

Abstract
The intermediate (IKCa) and small (SKCa) conductance Ca2+-sensitive K+ channels in endothelial cells (ECs) modulate vascular diameter through regulation of EC membrane potential. However, contribution of IKCa and SKCa channels to membrane current and potential in native endothelial cells remains unclear. In freshly isolated endothelial cells from mouse aorta dialyzed with 3 μM free [Ca2+]i and 1 mM free [Mg2+]i, membrane currents reversed at the potassium equilibrium potential and exhibited an inward rectification at positive membrane potentials. Blockers of large-conductance, Ca2+-sensitive potassium (BKCa) and strong inward rectifier potassium (Kir) channels did not affect the membrane current. However, blockers of IKCa channels, charybdotoxin (ChTX), and of SKCa channels, apamin (Ap), significantly reduced the whole-cell current. Although IKCa and SKCa channels are intrinsically voltage independent, ChTX- and Ap-sensitive currents decreased steeply with membrane potential depolarization. Removal of intracellular Mg2+ significantly increased these currents. Moreover, concomitant reduction of the [Ca2+]i to 1 μM caused an additional increase in ChTX- and Ap-sensitive currents so that the currents exhibited theoretical outward rectification. Block of IKCa and SKCa channels caused a significant endothelial membrane potential depolarization (≈11 mV) and decrease in [Ca2+]i in mesenteric arteries in the absence of an agonist. These results indicate that [Ca2+]i can both activate and block IKCa and SKCa channels in endothelial cells, and that these channels regulate the resting membrane potential and intracellular calcium in native endothelium.
INTRODUCTION
Blood flow is intimately linked to endothelial membrane potential and intracellular Ca2+ levels ([Ca2+]i). Endothelial Ca2+ influx appears to depend on the electrochemical gradient, and likely occurs through nonvoltage-dependent Ca2+ entry pathways, possibly transient receptor potential (TRP) channels. Therefore, hyperpolarization of the endothelium membrane elevates [Ca2+]i by an increase in Ca2+ influx that induces relaxation of the underlying smooth muscle (Luckhoff and Busse, 1990) through endothelium-derived hyperpolarizing factors (EDHFs) and the generation of nitric oxide and prostacyclin. Several types of potassium channels have been proposed to regulate endothelial membrane potential, including large conductance, calcium-sensitive potassium (BKCa) channels, inward rectifier (Kir) potassium channels, and small (SKCa) and intermediate (IKCa) conductance Ca2+-activated potassium channels (Hoger et al., 2002; Shimoda et al., 2002; Fang et al., 2005). SKCa and IKCa channels in vascular endothelium appear to have particularly prominent roles, since inhibition of these channels prevents EDHF-mediated vasodilation (Eichler et al., 2003; Weston et al., 2005; Feletou and Vanhoutte, 2006). The current view is that endothelial-dependent vasodilators such as acetylcholine, bradykinin, or substance P elevate intracellular calcium through calcium influx and release; this in turn activates SKCa and IKCa channels, which cause membrane potential hyperpolarization and further elevation of intracellular calcium through increases in the calcium electrochemical gradient. The SKCa- and IKCa-induced membrane hyperpolarization and elevation of intracellular calcium directly or indirectly induce membrane hyperpolarization and relaxation of the nearby vascular smooth muscle (Feletou and Vanhoutte, 2006). Therefore, SKCa and IKCa channels in vascular endothelial cells are thought to act as a positive feedback element, such that their activation causes membrane potential hyperpolarization and thus increased calcium entry (Garland et al., 1995; Marchenko and Sage, 1996; Eichler et al., 2003; Weston et al., 2005; Feletou and Vanhoutte, 2006). However, the role of these channels in the regulation of endothelial membrane potential and intracellular calcium in the absence of endothelial agonists is not known.
Although it is clear that SKCa and IKCa channels can affect endothelial membrane potential, it is unclear how membrane potential regulates currents through these channels. Expressed SKCa (KCa2.1-2.3) and IKCa (KCa3.1) channels lack an intrinsic voltage sensor. Yet, SKCa and IKCa currents in native endothelium as well as expressed KCa2.2, KCa2.3, and KCa3.1 channels appear to exhibit inward rectification, i.e., their conductance decreases with membrane potential depolarization (Kohler et al., 1996; Xia et al., 1998; Castle et al., 2003; Eichler et al., 2003; Joiner et al., 2003; Taylor et al., 2003; Si et al., 2006). Although intracellular divalent cations have recently been shown to block the pore of rat KCa2.2 channels exogenously expressed in oocytes (Soh and Park, 2001), evidence for a similar mechanism occurring with IKCa and SKCa in native endothelial cells is lacking.
Recent evidence indicates that SKCa and IKCa currents in native endothelium are through KCa2.3 and KCa3.1 channels, respectively (Taylor et al., 2003; Si et al., 2006; Kohler and Hoyer, 2007). KCa2.1-2.3 channels are blocked by the bee venom apamin (Ap), whereas KCa3.1 channels are blocked by scorpion toxin, charybdotoxin (ChTX), and by TRAM-34 (Ledoux et al., 2006). ChTX and iberiotoxin (IbTX) block BKCa channels, which are present in the vascular smooth muscle. Suppression of KCa2.3 channel expression eliminates Ap-sensitive potassium currents in the endothelium, but not IKCa currents (Taylor et al., 2003). This causes depolarization of the endothelium and vascular smooth muscle, and increases tone and blood pressure (Taylor et al., 2003). Furthermore, molecular evidence supports the idea that functional isoform of SKCa channels in vascular endothelium is the KCa2.3 (SK3) channel (Kohler et al., 2001; Burnham et al., 2002; Eichler et al., 2003; Taylor et al., 2003; Burnham et al., 2006; Kohler and Hoyer, 2007). Similarly, targeted disruption of gene for KCa3.1 channels eliminates TRAM-34–sensitive, but not Ap-sensitive, potassium currents, and increases vascular tone and blood pressure (Si et al., 2006). Therefore, both channel types appear to be important modulators of vascular function (Kohler and Hoyer, 2007).
One goal of the present study was to determine the properties of SKCa and IKCa channels in freshly isolated vascular endothelial cells, in particular their voltage dependence and possible block by intracellular Ca2+ and Mg2+ ions. Whole cell currents reversed at the potassium equilibrium potential (EK) and were composed of currents through IKCa and SKCa channels, as well as an intracellular Ca2+-insensitive component. However, BKCa channel and Kir channel blockers did not affect the membrane currents. A pronounced inward rectification was observed for the SKCa and IKCa currents positive to the potassium equilibrium potential, EK. Voltage-dependent block by intracellular Mg2+ and Ca2+ ions was responsible for this decrease of K+ efflux through SKCa and IKCa channels. The similar voltage dependence of SKCa and IKCa current block by Mg2+ and Ca2+ suggests that their binding sites within the two channel types share common features. Importantly, significant internal Ca2+ block occurred at physiological membrane potentials with potentially physiological ion concentrations. These findings suggest a novel negative feedback mechanism in endothelium by which intracellular Ca2+ ions would have opposing effect (activation and block) on SKCa and IKCa channels, depending on local Ca2+ levels. Finally, we found that SKCa and IKCa channels regulate the resting membrane potential and [Ca2+]i in intact endothelium in the absence of stimulation. Preliminary findings have been previously presented (Ledoux, J., and M.T. Nelson. 2005. FASEB J. 19:692.2).
MATERIALS AND METHODS
Endothelial Cell isolation and electrophysiology
Aortic endothelial cells were freshly isolated from C57BL6 female mice (3–4 mo old) as previously described (Taylor et al., 2003). Animal procedures used in this study were in accordance with institutional guidelines and approved by the Institutional Animal Care and Use committee of the University of Vermont. In brief, adult female mice were killed by intraperitoneal injection of sodium pentobarbital (150 mg/kg) followed by a thoracotomy. The aorta was removed, cleaned of connective tissue, and cut into small rings. The tissue pieces were then enzymatically digested using dispase (4 mg/ml) in physiological salt solution for 40 min at 35°C. Elastase (0.1 mg/ml) was then added to the enzyme cocktail and the tissue was further incubated for 10 min. After washing the aorta rings in fresh solution, the rings were cut open and gently triturated to dissociate the endothelial cells. Isolated cells were stored at 4°C and used within 6–8 h following isolation.
Electrophysiology
Membrane currents from freshly isolated endothelial cells were recorded using the conventional whole-cell patch clamp technique. Sampled at 2 kHz, endothelial currents were acquired in voltage-clamp mode with an axopatch 200A (Axon Instruments) and analyzed using the pClamp suite (Axon Instruments). The series resistances were ≈3–5 MΩ and the series resistance error was negligible due to the small amplitude of the recorded currents. Therefore, no compensation was applied. The capacitance error was also negligible in our conditions with a time constant (τ) of ≈50 μs. Isolated endothelial cells were identified by their characteristic round and rough shape.
Simultaneous Membrane Potential Recording and Ca2+ Imaging In Situ
Mesenteric arteries from mice were cleaned of connective tissue, cut open, and pinned on a sylgard block with the endothelium facing up. The endothelium was preferentially loaded with Fluo-4 (10 μM) for 45 min at 30°C in the presence of pluronic acid (2.5 μg/ml). Ca2+ imaging was then performed using a Solamere confocal system (Solamere Technologies) with a CCD camera on an upright Nikon microscope with a 60× water dipping objective (NA 1.0). Images were acquired at 30 frames/s with the QED acquisition software (Media Cybernetics). Ca2+ dye was excited using a krypton/argon laser (488 nm) and emission fluorescence was collected above 495 nm. The images were processed using custom designed software (A. Bonev), and the fractional fluorescence was evaluated by dividing the fluorescence of a region of interest (ROI) by an average fluorescence of 50 images without activity from the same ROI. Simultaneous measurement of the membrane potential was performed with the perforated configuration of the patch clamp technique at a sampling rate of 5 kHz by obtaining a gigaohm seal with a borosilicate micropipette (6–8 MΩ) on the facing endothelium. The membrane potential recordings (perforated patch) were performed without current injection (I = 0 mode). All in situ experiments were performed in the presence of nisoldipine (100 nM) to minimize the contraction of the smooth muscle to KCl and the impact of smooth muscle on endothelial membrane potential.
Solutions
For isolated cell experiments, the following extracellular solution was used (in mM): NaCl 134, KCl 6, glucose 10, HEPES 10 (pH 7.4), MgCl2 1, and CaCl2 2. For Fig. 1 B, extracellular KCl concentration was increased to 45 mM with an equivalent reduction of NaCl. The composition of the pipette solution used for the whole-cell experiments was (in mM): KCl 134, HEDTA 5, and HEPES 10 (pH 7.2). The amounts of MgCl2 and CaCl2 added were determined with the software WinMaxC (http://www.stanford.edu/~cpatton/maxc.html) to achieve the indicated free cation concentrations. For 1 mM Mg2+ and 3 μM Ca2+ (in mM): MgCl2 (5.53) and CaCl2 (0.207); for 1 mM Mg2+ and 1 μM Ca2+ (mM): MgCl2 (5.51) and CaCl2 (0.0683); for 0 Mg2+ and 3 μM Ca2+ (mM): MgCl2 (0) and CaCl2 (1.61); for 0 Mg2+ and 1 μM Ca2+ (mM): MgCl2 (0) and CaCl2 (1.36). For the experiments in the absence of intracellular Ca2+, the CaCl2 was omitted from the solution above (with 5 mM HEDTA) and MgCl2 was added to set the free Mg2+ to 1 mM. These experiments were performed at room temperature.
Figure 1.

Functional BKCa or Kir channels are not present in freshly isolated aortic endothelial cells. (Aa) Typical family of traces recorded from freshly isolated aortic endothelial cells dialyzed with a pipette solution containing 1 mM Mg2+ and 3 μM Ca2+ before (Control) or following application of 100 nM IbTX. From a holding potential of −50 mV, currents were evoked by 100-ms voltage steps from −100 to +100 mV in a 10-mV increment followed by a 100-ms repolarizing step to −50 mV. (Ab) Mean IbTX-sensitive current recorded from four experiments similar to the one shown in Aa. (B) Mean ramp currents in the absence (Control, black trace) and presence (Barium, red trace) of 0.5 mM barium (n = 5). These currents were elicited using 45 mM extracellular K+ to emphasize potential Kir currents. Currents were induced by a 200-ms voltage ramp protocol from −100 mV to +100 mV (HP = −60 mV).
For simultaneous membrane potential and Ca2+ imaging experiments in intact endothelium of cut-open mesenteric arteries, a physiological salt solution was used with the following constituents (in mM): NaCl 119, KCl 4.7, NaHCO3 24, KH2PO4 1.2, EDTA 0.0023, MgCl2 1.2, glucose 11, and CaCl2 1.6, and the following pipette solution for the simultaneous membrane potential recording (in mM): K-aspartate 110, NaCl 10, KCl 30, MgCl2 1, HEPES 10 (pH 7.2), and EGTA 0.05. Amphotericin (200 μg/ml) was used to achieve perforated patch. These experiments were performed at 30°C to minimize dye leakage.
Statistics
All data are the mean ± SEM of n cells. Data were analyzed using the paired or unpaired Student's t test as appropriate and considered significant with P values <0.05.
RESULTS
Electrophysiological Characteristics of Native Endothelial Cells
Freshly isolated aortic endothelial cells (AECs) were dialyzed with 3 μM Ca2+ to activate Ca2+-activated K+ channels using the conventional whole cell configuration of the patch clamp technique. The endothelial cells had a mean capacitance of 10.6 ± 0.8 pF (n = 31). Under these conditions, the relationship between membrane current and voltage was examined by applying voltage steps from −100 to + 100 mV from a holding potential of −50 mV. The whole cell currents exhibited inward rectification at membrane potentials positive to the potassium equilibrium potential (EK = −83 mV). This current/voltage (I/V) relationship is strikingly different from that observed in vascular smooth muscle, which exhibits marked increases in voltage-dependent potassium (Kv) channel and BKCa channel currents at positive voltages (Marijic et al., 2001; Rainbow et al., 2006). The presence of BKCa currents was probed by applying IbTX (100 nM), a specific inhibitor of BKCa channels (Fig. 1 A). IbTX did not alter endothelial currents (−100 mV, control −13 ± 8 pA/pF and IbTX −15 ± 11 pA/pF, n = 4; +100 mV, control 39 ± 20 pA/pF and IbTX 40 ± 24 pA/pF, n = 4) (Fig. 1 A, b). Aortic vascular smooth muscle cells patched in the same conditions showed a large IbTX-sensitive outwardly rectifying current (≈275 pA/pF at +100 mV). These data are consistent with previous reports from native endothelium (Gauthier et al., 2002; Eichler et al., 2003) and suggest that aortic endothelial cells do not have functional BKCa channels.
Inward rectifier K+ channels (Kir) have been reported in cultured endothelial cell (Forsyth et al., 1997; Fang et al., 2005), and in some types of native endothelial cell preparations (von Beckerath et al., 1996; Crane et al., 2003; Fang et al., 2006). To evaluate the presence of Kir currents in freshly isolated AECs, the cells were exposed to a high concentration (0.5 mM) of barium, a potent blocker of Kir channels. The K+ concentration of the superfusate was increased to 45 mM to elevate, if present, Kir inward current at membrane potentials negative to EK. Application of barium had no effect on the current evoked by a 200-ms voltage ramp from −100 to + 100 mV as illustrated by the mean traces in the absence (black trace) or presence (red trace) of 0.5 mM barium in Fig. 1 B (n = 5), suggesting that AECs do not express functional Kir channels.
SKCa and IKCa currents have been measured in native endothelial cells (Burnham et al., 2002; Bychkov et al., 2002; Eichler et al., 2003; Taylor et al., 2003; Si et al., 2006). To examine properties of IKCa channels, the IKCa (and BKCa) channel blocker, ChTX (100 nM) was applied to AECs. Currents were evoked by a 200-ms voltage ramp from −100 to + 100 mV (Fig. 2 A). The current in cells dialyzed with 3 μM Ca2+ and 1 mM Mg2+ exhibited inward rectification and reversed near EK (ERev = −79 ± 2 mV; EK = −83 mV), indicating that the membrane currents are largely carried by potassium ions. In contrast to IbTX, ChTX significantly reduced the currents (−25 ± 4% at + 80 mV, n = 6). The ChTX-sensitive current also exhibited marked inward rectification and reversed near EK (Fig. 2 C; −79 ± 4 mV). Addition of the selective blocker of SKCa (KCa2.1-2.3) channels, Ap (300 nM), further reduced the current (−17 ± 3% of the current in the presence of ChTX at + 80 mV, n = 5). The Ap-sensitive current also exhibited inward rectification and reversed near EK (Fig. 2 D; −83 ± 3 mV). The contribution of the ChTX-sensitive current, IKCa, to the total current in the presence of 3 μM Ca2+ and 1 mM Mg2+ was larger than the Ap-sensitive current, SKCa (4.2 ± 1.4 pA/pF and 2.4 ± 0.5 pA/pF at +80 mV for IKCa and SKCa currents, respectively; P < 0.05, n = 6 and 5).
Figure 2.

Contribution of Ca2+-dependent currents, SKCa and IKCa, to membrane currents in AECs. (A) Examples of ramp currents in the absence (black trace; Erev = −78.4 mV) or in presence of 100 nM ChTX, an IKCa inhibitor, alone (ChTX, red trace, Erev = −74.5 mV) or in combination with 300 nM Ap, a SKCa inhibitor (Ap, green trace, Erev = −75.6 mV). The AEC was dialyzed with a pipette solution containing 5 mM HEDTA with 1 mM free Mg2+ and 3 μM free Ca2+. (B) Recording of currents as in A except that the cell was dialyzed without addition of Ca2+. IKCa and SKCa currents from the experiment presented in A were obtained as the ChTX- (C, Erev = −75.4 mV) and Ap-sensitive (D, Erev = −76.2 mV) currents, respectively. Currents were evoked by a 200-ms voltage ramp protocol from −100 to + 100 mV (HP = −60 mV).
SKCa and IKCa channel activation require intracellular calcium (EC50 ≈ 500 nM; Kohler et al., 1996; Hirschberg et al., 1998). Therefore, the removal of intracellular calcium on endothelial currents was examined by dialyzing the cells with a Ca2+-free intracellular solution containing 5 mM HEDTA (Mg2+ was maintained at 1 mM) (Fig. 2, A and B). Removing intracellular calcium significantly reduced the membrane current, and this remaining current was insensitive to ChTX and Ap (9.8 ± 0.9 pA/pF and 9.6 ± 1.3 pA/pF at +80 mV in the absence and presence of ChTX and Ap, respectively; n = 4). The membrane current in cells dialyzed with the 0 Ca2+ solution was similar in amplitude to the currents in cells dialyzed with 3 μM Ca2+ in the presence of ChTX and Ap (10 ± 1 pA/pF at +80 mV, n = 5). Interestingly, in all cases, the currents reversed near EK, suggesting that a [Ca2+]i-insensitive potassium current contributes to the membrane current (Fig. 2, A and B). These results indicate that SKCa, IKCa, and Ca2+-insensitive K+ conductances contribute significantly to the overall membrane conductance of isolated AECs.
The Basis of the Apparent Voltage Dependence of IKCa and SKCa: Block by Intracellular Mg2+ and Ca2+
SKCa and IKCa channels do not exhibit intrinsic voltage dependence, and therefore, in a physiological potassium gradient, their current–voltage relationship should exhibit theoretical outward rectification. The decrease in current and increase in current noise at positive potentials suggest that intracellular cations are blocking IKCa and SKCa channels in a voltage-dependent manner (Fig. 2, C and D). To investigate the involvement of intracellular divalent cations in the apparent voltage dependence of IKCa and SKCa currents, AECs were dialyzed with pipette solutions containing different concentrations of free Mg2+ and Ca2+, and ChTX- and Ap-sensitive currents were analyzed. As the IKCa and SKCa currents are relatively small and intracellular calcium is required for activation, it was not possible to evaluate intracellular calcium block <1 μM. Therefore, SKCa and IKCa currents were compared with the predictions from the Goldman-Hodgkin-Katz (GHK) constant field equation. The relationship between any type of current, in this case a macroscopic K+ current, and the membrane potential can be described by the appropriately modified GHK constant field equation as follows:
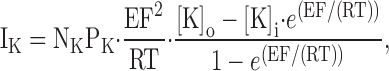 |
(1) |
where IK is the K+ current density in pA/pF, NK is the channel density (channels/pF), PK is the permeability of a channel to K+ ions in cm/s, [K]o/i is the concentration of K+ in the extracellular or intracellular compartments, E is the membrane potential in V, F is the Faraday's constant, R is the gas constant, and T the temperature in Kelvin. The permeability constants were calculated using a simplified GHK equation (Benham et al., 1986) and using a single channel conductance of 40 and 10 pS for IKCa and SKCa, respectively (Kohler et al., 1996; Ishii et al., 1997), in the presence of symmetrical K+ (120 mM). The permeability constants of IKCa and SKCa channels obtained were 8.7872 E − 14 and 2.1968 E − 14 cm/s, respectively. Based on the results presented below, the inward rectification of IKCa and SKCa is due to intracellular block of the pore by Ca2+ and Mg2+, and membrane potential depolarization drives the blocking ion into the intracellular side of the pore. Therefore, the inward component of the ramp-evoked current (i.e., negative to EK) is assumed not to experience much block. This region was fit with the Eq. 1 and then extrapolated up to + 100 mV to define the current–voltage relationship in the absence of blocking ions.
Removal of internal Mg2+ in the presence of 3 μM intracellular Ca2+ increased SKCa and IKCa currents but did not eliminate inward rectification (Fig. 3, A and C, solid lines). The absence of intracellular Mg2+ resulted in an approximate twofold increase in the outward current amplitude at +80 mV for both IKCa (4.4 ± 1.8 pA/pF with 1 mM Mg2+ and 7.4 ± 0.3 pA/pF without Mg2+, respectively; P < 0.05, n = 6 and 5) and SKCa (2.8 ± 0.5 pA/pF with 1 mM Mg2+ and 4.9 ± 0.8 pA/pF without Mg2+, P < 0.05, n = 5 and 5). These data support the concept that voltage-dependent block of SKCa and IKCa channels by intracellular Mg2+ is a mechanism contributing to the voltage-dependent decrease in potassium efflux through these channels.
Figure 3.

The inward rectification of IKCa and SKCa is caused by intracellular Mg2+ and Ca2+. (A and B) Mean IKCa (a) and SKCa (b) currents (filled lines) elicited by voltage ramp protocol in AECs dialyzed with 1 mM free Mg2+ and (A) 3 μM free Ca2+ (n = 6 and 5, respectively) or (B) 1 μM free Ca2+ (n = 5 and 5). Predictions from the GHK constant field equation (dashed lines) were obtained by fitting the mean inward currents of IKCa (a) and SKCa (b) of AECs (filled lines) in each condition. (C and D) Similar to A and B except that endothelial cells were dialyzed with a Mg2+-free ([Mg2+] = 0) pipette solution containing either (C) 3 μM free Ca2+ (n = 4 and 4) or (D) 1 μM Mg2+ (n = 4 and 5 for IKCa and SKCa, respectively). Predictions from the GHK constant field equation (dashed lines) were obtained by fitting the mean inward currents of IKCa (a) and SKCa (b) of AECs (filled lines) in each condition. Currents were evoked by a 200-ms voltage ramp protocol from −100 to + 100 mV (HP = −60 mV). [K]o, 6 mM; [K]i, 150 mM; and PK, 8.7872 E − 14 and 2.1968 E − 14 for IKCa and SKCa, respectively.
To examine the effect of intracellular Ca2+ on the rectification properties of IKCa and SKCa, pipette Ca2+ was lowered from 3 to 1 μM, which should still cause near-maximal activation of SKCa and IKCa channels. As depicted in Fig. 3 B, lowering intracellular Ca2+ from 3 to 1 μM, in the presence of 1 mM Mg2+, increased both ChTX- and Ap-sensitive currents, IKCa and SKCa, respectively. Similarly, lowering intracellular Ca2+ from 3 to 1 μM in the absence of internal Mg2+ dramatically increased SKCa and IKCa currents (Fig. 3 D, solid lines). The amplitude of IKCa and SKCa currents recorded at +80 mV in the presence of 1 μM Ca2+ are 14 ± 3 and 9 ± 2 pA/pF, respectively (n = 5 and 6). Under these conditions, both IKCa and SKCa currents now exhibited theoretical outward rectification. These results indicate that intracellular calcium between 1 and 3 μM caused substantial voltage-dependent block of IKCa and SKCa channels. Thus, it appears that intracellular calcium both activates, via calmodulin, and blocks IKCa and SKCa channels.
The theoretical currents (dashed) and measured currents (solid) for IKCa and SKCa are shown on Fig. 3 in the presence (A and B, with 3 and 1 μM Ca2+, respectively) and absence of Mg2+ (C and D, with 3 and 1 μM Ca2+, respectively). With free intracellular Ca2+ set to 3 μM, the current–voltage relationships of IKCa and SKCa were significantly smaller than that predicted from the GHK equation (Fig. 3 C, a and b). Since the physiological membrane potentials of endothelium spans from −55 to −35 mV (Chen and Cheung, 1992; Burnham et al., 2002; Taylor et al., 2003; Eichler et al., 2003; Weston et al., 2005), the impact of intracellular cations on SKCa and IKCa was further analyzed at −45 mV. Fig. 4 shows that in the presence of Mg2+ (1 mM) and elevated Ca2+ (3 μM), the currents were dramatically smaller compared with the predicted GHK at −45 mV (35 ± 4% and 25 ± 10% of the predicted GHK current for IKCa and SKCa, respectively; n = 6 and 5). Lowering intracellular calcium to 1 μM in the presence of Mg2+ (1 mM) increased IKCa and SKCa currents closer to the currents predicted by the GHK equation (Fig. 4). In 1 μM Ca2+ and in the absence of Mg2+, the currents exhibited almost theoretical outward rectification (Fig. 3 D). Under these conditions, IKCa and SKCa currents were 99 ± 10% and 80 ± 17%, respectively (n = 4 and 5) of the predicted GHK current at −45 mV (Fig. 4).
Figure 4.

Comparison between native and theoretical IKCa and SKCa currents. IKCa (A) and SKCa (B) currents at −45 mV in cells dialyzed with different pipette solutions normalized to the GHK predictions depicted in Fig. 3. For 1 mM Mg2+ and 3 μM Ca2+ (% of GHK fit): IKCa (35 ± 4) and SKCa (25 ± 10); for 0 Mg2+ and 1 μM Ca2+ (mM): IKCa (71 ± 9) and SKCa (55 ± 5); for 1 mM Mg2+ and 3 μM Ca2+ (mM): IKCa (71 ± 11) and SKCa (56 ± 13); for 0 Mg2+ and 1 μM Ca2+ (mM): IKCa (99 ± 10) and SKCa (80 ± 17). *, P < 0.05.
The numbers of IKCa and SKCa channels in a single endothelial cell are not known. The IKCa and SKCa channel density (NK) can be estimated by the fit of Eq. 1 to the macroscopic currents shown in Fig. 3. However, since the macroscopic current is a function of the open state probability (Po), the estimated NK was corrected for the Po of the channels. A maximal open state probability (Po(max)) of 0.6 was used, based on the measurements of Hirschberg et al. (1998). Using this value of Pomax, the density of IKCa and SKCa channels in a single endothelial cell was then estimated to be 9.4 ± 0.1 and 29.0 ± 0.3 channels/100 μm2, respectively. A single endothelial cell would then have ≈99 IKCa and ≈307 SKCa functional channels in its cell membrane.
Intracellular Block of Channel Pore
The inward rectification of IKCa and SKCa by Mg2+ and Ca2+ ions could be interpreted as divalent cations binding to a site within the pore, thus preventing passage of K+ ions. Using the simplest model, the binding site would be inside the pore and accessible from the cytoplasm. According to the Woodhull model (Woodhull, 1973), the blocking cation penetrates partway through the electrical field of the membrane. Therefore, depolarization will favor penetration of the blocking cations into the permeation pathway and thereby decrease K+ efflux. The blocking process is analogous to the well-described block of Kir channels by extracellular barium in smooth muscle (Quayle et al., 1993). The apparent Kd of the blocking intracellular divalent cation can be estimated using the following relation:
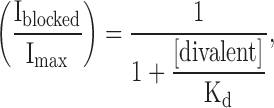 |
(2) |
where Kd is the apparent dissociation constant, [divalent] is the intracellular concentration of the blocking divalent cation, and Imax and Iblocked are the currents recorded with nominally zero and the highest intracellular concentration of the blocking cation, respectively. Since intracellular Ca2+ cannot be nominally zero, the current value from the GHK equation was used as Imax for Ca2+. The values obtained were then plotted as a function of the membrane potential as depicted in Fig. 5 for the SKCa channels. The apparent Kd is an exponential function of the membrane potential as expected for an ion binding site within the membrane voltage field. The relationship between the apparent Kd and the membrane potential between +40 and +90 mV was fit with the following relation:
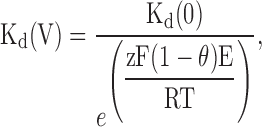 |
(3) |
where Kd(0) is the Kd at 0 mV, z is the valence of the ion, θ is the slope factor representing the sensitivity of the Kd to the applied voltage from the inside of the membrane, and F, E, R, and T have their usual meanings. The Kd(0) for Mg2+ obtained from the fitted lines are 2 and 3 mM for IKCa and SKCa with a slope factor of −0.10 and −0.20, respectively. For Ca2+, a lower Kd(0) was isolated from the fitted lines, 5 and 3 μM (θ = −0.17 and −0.20) for IKCa and SKCa, respectively. These results are consistent with our findings suggesting that Ca2+ is more effective at blocking the pore than Mg2+. Also, the slope factor values suggest that IKCa and SKCa channels have similar Mg2+ and Ca2+ binding sites within their channel pores.
Figure 5.

Voltage dependence of Mg2+ and Ca2+ block of SKCa. (A) Voltage dependence of the apparent Kd of Mg2+ calculated with Eq. 2. Dashed line was fitted to data with Eq. 3, with a Kd at 0 mV of 3 mM and θ (slope factor indicating voltage sensitivity of Kd) = −0.20. (B) Voltage dependence of the apparent Kd of Ca2+ calculated with Eq. 2. Dashed line was fitted to data with Eq. 3, with a Kd at 0 mV of 3 μM and θ = −0.20.
Ca2+-activated K+ Channels and their Impact on Resting Membrane Potential and Intracellular Ca2+ Levels
Membrane potential regulates Ca2+ entry, and hence intracellular Ca2+, in endothelial cells through changes in the Ca2+ electrochemical gradient (Luckhoff and Busse, 1990). Although previous studies have demonstrated the role of IKCa and SKCa channels in the regulation of endothelial [Ca2+]i following stimulation by ACh, bradykinin, or substance P (Eichler et al., 2003; Weston et al., 2005), the impact of these channels on membrane potential and [Ca2+]i under resting conditions remains unclear. To investigate the impact of IKCa and SKCa channels on the basal endothelial [Ca2+]i and membrane potential, intact endothelium preferentially loaded with the Ca2+ indicator Fluo-4 was used to simultaneously monitor the intracellular Ca2+ levels and membrane potential in cut-open mesenteric arteries (Fig. 6). The resting membrane potential of endothelium measured in current clamp mode of the perforated patch clamp technique was found to be −52 ± 2 mV (n = 6 vessels). Exposure to ChTX (100 nM) depolarized the endothelial membrane potential by 8 ± 1 mV (n = 6). Addition of Ap (300 nM) to the ChTX superfusate further depolarized the membrane potential by 3.1 ± 0.4 mV (n = 6). ChTX, alone, and ChTX + Ap significantly reduced Ca2+ fluorescence 22 ± 5% and 43 ± 8% (n = 6 and 6). The exposure of vessels to an extracellular solution containing 60 mM K+ depolarized the endothelium to a membrane potential of −17 ± 1 mV, close to EK (−20 mV), and was associated with a 78 ± 4% decrease in Ca2+ fluorescence (n = 5).
Figure 6.

IKCa and SKCa regulate intracellular Ca2+ of resting endothelium in mesenteric arteries. (A) Perforated patch clamp was used to record membrane potential of Fluo-4–loaded endothelium to simultaneously measure changes in intracellular Ca2+. Time course graph illustrating the membrane potential and relative fluorescence of resting endothelium exposed to ChTX alone (black bars), with the addition of Ap (+Ap; white bars), and following the addition of 60 mM KCl (KCl; gray bars). (B) Voltage dependence of intracellular Ca2+ concentration from nonstimulated endothelium. Intracellular Ca2+ concentrations in each condition were estimated with Eq. 4 using [Ca2+]Fo = 123 nM in the presence of 60 mM KCl (Knot et al., 1999) and associated with the corresponding membrane potential recorded. The value used for the Kd of Fluo-4 at 30°C was 370 nM (Woodruff et al., 2002). Solid line was fitted to data with the function Y = A*exp(−x/t) + B, where B = 120 ± 1 nM, A = 0.9 ± 0.4, and t = 13 ± 2.
The fluorescent Ca2+ indicator, Fluo-4, provides fractional changes in intracellular Ca2+. To provide a sense of changes in intracellular Ca2+, a baseline (Fo) value of intracellular Ca2+ previously measured with ratiometric Ca2+ indicator Fura-2 was used (Knot et al., 1999) in conjunction with the following equation (Jaggar et al., 1998):
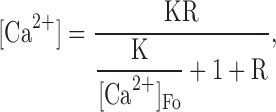 |
(4) |
where K is the Kd of Fluo-4 for Ca2+, R is the fractional change in fluorescence (F/Fo), [Ca2+]Fo is the Ca2+ concentration (measured with Fura-2; Knot et al., 1999) at Fo and [Ca2+] is the endothelial intracellular Ca2+. The [Ca2+]i previously measured in the presence of 60 mM extracellular K+ in rat endothelium with the ratiometric Ca2+ indicator Fura-2 was 123 nM (Knot et al., 1999). The same approach was used to estimate [Ca2+] in the presence of ChTX and Ap. This approach yielded a [Ca2+]i in physiological external potassium of 176 nM, which was similar to the value previously obtained with Fura-2 (174 nM) in rat coronary artery endothelium (Knot et al., 1999). Membrane depolarization induced by blocking IKCa and SKCa channels reduced [Ca2+]i (Fig. 6 B) to a similar level as obtained previously with elevations of external potassium (Knot et al., 1999). Thus, blocking IKCa and SKCa channels causes a membrane potential depolarization and a decrease in endothelial calcium in the absence of an agonist.
DISCUSSION
Endogenous Currents in Native Endothelium
Most studies on endothelial ion channels have been performed on cultured cells in which phenotypical changes might be induced by culture media (Kestler et al., 1998; Jow et al., 1999). Although the presence of Kir currents has been reported in cultured endothelium (Shimoda et al., 2002; Fang et al., 2005) and in endothelium isolated from some species (von Beckerath et al., 1996; Fang et al., 2006;Crane et al., 2003), we did not detect Kir currents in endothelial cells freshly isolated from mouse aorta. In agreement with Gauthier et al. (2002), we found that freshly isolated endothelial cells do not exhibit BKCa currents. Interestingly, BKCa channel expression was shown to be induced in human endothelial cells by culture passage (Kestler et al., 1998; Jow et al., 1999). However, BKCa current and channel expression have been reported in porcine and rabbit endothelium (Rusko et al., 1992; Papassotiriou et al., 2000), suggesting that the expression of BKCa channels may be species or vascular bed dependent.
The current–voltage relationship of AECs suggested that the majority of membrane current was carried by potassium channels. When cells were dialyzed with 3 μM Ca2+, ChTX and Ap, but not IbTX, inhibited a substantial fraction (44%) of the K+ current at +80 mV, indicating that a significant portion of the potassium conductance is carried by SKCa and IKCa channels. In the absence of intracellular Ca2+, the K+ currents were significantly reduced, and ChTX and Ap had no effect. Therefore, local Ca2+ influx in the absence of intracellular Ca2+ does not seem to be sufficient to activate a substantial number of ChTX- and Ap-sensitive channels. However, the remaining current reversed at the potassium equilibrium potential, indicating that another type of potassium conductance is also present in AECs, and that this current would be more prominent at low intracellular calcium levels. KATP and two-pore-domain K+ channels have been reported in endothelial cells (Janigro et al., 1993; Chatterjee et al., 2003; Garry et al., 2007) and are possible candidates for the Ca2+-independent potassium channel. In contrast, vascular myocytes have prominent BKCa currents, KV and voltage-dependent Ca2+ channels, and do not exhibit SKCa and IKCa currents (Thorneloe and Nelson, 2005; Ledoux et al., 2006).
Ca2+-activated K+ Currents in Aortic Endothelial Cells
It has been well established that the ChTX- and Ap-sensitive Ca2+-dependent K+ currents recorded in native endothelial cells (IKCa and SKCa currents) are carried by KCa3.1 and KCa2.3 channels, respectively (Kohler and Hoyer, 2007). Although the concentration of Ap used (300 nM) is sufficient to inhibit all three KCa2.x isoforms (Ledoux et al., 2006), the endothelial KCa2.x current has previously been shown using molecular biology, immunohistological, and electrophysiological approaches to result from the functional expression of KCa2.3 channels (Kohler et al., 2001; Burnham et al., 2002; Bychkov et al., 2002; Eichler et al., 2003; Taylor et al., 2003; Burnham et al., 2006; McNeish et al., 2006; Si et al., 2006; Sandow and Tare, 2007). Although well characterized in expression system, little information is available on endogenous endothelial KCa2.3 and KCa3.1 currents. The channel density of KCa3.1 and KCa2.3 in AECs was estimated to be ≈10 and ≈29 channels/100 μm2 or ≈99 and ≈307 channels/cell, respectively. Even with a lower channel density, KCa3.1 current amplitude appears to be ≈1.75-fold larger than the KCa2.3 current, which reflects the higher single channel conductance of KCa3.1 channels. In contrast with our findings, Si et al. (2006) reported that KCa2.3 and KCa3.1 currents from mouse AECs had similar amplitude. The inward-rectifying current–voltage relationship of the Ca2+-dependent currents in AECs is similar to what has been reported for heterologous expression of KCa3.1 and KCa2.3 (Kohler et al., 1996; Ishii et al., 1997; Xia et al., 1998; Castle et al., 2003) and in endothelium (Eichler et al., 2003; Taylor et al., 2003; Si et al., 2006). Inward rectification was also observed in AECs perforated patch cells (unpublished data), suggesting that KCa3.1 and KCa2.3 channels present the same characteristic I/V relationship with unaltered cytoplasm content.
Inward Rectification and Intracellular Divalent Cations
One aim of our study was to investigate the role of physiological intracellular divalent cations (Mg2+ and Ca2+) in the regulation of endothelial KCa3.1 and KCa2.3 channels in native endothelial cells, using physiological levels of potassium. Intracellular divalent cations (Ba2+, Sr2+, Mg2+, Ca2+) have been shown to block rat KCa2.2 channels exogenously expressed in Xenopus oocytes (Soh and Park, 2001, 2002). The authors found that the apparent dissociation constant (Kd) for intracellular Ca2+ and Mg2+ at +90 mV for KCa2.2 channels was 19.3 and 180 μM, respectively (Soh and Park, 2002). In contrast, our data would indicate an apparent Kd for Mg2+ at +90 mV of 1.2 and 0.7 mM for KCa3.1 and KCa2.3, respectively. Soh and Park (2001) reported that Ca2+ slightly reduces the apparent affinity for Mg2+ between 0.4 and 2 μM [Ca2+]i (Kd for Mg2+ from 130 to 180 μM in the presence of 0.4 and 2 μM Ca2+, respectively). To minimize the impact of Ca2+ on the apparent Kd for Mg2+, the experimental data used in the present study to calculate the apparent Kd for Mg2+ were obtained with solutions containing identical Ca2+ concentration (3 μM). Also at +90 mV, our results indicate an apparent Kd for Ca2+ (in the absence of Mg2+) of 1.3 and 0.99 μM for KCa3.1 and KCa2.3, respectively. The inhibitory binding site for Mg2+ and Ca2+ is within 10–20% of the electrical field sensed by the ion from the cytoplasmic side of the plasma membrane, a value similar to the reported δ for rat KCa2.2 (17%) (Soh and Park, 2001, 2002). Our findings are in agreement with the idea that the divalent ion binding site resides inside the channel pore closer to the cytoplasmic side (Soh and Park, 2002). Soh and Park (2002), based on functional analysis of mutations, identified residue Ser-359 in the pore region of KCa2.2 as being critical for rectification. Interestingly, Ser-359 appears to be conserved in KCa2.3 channels, but not KCa3.1 (Soh and Park, 2002).
Our results suggest that elevation of intracellular Ca2+ levels can considerably decrease the current at −45 mV, a physiological membrane potential (Chen and Cheung, 1992; Marchenko and Sage, 1993; Taylor et al., 2003). Indeed, decreasing [Ca2+]i from 3 to 1 μM significantly increased KCa3.1 and KCa2.3 currents, even though the channels should be maximally activated at 1 μM [Ca2+]i (EC50 ≈ 500 nM) (Kohler et al., 1996; Hirschberg et al., 1998). It is clear then that depending on their concentration, intracellular Ca2+ ions could potentially have opposing effects in the regulation of membrane potential by KCa3.1 and KCa2.3 channels. Furthermore, voltage-dependent block by intracellular Ca2+ and Mg2+ would modulate KCa3.1 and KCa2.3 currents, and thus their contributions to membrane conductance.
Following endothelial stimulation, KCa3.1 and KCa2.3 channel activation would increase Ca2+ influx through membrane potential hyperpolarization. This [Ca2+]i rise would further increase the activity of KCa3.1 and KCa2.3 channels, generating a positive feedback loop (Garland et al., 1995; Feletou and Vanhoutte, 2006; Ledoux et al., 2006). Membrane potential hyperpolarization would relieve intracellular Mg2+ and Ca2+ block, and thus increase contribution of KCa3.1 and KCa2.3 channels to the membrane conductance. However, the rise in intracellular calcium would block KCa3.1 and KCa2.3 channels, and thus decrease channel conductance. The degree of intracellular calcium block would therefore depend on the membrane potential and the level of calcium sensed by the channels. Indeed, recent evidence indicates that the endothelium has localized submembrane microdomains where the local Ca2+ level is higher at the cell membrane (Isshiki et al., 2004; Ledoux, J., A.D. Bonev, and M.T. Nelson. 2007. FASEB J. 21:745.18). Recent evidence also suggests a differential localization of KCa3.1 and KCa2.3 that might increase the degree of complexity in the regulation of the channels (Absi et al., 2007).
Regulation of Basal Membrane Potential and Intracellular Ca2+ by KCa3.1 and KCa2.3 Channels
In intact endothelium, the resting membrane potential, measured with microelectrodes (Chen and Cheung, 1992; Burnham et al., 2002; Taylor et al., 2003) or the patch clamp technique (this study; Burnham et al., 2002; Eichler et al., 2003; Weston et al., 2005), is −55 to −35 mV, and can be hyperpolarized to ≈−75 mV by KCa3.1 and KCa2.3 stimulators (Quignard et al., 2000; Burnham et al., 2002) or endothelial agonists (e.g., ACh, substance P, bradykinin) (Chen and Cheung, 1992; Marchenko and Sage, 1993; Quignard et al., 2000; Burnham et al., 2002; Eichler et al., 2003; Weston et al., 2005). The induced hyperpolarizations appear to be mediated by activation of KCa3.1 and KCa2.3 channels, since they are inhibited by blockers of these channels or targeted gene disruption (Kohler and Hoyer, 2007).
The resting membrane potential in intact endothelium is significantly positive to EK, indicating that sodium- or chloride-permeable channels also contribute to the membrane potential in the intact endothelium. This depolarizing current may be mediated by TRP (transient receptor potential) channels expressed in endothelial cells (Nilius et al., 2003; Yao and Garland, 2005). However, only K+ currents were detected in isolated AECs, suggesting that cell isolation or lower temperature affected the functionality of channels that mediate the depolarizing current.
Little is known about the role of KCa3.1 and KCa2.3 channels in the regulation of endothelium membrane potential and intracellular Ca2+ in the absence of agonists. Block of KCa2.3 channels by Ap or by silencing its gene caused a 15-mV depolarization of the membrane potential of mesenteric artery endothelial cells in a mouse model that conditionally overexpresses KCa2.3 channels (Taylor et al., 2003). Here, we show that blocking KCa3.1 channels with ChTX depolarizes the endothelium membrane by ≈8 mV, and blocking KCa2.3 channels causes an additional ≈3 mV depolarization (Fig. 6). Therefore, KCa3.1 and KCa2.3 channels likely regulate endothelial function in the absence and presence of agonists.
Acknowledgments
The authors are grateful to Drs. J. Brayden, H. Girouard, and S.V. Straub for their insightful comments for the manuscript.
This study was supported by Canadian Institutes for Health Research and Fonds de Recherche en Santé du Québec fellowships awarded to J. Ledoux and National Institutes of Health grants HL44455, HL63722, and HL77378, Totman Trust for Medical Research to M.T. Nelson.
Olaf S. Andersen served as editor.
Abbreviations used in this paper: AEC, aortic endothelial cell; Ap, apamin; BKCa, large conductance Ca2+-activated potassium channel; [Ca2+]i, intracellular Ca2+ level; ChTX, charybdotoxin; EC, endothelial cell; EDHF, endothelium-derived hyperpolarizing factor; EK, equilibrium potential; GHK, Goldman-Hodgkin-Katz; IbTX, iberiotoxin; IKCa, intermediate conductance Ca2+-activated potassium channel; Kir, inward rectifier potassium channel; Kv, voltage-dependent potassium channel; SKCa, small conductance Ca2+-activated potassium channel; TRP, transient receptor potential.
References
- Absi, M., M.P. Burnham, A.H. Weston, E. Harno, M. Rogers, and G. Edwards. 2007. Effects of methyl β-cyclodextrin on EDHF responses in pig and rat arteries; association between SK(Ca) channels and caveolin-rich domains. Br. J. Pharmacol. 151:332–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benham, C.D., T.B. Bolton, R.J. Lang, and T. Takewaki. 1986. Calcium-activated potassium channels in single smooth muscle cells of rabbit jejunum and guinea-pig mesenteric artery. J. Physiol. 371:45–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnham, M.P., R. Bychkov, M. Feletou, G.R. Richards, P.M. Vanhoutte, A.H. Weston, and G. Edwards. 2002. Characterization of an apamin-sensitive small-conductance Ca2+-activated K+ channel in porcine coronary artery endothelium: relevance to EDHF. Br. J. Pharmacol. 135:1133–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnham, M.P., I.T. Johnson, and A.H. Weston. 2006. Impaired small-conductance Ca2+-activated K+ channel-dependent EDHF responses in Type II diabetic ZDF rats. Br. J. Pharmacol. 148:434–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bychkov, R., M.P. Burnham, G.R. Richards, G. Edwards, A.H. Weston, M. Feletou, and P.M. Vanhoutte. 2002. Characterization of a charybdotoxin-sensitive intermediate conductance Ca2+-activated K+ channel in porcine coronary endothelium: relevance to EDHF. Br. J. Pharmacol. 137:1346–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castle, N.A., D.O. London, C. Creech, Z. Fajloun, J.W. Stocker, and J.M. Sabatier. 2003. Maurotoxin: a potent inhibitor of intermediate conductance Ca2+-activated potassium channels. Mol. Pharmacol. 63:409–418. [DOI] [PubMed] [Google Scholar]
- Chatterjee, S., A.B. Al Mehdi, I. Levitan, T. Stevens, and A.B. Fisher. 2003. Shear stress increases expression of a KATP channel in rat and bovine pulmonary vascular endothelial cells. Am. J. Physiol. Cell Physiol. 285:C959–C967. [DOI] [PubMed] [Google Scholar]
- Chen, G.F., and D.W. Cheung. 1992. Characterization of acetylcholine-induced membrane hyperpolarization in endothelial cells. Circ. Res. 70:257–263. [DOI] [PubMed] [Google Scholar]
- Crane, G.J., S.D. Walker, K.A. Dora, and C.J. Garland. 2003. Evidence for a differential cellular distribution of inward rectifier K channels in the rat isolated mesenteric artery. J. Vasc. Res. 40:159–168. [DOI] [PubMed] [Google Scholar]
- Eichler, I., J. Wibawa, I. Grgic, A. Knorr, S. Brakemeier, A.R. Pries, J. Hoyer, and R. Kohler. 2003. Selective blockade of endothelial Ca2+-activated small- and intermediate-conductance K+-channels suppresses EDHF-mediated vasodilation. Br. J. Pharmacol. 138:594–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, Y., G. Schram, V.G. Romanenko, C. Shi, L. Conti, C.A. Vandenberg, P.F. Davies, S. Nattel, and I. Levitan. 2005. Functional expression of Kir2.x in human aortic endothelial cells: the dominant role of Kir2.2. Am. J. Physiol. Cell Physiol. 289:C1134–C1144. [DOI] [PubMed] [Google Scholar]
- Fang, Y., E.R. Mohler III, E. Hsieh, H. Osman, S.M. Hashemi, P.F. Davies, G.H. Rothblat, R.L. Wilensky, and I. Levitan. 2006. Hypercholesterolemia suppresses inwardly rectifying K+ channels in aortic endothelium in vitro and in vivo. Circ. Res. 98:1064–1071. [DOI] [PubMed] [Google Scholar]
- Feletou, M., and P.M. Vanhoutte. 2006. Endothelium-derived hyperpolarizing factor: where are we now? Arterioscler. Thromb. Vasc. Biol. 26:1215–1225. [DOI] [PubMed] [Google Scholar]
- Forsyth, S.E., A. Hoger, and J.H. Hoger. 1997. Molecular cloning and expression of a bovine endothelial inward rectifier potassium channel. FEBS Lett. 409:277–282. [DOI] [PubMed] [Google Scholar]
- Garland, C.J., F. Plane, B.K. Kemp, and T.M. Cocks. 1995. Endothelium-dependent hyperpolarization: a role in the control of vascular tone. Trends Pharmacol. Sci. 16:23–30. [DOI] [PubMed] [Google Scholar]
- Garry, A., B. Fromy, N. Blondeau, D. Henrion, F. Brau, P. Gounon, N. Guy, C. Heurteaux, M. Lazdunski, and J.L. Saumet. 2007. Altered acetylcholine, bradykinin and cutaneous pressure-induced vasodilation in mice lacking the TREK1 potassium channel: the endothelial link. EMBO Rep. 8:354–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier, K.M., C. Liu, A. Popovic, S. Albarwani, and N.J. Rusch. 2002. Freshly isolated bovine coronary endothelial cells do not express the BK Ca channel gene. J. Physiol. 545:829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschberg, B., J. Maylie, J.P. Adelman, and N.V. Marrion. 1998. Gating of recombinant small-conductance Ca-activated K+ channels by calcium. J. Gen. Physiol. 111:565–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoger, J.H., V.I. Ilyin, S. Forsyth, and A. Hoger. 2002. Shear stress regulates the endothelial Kir2.1 ion channel. Proc. Natl. Acad. Sci. USA. 99:7780–7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii, T.M., C. Silvia, B. Hirschberg, C.T. Bond, J.P. Adelman, and J. Maylie. 1997. A human intermediate conductance calcium-activated potassium channel. Proc. Natl. Acad. Sci. USA. 94:11651–11656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isshiki, M., A. Mutoh, and T. Fujita. 2004. Subcortical Ca2+ waves sneaking under the plasma membrane in endothelial cells. Circ. Res. 95:e11–e21. [DOI] [PubMed] [Google Scholar]
- Jaggar, J.H., A.S. Stevenson, and M.T. Nelson. 1998. Voltage dependence of Ca2+ sparks in intact cerebral arteries. Am. J. Physiol. 274:C1755–C1761. [DOI] [PubMed] [Google Scholar]
- Janigro, D., G.A. West, E.L. Gordon, and H.R. Winn. 1993. ATP-sensitive K+ channels in rat aorta and brain microvascular endothelial cells. Am. J. Physiol. 265:C812–C821. [DOI] [PubMed] [Google Scholar]
- Joiner, W.J., S. Basavappa, S. Vidyasagar, K. Nehrke, S. Krishnan, H.J. Binder, E.L. Boulpaep, and V.M. Rajendran. 2003. Active K+ secretion through multiple KCa-type channels and regulation by IKCa channels in rat proximal colon. Am. J. Physiol. Gastrointest. Liver Physiol. 285:G185–G196. [DOI] [PubMed] [Google Scholar]
- Jow, F., K. Sullivan, P. Sokol, and R. Numann. 1999. Induction of Ca2+-activated K+ current and transient outward currents in human capillary endothelial cells. J. Membr. Biol. 167:53–64. [DOI] [PubMed] [Google Scholar]
- Kestler, H.A., S. Janko, U. Haussler, R. Muche, V. Hombach, M. Hoher, and J. Wiecha. 1998. A remark on the high-conductance calcium-activated potassium channel in human endothelial cells. Res. Exp. Med. (Berl.). 198:133–143. [DOI] [PubMed] [Google Scholar]
- Knot, H.J., K.M. Lounsbury, J.E. Brayden, and M.T. Nelson. 1999. Gender differences in coronary artery diameter reflect changes in both endothelial Ca2+ and ecNOS activity. Am. J. Physiol. 276:H961–H969. [DOI] [PubMed] [Google Scholar]
- Kohler, M., B. Hirschberg, C.T. Bond, J.M. Kinzie, N.V. Marrion, J. Maylie, and J.P. Adelman. 1996. Small-conductance, calcium-activated potassium channels from mammalian brain. Science. 273:1709–1714. [DOI] [PubMed] [Google Scholar]
- Kohler, R., and J. Hoyer. 2007. The endothelium-derived hyperpolarizing factor: insights from genetic animal models. Kidney Int. 72:145–150. [DOI] [PubMed] [Google Scholar]
- Kohler, R., S. Brakemeier, M. Kuhn, C. Behrens, R. Real, C. Degenhardt, H.D. Orzechowski, A.R. Pries, M. Paul, and J. Hoyer. 2001. Impaired hyperpolarization in regenerated endothelium after balloon catheter injury. Circ. Res. 89:174–179. [DOI] [PubMed] [Google Scholar]
- Ledoux, J., M.E. Werner, J.E. Brayden, and M.T. Nelson. 2006. Calcium-activated potassium channels and the regulation of vascular tone. Physiology (Bethesda). 21:69–78. [DOI] [PubMed] [Google Scholar]
- Luckhoff, A., and R. Busse. 1990. Calcium influx into endothelial cells and formation of endothelium-derived relaxing factor is controlled by the membrane potential. Pflugers Arch. 416:305–311. [DOI] [PubMed] [Google Scholar]
- Marchenko, S.M., and S.O. Sage. 1993. Electrical properties of resting and acetylcholine-stimulated endothelium in intact rat aorta. J. Physiol. 462:735–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchenko, S.M., and S.O. Sage. 1996. Calcium-activated potassium channels in the endothelium of intact rat aorta. J. Physiol. 492(Pt 1):53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marijic, J., Q. Li, M. Song, K. Nishimaru, E. Stefani, and L. Toro. 2001. Decreased expression of voltage- and Ca2+-activated K+ channels in coronary smooth muscle during aging. Circ. Res. 88:210–216. [DOI] [PubMed] [Google Scholar]
- McNeish, A.J., S.L. Sandow, C.B. Neylon, M.X. Chen, K.A. Dora, and C.J. Garland. 2006. Evidence for involvement of both IKCa and SKCa channels in hyperpolarizing responses of the rat middle cerebral artery. Stroke. 37:1277–1282. [DOI] [PubMed] [Google Scholar]
- Nilius, B., J. Prenen, G. Droogmans, T. Voets, R. Vennekens, M. Freichel, U. Wissenbach, and V. Flockerzi. 2003. Voltage dependence of the Ca2+-activated cation channel TRPM4. J. Biol. Chem. 278:30813–30820. [DOI] [PubMed] [Google Scholar]
- Papassotiriou, J., R. Kohler, J. Prenen, H. Krause, M. Akbar, J. Eggermont, M. Paul, A. Distler, B. Nilius, and J. Hoyer. 2000. Endothelial K+ channel lacks the Ca2+ sensitivity-regulating β subunit. FASEB J. 14:885–894. [PubMed] [Google Scholar]
- Quayle, J.M., J.G. McCarron, J.E. Brayden, and M.T. Nelson. 1993. Inward rectifier K+ currents in smooth muscle cells from rat resistance-sized cerebral arteries. Am. J. Physiol. 265:C1363–C1370. [DOI] [PubMed] [Google Scholar]
- Quignard, J.F., M. Feletou, G. Edwards, J. Duhault, A.H. Weston, and P.M. Vanhoutte. 2000. Role of endothelial cell hyperpolarization in EDHF-mediated responses in the guinea-pig carotid artery. Br. J. Pharmacol. 129:1103–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainbow, R.D., M.E. Hardy, N.B. Standen, and N.W. Davies. 2006. Glucose reduces endothelin inhibition of voltage-gated potassium channels in rat arterial smooth muscle cells. J. Physiol. 575:833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusko, J., F. Tanzi, C. van Breemen, and D.J. Adams. 1992. Calcium-activated potassium channels in native endothelial cells from rabbit aorta: conductance, Ca2+ sensitivity and block. J. Physiol. 455:601–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandow, S.L., and M. Tare. 2007. C-type natriuretic peptide: a new endothelium-derived hyperpolarizing factor? Trends Pharmacol. Sci. 28:61–67. [DOI] [PubMed] [Google Scholar]
- Shimoda, L.A., L.E. Welsh, and D.B. Pearse. 2002. Inhibition of inwardly rectifying K+ channels by cGMP in pulmonary vascular endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 283:L297–L304. [DOI] [PubMed] [Google Scholar]
- Si, H., W.T. Heyken, S.E. Wolfle, M. Tysiac, R. Schubert, I. Grgic, L. Vilianovich, G. Giebing, T. Maier, V. Gross, et al. 2006. Impaired endothelium-derived hyperpolarizing factor-mediated dilations and increased blood pressure in mice deficient of the intermediate-conductance Ca2+-activated K+ channel. Circ. Res. 99:537–544. [DOI] [PubMed] [Google Scholar]
- Soh, H., and C.S. Park. 2001. Inwardly rectifying current-voltage relationship of small-conductance Ca2+-activated K+ channels rendered by intracellular divalent cation blockade. Biophys. J. 80:2207–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh, H., and C.S. Park. 2002. Localization of divalent cation-binding site in the pore of a small conductance Ca2+-activated K+ channel and its role in determining current-voltage relationship. Biophys. J. 83:2528–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, M.S., A.D. Bonev, T.P. Gross, D.M. Eckman, J.E. Brayden, C.T. Bond, J.P. Adelman, and M.T. Nelson. 2003. Altered expression of small-conductance Ca2+-activated K+ (SK3) channels modulates arterial tone and blood pressure. Circ. Res. 93:124–131. [DOI] [PubMed] [Google Scholar]
- Thorneloe, K.S., and M.T. Nelson. 2005. Ion channels in smooth muscle: regulators of intracellular calcium and contractility. Can. J. Physiol. Pharmacol. 83:215–242. [DOI] [PubMed] [Google Scholar]
- von Beckerath, N., M. Dittrich, H.G. Klieber, and J. Daut. 1996. Inwardly rectifying K+ channels in freshly dissociated coronary endothelial cells from guinea-pig heart. J. Physiol. 491(Pt 2):357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston, A.H., M. Feletou, P.M. Vanhoutte, J.R. Falck, W.B. Campbell, and G. Edwards. 2005. Bradykinin-induced, endothelium-dependent responses in porcine coronary arteries: involvement of potassium channel activation and epoxyeicosatrienoic acids. Br. J. Pharmacol. 145:775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhull, A.M. 1973. Ionic blockage of sodium channels in nerve. J. Gen. Physiol. 61:687–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff, M.L., A.P. Sampath, H.R. Matthews, N.V. Krasnoperova, J. Lem, and G.L. Fain. 2002. Measurement of cytoplasmic calcium concentration in the rods of wild-type and transducin knock-out mice. J. Physiol. 542:843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, X.M., B. Fakler, A. Rivard, G. Wayman, T. Johnson-Pais, J.E. Keen, T. Ishii, B. Hirschberg, C.T. Bond, S. Lutsenko, et al. 1998. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature. 395:503–507. [DOI] [PubMed] [Google Scholar]
- Yao, X., and C.J. Garland. 2005. Recent developments in vascular endothelial cell transient receptor potential channels. Circ. Res. 97:853–863. [DOI] [PubMed] [Google Scholar]