

Abstract
Activation of polymorphonuclear leukocytes (PMNs) and adhesion to the endothelial lining is a major cause of edema formation. Although known to be dependent on the function of β2 integrins (CD11/CD18), the precise mechanisms by which adherent PMNs may impair endothelial barrier capacity remain unclear. Here, the role of transmembrane signaling by β2 integrins in PMN-induced alterations in tight junctional permeability of cultured endothelial cell (EC) monolayers was investigated. PMN activation, in the absence of proinflammatory stimuli, was accomplished through antibody cross-linking of CD11b/CD18, mimicking adhesion-dependent receptor engagement. CD18 cross-linking in PMNs added to the EC monolayer provoked a prompt increase in EC permeability that coincided with a rise in EC cytosolic free Ca2+ and rearrangement of actin filaments, events similar to those evoked by chemoattractant PMN activation. Cell-free supernatant obtained after CD18 cross-linking in suspended PMNs triggered an EC response indistinguishable from that induced by direct PMN activation, and caused clear-cut venular plasma leakage when added to the hamster cheek pouch in vivo preparation. The PMN-evoked EC response was specific to β2 integrin engagement inasmuch as antibody cross-linking of l-selectin or CD44 was without effect on EC function. Our data demonstrate a causal link between outside-in signaling by β2 integrins and the capacity of PMNs to induce alterations in vascular permeability, and suggest a paracrine mechanism that involves PMN-derived cationic protein(s) in the cellular crosstalk between PMNs and ECs.
Keywords: inflammation, leukocyte extravasation, endothelium, vascular permeability, integrin signaling
Introduction
Activation and local accumulation of PMNs is an initial event in the host defense against infectious and noxious stimuli. Circulating PMNs respond to locally formed chemotactic factors and emigrate to the surrounding tissue. As a consequence of PMN activation and extravasation, the endothelial cell (EC) barrier function is adjusted, leading to increased vascular permeability for macromolecules and plasma exudation 1 2. These events represent cardinal signs of inflammation, and they also contribute to the pathogenesis of inflammatory disorders. Leukocytic β2 integrins (CD11/CD18) are critical in the PMN-induced EC changes, because inhibition of PMN adhesion to ECs with anti–β2 integrin antibodies effectively prevents PMN-dependent plasma leakage 2 3 4. However, engagement of β2 integrins not only governs cell attachment, but may also influence PMN activity in various ways.
Two major functions have been attributed to β2 integrins: activation-induced ligand binding (inside-out signaling) that mediates stable adhesion to cells or matrices, and, consequent to ligand occupancy, signal transduction into the cell (outside-in signaling; references 5, 6). Adherence-dependent signaling via intracellular regulatory proteins is likely to be involved in PMN cytoskeletal reorganization and induction of cell motility, as well as triggering of secretory events and respiratory burst 7 8 9. Thus, chemoattractant stimulation of PMNs may, via activation of β2 integrins, initiate transmembrane signaling simultaneously with substrate adhesion. Because both events have obvious consequences for EC barrier function, clarifying their downstream effects may provide better understanding of the mechanisms behind PMN-induced changes in EC permeability. To this end, antibody cross-linking of cell surface receptors, considered to mimic natural ligand engagement, may be experimentally employed to induce a signaling cascade independently of the adhesive function of the receptor. Thus, antibody-induced engagement of CD18 has been shown to initiate a number of PMN activities, including mobilization of cytosolic free calcium 10 11 12, protein tyrosine phosphorylation 13 14, and actin polymerization and cytoskeletal rearrangement 12 15.
We have reported previously on the kinetics of PMN-induced changes in endothelial barrier function based on measurements of electrical resistance, protein flux, and PMN migration across EC monolayers cultured on permeable membranes 16. These and other data 17 indicate that increase in EC permeability induced by activated PMNs is linked to initial CD11/CD18–dependent adhesive events and is independent of subsequent transendothelial migration. However, the precise mechanisms by which activated PMNs trigger an increase in EC permeability remain unresolved. We now demonstrate a direct and causal association between adherence-dependent CD11/CD18 signaling and PMN-induced alterations in EC permeability. Thus, engagement of β2 integrins in quiescent PMNs through antibody receptor cross-linking is sufficient to provoke an active and instantaneous response of EC and impaired EC barrier capacity indistinguishable from that achieved by chemoattractant stimulation of the PMNs. Our data further suggest that, downstream to β2 integrin signaling, paracrine stimulation via PMN cationic protein(s) mediates the PMN-evoked EC responses.
Materials and Methods
Reagents.
Medium 199, RPMI 1640 (containing l-glutamine), FBS, trypsin-EDTA, PBS, and HBSS were from Life Technologies. Gelatin, collagenase, penicillin, streptomycin, catalase, FMLP, Evans blue dye, hexadecyltrimethylammonium bromide, and tetramethylbenzidine were from Sigma-Aldrich. H2O2 was from Merck KGaA, Biomatrix I was from Biomedical Technologies, Inc., and dextran 70 and Ficoll-Paque were from Amersham Pharmacia Biotech. The basal culture medium consisted of a 1:1 mixture of medium 199 and RPMI 1640 supplemented with 20% heat-inactivated FBS, penicillin (100 U/ml), and streptomycin (100 μg/ml). The following mouse mAbs against human antigens were used: mAb IB4 against the common β chain (CD18) of β2 integrins; mAb 60.1 recognizing integrin αm chain (CD11b); mAb DREG 200 and mAb SK11 against l-selectin (CD62L); and mAb G44-26 against CD44. Sheep IgG against human elastase was from The Binding Site, and rabbit IgG against human cathepsin G was from Athens Research and Technology, Inc. mAbs to human elastase and cathepsin G were from Research Diagnostics, Inc. and Chemicon International, respectively. F(ab′)2 fragment of goat anti–mouse IgG was purchased from Jackson ImmunoResearch Laboratories, Inc., and FITC-conjugated F(ab′)2 fragment of rabbit anti–mouse IgG was from Dako A/S.
ECs.
Bovine aorta ECs (BAECs) and human umbilical vein ECs (HUVECs) were isolated and cultured as described previously 16. BAECs or HUVECs in the first to fifth passages were detached by brief (2-min) trypsin-EDTA treatment (0.25% trypsin/0.01% EDTA) and replated onto 3.0-μm pore size polycarbonate filters (tissue culture inserts, 10 mm; Nalge Nunc International) or, for the purpose of visual inspection, 0.2 μm-pore size Anopore inorganic membrane (transparent when wet; Nalge Nunc International). To promote cellular differentiation and enhance attachment of ECs, the filters were pretreated with 50 μl Biomatrix I (167 μg/ml) and air dried. ECs were seeded at a density of 2 × 105 cells per filter and incubated in culture medium at 37°C in a humidified atmosphere of 5% CO2 in air. The ECs were grown to confluent monolayers, as assessed by daily microscopic observation in transparent 0.2-μm pore size filters and measurement of the electrical resistance across the monolayer 16. Under these experimental conditions, maximal resistance is obtained at day 7 after seeding 16, at which day the EC monolayers were then used in the experiments.
Measurement of EC Barrier Function.
Two different approaches were used for assessment of stimulation-induced changes in EC permeability. For measurement of transendothelial electrical resistance (TEER), the filter insert with ECs was transferred to a resistance measurement chamber 16. The chamber (lower abluminal compartment) with the filter insert (upper luminal compartment), filled with 2 ml and 400 μl culture medium, respectively, was placed in a cell culture incubator. Electrical resistance across the EC monolayer was measured at 37°C with an electrode in each compartment exactly positioned in relation to each other and to the monolayer. The electrical resistance of individual EC monolayers was obtained by subtracting the resistance of the corresponding naked filter coated with Biomatrix as measured before seeding of the ECs. TEER values so achieved represent the resistance of the EC monolayer alone and depend on the development of tight junctions between individual ECs 16.
In parallel experiments, Evans blue dye–conjugated albumin (EBA) was used as a marker for macromolecular permeability of the EC monolayers. Before stimulation of ECs, the culture medium in the upper compartment was exchanged for medium containing EBA (culture medium containing 4% BSA mixed with Evans blue dye at a final concentration of 0.67 mg/ml). The EBA concentration in fluid samples of the upper and lower compartment was determined spectrophotometrically through measurement of absorbency at 620 nm (Titertek Multiskan MCC; Flow Laboratories). Albumin clearance was calculated according to the relationship V1 = A2 × V2 × 1/A1, where V1 is the clearance volume (i.e., the theoretical volume of the luminal medium cleared from albumin by diffusion to the abluminal compartment), V2 is the abluminal volume, and A1 and A2 are the absorbencies of the luminal and abluminal medium, respectively. The baseline clearance in absence of any stimulus, 0.08 ± 0.03 μl/min, was subtracted from the clearance volume obtained in response to respective specific stimulus.
Preparation and Quantification of PMNs.
Human PMNs were isolated from leukocyte-rich plasma as described 16. Purity of PMNs was >98%, and viability by Trypan blue exclusion was >95%. Before being added to the EC monolayer, the PMNs were pretreated with mAbs against cell surface molecules for subsequent cross-linking with secondary antibody (see below). After incubation with primary mAb (3 μg per 106 PMNs) for 30 min at room temperature, the cells were washed twice to remove unbound antibody and were resuspended in HBSS. PMNs not subjected to antibody treatment were prepared according to the same protocol, using vehicle instead of antibody. In some cases, cell surface expression of CD11/CD18 was upregulated through temperature transition of the PMNs before use. PMNs in the presence of anti-CD18 mAb IB4 were incubated at 4°C for 10 min, transferred to 37°C for 10 min, and then returned to 4°C for another 10 min, followed by two washes. There was no difference in the EC responses to stimulation with these cells compared with PMNs prepared according to the standard procedure.
PMN adhesion to ECs and transendothelial migration were quantified by assaying the PMN-specific enzyme myeloperoxidase (MPO). In brief, PMNs were lysed in 0.5% hexadecyltrimethylammonium bromide and centrifuged, and the enzyme activity in the supernatant was determined spectrophotometrically as the change in absorbency at 650 nm that occurs in the redox reaction of H2O2-tetramethylbenzidine catalyzed by MPO 18. MPO activity of adherent and transmigrated PMNs, respectively, was related to that of the total number of PMNs added to the EC monolayer.
Stimulation of ECs by Activated PMNs.
Before transferring the filter insert to the resistance measurement chamber (kept at 37°C), the medium in the insert and in the chamber was replaced with fresh culture medium (37°C) containing 20% FBS and supplemented with 10 mM Hepes. PMNs (2 × 106) with or without mAbs bound to cell surface molecules were added to the upper compartment (PMN/EC ratio = 10:1) and allowed to sediment for 10 min onto the EC monolayer. In separate experiments, dextran sulfate (mol wt: 500,000; Amersham Pharmacia Biotech) was added to the upper compartment at a final concentration of 1 mg/ml before PMN addition. Activation of PMN was induced either with FMLP (10−7 M) added to the upper compartment or through antibody cross-linking of cell surface molecules by secondary goat anti–mouse IgG F(ab′)2 fragments (1:20). In each single experiment, TEER was measured before and every minute after start of stimulation until a plateau phase in the resistance change was reached, and then at 5-min intervals. In experiments where albumin permeability and PMN migration across the EC monolayer were studied, the medium in the insert contained EBA. After challenge, the filter insert was moved at 5–10-min intervals to new wells containing fresh medium. All incubations were performed at 37°C. At the end of the experiment, the lower wells were centrifuged at 425 g and 4°C for 20 min, and the supernatant was analyzed for EBA content. PMNs that had migrated across the monolayer were quantified through analysis of MPO activity in the pellet remaining in the well. The medium in the filter insert was aspirated for quantification of nonadherent PMNs, and the filter membrane with ECs was removed from the insert to quantify the adherent fraction of PMNs.
CD18 Cross-Linking–induced PMN Secretion.
Purified PMNs in suspension (5 × 107 cells/ml) pretreated with anti-CD18 mAb IB4 according to the protocol described above were incubated with or without goat anti–mouse F(ab′)2 for 10 min at 37°C. The PMNs were sedimented by centrifugation at 300 g for 15 min at room temperature, and the cell-free supernatant containing PMN secretion products was decanted for use in experiments with the EC monolayers or in vivo (see below). The PMN postsecretory supernatant was examined for the presence of proteins by SDS-PAGE analysis. Proteins were separated on a 12% SDS polyacrylamide gel (Bio-Rad) under standard conditions and visualized in gel by Coomassie blue staining (Bio-Rad). In addition, samples of the postsecretory supernatant were applied to a cation exchange column (HiTrap SP; Amersham Pharmacia Biotech) according to the manufacturer's instructions in order to remove positively charged molecules from the supernatant.
To test the activity of the PMN postsecretory supernatant on EC barrier function, the medium in the filter/EC insert was exchanged for 400 μl PMN secretion–containing medium supplemented with 20% FBS, and effects on TEER and albumin permeability were recorded as described. The amount of supernatant added to each monolayer thus represented 10 times the number of PMNs that were added to ECs in the case of direct CD18 cross-linking, as described above. In some cases, the postsecretory supernatant was heat incubated (80°C for 15 min) before treatment of ECs. The use of the PMN postsecretory supernatant to stimulate ECs was combined in additional experiments with pretreatment of the monolayer with either the tyrosine kinase inhibitor herbimycin A (1 μM; Sigma-Aldrich) or the cell-permeant Ca2+ chelator BAPTA-AM (5 μM; Molecular Probes). EC monolayers were incubated with the reagents for 30 min at 37°C and washed twice before use. Stimulation of ECs with the postsecretory supernatant was also induced in the presence of negatively charged dextran sulfate. Dextran sulfate (mol wt: 500,000) was added to the upper compartment at a final concentration of 1 mg/ml before stimulation.
PMN granule proteins cathepsin G and elastase have been suggested to be capable of mediating changes in EC barrier capacity. In an attempt to elucidate the potential involvement of cathepsin G and/or elastase in the PMN-evoked effects on EC permeability, it was sought to deplete the postsecretory supernatant of these proteins by immunoadsorption. For this purpose, anti–cathepsin G IgG or antielastase IgG was mixed with a slurry of protein A sepharose CL-4B (Amersham Pharmacia Biotech) according to the manufacturer's instructions. The PMN postsecretory supernatant obtained as above was incubated under gentle rotation for 15 min, first with anti–cathepsin G and then with antielastase-coupled protein A sepharose (400 μg IgG/ml supernatant). After each incubation step, the sepharose beads were sedimented by centrifugation at 425 g for 15 min, and the supernatant was decanted for further use. The efficacy of this procedure to deplete the supernatant of these proteins was analyzed by sandwich ELISA. In brief, microtiter plates were coated with 200 μl/well of 0.25 μg/ml mAb to human cathepsin G or human elastase at 4°C, washed five times with PBS containing 0.05% Tween 20, and blocked with 2% BSA (wt/vol) in PBS for 30 min at 37°C. The plates were then incubated with a serial dilution (2n) of native and depleted supernatant, respectively, for 1 h at 37°C. After washing, plates were incubated with rabbit anti–cathepsin G antibody or sheep antielastase antibody (dilution 1:1,000) for 1 h at 37°C. Bound antibody was detected by a horseradish peroxidase–labeled secondary antibody to rabbit IgG and goat IgG, respectively, and the chromogenic substrate diammonium-2,2′-azino-bis-(3-ethyl-2,3-dihydrobenzthiazoline)-6-sulfonate/H2O2. The relative protein content was spectrophotometrically determined.
Determination of EC Intracellular [Ca2+] and F-actin Formation.
The Ca2+-sensitive fluorescent probe fluo-3/AM (Molecular Probes) was used for determination of changes in EC intracellular free Ca2+ ([Ca2+ ]i). Confluent BAEC monolayers on filters were incubated for 30 min at 37°C with fluo-3/AM (3 μM in HBSS containing 2% FBS and 10 mM Hepes) added to both the luminal and abluminal surface. The monolayers were washed three times and incubated with fresh HBSS in the dark for 20 min at room temperature to allow complete hydrolysis of the dye ester. Anti-CD18 or anti–l-selectin mAb–treated PMNs were added to ECs and allowed to settle for 10 min before receptor cross-linking was accomplished with secondary antibody as described above. Alternatively, mAb-treated PMNs were stimulated with FMLP (10−7 M). In separate experiments, PMN secretion–containing medium (described above) was used to stimulate ECs in the absence of PMNs. Changes in EC [Ca2+]i in response to these stimuli (the latter also combined with BAPTA pretreatment of ECs) were measured through continuous registration of fluorescence intensity with a laser-scanning confocal imaging system (Insight Plus; Meridian Instruments).
For analysis of F-actin formation in ECs in response to PMN activation, confluent EC monolayers grown on Biomatrix-coated coverslips were incubated for 15 min at 37°C with either PMN subjected to CD18 antibody cross-linking or with PMN secretion–containing medium identical to the procedures described above. Incubation of ECs with IB4-treated PMN served as control. The monolayers were fixed in 3.7% formaldehyde in PBS for 10 min at room temperature, washed twice, and permeabilized with 0.2% Triton X-100 in PBS (1 min at 4°C). The cells were washed twice and stained for actin filaments with FITC-conjugated phalloidin (Sigma-Aldrich) for 20 min at 37°C. After three additional washes, the ECs were viewed in a laser-scanning confocal imaging system (Insight Plus; Meridian Instruments).
Visualization of β 2 Integrin Cell Surface Distribution.
PMNs, preincubated with anti-CD18 mAb IB4 according to the protocol described above, were subjected to antibody cross-linking with FITC-conjugated rabbit anti–mouse IgG F(ab′)2 fragments for 2 min at room temperature. The reaction was stopped by fixation in 3.7% formaldehyde for 10 min. Control PMNs were fixed in the same way before incubation with secondary FITC-conjugated F(ab′)2. The PMNs were washed twice and viewed with laser-scanning confocal microscopy to visualize β2 integrin receptor distribution on the PMN surface. Cell shape changes and spreading of PMNs were observed with phase-contrast light microscopy 15 min after induction of CD18 cross-linking.
In Vivo Determination of Microvessel Permeability Changes.
To study the effect of PMN secretion products on microvessel permeability in vivo, intravital microscopy of the hamster cheek pouch microcirculation was applied. As described previously in detail 19, the left cheek pouch of anesthetized Syrian golden hamsters was everted and prepared for microscopic observation under continuous superfusion with a bicarbonate buffer maintaining physiological levels of temperature, pH, and gas tensions. FITC-conjugated dextran (mol wt: 150,000) injected intravenously (250 mg/kg body weight) was used as plasma tracer to permit visualization of changes in vascular permeability to macromolecules. The hamsters were treated with fucoidin (10 mg/kg intravenously; Sigma-Aldrich) to prevent leukocyte adhesion in the microvessels 20 and in that way eliminate influences of potential activation of endogenous leukocytes. The microvasculature in the exposed cheek pouch was observed under low magnification in fluorescent light with a Leitz Orthoplan microscope (Leica) and recorded on video. After control recordings, superfusion was stopped and 1 ml of medium containing CD18 cross-linking–evoked PMN secretion at 37°C was topically applied to the cheek pouch. In parallel experiments, medium of anti-CD18–treated PMNs not subjected to cross-linking was used. The animal experiments were approved by the regional ethical committee for animal experimentation.
Results
Previously, we have shown that there is no qualitative difference between responses of BAEC monolayers and those of HUVECs to several experimental maneuvers, although the results with BAECs are more consistent 16. Accordingly, the data presented in this communication refer to results obtained with BAECs, but confirmed in experiments using HUVECs.
CD18 Cross-Linking in Quiescent PMNs Elicits a Prompt Increase in EC Monolayer Permeability in the Absence of PMN Adhesion and Transmigration.
PMNs (2 × 106) pretreated with anti-CD18 mAb (IB4) were added to the luminal side of the EC/filter insert (upper compartment of the resistance chamber) and allowed to settle for 10 min. CD18 cross-linking with secondary goat anti–mouse F(ab′)2 elicited a marked fall in TEER from a control value of 18.0 ± 1.1 ohm×cm2 to 6.5 ± 2.3 ohm·cm2 (means ± SD; n = 20). The change in resistance was manifested within 1 min, reached its lowest value (36 ± 13% of control) after 15 min, and remained at this level throughout the observation period (Fig. 1). Treatment of PMNs with IB4 alone or with secondary antibody in the absence of IB4 caused no change in TEER (data not shown). PMNs were routinely added to ECs at a ratio of 10:1. In pilot experiments, a ratio of 1:1 was also used, but without causing any change in resistance.
Figure 1.

Changes in TEER and macromolecular permeability (albumin clearance) of BAEC monolayers, and in PMN transmigration in response to antibody cross-linking of leukocytic CD18. PMNs were preincubated with anti-CD18 mAb for 30 min before being added to ECs. Cross-linking with secondary F(ab′)2 was induced at time zero. Mean ± SD; n = 9–20.
The capacity of CD18 cross-linking in causing increased macromolecular flux and PMN migration across the EC monolayer was studied in parallel experiments. PMNs pretreated with saturating concentration of IB4 were added to the EC/filter insert and allowed to settle for 10 min in medium containing EBA. CD18 cross-linking was then induced with goat anti–mouse F(ab′)2, and the PMN/EBA–containing filter inserts were moved at regular intervals to new wells containing fresh medium. In the absence of secondary antibody, there was a minute and continuous clearance of albumin, 0.08 ± 0.03 μl·min−1 (mean ± SD; n = 10), which did not change with time, and which exactly matched the spontaneous clearance rate found in filter inserts containing untreated PMNs (data not shown). In contrast, activation of the PMNs by CD18 cross-linking elicited augmented albumin clearance that increased progressively with time (Fig. 1). The net increase in albumin clearance rose from 0.5 ± 0.1 μl during the first 5-min period to a total of 20.0 ± 4.5 μl (mean ± SD; n = 10) in 60 min. Distinct from increased albumin clearance, CD18 cross-linking caused no measurable PMN adhesion to ECs or transendothelial migration (Fig. 1). Analysis of MPO activity in the two compartments 60 min after CD18 cross-linking revealed that 91.0 ± 6.0% (mean ± SD; n = 9) of added PMNs were recovered in the medium of the upper compartment, whereas no significant MPO activity was detected in the EC/filter unit or in the medium of the lower compartment. In separate experiments, direct microscopic observation of transparent 0.2-μm pore size filters washed at the end of the experiment confirmed complete absence of PMNs adherent to the EC monolayer.
As opposed to treatment with anti-CD18 mAb alone, cross-linking with secondary antibody induced prompt clustering of β2 integrins on the cell surface, and as a consequence of cell activation, pronounced shape changes and spreading of the PMNs (Fig. 2). Notably, even though PMNs were in close apposition to ECs, these responses occurred in the absence of firm attachment of PMNs to the monolayer, because this was prevented by CD18 mAb treatment.
Figure 2.

a and b show confocal microscopic images of PMNs pretreated with anti-CD18 mAb and incubated with FITC-conjugated secondary F(ab′)2. The cells were fixed in formaldehyde either before or after 2-min incubation with the secondary antibody. (a) Staining for CD18 was weak and uniformly distributed in prefixed cells, whereas (b) redistribution and clustering of β2 integrin receptors was evident in cells that were fixed 2 min after CD18 cross-linking was induced. c and d show light microscopic images of PMN pretreated with anti-CD18 mAb, before (c) and after (d) incubation with secondary F(ab′)2 for 15 min. Cell spreading and pseudopod formation were clearly visible after CD18 cross-linking. Micrographs are representative of >10 separate analyses. Bar indicates 5 μm.
PMN-evoked Alteration in EC Permeability Is Specific to CD11/CD18 Cross-Linking.
Treatment of the PMNs with IB4 prevented PMN adhesion to ECs and the change in TEER in response to stimulation with FMLP (10−7 M) added to the upper compartment (Fig. 3 a). On the other hand, subsequent cross-linking with secondary antibody caused a change in TEER (from 19.0 ± 1.5 ohm×cm2 to 7.0 ± 1.5 ohm×cm2; means ± SD; n = 12) that was virtually identical to the response induced by FMLP stimulation of untreated PMNs (Fig. 3 a). A similar decrease in TEER to that found for CD18 cross-linking was observed after cross-linking the αm chain (CD11b) of β2 integrins in PMNs pretreated with the anti-CD11b mAb 60.1 (Fig. 4). To check for specificity of signaling via CD11/CD18 in causing permeability changes in ECs, experiments were performed with PMNs pretreated with mAb against l-selectin (DREG-200 or SK11) or CD44 (G44-26). In this case, cross-linking of l-selectin with goat anti–mouse F(ab′)2 failed to induce any change in TEER. On the other hand, subsequent chemoattractant stimulation with FMLP (10− 7M) activated the PMNs and elicited a decrease in TEER (from 19.4 ± 2.0 ohm×cm2 to 5.5 ± 1.5 ohm×cm2; means ± SD; n = 6), that was indistinguishable from that of FMLP challenge of untreated PMNs (Fig. 3 b). Also, antibody cross-linking of CD44 had no effect on TEER (Fig. 4), whereas subsequent PMN activation with FMLP caused substantial decrease in TEER.
Figure 3.
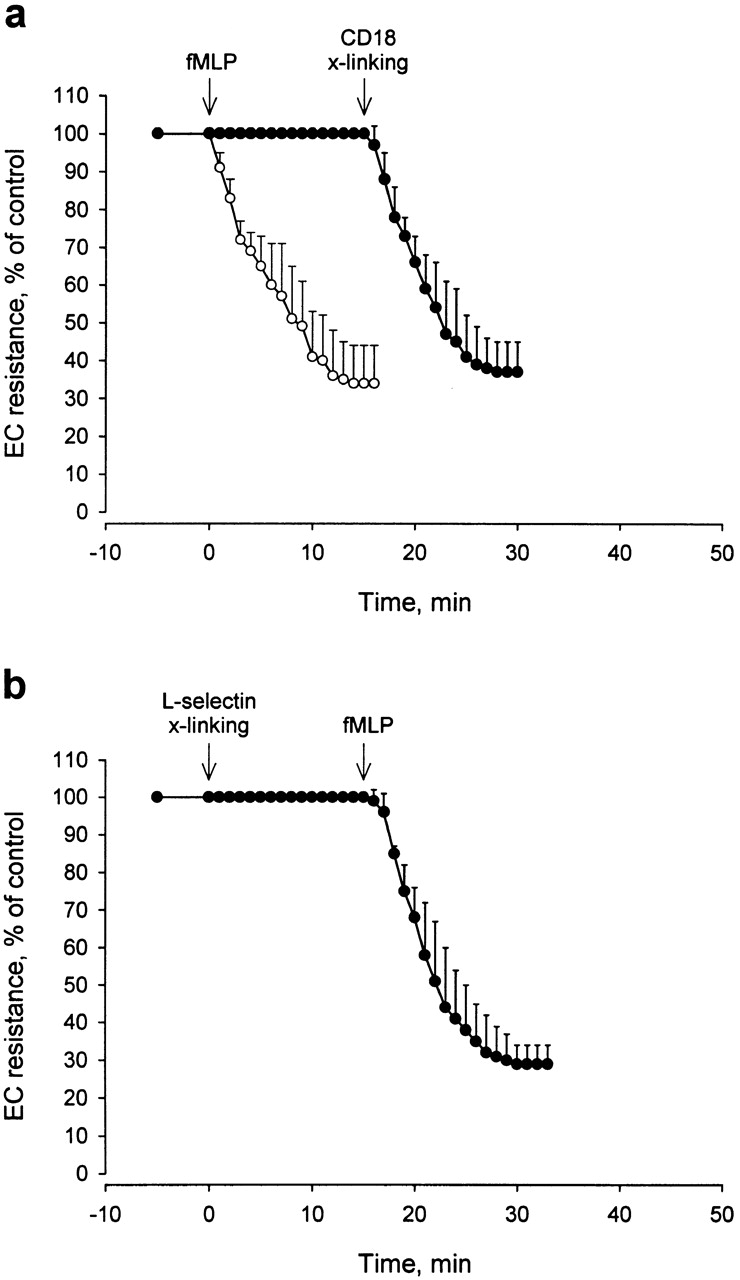
Kinetics of change in electrical resistance across BAEC monolayers in response to PMN activation. (a) PMNs, preincubated with anti-CD18 mAb and added to ECs, first were challenged with FMLP (10−7 M) and 15 min later were subjected to antibody cross-linking (x-linking) of CD18 with secondary F(ab′)2 (•). Mean ± SD; n = 12. Note lack of EC response to FMLP stimulation of the IB4-coated PMNs compared with the response to FMLP stimulation (10−7 M) of untreated PMNs obtained in a separate set of experiments (○). Mean ± SD; n = 8. (b) PMNs, preincubated with anti–l-selectin mAb, were added to ECs. Cross-linking (x-linking) of l-selectin with secondary F(ab′)2 was induced at time zero, followed by stimulation with FMLP (10−7 M) at 15 min (mean ± SD; n = 6).
Figure 4.

Magnitude of TEER changes in BAEC monolayers in response to different modes of PMN activation according to protocols described in Materials and Methods. Antibody cross-linking (x-linking) of either CD11b or CD18 β2 integrin subunit resulted in a decrease in EC electrical resistance of the same magnitude as that induced by FMLP stimulation, whereas FMLP stimulation of IB4-coated PMNs (FMLP + anti-CD18) and cross-linking (x-linking) of l-selectin or CD44 failed to induce any change in resistance. Data are means ± SD based on 6–20 experiments in each group.
Alteration of EC Permeability in Response to CD18 Cross-Linking Coincides with Increase in EC Cytosolic Free Calcium and F-actin Content.
In separate experiments, monolayers of ECs were labeled with the Ca2+-sensitive fluorophore fluo-3, and stimulus-induced changes in EC [Ca2+]i were monitored by laser-scanning confocal microscopy. Addition of anti-CD18–pretreated PMNs to ECs and sedimentation onto the monolayer, rendering PMNs in loose contact with ECs, caused no change in EC [Ca2+]i. However, CD18 cross-linking with secondary antibody elicited a rapid increase in EC cytosolic free [Ca2+], which peaked within 100–200 s and then slowly subsided (Fig. 5). Antibody cross-linking of l-selectin in an equivalent manner failed to induce any change in EC [Ca2+]i (data not shown). A Ca2+ response similar to that induced by CD18 cross-linking was seen after stimulation of untreated PMNs with FMLP 16, whereas chemoattractant stimulation of IB4-coated PMNs was unable to cause a rise in EC [Ca2+]i (Fig. 5), reflecting the necessity of β2 integrin engagement (adhesion dependent or antibody induced) in signaling PMN-evoked EC calcium responses.
Figure 5.

Effect of PMN activation on EC cytosolic free [Ca2+]. PMNs, preincubated with anti-CD18 mAb, were layered on fluo-3–loaded BAEC monolayers. Antibody cross-linking of CD18 with secondary F(ab′)2 was induced at time zero. Tracings show fluorescence intensity changes in four individual ECs of the same monolayer normalized to the average fluorescence intensity before stimulation. Dotted curve illustrates response to FMLP stimulation of anti-CD18 mAb–treated PMNs. Tracings are representative of responses in 10 separate monolayers.
In further experiments, EC monolayers were incubated with PMN and then stained for actin filaments with FITC-conjugated phalloidin. Laser-scanning confocal microscopy revealed that ECs incubated with IB4-treated PMNs in the absence of secondary antibody exhibited few stress fibers and a thin band of actin along cell margins in direct apposition to adjacent cells (Fig. 6 a). On the other hand, ECs incubated with PMNs subjected to CD18 cross-linking presented a marked increase in the number and density of F-actin stress fibers (Fig. 6 b).
Figure 6.
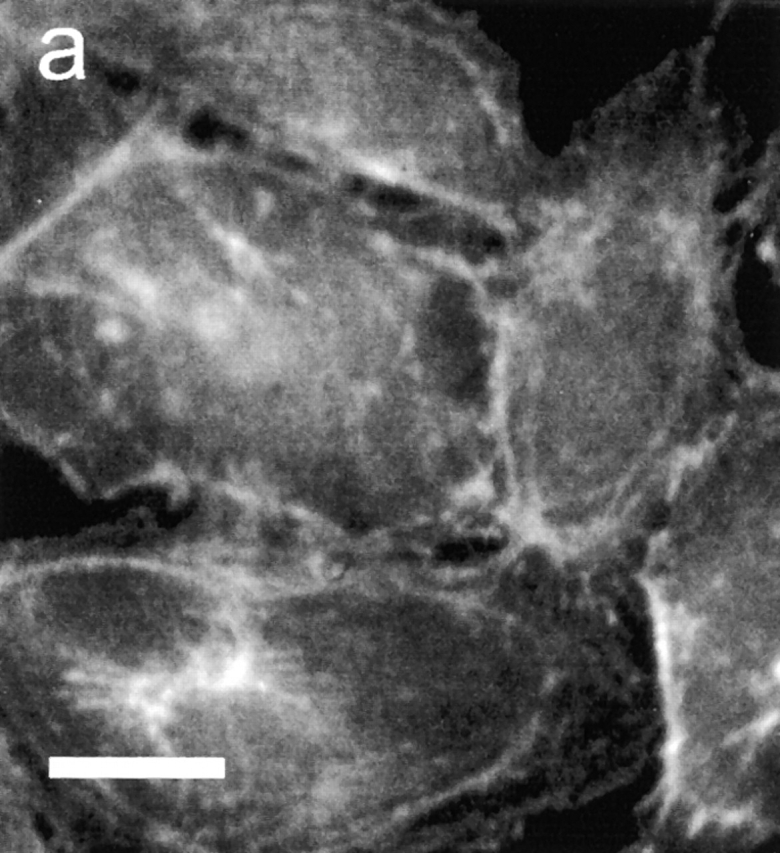
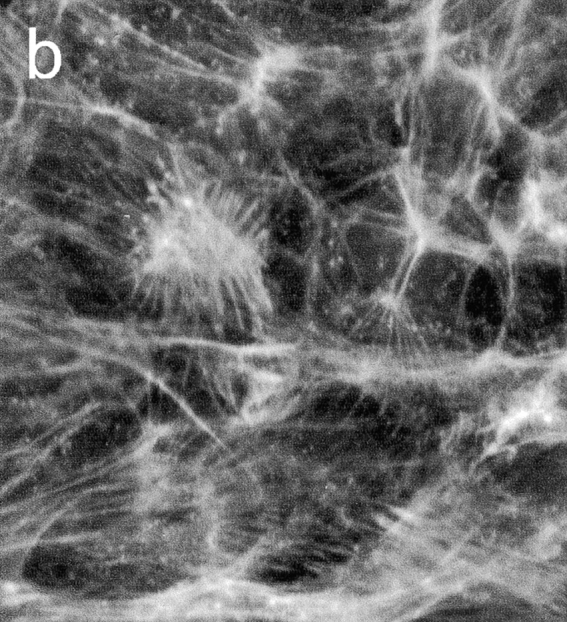
Effect of PMN activation on EC F-actin formation. PMNs, pretreated with anti-CD18 mAb, were added to BAEC monolayers and incubated with or without secondary F(ab′)2. (a) Staining for actin filaments with FITC-conjugated phalloidin revealed few stress fibers and a thin band of actin along cell margins in ECs incubated with PMNs in absence of secondary antibody. (b) In contrast, ECs exposed to PMNs subjected to antibody cross-linking of CD18 exhibited a marked increase in F-actin and density of stress fibers spanning the cells. Micrographs are representative of >20 separate experiments. Bar indicates 25 μm.
CD18 Cross-Linking Triggers PMN Secretion that Mediates Alterations in EC Permeability.
The finding that PMN activation through antibody cross-linking of CD18 altered the characteristics of the endothelium, although there was no PMN attachment to the monolayer, prompted us to investigate if factor(s) secreted by the PMNs mediated the response ultimately leading to increased EC permeability. Thus, PMNs pretreated with anti-CD18 mAb IB4 and washed to remove unbound antibody were incubated with goat anti–mouse F(ab′)2 for 30 min, then spun down by gentle centrifugation. Analysis of the cell-free supernatant by SDS-PAGE and Coomassie staining indicated a prominent fraction of proteins with apparent molecular mass in the range of 25–30 kD (Fig. 7). Presumably, the majority of these proteins were positively charged because the corresponding bands disappeared after running the supernatant through a cation exchange column (Fig. 7).
Figure 7.
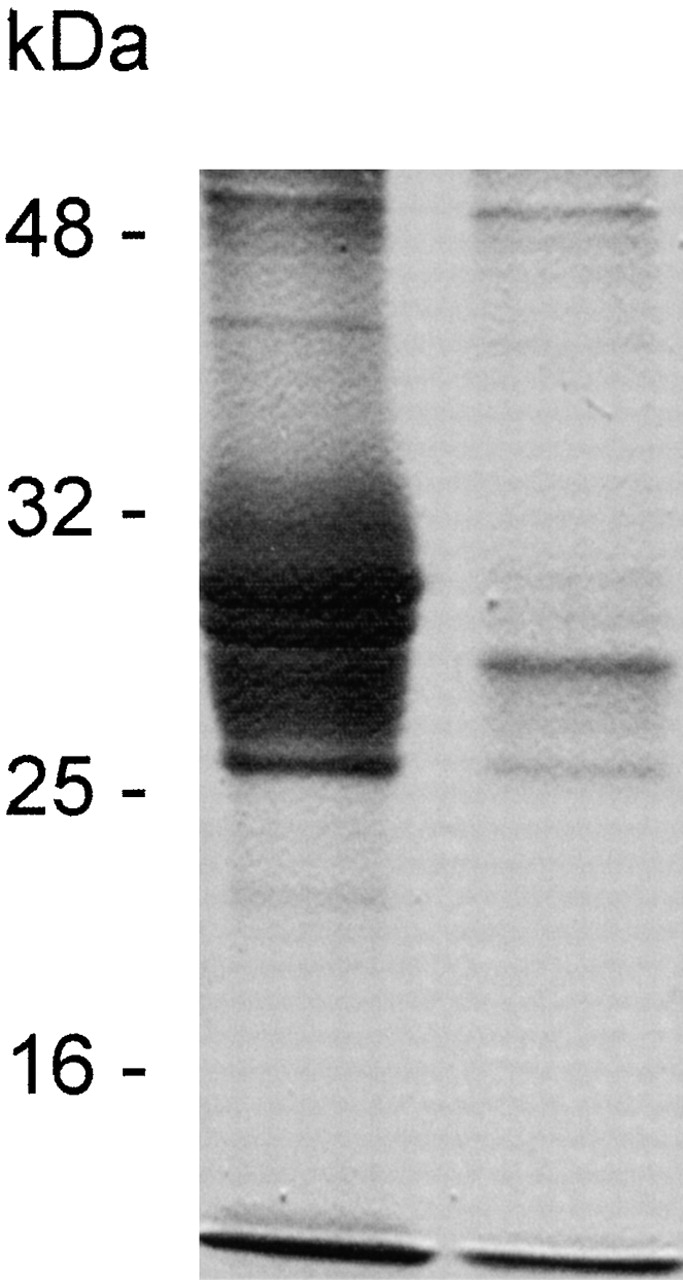
SDS-PAGE analysis of cell-free supernatant obtained after CD18 cross-linking in suspended PMNs. Coomassie blue staining revealed multiple bands corresponding to proteins with apparent molecular mass of 25–30 kD (left lane). After running the supernatant through a cation exchange column, the majority of these bands were missing (right lane). Molecular mass is indicated on the left.
Addition of 400 μl cell-free supernatant to the EC monolayer, corresponding to a PMN/EC ratio of 100:1, elicited a fall in TEER (from 17.8 ± 1.0 ohm×cm2 to 5.1 ± 2.1 ohm×cm2; means ± SD; n = 10) and an increase in albumin clearance, both of which had the same magnitude and time course as after CD18 cross-linking in PMNs added to ECs (Fig. 8 a). These responses were tyrosine kinase dependent, inasmuch as pretreatment of ECs with herbimycin A significantly attenuated the permeability change (Fig. 8 b). Also, the supernatant caused changes in EC [Ca2+]i and in F-actin content and distribution, which closely resembled those documented in Fig. 5 and Fig. 6 for direct PMN stimulation of ECs. A causal link between changes in intracellular Ca2+ activity and the functional response of ECs to PMN activation was evidenced by experiments where ECs were pretreated with the calcium-chelating agent BAPTA-AM. This treatment completely inhibited both the rise in EC [Ca2+]i (data not shown) and the change in TEER in response to CD18 cross-linking (Fig. 8 b).
Figure 8.
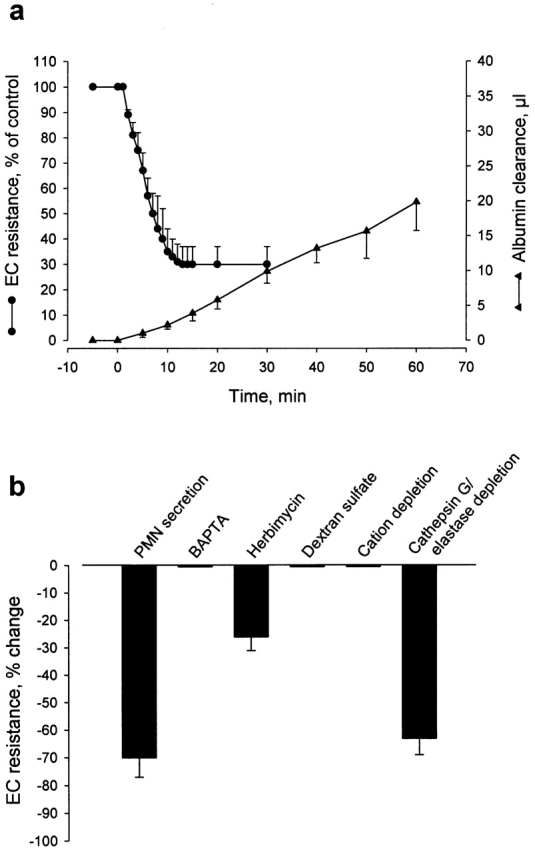
(a) Kinetics of changes in TEER and macromolecular permeability (albumin clearance) of BAEC monolayers stimulated with cell-free supernatant obtained after CD18 cross-linking in suspended PMNs. The medium in the filter/EC insert was exchanged at time zero for medium containing CD18 cross-linking–evoked PMN secretion. Means ± SD; n = 10). (b) Magnitude of TEER changes in response to stimulation of EC monolayers with the PMN postsecretory supernatant. Indicated in the figure are effects of the PMN-derived secretion on ECs pretreated with calcium-chelator BAPTA-AM or tyrosine-kinase inhibitor herbimycin A, and of challenge with PMN secretion in the presence of dextran sulfate. Also shown is the loss of activity of postsecretory supernatant after cation depletion, whereas specific immunoabsorption of cathepsin G and elastase does not significantly attenuate the TEER response. Data are means ± SD based on 6–10 experiments in each group.
The presence of cationic proteins in the secreted material prompted us to examine whether the permeability-increasing activity was in fact due to this protein fraction. This was clearly suggested to be the case, as there was no change in TEER in response to stimulation with supernatant depleted of cations (Fig. 8 b). To test the hypothesis that the activity of the postsecretory supernatant could potentially be due to charge-related effects of the neutrophil-derived cationic proteins, we sought to neutralize such activity through addition of the polyanion dextran sulfate to the medium of the EC monolayer. Indeed, in the presence of dextran sulfate (1 mg/ml), stimulation with the postsecretory supernatant proved ineffective in inducing EC permeability changes (Fig. 8 b). The ability of dextran sulfate to prevent the EC response to stimulation with cell-free supernatant raised the question of whether this treatment could also inhibit the response to direct PMN activation. This was confirmed in experiments where PMNs sedimented on ECs were activated either by antibody cross-linking of CD18 (IB4-coated PMNs) or by FMLP (untreated PMNs) in the presence of dextran sulfate (1 mg/ml). In both cases, the fall in TEER was almost completely prevented by dextran sulfate (TEER decreased only by 3 ± 5% and 5 ± 4%, respectively; means ± SD; n = 6).
Having demonstrated that the PMN-derived secretion exerted its effect on EC permeability because its polycationic nature, we went on by examining whether neutrophil-derived cathepsin G and/or elastase could be responsible for communicating this effect, as both these proteins, potentially released from the activated PMN, have been shown previously to be capable of inducing EC permeability changes. By specific immunoadsorption, there was complete removal of cathepsin G and elastase from the postsecretory supernatant, as verified by sandwich ELISA for the two proteins, respectively (data not shown). However, functional analysis revealed that the activity of the PMN-derived secretion was independent on these proteins, because postsecretory supernatant depleted of both proteins induced a fall in TEER of the same magnitude as that evoked by stimulation with native supernatant (Fig. 8 b).
CD18 Cross-Linking–evoked PMN Secretion Induces Venular Plasma Leakage In Vivo.
Topical administration of the PMN postsecretory supernatant to the hamster cheek pouch in vivo preparation elicited prompt leakage of plasma from postcapillary and small venules (Fig. 9). Leaky spots were observed within 3 min after application, and increased progressively in number and intensity with time. After wash of the tissue with buffer, the leakage slowly subsided. This effect on microvessel permeability was lost after heat treatment of the PMN-derived secretion, indicating that a heat-sensitive factor(s) was responsible for its permeability-increasing activity. Application to the cheek pouch of supernatant of IB4-coated PMN, not subjected to cross-linking, did not cause any visible plasma leakage (data not shown).
Figure 9.
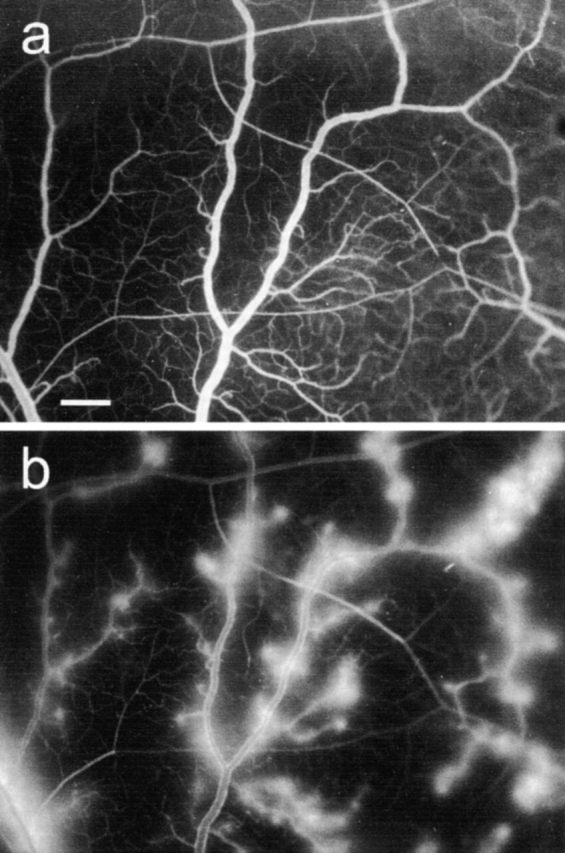
In vivo fluorescence micrographs of the hamster cheek pouch microcirculation before (a) and 10 min after (b) topical application of medium containing CD18 cross-linking–evoked PMN secretion, at 37°C. FITC-dextran injected intravenously served as plasma tracer. Leakage of plasma from postcapillary and small venules was evident within 3 min after stimulation and increased progressively. After wash of the tissue with buffer, the leaky spots disappeared slowly. Bar indicates 200 μm.
Discussion
PMN adhesion to the endothelial lining and recruitment to extravascular tissue in acute inflammation is critically dependent on the function of β2 integrins. Increased vascular permeability for macromolecules is induced in conjunction with this cellular response, leading to plasma exudation and edema formation. Here, we provide evidences for a causal connection between transmembrane signaling by the β2 integrins in PMNs and the capacity of these cells to induce alterations in EC barrier function. We show that engagement of β2 integrins initiates PMN activation and secretion that triggers an active tyrosine kinase–dependent response of ECs, involving cytoskeletal reorganization and intercellular gap formation that gives rise to increased EC permeability.
Increased vascular permeability for plasma components is a cardinal feature of acute inflammation. Besides being induced by directly acting agonists, plasma leakage is causally linked to the activation and tissue recruitment of circulating PMNs. Because of the important homeostatic function of EC as a selective barrier between blood and tissue, PMN-induced EC barrier dysfunction has been implicated in the pathogenesis of a number of inflammatory disorders. β2 integrin (CD11/CD18)–mediated PMN attachment to ECs is required for the activated PMNs to induce changes in EC integrity 2 21 22 23. Although dependent on this early physical interaction, the EC response appears unrelated, at least initially, to subsequent PMN transmigration (16, 17). To explore mechanisms by which adherent PMNs may transfer signals to ECs in this process, we simulated PMN–EC interactions and adhesion-dependent engagement of β2 integrins through antibody cross-linking of CD11/CD18. Although engagement of β2 integrins in the physiological setting requires agonist-induced PMN activation and adhesion to a biological surface, antibody cross-linking of the common β chain or the distinct α chains of β2 integrins provides a means to explore signaling properties of β2 integrins in PMN functional responses. These include, for example, mobilization of [Ca2+]i, cytoskeletal rearrangement, oxidative burst, and release of granule components. However, to date there has been no demonstration implying a causal association between PMN activation via this mode of stimulation and effects on EC barrier function. We show that antibody-induced ligation and clustering of β2 integrins triggers an increase in EC permeability matching that seen after chemoattractant-induced PMN adhesion to ECs. What is more, the cellular crosstalk initiated by CD18 cross-linking occurred in the absence of physical binding of PMNs to ECs (prevented by anti-CD18 mAb), indicating that the signal transfer from leukocyte to endothelium is not effected via a structural linkage between CD11/CD18 and counterreceptors on the EC surface. Instead, signaling is likely to be accomplished via PMN-derived secretion. Such a paracrine mechanism is further substantiated by the capacity of the cell-free supernatant, obtained after antibody cross-linking of CD18 in suspended PMN to induce EC hyperpermeability. On the other hand, mere activation of the leukocyte β2 integrins by FMLP is insufficient to elicit the secreted factor. Cell-free supernate recovered from such PMNs activated in suspension failed to induce changes in EC permeability (21; our unpublished data). It thus appears that both activation and engagement of the integrins with the endothelial surface (the latter mimicked experimentally by cross-linking the anti-CD18 antibody) is required for secretion of this factor(s).
The capacity of PMN cell surface molecules to signal alterations in EC barrier function was restricted to the β2 integrins. Antibody cross-linking of l-selectin or CD44 did not result in any functional response of ECs, and blockade of these receptor functions did not influence the EC responsiveness to chemoattractant stimulation. Interestingly, l-selectin cross-linking in PMN has been reported to initiate intracellular activation events, as indicated by increased levels of cytosolic free Ca2+ and induction of oxidative burst 24 25. In line with these observations, we have found that l-selectin cross-linking causes elevations in PMN [Ca2+]i that are similar to those obtained with CD18 cross-linking (our unpublished data). Yet, the divergent responses with regard to effect on EC barrier function indicate that distinct signaling mechanisms are elicited by engagement of l-selectin and CD11/CD18, respectively. The signal transduction pathways initiated by integrin ligation are highly complex, and the contribution of various intracellular regulatory proteins to the PMN response ultimately leading to secretion is unknown. Nevertheless, our data demonstrate that the signaling cascade initiated by ligation and clustering of β2 integrins is not only essential, but also is sufficient for triggering PMN responses that lead to alterations in EC permeability.
Antibody-induced engagement of CD11/CD18 led to secretion of PMN proteins, with apparent molecular mass in the range of 25–30 kD and seemingly responsible for mediating the PMN-evoked change in EC permeability. A vast number of proteins are kept in different storage organelles of the PMN and may be mobilized upon cell activation 26. For example, azurophil granules contain a family of antimicrobial cationic glycoproteins that have extensive homology to one another and have molecular weights similar to that found for the secreted proteins in our experiments. Members of this family, cathepsin G and elastase, have in various model systems been reported to induce EC barrier dysfunction 27 28 29 30. Presumably, this is because of their positive surface charge, as neutralization with polyanions (e.g., heparin, dextran sulfate) inhibits their capability, as well as that of cationic polyamino acids, to induce EC hyperpermeability 28 31. However, the finding that the permeability-increasing activity of the PMN-derived secretion remained after immunoabsorption of potentially released cathepsin G and/or elastase refutes the involvement of these proteins in the EC responses to PMN activation. Nevertheless, our findings that PMN-induced alterations in EC barrier function were totally prevented in the presence of dextran sulfate, combined with the observation that cation-depleted PMN secretion lost its permeability-increasing activity, suggest a mechanistic basis for PMN–EC intercellular crosstalk that involves paracrine signaling via PMN-derived cationic protein(s).
PMN activation through antibody engagement of β2 integrins initiated an active response in adjacent EC, as evidenced by rapid mobilization of cytosolic free Ca2+ and redistribution of actin filaments, leading to structural rearrangement of the EC cytoskeleton and impaired barrier function. The EC responses to CD18 cross-linking in PMNs resembled those evoked by chemoattractant-induced PMN activation 16 17, suggesting a basic principle by which activated PMNs may regulate EC barrier function. Increase in EC permeability has been shown to be associated with calcium-dependent conformational changes of the EC cytoskeleton, leading to cell contraction and intercellular gap formation 32 33. Reorganization of actin filaments is essential for these cell shape changes, and formation of F-actin may thus be an important determinant of increased EC permeability 34. We found that stimulation of ECs through PMN activation resulted in loss of peripheral actin bands and increased stress fiber density similar to what has been shown to be associated with agonist-induced (e.g., histamine, thrombin) increases in EC permeability 34 35. EC shape changes due to actin polymerization and cytoskeletal reorganization are dependent on tyrosine kinase activity 36 37. Because pretreatment of ECs with herbimycin A largely attenuated the EC permeability response to PMN activation, a coupling is provided between the PMN-derived stimulus and phosphorylation of cytoskeletal proteins, leading to impaired barrier capacity. This is in line with recent observations on activation of EC myosin light chain kinase by chemoattractant-stimulated PMNs 38 39. The critical role of calcium-dependent active processes in ECs for manifestation of the PMN-induced permeability change was further indicated by the finding that chelation of EC intracellular Ca2+ totally prevented the increase in EC permeability.
In conclusion, as demonstrated in vitro and confirmed in vivo, antibody cross-linking of β2 integrins (CD11/CD18) initiates a functional response in quiescent PMN that provokes cytoskeletal rearrangement in adjacent ECs and increase in EC permeability, which is identical to the response seen after chemoattractant-induced PMN activation. Our data suggest, with reference to the inflammatory reaction in vivo, that stimulus-induced PMN adhesion to the vascular endothelium initiates PMN secretion consequent to β2 integrin signaling, which capacitates changes in endothelial barrier function that are independent of PMN transmigration. These findings provide novel information with regard to the role of β2 integrins in PMN-evoked effects on vessel wall permeability in conjunction with neutrophil trafficking in inflammation.
Acknowledgments
This study was supported by the Swedish Medical Research Council (grants 14X-4342 and 04P-10738), the Swedish Foundation for Health Care Sciences and Allergy Research (grant A98110), the Swedish National Board for Laboratory Animals, the IngaBritt and Arne Lundbergs Foundation, and the Karolinska Institutet.
Footnotes
Abbreviations used in this paper: BAEC, bovine aorta EC; [Ca2+]i, intracellular free Ca2+; EBA, Evans blue dye-conjugated albumin; EC, endothelial cell; HUVEC, human umbilical vein endothelial cell; TEER, transendothelial electrical resistance.
References
- Wedmore C.V., Williams T.J. Control of vascular permeability by polymorphonuclear leukocytes in inflammation. Nature. 1981;289:646–650. doi: 10.1038/289646a0. [DOI] [PubMed] [Google Scholar]
- Arfors K.E., Lundberg C., Lindbom L., Lundberg K., Beatty P.G., Harlan J.M. A monoclonal antibody to the membrane glycoprotein complex CD18 inhibits polymorphonuclear leukocyte accumulation and plasma leakage in vivo. Blood. 1987;69:338–340. [PubMed] [Google Scholar]
- Kaslovsky R.A., Horgan M.J., Lum H., McCandless B.K., Gilboa N., Wright S.D., Malik A.B. Pulmonary edema induced by phagocytosing neutrophils. Protective effect of monoclonal antibody against phagocyte CD18 integrin. Circ. Res. 1990;67:795–802. doi: 10.1161/01.res.67.4.795. [DOI] [PubMed] [Google Scholar]
- Lindbom L., Lundberg C., Prieto J., Raud J., Nortamo P., Gahmberg C.G., Patarroyo M. Rabbit leukocyte adhesion molecules CD11/CD18 and their participation in acute and delayed inflammatory responses and leukocyte distribution in vivo. Clin. Immunol. Immunopathol. 1990;57:105–119. doi: 10.1016/0090-1229(90)90026-m. [DOI] [PubMed] [Google Scholar]
- Hynes R.O. Integrinsversatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- Lowell C.A., Berton G. Integrin signal transduction in myeloid leukocytes. J. Leukoc. Biol. 1999;65:313–320. doi: 10.1002/jlb.65.3.313. [DOI] [PubMed] [Google Scholar]
- Nathan C., Srimal S., Farber C., Sanchez E., Kabbash L., Asch A., Gailit J., Wright S.D. Cytokine-induced respiratory burst of human neutrophilsdependence on extracellular matrix proteins and CD11/CD18 integrins. J. Cell Biol. 1989;109:1341–1349. doi: 10.1083/jcb.109.3.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaconi M.E., Theler J.M., Schlegel W., Appel R.D., Wright S.D., Lew P.D. Multiple elevations of cytosolic-free Ca2+ in human neutrophilsinitiation by adherence receptors of the integrin family. J. Cell Biol. 1991;112:1249–1257. doi: 10.1083/jcb.112.6.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berton G., Yan S.R., Fumagalli L., Lowell C.A. Neutrophil activation by adhesionmechanisms and pathophysiological implications. Int. J. Clin. Lab. Res. 1996;26:160–177. doi: 10.1007/BF02592978. [DOI] [PubMed] [Google Scholar]
- Ng-Sikorski J., Andersson R., Patarroyo M., Andersson T. Calcium signaling capacity of the CD11b/CD18 integrin on human neutrophils. Exp. Cell Res. 1991;195:504–508. doi: 10.1016/0014-4827(91)90402-g. [DOI] [PubMed] [Google Scholar]
- Petersen M., Williams J.D., Hallett M.B. Cross-linking of CD11b or CD18 signals release of localized Ca2+ from intracellular stores in neutrophils. Immunology. 1993;80:157–159. [PMC free article] [PubMed] [Google Scholar]
- Walzog B., Seifert R., Zakrzewicz A., Gaehtgens P., Ley K. Cross-linking of CD18 in human neutrophils induces an increase of intracellular free Ca2+, exocytosis of azurophilic granules, quantitative up-regulation of CD18, shedding of l-selectin, and actin polymerization. J. Leukoc. Biol. 1994;56:625–635. doi: 10.1002/jlb.56.5.625. [DOI] [PubMed] [Google Scholar]
- Berton G., Fumagalli L., Laudanna C., Sorio C. Beta 2 integrin–dependent protein tyrosine phosphorylation and activation of the FGR protein tyrosine kinase in human neutrophils. J. Cell Biol. 1994;126:1111–1121. doi: 10.1083/jcb.126.4.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L., Sjolander A., Eckerdal J., Andersson T. Antibody-induced engagement of beta 2 integrins on adherent human neutrophils triggers activation of p21ras through tyrosine phosphorylation of the protooncogene product Vav. Proc. Natl. Acad. Sci. USA. 1996;93:8431–8436. doi: 10.1073/pnas.93.16.8431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lofgren R., Ng-Sikorski J., Sjolander A., Andersson T. Beta 2 integrin engagement triggers actin polymerization and phosphatidylinositol trisphosphate formation in non-adherent human neutrophils. J. Cell Biol. 1993;123:1597–1605. doi: 10.1083/jcb.123.6.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam N., Hedqvist P., Lindbom L. Kinetics of leukocyte-induced changes in endothelial barrier function. Br. J. Pharmacol. 1998;125:1109–1114. doi: 10.1038/sj.bjp.0702186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang A.J., Manning J.E., Bandak T.M., Ratau M.C., Hanser K.R., Silverstein S.C. Endothelial cell cytosolic free calcium regulates neutrophil migration across monolayers of endothelial cells. J. Cell Biol. 1993;120:1371–1380. doi: 10.1083/jcb.120.6.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K., Ota H., Sasagawa S., Sakatani T., Fujikura T. Assay method for myeloperoxidase in human polymorphonuclear leukocytes. Anal. Biochem. 1983;132:345–352. doi: 10.1016/0003-2697(83)90019-2. [DOI] [PubMed] [Google Scholar]
- Raud J., Lindbom L. Studies by intravital microscopy of basic inflammatory reactions and acute allergic inflammation. In: Brain S., editor. The Handbook of Immunopharmacology. Academic Press; London: 1994. pp. 127–170. [Google Scholar]
- Lindbom L., Xie X., Raud J., Hedqvist P. Chemoattractant-induced firm adhesion of leukocytes to vascular endothelium in vivo is critically dependent on initial leukocyte rolling. Acta Physiol. Scand. 1992;146:415–421. doi: 10.1111/j.1748-1716.1992.tb09442.x. [DOI] [PubMed] [Google Scholar]
- Harlan J.M., Schwartz B.R., Reidy M.A., Schwartz S.M., Ochs H.D., Harker L.A. Activated neutrophils disrupt endothelial monolayer integrity by an oxygen radical-independent mechanism. Lab. Invest. 1985;52:141–150. [PubMed] [Google Scholar]
- Lum H., Gibbs L., Lai L., Malik A.B. CD18 integrin-dependent endothelial injuryeffects of opsonized zymosan and phorbol ester activation. J. Leukoc. Biol. 1994;55:58–63. doi: 10.1002/jlb.55.1.58. [DOI] [PubMed] [Google Scholar]
- Del Maschio A., Zanetti A., Corada M., Rival Y., Ruco L., Lampugnani M.G., Dejana E. Polymorphonuclear leukocyte adhesion triggers the disorganization of endothelial cell-to-cell adherens junctions. J. Cell Biol. 1996;135:497–510. doi: 10.1083/jcb.135.2.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crockett-Torabi E., Sulenbarger B., Smith C.W., Fantone J.C. Activation of human neutrophils through l-selectin and Mac-1 molecules. J. Immunol. 1995;154:2291–2302. [PubMed] [Google Scholar]
- Waddell T.K., Fialkow L., Chan C.K., Kishimoto T.K., Downey G.P. Signaling functions of l-selectin. Enhancement of tyrosine phosphorylation and activation of MAP kinase. J. Biol. Chem. 1995;270:15403–15411. doi: 10.1074/jbc.270.25.15403. [DOI] [PubMed] [Google Scholar]
- Borregaard N., Cowland J.B. Granules of the human neutrophilic polymorphonuclear leukocyte. Blood. 1997;89:3503–3521. [PubMed] [Google Scholar]
- Smedly L.A., Tonnesen M.G., Sandhaus R.A., Haslett C., Guthrie L.A., Johnston R.B.J., Henson P.M., Worthen G.S. Neutrophil-mediated injury to endothelial cells. Enhancement by endotoxin and essential role of neutrophil elastase. J. Clin. Invest. 1986;77:1233–1243. doi: 10.1172/JCI112426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson M.W., Stone P., Shasby D.M. Cationic neutrophil proteins increase transendothelial albumin movement. J. Appl. Physiol. 1987;62:1521–1530. doi: 10.1152/jappl.1987.62.4.1521. [DOI] [PubMed] [Google Scholar]
- Peterson M.W., Gruenhaupt D., Shasby D.M. Neutrophil cathepsin G increases calcium flux and inositol polyphosphate production in cultured endothelial cells. J. Immunol. 1989;43:609–616. [PubMed] [Google Scholar]
- Carden D., Xiao F., Moak C., Willis B.H., Robinson-Jackson S., Alexander S. Neutrophil elastase promotes lung microvascular injury and proteolysis of endothelial cadherins. Am. J. Physiol. 1998;275:H385–392. doi: 10.1152/ajpheart.1998.275.2.H385. [DOI] [PubMed] [Google Scholar]
- Rosengren S., Arfors K.E. Polycations induce microvascular leakage of macromolecules in hamster cheek pouch. Inflammation. 1991;15:159–172. doi: 10.1007/BF00918643. [DOI] [PubMed] [Google Scholar]
- Lum H., Malik A.B. Regulation of vascular endothelial barrier function. Am. J. Physiol. 1994;267:L223–241. doi: 10.1152/ajplung.1994.267.3.L223. [DOI] [PubMed] [Google Scholar]
- Garcia J.G., Schaphorst K.L. Regulation of endothelial cell gap formation and paracellular permeability. J. Investig. Med. 1995;43:117–126. [PubMed] [Google Scholar]
- Phillips P.G., Lum H., Malik A.B., Tsan M.F. Phallacidin prevents thrombin-induced increases in endothelial permeability to albumin. Am. J. Physiol. 1989;257:C562–567. doi: 10.1152/ajpcell.1989.257.3.C562. [DOI] [PubMed] [Google Scholar]
- Ehringer W.D., Edwards M.J., Miller F.N. Mechanisms of alpha-thrombin, histamine, and bradykinin induced endothelial permeability. J. Cell. Physiol. 1996;167:562–569. doi: 10.1002/(SICI)1097-4652(199606)167:3<562::AID-JCP20>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Yuan Y., Meng F.Y., Huang Q., Hawker J., Wu H.M. Tyrosine phosphorylation of paxillin/pp125FAK and microvascular endothelial barrier function. Am. J. Physiol. 1998;275:H84–93. doi: 10.1152/ajpheart.1998.275.1.H84. [DOI] [PubMed] [Google Scholar]
- van Nieuw Amerongen G.P., Draijer R., Vermeer M.A., van Hinsbergh V.W. Transient and prolonged increase in endothelial permeability induced by histamine and thrombinrole of protein kinases, calcium, and RhoA. Circ. Res. 1998;83:1115–1123. doi: 10.1161/01.res.83.11.1115. [DOI] [PubMed] [Google Scholar]
- Hixenbaugh E.A., Goeckeler Z.M., Papaiya N.N., Wysolmerski R.B., Silverstein S.C., Huang A.J. Stimulated neutrophils induce myosin light chain phosphorylation and isometric tension in endothelial cells. Am. J. Physiol. 1997;273:H981–988. doi: 10.1152/ajpheart.1997.273.2.H981. [DOI] [PubMed] [Google Scholar]
- Garcia J.N., Verin A.D., Herenyiova M., English D. Adherent neutrophils activate endothelial myosin light chain kinaserole in transendothelial migration. J. Appl. Physiol. 1998;84:1817–1821. doi: 10.1152/jappl.1998.84.5.1817. [DOI] [PubMed] [Google Scholar]