

Abstract
Purpose of review
The deciphering of the human genome sequence has enabled the identification of genetic polymorphisms that are responsible for inter-individual variation in the response to drug therapy. This field is referred to as pharmacogenetics. We review the impact of pharmacogenetics on therapy in diseases of the colon using three common variant enzyme systems as examples.
Recent findings
Many enzyme systems impact the treatment of diseases of the colon. Examples include thiopurine S-methyltransferase, dihydropyrimidine dehydrogenase and flavin monooxygenase 3. They affect the management of inflammatory bowel disease, colorectal cancer and the chemoprevention of colorectal adenoma by influencing the metabolism of their respective substrates, azathioprine/6-mercaptopurine, 5-fluorouracil and sulindac. Recent studies have implicated the significance of genetic polymorphisms in each of the three drug-metabolizing enzymes, which impacts on the therapeutic outcome of the stated diseases. These studies highlight the potential role of pharmacogenetics in the design of a therapeutic plan which would increase efficacy and limit toxicity.
Summary
Pharmacogenetics of drug-metabolizing systems continues to gain significance in the drug therapy of a variety of disease states including those of the gastrointestinal tract.
Keywords: colorectal cancer, dihydropyrimidine dehydrogenase, flavin monooxygenase 3, inflammatory bowel disease, pharmacogenetics, thiopurine S-methyltransferase
Introduction
The therapeutic response to a drug depends on both genetic and environmental factors. The determinants of response can be better understood by defining two categories – pharmacodynamics and pharmacokinetics. Pharmacodynamics is the study of the biochemical and physiological effects of drugs, the mechanism of drug action, and the relationship between drug concentrations and therapeutic effects. Pharmacokinetics is the study of the therapeutic response to drugs as determined by drug absorption, distribution, metabolism and excretion by a variety of pathways. In layman's terms, pharmacodynamics is the study of what a drug does to the body, whereas pharmacokinetics is the study of what the body does to a drug. An understanding of the pharmacokinetics and pharmacodynamics of drugs is particularly important for those drugs that have a narrow therapeutic index.
Recent advances in deciphering the human genome sequence have introduced the additional concepts of pharmacogenetics and pharmacogenomics. The terms pharmacogenomics and pharmacogenetics tend to be used interchangeably and a precise, consensus definition of either remains elusive. In general, pharmacogenetics refers to the study of genetic variations that give rise to differing response to drugs, while pharmacogenomics is the broader application of genomic technologies to new drug discovery and further characterization of older drugs (see earlier articles [1–5] that summarize the development of pharmacogenetics and pharmacogenomics, describe the important application of pharmacogenetics using cancer therapy as a clinical model, and review the pharmacogenetics of phase I and II drug metabolism and reinforce the significance of the genotype to phenotype relation in pharmacogenetics). Much of current clinical interest is at the level of pharmacogenetics that examines variation in genes involved in drug metabolism with a particular emphasis on improving drug efficacy and safety.
Single-nucleotide polymorphisms (SNPs) account for greater than 90% of the variation in the human genome. The remaining variations are accounted for by insertions and deletions, tandem repeats, and microsatellites [6]. An estimated 1.42 million SNPs are distributed throughout the human genome, providing an average density on available sequence of one SNP every 1.9 kb. Approximately 60 000 SNPs fall within exons (the coding and untranslated regions of a gene). Eight-five percent of exons are within 5 kb of the nearest SNP [7]. Several methods have been devised to study these polymorphisms, including assays that use allele-specific oligonucleotide probes or nucleic acid-modifying enzymes as genotyping tools and assays that use primer extension catalyzed by a DNA polymerase or DNA ligation or cleavage by endonucleases [8].
The impact of genetic polymorphisms/SNPs on the therapeutics of a variety of medical disorders has improved our understanding of drug benefit/toxicity ratio. As such, pharmacogenetics is being more consistently applied to clinical practice as a tool for selecting appropriate drugs or dosages for individualized therapeutic plans. Genetic variations in several enzyme systems involved in the metabolism of drugs utilized for the treatment of a variety of gastrointestinal disorders have been well documented. Table 1 illustrates some of the commonly studied drug-metabolizing enzymes that have an impact on the therapeutic efficacies in specific gastrointestinal diseases. We review three specific drug-metabolizing systems, thiopurine S-methyltransferase (TPMT), dihydropyrimidine dehydrogenase (DPD) and flavin monooxygenase 3 (FMO3). These systems are involved in the metabolism of thiopurines [azathioprine (AZA) and 6-mercaptopurine (6-MP)], 5-fluorouracil (5-FU) and sulindac sulfide, respectively. These drugs are utilized in the management of inflammatory bowel disease, colorectal cancer and the chemoprevention of colorectal adenoma, respectively. Variations in the activity of these enzyme systems illustrate the application of the principle of pharmacogenetics towards the design of appropriate therapeutic interventions.
Table 1. Examples of gastrointestinal diseases with the therapeutic medications used and the polymorphisms in their metabolizing enzymes or targets.
| Disease condition | Medication | Enzyme system |
|---|---|---|
| Inflammatory bowel disease | azathioprine | thiopurine S-methyltransferase |
| 6-mercaptopurine | thiopurine S-methyltransferase | |
| Colorectal cancer | 5-fluorouracil | dihydropyrimidine dehydrogenase/thymidylate synthase/xeroderma pigmentosum, complementation group D |
| irinotecan | uridine diphosphate glucuronosyltransferase/carboxylesterase | |
| oxaliplatin | xeroderma pigmentosum, complementation group D/excision-repair, complementing defective, in Chinese hamster-1/glutathione S-transferase π-1 | |
| Colorectal adenoma | sulindac sulfide | flavin monooxygenase 3 |
| aspirin | uridine diphosphate glucuronosyltransferase 1A6 |
Thiopurine S-methyltransferase
Crohn's disease is a chronic inflammatory disease of the gastrointestinal tract. The precise etiology of Crohn's disease is unknown, although a genetic susceptibility to innocuous environmental or infectious stimuli is suspected [9]. The medical management of Crohn's disease includes the use of several classes of drugs including 5-aminosalicylic acid-containing compounds such as sulfasalazine, corticosteroids, antibiotics, anti-tumor necrosis factor-α and immunomodulators or immunosuppressants. Among the immunomodulators, the most widely used are the thiopurines, which include AZA and 6-MP. Azathioprine is the prodrug form of 6-MP and is nonenzymatically converted in tissues to 6-MP. It is widely used as an adjunctive immunosuppressive and steroid-sparing agent in the management of Crohn's disease. It is also utilized in rheumatology, dermatology, solid organ transplant recipients and childhood leukemia. AZA and 6-MP are rapidly absorbed, evenly distributed and do not cross the blood–brain barrier [10].
After the conversion of AZA to 6-MP, 6-MP is further metabolized by three different enzymatic pathways (Fig. 1). The major pathway converts 6-MP to the inactive 6-methyl mercaptopurine via the enzyme TPMT. The second pathway converts 6-MP to 6-thiouric acid via the enzyme xanthine oxidase. The last pathway converts 6-MP to 6-thioinosine 5-monophosphate via the enzyme hypoxanthine guanine phosphoribosyl transferase. 6-Thioinosine 5-monophosphate can either be metabolized to 6-methyl mercaptopurine by TPMT or to the biologically active metabolites 6-thioguanine nucleotides (6-TGN) by inosine monophosphate dehydrogenase/guanine monophosphate synthase pathway. Erythrocyte 6-TGN levels are associated with therapeutic response to 6-MP in patients with inflammatory bowel disease [11]. A reduced activity in TPMT would shift the pathways toward the generation of 6-TGN. Excessive accumulation of 6-TGN has been reported to result in life-threatening leucopenia [12–15].
Figure 1. Pathways of azathioprine and 6-mercaptoprine metabolism.
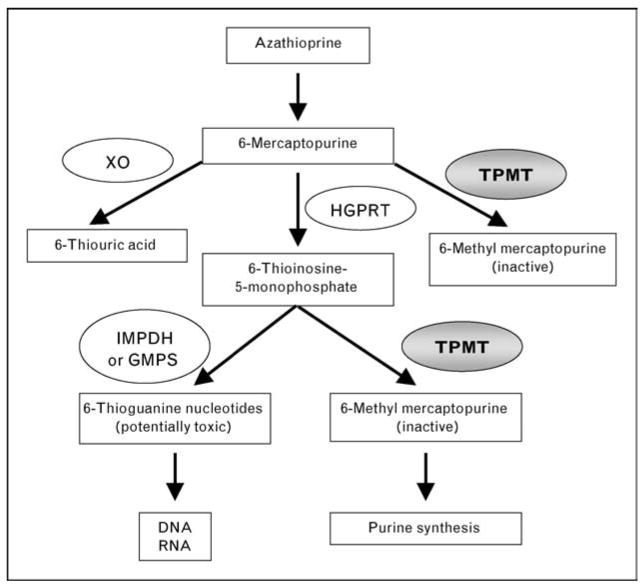
TPMT, thiopurine S-methyltransferase; XO, xanthine oxidase; HGPRT, hypoxanthine guanine phosphoribosyl transferase; IMPDH, inosine monophosphate dehydrogenase; GMPS, guanine monophosphate synthase.
TPMT activity can be measured in erythrocytes. There is a positive correlation between activity levels in erythrocytes and other tissues [16–18]. It is clear from the pathways illustrated in Fig. 1 that TPMT activity is important in determining both the rate of metabolism and formation of particular metabolic products of AZA and 6-MP. Variation in the serum levels of AZA and 6-MP can have several clinical consequences, both desirable and undesirable. The toxicity results from either excessive accumulation of the parent compounds or the metabolic byproducts. It has been estimated that significant drug toxicity leads to cessation of therapy in 9–25% of patients and to failure in achieving the desired therapeutic dosage in approximately 15% of cases [14]. Patients with significantly low TPMT activity are at risk of excessive accumulation of the active metabolites, 6-thioguanine nucleotides (6-TGN), with resultant myelosuppression [19,20]. This is due to the fact that the metabolism will be shifted towards the inosine monophosphate dehydrogenase/guanine monophosphate synthase pathway (Fig. 1). Patients with intermediate TPMT activity have an intermediate risk for toxicity. The importance of assessing the activity of TPMT has been confirmed in studies involving other drugs metabolized primarily by it. There is an association between low activity and the development of secondary brain tumors, myelodysplasia and acute myeloid leukemia in children treated with etoposide [21].
The TPMT gene is localized to chromosome band 6p22.3. It encodes a protein with 245 amino acid residues with a predicted molecular weight of 35 kDa [22]. The identification of SNPs that result in a significant reduction of TPMT activity has assisted in deciding on the appropriate dosing of immunomodulatory therapy. This leads to an optimization of the therapeutic benefit/toxic effects ratio. Ninety percent of individuals have high TPMT activity, 10% have an intermediate activity due to heterozygous mutations and 0.3% have no activity due to homozygous polymorphisms (mutations) [23,24]. The first SNP identified in the TPMT gene was designated as TPMT*2 (G238C). It involves the substitution of a proline for alanine in amino acid position 80 (A80P) [25]. This results in a decreased activity level. Transitions at another allele, TPMT*3A (G460A and A719G), are also associated with decreased activity [26]. Population studies have demonstrated that TPMT*3A mutations are the most common cause of reduced activity in American (allelic frequency of 3.2%), British (4.5%), and French (5.7%) Caucasians, while TPMT*3C (A719G) is more common in African Americans (2.4%), Kenyans (5.4%), Ghanians (7.6%), Chinese (2.3%) and Japanese (1.5%) [27,28]. Several additional polymorphic alleles have been described (Table 2).
Table 2. Allelic polymorphisms of thiopurine S-methyltransferse and their representative phenotypes.
| Allele | Genotype | Phenotype |
|---|---|---|
| 1 | wild-type | high metabolizer |
| 1A | C178T | high metabolizer |
| 1S | T474C | high metabolizer |
| 2 | G238C | intermediate metabolizer |
| 3A | G460A + A719G | intermediate metabolizer |
| 3B | G460A | intermediate metabolizer |
| 3C | A719G | intermediate metabolizer |
| 3D, 4–8, 10 | N/A | minimal effect |
TPMT genotyping is primarily a DNA-based polymerase chain reaction amplification of genomic DNA followed by detection of individual SNPs by various methods including restriction fragment length polymorphism, allele-specific amplification [6], direct sequencing, single-strand conformation polymorphism Yates et al. [24] showed a high degree of concordance between a genotype and its phenotypic expression with polymerase chain reaction-based genotyping methods. A pharmacoeconomic model developed to analyze the potential cost of screening for TPMT polymorphisms prior to AZA therapy showed that it improves outcomes and reduces the total cost of care for steroid-treated chronically active Crohn's disease patients [30]. In addition, compliance and bioavailability are additional factors which could cause myelosuppression independent of TPMT activity in patients receiving immunomodulatory therapy. As a result, some in the gastrointestinal community recommend TPMT genotyping or enzymatic activity determination prior to instituting AZA therapy in patients with inflammatory bowel disease [31].
Dihydropyrimidine dehydrogenase
5-FU, an antimetabolite that inhibits thymidylate synthase, is the most commonly used drug in the treatment of gastrointestinal malignancies, including gastric and colorectal carcinomas. It is metabolically degraded by a rate-limiting enzyme, DPD [32]. DPD deficiency has been recognized as an important risk factor in predisposing patients to the development of severe 5-FU-associated toxicity. The therapeutic effect and the toxicities related to 5-FU are also determined by factors such as age, high serum alkaline phosphatase, the elapsed time from infusion and whether the medication is administered as a bolus or a continuous infusion [33]. There are inter-individual and inter-ethnic differences in the levels of DPD activity [34]. Mechanisms such as gene deletion, exon skipping and missense mutations may account for this difference in activity [35–38].
The human DPD gene has been mapped to chromosome 1p22. It consists of 23 exons, is 950 kb in length and includes 3 kb of coding sequences [39]. DPD is expressed at high levels by both hepatocytes and mononuclear cells. Three percent of Caucasians are partially deficient in DPD activity with no clinical consequences [40]. They are, however, at risk for developing toxicity if exposed to 5-FU due to its narrow therapeutic index. DPD activity is inversely correlated with clearance of 5-FU. Tumors expressing low levels of DPD as reflected by low mRNA levels or DPD activity show a significantly better response to 5-FU compared to tumors with high DPD activity or high mRNA levels [41,42]. In addition, low intra-tumoral DPD gene expression appears to be associated with a much longer survival period for patients [42].
Fourteen SNPs have been identified in the DPD gene. The most common polymorphism is a G→A point mutation within the 5′-splicing site of intron 14 (exon skipping mutation, IVS14+1G→A) [43]. Heterozygote and homozygote mutations result in reduced DPD levels/activity and can lead to severe 5-FU-induced toxicity. The onset of 5-FU toxicity appears twice as rapidly in patients with a partial DPD deficiency as compared to patients with normal DPD activity [44].
Mutations in the DPD gene result in clinically significant differences in the metabolism of 5-FU. This causes an alteration in the benefit/toxicity ratio associated with this medication. Since 5-FU-based chemotherapy is important in treating gastrointestinal malignancies and given the high prevalence of DPD mutations, genetic screening for the presence of these mutations may be warranted prior to the administration of 5-FU [45••].
Flavin monooxygenase 3
FMO3 is a hepatic microsomal enzyme that is involved in the metabolism of a host of heteroatom-containing chemicals and drugs, such as trimethylamine, clonazapine, nicotine and ranitidine [46,47] (the latter reference defines the known FMO3 polymorphisms and discusses the ones that are associated with a significant impact on the enzyme activity). Studies indicate that there are substantial inter-individual and inter-ethnic differences in FMO3 activities [48]. Much of this variation is contributed by SNPs, of which 26 have been identified to date [49]. Most of these polymorphisms result in variant enzymes with amino acid substitutions, some of which lead to decreased enzyme activity [47]. The identification of these polymorphisms defined the genetic etiology of trimethylaminuria [49], also called fish odor syndrome. It also facilitated the understanding of the variation in responses of individuals to different drugs metabolized by FMO3.
Sulindac sulfide, a nonsteroidal antiinflammatory drug, is metabolized by FMO3 [50]. Sulindac has a chemopreventive effect, causing a reduction in the development of adenomatous polyps in the hereditary disorder, familial adenomatous polyposis (FAP) [51]. Oral sulindac is a prodrug that contains a racemic mixture of a sulfoxide moiety. This moiety is reduced by the gut flora to the active sulfide form prior to its absorption. In turn, the sulfide form is re-oxidized by FMO3 to the sulfoxide form. Further oxidation results in the sulfone form. The latter two compounds (sulfoxide and sulfone) are relatively inactive when compared with the sulfide form [52] (Fig. 2). Thus, the activity of FMO3 is a potential determinant of the levels of the active sulindac sulfide in the body.
Figure 2. Pathways of sulindac sulfide metabolism.

The figure shows the oxidative and reductive biotransformations of sulindac in vivo. Racemic sulindac sulfoxide, a prodrug, is reversibly reduced to the active sulfide metabolite by the gut flora (Step 1), which is re-oxidized to either the R- or S-sulfoxide enantiomer (Step 2). The sulfoxides are also irreversibly oxidized further to a sulfone metabolite (Step 3). Both oxidation steps are mediated by flavin monooxygenase 3.
Randomized, placebo-controlled trials have demonstrated that sulindac could reduce the polyp burden (measured as number and size) in patients with FAP [51,53]. A survey of seven established FMO3 polymorphisms in these patients suggests that two particular alleles, E158K and E308G, are associated with a more favorable response to sulindac, showing a significant reduction in tissue prostaglandin levels and response to primary chemoprevention with sulindac therapy in FAP patients [54]. Both of the polymorphic alleles are associated with reduced FMO3 activity [47]. This may then account for the increased efficacy of sulindac in regressing polyposis. This observation is supported by the finding of an independent study that demonstrated an association between the E158K and E308G alleles and a primary chemopreventive effect of sulindac in FAP [55,56]. The study showed that patients with the two FMO3 alleles had significantly reduced levels of prostaglandin metabolites in their rectal mucosa in response to sulindac therapy [55]. This was correlated with a more favorable response to the drug [57]. These findings are therefore highly suggestive that polymorphisms at the E158K and E308G alleles may reduce FMO3's ability to inactivate sulindac sulfide, which then results in an increased efficacy in the prevention of polyposis in FAP.
Conclusion
The current application of pharmacogenetics is limited to certain drug-metabolizing enzyme systems. Widespread application of this concept of ‘individualized medicine’ will likely become a matter of practice in the near future. As this review has shown, the principal of pharmacogenetics has an impact on the practice of gastroenterology. For example, TPMP genotyping or enzymatic activity determination is currently recommended prior to instituting AZA therapy and has shown to have a positive impact on disease management. It is expected that the use of pharmacogenetics will only grow as the field develops, the sensitivity and specificity of the tests improve, and economies of scale are applied. This will have the benefit of targeting selected patients with the proper medication/dosages and, above all, will reduce the risk for unnecessary toxicities.
Acknowledgments
Sponsorship: This work was in part supported by grants from the National Institutes of Health (DK52230 and CA84197). V.W.Y. is the recipient of a Georgian Cancer Coalition Distinguished Cancer Clinician Scientist Award.
Abbreviations
- 5-FU
5-fluorouracil
- 6-MP
6-mercaptopurine
- 6-TGN
6-thioguanine nucleotides
- AZA
azathioprine
- DPD
dihydropyrimidine dehydrogenase
- FAP
familial adenomatous polyposis
- FMO3
flavin monooxygenase 3
- SNP
single-nucleotide polymorphism
- TPMT
thiopurine S-methyltransferase
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
Additional references related to this topic can also be found in the Current World Literature section in this issue (pp. 86–89).
- 1.Weinshilboum R, Wang L. Pharmacogenomics: bench to bedside. Nat Rev Drug Discov. 2004;3:739–748. doi: 10.1038/nrd1497. [DOI] [PubMed] [Google Scholar]
- 2.Goetz MP, Ames MM, Weinshilboum RM. Primer on medical genomics. Part XII: pharmacogenomics – general principles with cancer as a model. Mayo Clin Proc. 2004;79:376–384. doi: 10.4065/79.3.376. [DOI] [PubMed] [Google Scholar]
- 3.Roden DM, George AL., Jr The genetic basis of variability in drug responses. Nat Rev Drug Discov. 2002;1:37–44. doi: 10.1038/nrd705. [DOI] [PubMed] [Google Scholar]
- 4.Weinshilboum R. Inheritance and drug response. N Engl J Med. 2003;348:529–537. doi: 10.1056/NEJMra020021. [DOI] [PubMed] [Google Scholar]
- 5.Krynetski EY, Evans WE. Pharmacogenetics of cancer therapy: getting personal. Am J Hum Genet. 1998;63:11–16. doi: 10.1086/301941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 7.Sachidanandam R, Weissman D, Schmidt SC, et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 2001;409:928–933. doi: 10.1038/35057149. [DOI] [PubMed] [Google Scholar]
- 8.Syvanen AC. Accessing genetic variation: genotyping single nucleotide polymorphisms. Nat Rev Genet. 2001;2:930–942. doi: 10.1038/35103535. [DOI] [PubMed] [Google Scholar]
- 9.Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347:417–429. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- 10.El-Azhary RA. Azathioprine: current status and future considerations. Int J Dermatol. 2003;42:335–341. doi: 10.1046/j.1365-4362.2003.01823.x. [DOI] [PubMed] [Google Scholar]
- 11.Dubinsky MC, Lamothe S, Yang HY, et al. Pharmacogenomics and metabolite measurement for 6-mercaptopurine therapy in inflammatory bowel disease. Gastroenterology. 2000;118:705–713. doi: 10.1016/s0016-5085(00)70140-5. [DOI] [PubMed] [Google Scholar]
- 12.Duley JA, Florin TH. Thiopurine therapies: problems, complexities, and progress with monitoring thioguanine nucleotides. Ther Drug Monit. 2005;27:647–654. doi: 10.1097/01.ftd.0000169061.52715.3e. [DOI] [PubMed] [Google Scholar]
- 13.Coulthard S, Hogarth L. The thiopurines: an update. Invest New Drugs. 2005;23:523–532. doi: 10.1007/s10637-005-4020-8. [DOI] [PubMed] [Google Scholar]
- 14.Gearry RB, Barclay ML. Azathioprine and 6-mercaptopurine pharmacogenetics and metabolite monitoring in inflammatory bowel disease. J Gastroenterol Hepatol. 2005;20:1149–1157. doi: 10.1111/j.1440-1746.2005.03832.x. [DOI] [PubMed] [Google Scholar]
- 15.Gearry RB, Barclay ML, Roberts RL, et al. Thiopurine methyltransferase and 6-thioguanine nucleotide measurement: early experience of use in clinical practice. Intern Med J. 2005;35:580–585. doi: 10.1111/j.1445-5994.2005.00904.x. [DOI] [PubMed] [Google Scholar]
- 16.McLeod HL, Relling MV, Liu Q, et al. Polymorphic thiopurine methyltransferase in erythrocytes is indicative of activity in leukemic blasts from children with acute lymphoblastic leukemia. Blood. 1995;85:1897–1902. [PubMed] [Google Scholar]
- 17.Szumlanski CL, Honchel R, Scott MC, Weinshilboum RM. Human liver thiopurine methyltransferase pharmacogenetics: biochemical properties, liver-erythrocyte correlation and presence of isozymes. Pharmacogenetics. 1992;2:148–159. [PubMed] [Google Scholar]
- 18.Woodson LC, Dunnette JH, Weinshilboum RM. Pharmacogenetics of human thiopurine methyltransferase: kidney-erythrocyte correlation and immunotitration studies. J Pharmacol Exp Ther. 1982;222:174–181. [PubMed] [Google Scholar]
- 19.Black AJ, McLeod HL, Capell HA, et al. Thiopurine methyltransferase genotype predicts therapy-limiting severe toxicity from azathioprine. Ann Intern Med. 1998;129:716–718. doi: 10.7326/0003-4819-129-9-199811010-00007. [DOI] [PubMed] [Google Scholar]
- 20.Relling MV, Hancock ML, Rivera GK, et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst. 1999;91:2001–2008. doi: 10.1093/jnci/91.23.2001. [DOI] [PubMed] [Google Scholar]
- 21.Relling MV, Yanishevski Y, Nemec J, et al. Etoposide and antimetabolite pharmacology in patients who develop secondary acute myeloid leukemia. Leukemia. 1998;12:346–352. doi: 10.1038/sj.leu.2400928. [DOI] [PubMed] [Google Scholar]
- 22.Szumlanski C, Otterness D, Her C, et al. Thiopurine methyltransferase pharmacogenetics: human gene cloning and characterization of a common polymorphism. DNA Cell Biol. 1996;15:17–30. doi: 10.1089/dna.1996.15.17. [DOI] [PubMed] [Google Scholar]
- 23.Weinshilboum RM, Sladek SL. Mercaptopurine pharmacogenetics: monogenic inheritance of erythrocyte thiopurine methyltransferase activity. Am J Hum Genet. 1980;32:651–662. [PMC free article] [PubMed] [Google Scholar]
- 24.Yates CR, Krynetski EY, Loennechen T, et al. Molecular diagnosis of thiopurine S-methyltransferase deficiency: genetic basis for azathioprine and mercaptopurine intolerance. Ann Intern Med. 1997;126:608–614. doi: 10.7326/0003-4819-126-8-199704150-00003. [DOI] [PubMed] [Google Scholar]
- 25.Krynetski EY, Schuetz JD, Galpin AJ, et al. A single point mutation leading to loss of catalytic activity in human thiopurine S-methyltransferase. Proc Natl Acad Sci U S A. 1995;92:949–953. doi: 10.1073/pnas.92.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tai HL, Krynetski EY, Yates CR, et al. Thiopurine S-methyltransferase deficiency: two nucleotide transitions define the most prevalent mutant allele associated with loss of catalytic activity in Caucasians. Am J Hum Genet. 1996;58:694–702. [PMC free article] [PubMed] [Google Scholar]
- 27.Tai HL, Krynetski EY, Schuetz EG, et al. Enhanced proteolysis of thiopurine S-methyltransferase (TPMT) encoded by mutant alleles in humans (TPMT*3A, TPMT*2): mechanisms for the genetic polymorphism of TPMT activity. Proc Natl Acad Sci U S A. 1997;94:6444–6449. doi: 10.1073/pnas.94.12.6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coulthard SA, Hall AG. Recent advances in the pharmacogenomics of thiopurine methyltransferase. Pharmacogenomics J. 2001;1:254–261. doi: 10.1038/sj.tpj.6500066. [DOI] [PubMed] [Google Scholar]
- 29.Evans WE. Pharmacogenetics of thiopurine S-methyltransferase and thiopurine therapy. Ther Drug Monit. 2004;26:186–191. doi: 10.1097/00007691-200404000-00018. [DOI] [PubMed] [Google Scholar]
- 30.Dubinsky MC, Reyes E, Ofman J, et al. A cost-effectiveness analysis of alternative disease management strategies in patients with Crohn's disease treated with azathioprine or 6-mercaptopurine. Am J Gastroenterol. 2005;100:2239–2247. doi: 10.1111/j.1572-0241.2005.41900.x. [DOI] [PubMed] [Google Scholar]
- 31.Gearry RB, Barclay ML, Roberts RL, et al. Thiopurine methyltransferase and 6-thioguanine nucleotide measurement: early experience of use in clinical practice. Intern Med J. 2005;35:580–585. doi: 10.1111/j.1445-5994.2005.00904.x. [DOI] [PubMed] [Google Scholar]
- 32.Kubota T. 5-Fluorouracil and dihydropyrimidine dehydrogenase. Int J Clin Oncol. 2003;8:127–131. doi: 10.1007/s10147-003-0319-7. [DOI] [PubMed] [Google Scholar]
- 33.Etienne MC, Chatelut E, Pivot X, et al. Co-variables influencing 5-fluorouracil clearance during continuous venous infusion. A NONMEM analysis. Eur J Cancer. 1998;34:92–97. doi: 10.1016/s0959-8049(97)00345-6. [DOI] [PubMed] [Google Scholar]
- 34.Lu Z, Zhang R, Diasio RB. Population characteristics of hepatic dihydropyrimidine dehydrogenase activity, a key metabolic enzyme in 5-fluorouracil chemotherapy. Clin Pharmacol Ther. 1995;58:512–522. doi: 10.1016/0009-9236(95)90171-X. [DOI] [PubMed] [Google Scholar]
- 35.Meinsma R, Fernandez-Salguero P, Van Kuilenburg AB, et al. Human polymorphism in drug metabolism: mutation in the dihydropyrimidine dehydrogenase gene results in exon skipping and thymine uracilurea. DNA Cell Biol. 1995;14:1–6. doi: 10.1089/dna.1995.14.1. [DOI] [PubMed] [Google Scholar]
- 36.Van Kuilenburg AB, Vreken P, Abeling NG, et al. Genotype and phenotype in patients with dihydropyrimidine dehydrogenase deficiency. Hum Genet. 1999;104:1–9. doi: 10.1007/pl00008711. [DOI] [PubMed] [Google Scholar]
- 37.Vreken P, Van Kuilenburg AB, Meinsma R, van Gennip AH. Identification of novel point mutations in the dihydropyrimidine dehydrogenase gene. J Inherit Metab Dis. 1997;20:335–338. doi: 10.1023/a:1005357307122. [DOI] [PubMed] [Google Scholar]
- 38.Vreken P, Van Kuilenburg AB, Meinsma R, van Gennip AH. Dihydropyrimidine dehydrogenase (DPD) deficiency: identification and expression of missense mutations C29R, R886H and R235W. Hum Genet. 1997;101:333–338. doi: 10.1007/s004390050637. [DOI] [PubMed] [Google Scholar]
- 39.Wei X, Elizondo G, Sapone A, et al. Characterization of the human dihydropyrimidine dehydrogenase gene. Genomics. 1998;51:391–400. doi: 10.1006/geno.1998.5379. [DOI] [PubMed] [Google Scholar]
- 40.Etienne MC, Lagrange JL, Dassonville O, et al. Population study of dihydropyrimidine dehydrogenase in cancer patients. J Clin Oncol. 1994;12:2248–2253. doi: 10.1200/JCO.1994.12.11.2248. [DOI] [PubMed] [Google Scholar]
- 41.Ishikawa Y, Kubota T, Otani Y, et al. Dihydropyrimidine dehydrogenase activity and messenger RNA level may be related to the antitumor effect of 5-fluorouracil on human tumor xenografts in nude mice. Clin Cancer Res. 1999;5:883–889. [PubMed] [Google Scholar]
- 42.Salonga D, Danenberg KD, Johnson M, et al. Colorectal tumors responding to 5-fluorouracil have low gene expression levels of dihydropyrimidine dehydrogenase, thymidylate synthase, and thymidine phosphorylase. Clin Cancer Res. 2000;6:1322–1327. [PubMed] [Google Scholar]
- 43.Van Kuilenburg AB, Muller EW, Haasjes J, et al. Lethal outcome of a patient with a complete dihydropyrimidine dehydrogenase (DPD) deficiency after administration of 5-fluorouracil: frequency of the common IVS14+1G>A mutation causing DPD deficiency. Clin Cancer Res. 2001;7:1149–1153. [PubMed] [Google Scholar]
- 44.Arellano M, Malet-Martino M, Martino R, Gires P. The anticancer drug 5-fluorouracil is metabolized by the isolated perfused rat liver and in rats into highly toxic fluoroacetate. Br J Cancer. 1998;77:79–86. doi: 10.1038/bjc.1998.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45 ••.Van Kuilenburg AB. Screening for dihydropyrimidine dehydrogenase deficiency: to do or not to do, that's the question. Cancer Invest. 2006;24:215–217. doi: 10.1080/07357900500524702. [DOI] [PubMed] [Google Scholar]; Review assessing the need for DPD genetic polymorphism screening prior to initiating 5-FU therapy.
- 46.Ziegler DM. Recent studies on the structure and function of multisubstrate flavin-containing monooxygenases. Annu Rev Pharmacol Toxicol. 1993;33:179–199. doi: 10.1146/annurev.pa.33.040193.001143. [DOI] [PubMed] [Google Scholar]
- 47.Cashman JR. The implications of polymorphisms in mammalian flavin-containing monooxygenases in drug discovery and development. Drug Discov Today. 2004;9:574–581. doi: 10.1016/S1359-6446(04)03136-8. [DOI] [PubMed] [Google Scholar]
- 48.Cashman JR, Zhang J. Interindividual differences of human flavin-containing monooxygenase 3: genetic polymorphisms and functional variation. Drug Metab Dispos. 2002;30:1043–1052. doi: 10.1124/dmd.30.10.1043. [DOI] [PubMed] [Google Scholar]
- 49.Hernandez D, Addou S, Lee D, et al. Trimethylaminuria and a human FMO3 mutation database. Hum Mutat. 2003;22:209–213. doi: 10.1002/humu.10252. [DOI] [PubMed] [Google Scholar]
- 50.Hamman MA, Haehner-Daniels BD, Wrighton SA, et al. Stereoselective sulfoxidation of sulindac sulfide by flavin-containing monooxygenases. Comparison of human liver and kidney microsomes and mammalian enzymes. Biochem Pharmacol. 2000;60:7–17. doi: 10.1016/s0006-2952(00)00301-4. [DOI] [PubMed] [Google Scholar]
- 51.Giardiello FM, Hamilton SR, Krush AJ, et al. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N Engl J Med. 1993;328:1313–1316. doi: 10.1056/NEJM199305063281805. [DOI] [PubMed] [Google Scholar]
- 52.Etienne F, Resnick L, Sagher D, et al. Reduction of Sulindac to its active metabolite, sulindac sulfide: assay and role of the methionine sulfoxide reductase system. Biochem Biophys Res Commun. 2003;312:1005–1010. doi: 10.1016/j.bbrc.2003.10.203. [DOI] [PubMed] [Google Scholar]
- 53.Giardiello FM, Offerhaus JA, Tersmette AC, et al. Sulindac induced regression of colorectal adenomas in familial adenomatous polyposis: evaluation of predictive factors. Gut. 1996;38:578–581. doi: 10.1136/gut.38.4.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hisamuddin IM, Wehbi MA, Schmotzer B, et al. Genetic polymorphisms of flavin monooxygenase 3 in sulindac-induced regression of colorectal adenomas in familial adenomatous polyposis. Cancer Epidemiol Biomarkers Prev. 2005;14:2366–2369. doi: 10.1158/1055-9965.EPI-05-0312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hisamuddin IM, Wehbi MA, Chao A, et al. Genetic polymorphisms of human flavin monooxygenase 3 in sulindac-mediated primary chemoprevention of familial adenomatous polyposis. Clin Cancer Res. 2004;10:8357–8362. doi: 10.1158/1078-0432.CCR-04-1073. [DOI] [PubMed] [Google Scholar]
- 56.Giardiello FM, Yang VW, Hylind LM, et al. Primary chemoprevention of familial adenomatous polyposis with sulindac. N Engl J Med. 2002;346:1054–1059. doi: 10.1056/NEJMoa012015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Giardiello FM, Casero RA, Jr, Hamilton SR, et al. Prostanoids, ornithine decarboxylase, and polyamines in primary chemoprevention of familial adenomatous polyposis. Gastroenterology. 2004;126:425–431. doi: 10.1053/j.gastro.2003.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]