was critical for the induction of autoimmunity.
To understand the parameters that lead to the induction of tolerance versus autoimmunity, we assessed the status of T cell activity in the spleens of animals given peptide and anti-CD40 or control antibody. T cell activity was measured by the upregulation of T cell activation markers, the induction of effector function, and the infiltration of the pancreas. The induction of activation markers as well as cytotoxic activity was identical in both groups (data not shown). Treatment with peptide alone induced mild pancreatic infiltration (Fig. 2b and Fig. c); however, the severity of infiltration was insufficient to induce disease. In contrast, the combination of peptide and anti-CD40 antibody induced severe insulitis (Fig. 2b and Fig. d).
Activation of APCs via CD40 has been shown to lead to increased production of IL-12, which promotes the release of IFN-γ 15 19. Treatment of double-transgenic mice with peptide and control antibody did not promote the production of measurable levels of circulating IFN-γ. However, the addition of anti-CD40 to the peptide treatment induced levels of IFN-γ that were detectable in the serum (Fig. 3 a). Moreover, serum IFN-γ levels correlated with increased expression of class I in the pancreatic islets (Fig. 3b and Fig. c). As lymphocytes normally express MHC class I at high levels, the degree of class I expression was examined in islets that had relatively few CD8+ T cell infiltrates (Fig. 3 d). Therefore, anti-CD40 treatment led to the activation of APCs in vivo, with an enhanced production of IFN-γ and enhanced pancreatic islet expression of class I MHC. This may contribute to enhanced CTL infiltration of the pancreas and the onset of diabetes.
Figure 3.




Activation of APCs contributes to autoimmunity by inducing IFN-γ production and class I MHC expression. 3 and 5 d after peptide and antibody treatment, serum samples were collected from animals given gp33 and control antibody or gp33 and anti-CD40 and assessed by ELISA for IFN-γ (a) (dotted line = detection limit of the assay; n = 2 per group; variance <10%; data represent one of two experiments). Day 3 pancreata of gp33 and control antibody– (b) or anti-CD40–treated animals (c, d) was assessed by immunohistochemistry for MHC class I (b, c) and CD8 (d) expression (data representative of four mice per group, two nonserial pancreatic sections per mouse).
CD40-mediated APC activation has also been shown to upregulate the costimulatory molecules B7-1 and B7-2 and thereby also enhance T cell function 13 20. To determine whether the induction of autoimmunity was dependent upon engagement of costimulatory ligands, the P14 and RIP-gp transgenes were bred onto a CD28-deficient background 21. Immunization of RIP-gp/P14/CD28-negative mice with gp33 and anti-CD40 resulted in the induction of diabetes with the same incidence and kinetics as seen in CD28-competent mice (data not shown). Therefore, CD28/B7 interactions were not crucial for the induction of CD8-mediated autoimmunity in this model.
To examine whether the activation of APCs influenced deletion of T cells, P14 TCR–transgenic mice were treated intravenously with gp33 to induce deletion of the transgenic T cells. In addition, the mice received FGK45 or the isotype-matched control antiserum. As shown in Fig. 4, treatment with the activating anti-CD40 antibody dramatically inhibited the specific deletion of the CD8+Vα2+ transgenic T cells. Together, these studies support a model where T cell activation versus tolerance is governed by APCs.
Figure 4.

Treatment of P14-transgenic mice with anti-CD40 inhibits gp33-induced T cell deletion. P14-transgenic mice were treated three times, 3 d apart, starting on day 0, with 5 μg of gp33 intravenously. 2 d after each peptide treatment, mice were given 100 μg of activating anti-CD40 antibody or a rat polyclonal antiserum intravenously. Splenic numbers of CD8+Vα2+ transgenic T cells (a) or CD4+ T cells (b) were determined in different cohorts of mice over time by flow cytometry (n = 2–3 mice per group per time point). Data is presented as mean ± SEM and represents one of two experiments.
In this study, disease was actively induced in RIP-gp/P14 mice upon the administration of peptide and the activation of APCs with anti-CD40. However, in other transgenic models of autoimmunity, disease has often been shown to occur spontaneously 22 23 24 25. Spontaneous disease may be due to infection with pathogens present in the local environment, which could potentially induce the activation of APCs. This, together with cross-presentation of self-antigen, which is normally presented by resting APCs to induce tolerance, may promote the induction of autoimmunity 22. Alternatively, the induction of spontaneous autoimmunity may be due to the transient activation of a high number of transgenic T cells. Studies have shown that self-antigen that is shed can be cross-presented by bone marrow–derived APCs to induce peripheral tolerance 7. In addition, studies have shown that peripheral T cell tolerance induction is often preceded by T cell activation 6 7 8 10 26. Thus, if self-antigen is shed in a non-TCR–transgenic mouse, the numbers of self-specific T cells being transiently activated and tolerized are probably not sufficient to cause extensive tissue-specific damage. In a TCR-transgenic mouse, however, the number of self-specific T cells is high. If adequate amounts of self-antigen are shed to induce tolerance, the number of transiently activated T cells may be sufficient to induce tissue damage, thus promoting disease pathogenesis. However, if little to no self-antigen is shed, as in the RIP-gp/P14 model (data not shown), despite the high numbers of antigen-specific T cells, significant tolerance and activation are not induced and the T cells remain ignorant of their ligand.
Previous studies have reported that peripheral tolerance can be inhibited by the presence of bacterial or viral infections 27 28. The induction of T cell immunity in the presence of infectious organisms may be the result of proinflammatory agents produced by the organisms, such as LPS. Early reports demonstrated that bacterial LPS could interfere with the induction of B cell unresponsiveness to γ-globulin 29. Later studies revealed that LPS could also prevent the induction of CD4+ T cell tolerance 30 31. More recent data has shown that LPS can inhibit the induction of tolerance in adoptively transferred transgenic T cells into antigen-bearing mice and promote the induction of autoimmunity 28. In our LCMV double-transgenic model, the administration of gp33 and LPS can induce diabetes (data not shown). Such events involve APC activation, as it has been shown that the injection of LPS leads to dendritic cell (DC) migration from the marginal zones of the spleen to the T cell areas of splenic white pulp as well as to DC maturation and activation 32 33. Thus, infectious agents may bias a T cell response from tolerance toward immunity through the activation of APCs.
Additional reports have demonstrated that activation of APCs with anti-CD40 can prevent tolerance to tumors 34 35 and delay deletion induced by staphylococcal enterotoxin B 36. The activation and expansion of DCs with Flt3 ligand has also been shown to inhibit tolerance induction of adoptively transferred transgenic T cells into antigen-bearing mice 37. Collectively, studies using infectious agents, LPS, and direct activation of APCs demonstrate that the activation of APCs can break tolerance.
Our studies have made an important contribution to understanding the mechanisms that discriminate between the induction of tolerance versus autoimmunity. This study suggests that T cell interactions with peptide presented in a “resting” or naive environment are not sufficient to lead to autoimmunity. However, activation of APCs via anti-CD40 leads to enhanced cytokine production, contributing to a proinflammatory environment and autoimmune diabetes. Although this is an experimental model that examines the induction of immunity and autoimmunity, these findings suggest that the activation status of APCs plays a significant role in the mechanisms of clinical autoimmune disease. It is possible that the genetic predisposition to autoimmune disease involves factors that enhance or lead to chronic activation of APCs. Susceptible individuals may have a defect in peripheral tolerance where self-antigen shedding occurs in the presence of activated APCs. The activation of specific T cells in the presence of chronically activated APCs could thus bias T cell interactions from tolerance to autoimmunity.
The underlying mechanisms that lead to tolerance versus immunity have been a central issue for decades. Over the years, models have identified the importance of the TCR-specific signal 1 and a costimulatory signal 2 and also have identified the important role of resident APCs in the “passenger leukocyte” concept 1 2 38. More recent models have focused on the role of the APC in defining the outcome of tolerance and immunity, which incorporates the importance of costimulatory molecules that become upregulated after APC activation 5. Our studies provide clear evidence that resting versus activated APCs are crucial in determining the outcome of T cell–specific interactions. Many models have provided evidence for physiological events that can trigger APC maturation and activation, including the ability of the adaptive immune system to sense “danger” or to communicate with the innate immune system to discern infectious from noninfectious self 39 40. Further studies are still required to identify the key events that regulate APC function and the induction of tolerance versus immunity.
Acknowledgments
This work was supported by the Medical Research Council of Canada and the National Cancer Institute of Canada (to P.S. Ohashi) and the American Cancer Society (to S.P. Schoenberger). K.M. Garza was supported by the Medical Research Council of Canada and the National Science Foundation. P.S. Ohashi is a Medical Research Council Scientist.
Footnotes
K.M. Garza and S. Chan contributed equally to this work.
References
- Schwartz R.H. Models of T cell anergyis there a common molecular mechanism? J. Exp. Med. 1996;184:1–8. doi: 10.1084/jem.184.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill R.G., Coulombe M., Lafferty K.J. Pancreatic islet allograft immunity and tolerancethe two-signal hypothesis revisited. Immunol. Rev. 1996;149:75–96. doi: 10.1111/j.1600-065x.1996.tb00900.x. [DOI] [PubMed] [Google Scholar]
- Shortman K., Wu L., Süss G., Kronin V., Winkel K., Saunders D., Vremec D. Dendritic cells and T lymphocytesdevelopmental and functional interactions. Ciba Found. Symp. 1997;204:130–141. doi: 10.1002/9780470515280.ch9. [DOI] [PubMed] [Google Scholar]
- Fazekas de St. Groth B. The evolution of self-tolerancea new cell arises to meet the challenge of self-reactivity. Immunol. Today. 1998;19:448–454. doi: 10.1016/s0167-5699(98)01328-0. [DOI] [PubMed] [Google Scholar]
- Banchereau J., Steinman R.M. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Webb S., Morris C., Sprent J. Extrathymic tolerance of mature T cellsclonal elimination as a consequence of immunity. Cell. 1990;63:1249–1256. doi: 10.1016/0092-8674(90)90420-j. [DOI] [PubMed] [Google Scholar]
- Kurts C., Kosaka H., Carbone F.R., Miller J.F.A.P., Heath W.R. Class I–restricted cross-presentation of exogenous self-antigens leads to deletion of autoreactive CD8+ T cells. J. Exp. Med. 1997;186:239–245. doi: 10.1084/jem.186.2.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander-Miller M.A., Leggatt G.R., Sarin A., Berzofsky J.A. Role of antigen, CD8, and cytotoxic T lymphocyte (CTL) avidity in high dose antigen induction of apoptosis of effector CTL. J. Exp. Med. 1996;184:485–492. doi: 10.1084/jem.184.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi P.S., Oehen S., Bürki K., Pircher H., Ohashi C.T., Odermatt B., Malissen B., Zinkernagel R., Hengartner H. Ablation of “tolerance” and induction of diabetes by virus infection in viral antigen transgenic mice. Cell. 1991;65:305–317. doi: 10.1016/0092-8674(91)90164-t. [DOI] [PubMed] [Google Scholar]
- Kyburz D., Aichele P., Speiser D.E., Hengartner H., Zinkernagel R.M., Pircher H. T cell immunity after a viral infection versus T cell tolerance induced by soluble viral peptides. Eur. J. Immunol. 1993;8:1956–1962. doi: 10.1002/eji.1830230834. [DOI] [PubMed] [Google Scholar]
- Pircher H., Bürki K., Lang R., Hengartner H., Zinkernagel R. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature. 1989;342:559–561. doi: 10.1038/342559a0. [DOI] [PubMed] [Google Scholar]
- Rolink A., Melchers F., Andersson B. the SCID but not the RAG-2 gene product is required for S mu-S epsilon heavy chain class switching. Immunity. 1996;5:319–330. doi: 10.1016/s1074-7613(00)80258-7. [DOI] [PubMed] [Google Scholar]
- Grewal I.S., Flavell R.A. CD40 and CD154 in cell-mediated immunity. Annu. Rev. Immunol. 1998;16:111–135. doi: 10.1146/annurev.immunol.16.1.111. [DOI] [PubMed] [Google Scholar]
- Caux C., Massacrier C., Vanbervliet B., Dubois B., Van Kooten C., Durand I., Banchereau J. Activation of human dendritic cells through CD40 crosslinking. J. Exp. Med. 1994;180:1263–1272. doi: 10.1084/jem.180.4.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cella M., Sheidegger D., Palmer-Lehmann K., Lane P., Lanzavecchia A., Alber G. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacityT-T help via APC activation. J. Exp. Med. 1996;184:747–752. doi: 10.1084/jem.184.2.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridge J.P., Di Rosa F., Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 1998;393:474–478. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- Bennet S.R.M., Carbone F.R., Karamalis F., Flavell R.A., Miller J.F.A.P., Heath W.R. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 1998;393:478–480. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- Schoenberger S.P., Toes R.E.M., van der Voort E.I.H., Offringa R., Melief C.J.M. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- Ohteki T., Fukao T., Suzue K., Maki C., Ito M., Nakamura M., Koyasu S. Interleukin 12–dependent interferon γ production by CD8α1 lymphoid dendritic cells. J. Exp. Med. 1999;189:1981–1986. doi: 10.1084/jem.189.12.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Wilson J.M. CD40 ligand-dependent T cell acivationrequirement of B7-CD28 signaling through CD40. Science. 1996;273:1862–1864. doi: 10.1126/science.273.5283.1862. [DOI] [PubMed] [Google Scholar]
- Shahinian A., Pfeffer K., Lee K.P., Kundig T.M., Kishihara K., Wakeham A., Kawai K., Ohashi P.S., Thompson C.B., Mak T.W. Differential T cell costimulatory requirements in CD28-deficient mice. Science. 1993;261:609–612. doi: 10.1126/science.7688139. [DOI] [PubMed] [Google Scholar]
- Goverman J., Woods A., Larson L., Weiner L.P., Hood L., Zaller D.M. Transgenic mice that express a myelin basic protein specific T cell receptor develop spontaneous autoimmunity. Cell. 1993;72:551–560. doi: 10.1016/0092-8674(93)90074-z. [DOI] [PubMed] [Google Scholar]
- Katz J.D., Wang B., Haskins K., Benoist C., Mathis D. Following a diabetogenic T cell from genesis through pathogenesis. Cell. 1993;74:1089–1100. doi: 10.1016/0092-8674(93)90730-e. [DOI] [PubMed] [Google Scholar]
- Lafaille J.J., Nagashima K., Katsuki M., Tonegawa S. High incidence of spontaneous autoimmune encephalomyelitis in immunodeficient anti-myelin basic protein T cell receptor transgenic mice. Cell. 1994;78:399–408. doi: 10.1016/0092-8674(94)90419-7. [DOI] [PubMed] [Google Scholar]
- Matsumoto I., Staub A., Benoist C., Mathis D. Arthritis provoked by linked T and B cell recognition of glycolytic enzyme. Science. 2000;286:1732–1735. doi: 10.1126/science.286.5445.1732. [DOI] [PubMed] [Google Scholar]
- Kurts C., Heath W.R., Carbone F.R., Allison J., Miller J.F.A.P., Kosaka H. Constitutive class I–restricted exogenous presentation of self antigens in vivo. J. Exp. Med. 1996;184:923–930. doi: 10.1084/jem.184.3.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocken M., Urban J.F., Shevach E.M. Infection breaks T-cell tolerance. Nature. 1992;359:79–82. doi: 10.1038/359079a0. [DOI] [PubMed] [Google Scholar]
- Ehl S., Hombach J., Aichele P., Rulicke T., Odermatt B., Hengartner H., Zinkernagel R., Pircher H. Viral and bacterial infections interfere with peripheral tolerance induction and activate CD8+ T cells to cause immunopathology. J. Exp. Med. 1998;187:763–774. doi: 10.1084/jem.187.5.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiller J.M., Weigle W.O. Termination of tolerance to human gamma globulin in mice by antigen and bacterial lipopolysaccharide (endotoxin) J. Exp. Med. 1973;137:740–750. doi: 10.1084/jem.137.3.740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks D.E., Walker S.M., Weigle W.O. Bacterial lipopolysaccharide (endotoxin) interferes with the induction of tolerance and primes thymus-derived lymphocytes. J. Immunol. 1981;126:938–942. [PubMed] [Google Scholar]
- Vella A.T., McCormack J.E., Linsley P.S., Kappler J.W., Marrack P. Lipopolysaccharide interferes with the induction of peripheral T cell death. Immunity. 1995;2:261–270. doi: 10.1016/1074-7613(95)90050-0. [DOI] [PubMed] [Google Scholar]
- De Smedt T., Pajak B., Muraille E., Lespagnard L., Heinen E., De Baetselier P., Urbain J., Leo O., Moser M. Regulation of dendritic cell numbers and maturation by lipopolysaccharide in vivo. J. Exp. Med. 1996;184:1413–1424. doi: 10.1084/jem.184.4.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa C.R., Germain R. Analysis of adjuvant function by direct visualization of antigen presentation in vivoendotoxin promotes accumulation of antigen-bearing dendritic cells in the T cell areas of lymphoid tissue. J. Immunol. 1999;162:6552–6561. [PubMed] [Google Scholar]
- Sotomayor E.M., Borrello I., Tubb E., Rattis F.M., Bien H., Lu Z., Schoenberger S., Levitsky H.I. Conversion of tumor-specific CD4+ T-cell tolerance to T-cell priming through in vivo ligation of CD40. Nat. Med. 1999;5:780–787. doi: 10.1038/10503. [DOI] [PubMed] [Google Scholar]
- Diehl L., den Boer A., Schoenberger S., van der Voort E.I.H., Schumacher T.N.M., Melief C.J.M., Offringa R., Toes R.E.M. CD40 activation in vivo overcomes peptide-induced peripheral cytotoxic T-lymphocyte tolerance and augments anti-tumor vaccine efficacy. Nat. Med. 1999;5:774–779. doi: 10.1038/10495. [DOI] [PubMed] [Google Scholar]
- Maxwell J.R., Campbell J.D., Kim C.H., Vella A.T. CD40 activation boosts T cell immunity in vivo by enhancing T cell clonal expansion and delaying peripheral T cell deletion. J. Immunol. 1999;162:2024–2034. [PubMed] [Google Scholar]
- Bulendran B., Smith J.L., Jenkins M., Schoenborn M., Maraskovsky E., Maliszewski C.R. Prevention of peripheral tolerance by a dendritic cell growth factorFlt3 ligand as an adjuvant. J. Exp. Med. 1998;11:2075–2082. doi: 10.1084/jem.188.11.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafferty K.J., Prowse S.J., Simeonovic C.J. Immunobiology of tissue transplantationa return to the passenger leukocyte concept. Annu. Rev. Immunol. 1983;1:143–173. doi: 10.1146/annurev.iy.01.040183.001043. [DOI] [PubMed] [Google Scholar]
- Matzinger P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- Janeway C.A., Jr. The immune system evolved to discriminate infectious nonself to noninfectious self. Immunol. Today. 1992;13:11–16. doi: 10.1016/0167-5699(92)90198-G. [DOI] [PubMed] [Google Scholar]