

Abstract
Like Bcl-2, Mcl-1 is an important survival factor for many cancers, its expression contributing to chemoresistance and disease relapse. However, unlike other prosurvival Bcl-2–like proteins, Mcl-1 stability is acutely regulated. For example, the Bcl-2 homology 3 (BH3)–only protein Noxa, which preferentially binds to Mcl-1, also targets it for proteasomal degradation. In this paper, we describe the discovery and characterization of a novel BH3-like ligand derived from Bim, BimS2A, which is highly selective for Mcl-1. Unlike Noxa, BimS2A is unable to trigger Mcl-1 degradation, yet, like Noxa, BimS2A promotes cell killing only when Bcl-xL is absent or neutralized. Furthermore, killing by endogenous Bim is not associated with Mcl-1 degradation. Thus, functional inactivation of Mcl-1 does not always require its elimination. Rather, it can be efficiently antagonized by a BH3-like ligand tightly engaging its binding groove, which is confirmed here with a structural study. Our data have important implications for the discovery of compounds that might kill cells whose survival depends on Mcl-1.
Introduction
The control of apoptosis is governed largely by interactions between the prosurvival and prodeath members of the Bcl-2 protein family (Cory et al., 2003). The process is initiated when the proapoptotic Bcl-2 homology 3 (BH3)–only proteins dock their α-helical BH3 domains into a hydrophobic groove on their prosurvival targets (Bcl-2, Bcl-xL, Bcl-w, Mcl-1, and A1; Petros et al., 2000; Liu et al., 2003; Czabotar et al., 2007). This binding event results in activation of the essential death mediators Bax and Bak, thereby committing cells to apoptosis (Cheng et al., 2001; Zong et al., 2001; Willis and Adams, 2005).
It has recently become apparent that many BH3-only proteins show preferential binding to their prosurvival Bcl-2–like targets, the selective interactions being a central feature of apoptotic regulation (Chen et al., 2005; Kuwana et al., 2005; Willis et al., 2005, 2007; Certo et al., 2006; Kim et al., 2006). Although certain BH3-only proteins such as Bim and Puma bind to all prosurvival proteins tightly, others only bind to certain subsets: Bad engages Bcl-2, Bcl-xL, and Bcl-w tightly, whereas Noxa preferentially binds to Mcl-1 and A1. The molecular basis that underpins this selectivity is currently unclear despite the availability of several structures of BH3 domain–prosurvival protein complexes (Sattler et al., 1997; Petros et al., 2000; Liu et al., 2003; Czabotar et al., 2007). Such information will probably be invaluable for the design of ligands (including small-molecule BH3 mimetic compounds) that are highly specific. These ligands will be useful for probing aspects of apoptotic regulation controlled by the Bcl-2 pathway and may also provide clues for the development of drugs that target particular prosurvival proteins overexpressed within tumors. Highly specific, tailored therapies may have fewer side effects than those that act on a broad range of targets.
A promising BH3 mimetic anticancer drug is ABT-737 (Oltersdorf et al., 2005). Although ABT-737 causes the regression of some tumors in mouse xenograft models (Oltersdorf et al., 2005), its efficacy as a single agent against many cancers is limited because it only binds to Bcl-2, Bcl-xL, and Bcl-w with high affinity but not to Mcl-1 (van Delft et al., 2006). As inactivating Mcl-1 is critical for cell death to proceed (Chen et al., 2005), strategies that target Mcl-1 sensitize many cell types to ABT-737 (Konopleva et al., 2006; van Delft et al., 2006; Chauhan et al., 2007; Chen et al., 2007; Lin et al., 2007). Physiologically, the elimination of Mcl-1 in response to cytotoxic signals is also thought to be a critical step in cell death (Craig, 2002; Cuconati et al., 2003; Nijhawan et al., 2003; Willis et al., 2005; Brunelle et al., 2007). Thus, small-molecule drugs that specifically target Mcl-1, which are yet to be developed, may prove useful to complement the activity of ABT-737. However, it is currently not determined whether such molecules also need to target Mcl-1 for degradation to be effective.
To gain insight into the molecular basis of BH3 domain selectivity, we performed an extensive structure-function study to identify the functional epitopes on the promiscuous binding BH3 domain from Bim. Unexpectedly, this analysis enabled the design of a novel BimBH3 variant highly specific for Mcl-1 that enabled us to investigate the requirements for the neutralization of Mcl-1 prosurvival activity. Our data demonstrate that degradation is not essential for Mcl-1 antagonism and that ligands that merely engage its hydrophobic groove with high affinity are sufficient. Consistent with this finding, we also demonstrate that apoptotic stimuli that result in the induction of endogenous Bim kill cells without eliminating Mcl-1, which is in contrast to DNA-damaging and certain other stimuli. In addition, our biochemical and structural analyses provide new insights into how BH3 ligands engage their targets.
Results
Alanine-scanning mutagenesis reveals the unique way Bim engages Mcl-1
We postulated that identifying the determinants on BH3 domains for binding prosurvival proteins would provide clues for the design of variants with novel selectivity profiles. Previously, we have demonstrated that phage display could be applied to studying interactions between BH3 domains and prosurvival proteins (Kvansakul et al., 2007; Lee et al., 2007). In one study, a 26-residue peptide encompassing the BimBH3 domain was expressed on M13 phage as a fusion to the minor coat protein encoded by gene 3 (Sidhu et al., 2000) and was found to bind to four prosurvival proteins with IC50 values that closely matched those obtained with synthetic peptides (Lee et al., 2007), as measured in solution competition assays using the optical biosensor (Chen et al., 2005) or by isothermal titration calorimetry (ITC; unpublished data). In this study, we have mutated every amino acid residue within the 26-residue sequence to alanine except for wild-type alanine or glycine, which were instead replaced with glutamic acid. The sequences also had a FLAG epitope fused to their N terminus to ensure that all mutants expressed comparably in binding assays with an anti-FLAG antibody. Indeed, very similar expression levels were observed for all mutants (unpublished data), allowing us to proceed with a detailed analysis of prosurvival protein binding.
Significant differences were observed in the effect of BimBH3 mutations on binding to different prosurvival proteins. Bcl-xL and Bcl-w binding were comparably affected, with the same five BimBH3 mutations significantly affecting the interactions (Fig. 1 A and Fig. S1 A, available at http://www.jcb.org/cgi/content/full/jcb.200708096/DC1). Three mutations (A59E, G66E, and D67A) reduced binding to Bcl-xL or Bcl-w below detectable levels in our assay, whereas two others (L62A and F69A) had partial effects, reducing the affinity of BimBH3 for Bcl-xL and Bcl-w 18–45-fold. In contrast, the interactions of these mutants with Bcl-2 differed in two respects (Fig. 1 B). The L62A mutant had a greater impact, whereas F69A decreased Bcl-2 binding only by twofold, which is in contrast to the greater loss seen for its binding to Bcl-xL or Bcl-w.
Figure 1.

Identifying residues in the BH3 region of Bim critical for binding prosurvival Bcl-2 proteins. (A–C) Individual residues on the phage-displayed BimBH3 (amino acids 51–76 of human BimS) were mutated to alanine except for native alanine or glycine, which were replaced instead with glutamic acid. Their affinities (in IC50) for Bcl-xL (A), Bcl-2 (B), and Mcl-1 (C) were determined by solution competition ELISAs (Kvansakul et al., 2007; Lee et al., 2007). The data represent the IC50 (left) and fold reduction in binding compared with wild-type BimBH3 (right). The upper limits on the y axes were set to 1,000 nM (left) or at least a 40-fold reduction based on the IC50 of a nonbinding mutant (G66E) independently confirmed in solution competition assays using an optical biosensor (Chen et al., 2005). (D–F) Validation of in vitro binding assays. Interactions between FLAG-tagged prosurvival Bcl-xL (D), Bcl-2 (E), or Mcl-1 (F; black arrowheads) and HA-tagged BimS or selected BH3 mutants of it (white arrowheads) were assessed by coimmunoprecipitation. Equivalent 35S-labeled lysates harvested from transiently transfected 293T cells were immunoprecipitated with antibodies to the FLAG (FL), HA, or control (C) tags. Wild-type BimS or mutants that bound with wild-type affinities (in A–C) to the prosurvival proteins are indicated by boxes. A nonbinding BimS mutant (BimS4E; Chen et al., 2005) served as a negative control. Data in A–C represent means ± SD (error bars) of three experiments; those in D–F are from representative experiments.
The most striking difference was observed with Mcl-1 (Fig. 1 C). Only the G66E and D67A mutants had significant effects (28-fold and ninefold decreases, respectively) on Mcl-1 binding. Therefore, Mcl-1 is more tolerant of mutations in Bim, as no significant effects were observed with mutations (A59E, L62A, and F69A) that drastically affected binding to Bcl-xL, Bcl-w, or Bcl-2 (Figs. 1, A–C; and S1 A). Together, these results illustrate differences in the way Bim engages the various prosurvival proteins and highlights the unique nature of Bim binding to Mcl-1.
Binding of phage-displayed peptides predicts the association of intact proteins in cells
As the alanine scanning data (Fig. 1, A–C) reflects the binding of an isolated BH3 domain expressed on phage particles to C-terminally truncated recombinant proteins, we next tested several full-length BimBH3 domain mutants for their ability to interact with full-length prosurvival proteins in mammalian cells to determine the potential physiological relevance of these results. In all cases, we observed a close correlation between in vitro binding (Fig. 1, A–C) and the ability of full-length BimS to interact with prosurvival proteins in cells (Fig. 1, D–F).
For example, BimBH3 mutants that adversely affected Bcl-xL binding (i.e., A59E, L62A, G66E, D67A, and F69A; Fig. 1 A) had reduced ability to associate with wild-type Bcl-xL when introduced into full-length BimS (Fig. 1 D). Similar observations were obtained with Bcl-2 (Fig. 1, compare B with E). In this case, the BimSF69A mutant retained Bcl-2 binding (Fig. 1 E), which is consistent with the small (twofold) decrease seen with the phage-displayed mutant (Fig. 1 B). Finally, Mcl-1 bound to BimSA59E, L62A, and F69A but not to any significant extent to G66E or D67A (Fig. 1 F), which is in agreement with the phage data (Fig. 1 C).
An Mcl-1–specific Bim variant
The distinct binding profile of BimBH3 mutants for Mcl-1 (Fig. 1) suggested that it might be possible to target it specifically because two residues, L62 and F69, are important for binding Bcl-xL, Bcl-w, or Bcl-2 (Figs. 1, A and B; and S1 A) but not Mcl-1 (Fig. 1 C). Initial experiments with a phage-displayed BimBH3 L62A/F69A double mutant showed tight binding only to Mcl-1 (unpublished data). To accurately quantify its affinity, we tested a synthetic peptide harboring both mutations (hereafter referred to as BimBH3 2A) in solution competition assays using an optical biosensor. At peptide concentrations up to 10 μM, no binding to Bcl-xL, Bcl-w, or Bcl-2 was detected, and affinity for A1 was also reduced (Fig. 2 A). However, consistent with the phage experiments, its affinity for Mcl-1 was indistinguishable from the wild-type peptide. Binding selectivity with full-length proteins was confirmed in cells, as BimS2A (i.e., full-length BimS containing the L62A/F69A mutations) bound to Mcl-1 like its wild-type parent but not to Bcl-xL (Fig. 2 B).
Figure 2.

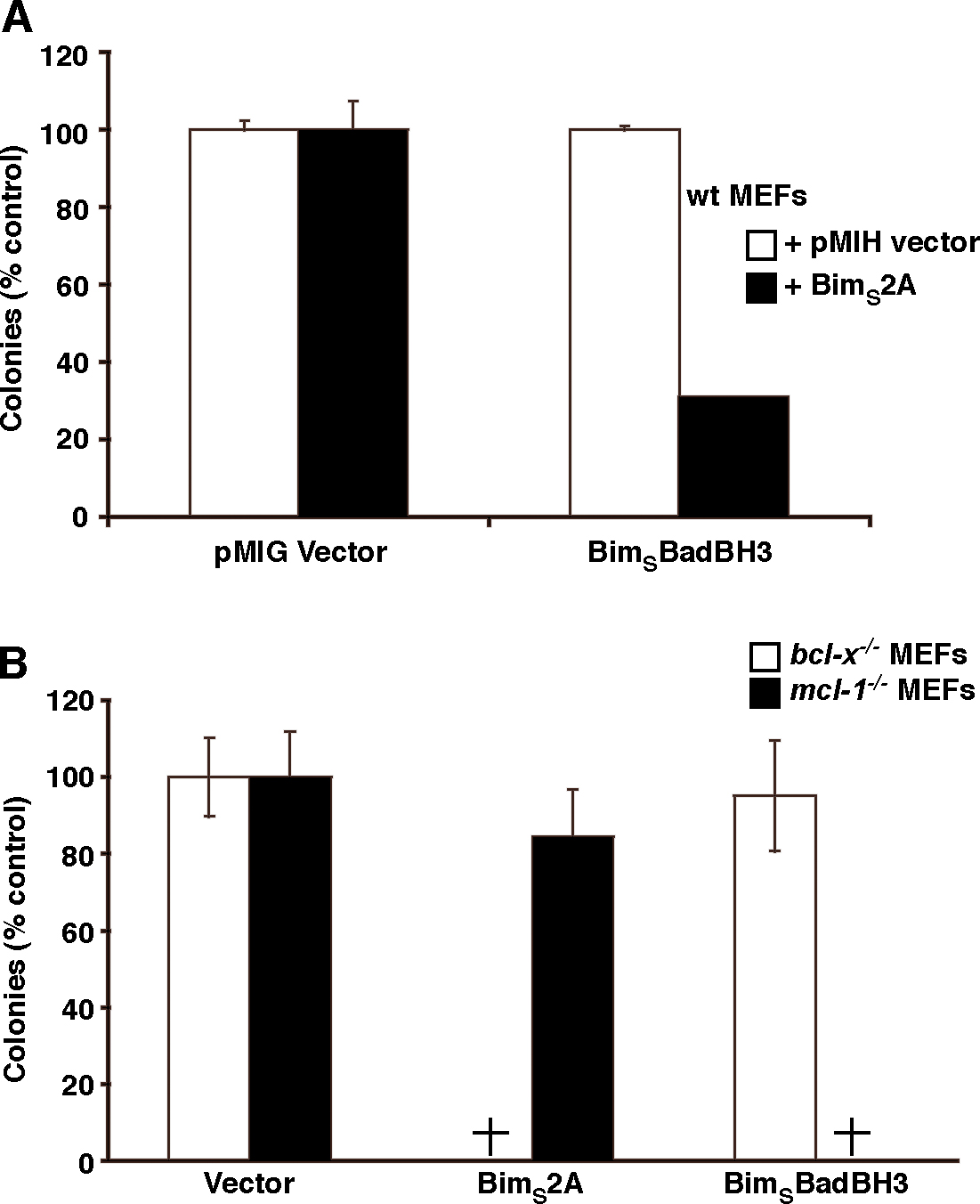
Characterization of a novel Mcl-1–specific Bim variant. (A) A BimBH3 mutant that retains significant binding only to Mcl-1. The relative affinities of wild-type or the BimBH3 mutant 2A (L62A/F69A) peptides for prosurvival proteins were determined in solution competition assays using an optical biosensor. Compared with the wild-type sequence, the mutant 2A has reduced affinity for all prosurvival proteins except for Mcl-1. (B) Full-length BimS2A (top) interacts with Mcl-1 in cells (right), but, unlike wild-type BimS (bottom), it does not bind Bcl-xL (left), which is consistent with the in vitro binding assays using purified components (A). Prosurvival proteins are indicated by black arrowheads, and BimS/BimS2A is indicated by white arrowheads. (C) Functional cooperation between the Mcl-1–specific BimS2A variant and Bad. Stable pools of wild-type MEFs were generated by hygromycin selection after retroviral infection with the parental pMIH vector or one expressing the full-length Mcl-1–specific BimS2A mutant. Long-term colony formation was assessed when these pools were reinfected with the control pMIG retrovirus or one expressing BimSBadBH3 (a BimS variant with its BH3 replaced with that of Bad; Chen et al., 2005). BimS2A was inert on its own, but there was a striking reduction in colonies formed if BimSBadBH3, which targets the prosurvival proteins (Bcl-xL, Bcl-2, and Bcl-w) that BimS2A does not bind, is coexpressed. (D) The Mcl-1–specific BH3 ligands, full-length BimSNoxaBH3 or BimS2A, potently kill only when Bcl-xL is absent, whereas BimSBadBH3 only kills the cells deficient in Mcl-1. Cell viability was assessed 24 h after infection with the indicated retroviruses. (E) HA-tagged wild-type BimS but neither the BimS2A nor the inert BimS mutant (BimS4E) coimmunoprecipitated with endogenous Bax. (F) FLAG-tagged Bak (middle) coimmunoprecipitates with HA-tagged Bcl-xL and Mcl-1 but not with HA-tagged BimS4E, BimS, or BimS2A (top). Asterisks are the remnant signals from reprobing of the FLAG immunoprecipitation (top) with the FLAG antibody. Data in D represent means ± SD (error bars) of at least three experiments; those in A–C, E, and F) are from representative experiments. WCL, whole cell lysate.
As BimS2A was derived from a promiscuously binding BH3-only protein, it was essential to test whether its biological action matches the binding profile. Because deletion or inactivation of Mcl-1 alone, such as that seen with the BH3-only protein Noxa, is not sufficient to kill mouse embryonic fibroblasts (MEFs; Chen et al., 2005; Chen et al., 2007), we anticipated that BimS2A would be inert, and, as expected, its high level expression was tolerated in long-term assays (Fig. 2 C and Fig. S2 A, available at http://www.jcb.org/cgi/content/full/jcb.200708096/DC1). Because Bad (which targets Bcl-xL, Bcl-w, and Bcl-2) cooperates with Noxa (targeting Mcl-1) for killing cells (Chen et al., 2005), we further anticipated that Bad should also complement BimS2A. Indeed, potent killing was observed with this combination (Figs. 2 C and S2 A). Our recent experiments (Willis et al., 2005) also suggest that only Mcl-1 and Bcl-xL have to be inactivated for Bak-mediated apoptosis. Therefore, in cells that express Bak but are deficient in Bcl-xL, only Mcl-1 needs to be targeted for cell death (Chen et al., 2005). Accordingly, killing of Bcl-xL–deficient MEFs by BimS2A was observed (Figs. 2 D and S2 B), which is comparable with killing by a BimS variant with a Noxa BH3 domain replacement that makes it relatively selective for Mcl-1 (Chen et al., 2005; Willis et al., 2005). No killing of Mcl-1–deficient MEFs by BimS2A was observed, supporting its specificity for Mcl-1.
Importantly, unlike wild-type BimS, BimS2A was unable to bind to Bax in immunoprecipitation experiments (Fig. 2 E), nor was any interaction between Bak and BimS or BimS2A detected (Fig. 2 F). Therefore, collectively, these data suggest that this novel Mcl-1–specific Bim variant neutralizes only one Bcl-2 family member, Mcl-1, and can cause killing, provided Bcl-xL is also inactivated.
The Mcl-1–specific ligand BimS2A stabilizes Mcl-1
Although the expression of Noxa causes the degradation of Mcl-1 (Willis et al., 2005; Czabotar et al., 2007), neither BimS nor BimS2A caused any reduction in basal Mcl-1 levels (Fig. 3 A). Instead, basal levels of Mcl-1 appear to be stabilized in BimS- or BimS2A-expressing cells. This apparent stabilization may be caused by the BimS ligand–inhibiting binding of the BH3 domain–containing E3 ubiquitin ligase, Mule, which appears to be important for regulating basal levels of Mcl-1 by targeting it for proteasomal degradation (Zhong et al., 2005).
Figure 3.

The Mcl-1–specific Bim variant does not trigger Mcl-1 degradation. (A) Immunoblots of equivalent lysates prepared from MEFs after infection with parental retroviral vector or ones expressing wild-type BimS, BimS4E, BimS2A, BimSNoxaBH3, or Noxa itself were probed with antibodies to Mcl-1 (top), Bcl-2 (middle), or HSP70 (loading control; bottom). Levels of Mcl-1 were significantly lower if Noxa or BimSNoxaBH3 (BimS with its BH3 domain replaced with that from Noxa) is expressed. In contrast, the Mcl-1–specific BimS2A variant stabilizes Mcl-1 levels. The images were assembled and cropped from a single gel. (B) BimS2A counters Mcl-1 degradation triggered by Noxa. Equivalent lysates were prepared from Noxa- or BimS4E-overexpressing MEFs after reinfection with the inert BimS4E, Noxa, or BimS2A retroviruses, and the blots were probed for Mcl-1 or HSP70 (loading control). Note that Mcl-1 levels are significantly higher in cells expressing the Mcl-1–specific BimS2A. (C) ITC analysis of BimBH3 2A and NoxaBH3 binding to Mcl-1. BimBH3 2A binds to Mcl-1 tighter than NoxaBH3.
This prompted us to examine the possibility that BimS2A could act as a specific inhibitor of Mcl-1 degradation. As both Noxa and Bim bind within the same hydrophobic groove on Mcl-1 (Czabotar et al., 2007), it would be predicted that BimS2A could compete with Noxa for Mcl-1 binding, thereby blocking the degradation triggered by Noxa. Indeed, our data support this hypothesis, as the enforced expression of BimS2A in cells stably expressing Noxa results in higher levels of Mcl-1 compared with Noxa-expressing cells, in which the inert BimS variant BimS4E was expressed (Fig. 3 B). The reintroduction of Noxa does not lead to a further significant decrease in Mcl-1 levels, suggesting that Noxa is not at limiting concentrations in these cells (Fig. 3 B). The ability of BimS2A to out-compete Noxa is supported by ITC analysis that shows that the actual dissociation constant of BimBH3 2A peptide binding to Mcl-1 is ninefold tighter than NoxaBH3 (Fig. 3 C).
Mcl-1 neutralization is as effective as degradation for cell killing
As the killing activity of BimS2A is not associated with Mcl-1 degradation (unlike Noxa), it provided an excellent reagent to address the question of whether ligands that simply engage the binding groove on Mcl-1 but do not also target it for degradation are as effective at killing cells as those that function more like Noxa. This has important consequences for the development of small-molecule drugs targeting Mcl-1, as these will most likely work in a manner like BimS2A.
Therefore, we compared the ability of BimS2A with that of Noxa or BimS with its BH3 domain replaced with that from Noxa (BimSNoxaBH3; both of which cause Mcl-1 degradation when overexpressed in cells) to cooperate with ABT-737 to kill cells. As shown previously, ABT-737 is a Bad BH3 mimetic (targeting Bcl-xL, Bcl-2, and Bcl-w) and, therefore, is unable to kill many cells, including MEFs, on its own (Fig. 4 A; Chen et al., 2005; Oltersdorf et al., 2005; Willis et al., 2005; Konopleva et al., 2006; van Delft et al., 2006; Lin et al., 2007). However, when combined with strategies that reduced Mcl-1 levels, ABT-737 is a very potent killer (Konopleva et al., 2006; van Delft et al., 2006; Lin et al., 2007; Trudel et al., 2007). By performing a dose-response experiment with ABT-737 on cells expressing either BimS2A or Noxa/BimSNoxaBH3, we observed that BimS2A is just as effective at sensitizing cells to the killing activity of ABT-737 as ligands that also cause Mcl-1 degradation (Fig. 4 A). Furthermore, the kinetics of killing with a fixed concentration of ABT-737 are very similar, with maximal cell death observed within 4–6 h after treatment with ABT-737 regardless of whether Mcl-1 was degraded or not (Fig. 4 B).
Figure 4.

BimS2A efficiently inactivates Mcl-1 to induce cell killing. (A) BimS2A sensitizes cells to ABT-737, a BadBH3 mimetic. The viability of MEFs stably infected with the vector control or ones expressing BimS2A, BimSNoxaBH3, or Noxa was determined 24 h after treatment with 0–10 μM ABT-737. Although ABT-737, which targets Bcl-xL, Bcl-2, and Bcl-w, was inert on its own, the three Mcl-1–selective BH3-only proteins were equipotent at cooperating with ABT-737 to efficiently kill cells. (B) The kinetics of killing with 1 μM ABT-737 were comparable regardless of whether Mcl-1 was degraded (with Noxa or BimSNoxaBH3) or neutralized (with BimS2A). (C) BimS2A promotes Bax/Bak-mediated cytochrome c release only when combined with ABT-737. Equivalent lysates prepared from wild-type (top) or bax−/−bak−/− (bottom) MEFs stably expressing the inert BimS4E or the Mcl-1–specific BimS2A were fractionated after incubation in vitro with 5 μM ABT-737 (+). Only the combination of BimS2A and ABT-737 caused cytochrome c release, which was abrogated in the absence of Bax and Bak (bottom). Blots for Bcl-2 (pellet fraction; p) and Apaf-1 (soluble fraction; s) served as markers for the subcellular fractionation. (D) Only when combined with ABT-737 does the Mcl-1–specific BimS2A kill cells, either through Bax or Bak without the need for the putative activator BH3-only proteins (Bim and Bid). Data in A, B, and D are means ± SD (error bars) of at least three experiments, whereas C is from a representative experiment.
As Bax/Bak-dependent cytochrome c release is a hallmark of apoptosis, this was used as a marker of the ability of BimS2A to sensitize MEFs to ABT-737 in vitro. No cytochrome c release was observed from mitochondria derived from BimS4E-expressing cells (or bax−/−bak−/− MEFs expressing either BimS4E or BimS2A) in response to ABT-737, but significant release was seen with mitochondria from BimS2A-expressing cells (Fig. 4 C). Previously, the Noxa/ABT-737 combination was shown to kill MEFs via both Bax or Bak (van Delft et al., 2006). Similarly, the BimS2A/ABT-737 combination was able to kill both bax−/− and bak−/− MEFs (Fig. 4 D) but not bax−/−bak−/− MEFs (not depicted), demonstrating that Mcl-1 degradation is not required for either Bak- or Bax-mediated cell death. The clear-cut results with cells expressing only Bax or Bak prove genetically that either can function to mediate killing triggered by the BimS2A/ABT-737 combination.
As shown, BimS2A is unable to directly engage Bax or Bak (Fig. 2, E and F); nevertheless, it may be exerting its proapoptotic activity by acting as a sensitizer BH3-only protein, releasing activator BH3-only proteins (such as Bim and/or Bid) sequestered by prosurvival proteins (Letai et al., 2002; Kuwana et al., 2005; Certo et al., 2006). This did not appear to be the case, however, as BimS2A and ABT-737 (or BimSBadBH3) were able to synergize and kill MEFs lacking both of the putative direct activators, Bim and Bid (Fig. 4 D and Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200708096/DC1). This suggests that the killing observed with this combination is caused by an indirect mechanism of Bax/Bak activation whereby multiple prosurvival proteins are neutralized (Willis et al., 2005, 2007) but not necessarily degraded (in the case of Mcl-1).
Finally, to see whether some of the aforementioned observations could be made for cells other than MEFs, we also expressed BimS2A in FDC-P1 myelomonocytic cells. Consistent with the results for MEFs, these cells did not die in response to ABT-737 alone but were killed in response to ABT-737 after the enforced expression of BimS2A (Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200708096/DC1).
The aforementioned results demonstrate that cells previously resistant to ABT-737 can be sensitized when treated in combination with ligands that neutralize but not necessarily degrade Mcl-1. Therefore, we envisage that small-molecule drugs targeting Mcl-1 should only need to engage the hydrophobic groove, as targeting the protein for destruction is not required.
Long-term neutralization of Mcl-1 is not well tolerated
Although the expression of BimS2A and Noxa appears to be tolerated in cell lines that express them under antibiotic selection pressure, we next examined the effect of long-term neutralization on an unselected population of cells. Thus, we infected wild-type or bax−/−bak−/− MEFs with retroviruses in which the expression of BimS2A, Noxa, Bad, or BimS4E was linked to GFP expression through an internal ribosome entry site (IRES). The population of viable cells (propidium iodide negative) that remained GFP+ve was then monitored over a period of ∼40 d. Essentially, >90% of all cells remained GFP+ve regardless of the construct expressed in the bax−/−bak−/− MEFs (Fig. 5 B). Similarly, wild-type MEFs expressing the inert BimS4E or Bad remained mostly (>80%) GFP+ve (Fig. 5 A). However, in cells in which Mcl-1 was neutralized by the enforced expression of either Noxa or BimS2A, there was a progressive loss of GFP+ve cells over the time period, with the loss somewhat more rapid in the BimS2A cells (Fig. 5 A). These data suggest that long-term neutralization of just Mcl-1 may not be well tolerated.
Figure 5.

Long-term neutralization of Mcl-1 is not well tolerated. Wild-type MEFs (A) stably expressing BimS4E (black triangles), BimS2A (red circles), Noxa (blue diamonds), or Bad (green squares) were generated. Expression of the BH3-only proteins was linked to GFP expression through an IRES. Long-term expression of the inert BimS4E or Bad was maintained for nearly 40 d, as indicated by the proportion of cells that remained GFP+ve. In contrast, targeting Mcl-1 (either with BimS2A or Noxa) was poorly tolerated unless the downstream mediators of apoptosis (Bax and Bak) were absent (B).
Mcl-1 binds BimBH3 2A similarly to wild-type BimBH3
Analysis of the three-dimensional structures of various BH3 domain peptides in complex with prosurvival proteins (including BimBH3–Mcl-1; Sattler et al., 1997; Petros et al., 2000; Liu et al., 2003; Czabotar et al., 2007) suggests that the four conserved hydrophobic residues that partly define the BH3 domain sequence motif are important for engaging their prosurvival counterparts, as they are usually buried within hydrophobic pockets along the prosurvival protein-binding groove. Thus, an outstanding question regarding the BimS2A mutant is how it engages Mcl-1 given that two of the defining features of a BH3 domain (the leucine at position h2 and a large hydrophobic residue at h4) are absent (Fig. 6 A). Therefore, we determined the crystal structure of a BimBH3 2A 26-mer synthetic peptide in complex with a human/mouse Mcl-1 chimera (hMcl-1) to 2.0-Å resolution and compared it with the wild-type BimBH3–hMcl-1 complex (Czabotar et al., 2007). This chimeric Mcl-1 had a very similar BimBH3 alanine scan binding profile (Fig. S1 B) to the mouse Mcl-1 used in the aforementioned studies (Fig. 1 C). The crystals grew in identical conditions to the wild-type BimBH3–Mcl-1 complex and had similar unit cell dimensions (Table S1; available at http://www.jcb.org/cgi/content/full/jcb.200708096/DC1; Czabotar et al., 2007).
Figure 6.

The Mcl-1–specific Bim variant binds Mcl-1 like wild-type Bim. (A) Alignment of sequences of representative BH3 domains from BH3-only proteins or the Mcl-1–specific BimS variant BimS2A. The numbers in parentheses indicate the positions of residues in full-length proteins, and numbers in orange at the top indicate the positions in full-length BimS. The four conserved hydrophobic residues are boxed (blue) and labeled as h1–h4. Residues shaded in black are conserved in all proapoptotic BH3 domains. (B) Overlay of the crystal structures of human Mcl-1 (yellow) in complex with wild-type BimBH3 (orange) and that of human Mcl-1 (cyan) complexed with BimBH3 2A (blue). (C–E) Minor structural differences between the wild-type and mutant BimBH3 complexes were noted in the Mcl-1 regions proximal to L62 (C), F69 (D), and around residues Y72 and Y73 (E) in the BimBH3 peptide. (F) The effect on Mcl-1 binding of additional mutations (bold) in the context of the phage-displayed BimBH3 2A variant. The fold decrease in binding with respect to BimBH3 2A is shown. Residue I65 (position h3) but not I58 (position h1) appears critical for the binding of BimBH3 2A to Mcl-1.
Surprisingly, the BimBH3 2A peptide binds to Mcl-1 in an almost identical fashion to the wild-type peptide (Fig. 6 B). Only minor differences were observed in the orientations of side chains on Mcl-1 close to the sites of the mutations in the BimBH3 2A peptide. For example, the side chains of F228 and M231, which form part of the h2 binding pocket, have moved slightly so as to fill the void introduced by the L62A substitution (Fig. 6 C). Similar subtle reorientations of the side chains of V216 and V220 on Mcl-1 in the vicinity of the F69A mutation were also observed (Fig. 6 D). The only differences on the peptide itself were in the side chains of Y72 and Y73, which now tilt more toward the h4 binding pocket (Fig. 6 E). Although the changes observed are small, the structures have been determined at high resolution, and the differences observed here are unambiguous.
Binding energy hotspots in the BimBH3 2A–Mcl-1 complex
The aforementioned structure demonstrates that even peptides without a canonical BH3 sequence can still bind within the hydrophobic groove on Mcl-1 with high affinity. However, it does not clarify the sites on the mutant peptide that are critical for the interaction. Therefore, we performed additional site-directed mutagenesis, this time using BimBH3 2A as the template sequence.
The crystal structure indicated several potentially important interacting amino acids on BimBH3 2A, particularly the hydrophobic residues at the h1 and h3 positions (I58 and I65, respectively; Fig. 6 A). Indeed, simultaneous replacement of all four conserved hydrophobic residues (h1–h4) caused a total loss of detectable binding, although, remarkably, replacement of h1 (I58) with alanine in combination with the L62A/F69A mutations had no discernable effect on the interaction as measured either in a competition ELISA (Fig. 6 F) or by ITC using a mutant synthetic peptide (not depicted). However, mutation of the h3 site (I65) to alanine in the L62A/F69A peptide ablated binding (Fig. 6 F). This demonstrates that just a single large hydrophobic residue (I65) within BimBH3 2A is required for a high affinity interaction with Mcl-1. Interestingly, this h3 position is also important in the context of the L62A single mutation (Fig. 6 F). However, essentially no effect was observed when I65 was mutated on its own (Fig. S1 B) or in combination with F69 (Fig. 6 F). Evidently, the small changes observed in Mcl-1 in the L62A region of the mutant complex are incompatible with maintaining wild-type binding affinity for the L62A/I65A combination. In contrast, the accommodating changes in Mcl-1 around the F69A site did not have a comparable knock-on effect. These results provide an example of the sensitivity of interactions between BH3 domains and prosurvival proteins, with minor structural alterations in the receptor affecting the contribution of a single amino acid residue on the ligand to the binding affinity.
Bim-induced apoptosis does not necessarily result in degradation of Mcl-1
Our biochemical and cellular studies with Bim variants in vitro suggest that Bim can efficiently inactivate Mcl-1 without its degradation (Figs. 2–4). However, as Mcl-1 degradation appears to be required for, or at least associated with, apoptosis caused by diverse stimuli (Cuconati et al., 2003; Nijhawan et al., 2003; Opferman, 2006), we were prompted to further investigate the physiological requirement for Mcl-1 degradation in Bim-induced apoptosis. As Bim is the primary BH3-only protein induced by calcium flux (Cante-Barrett et al., 2006), various apoptotic agents (e.g., thapsigargin and ionomycin) that elevate intracellular Ca2+ concentrations were used to specifically address this question.
Significant cell death was observed when wild-type breast epithelial carcinoma MCF-7 cells were treated with thapsigargin (Fig. 7 A), presumably as a result of the induction of the endogenous Bim isoforms (Fig. 7 B and Fig. S5, available at http://www.jcb.org/cgi/content/full/jcb.200708096/DC1; Puthalakath et al., 2007), as the same cells in which Bim expression was reduced by short hairpin RNA (shRNA; Fig. 7 B) were relatively insensitive to this death stimulus (Fig. 7 A). Importantly, levels of Mcl-1 were not reduced after treatment with thapsigargin, even after 48 h, but instead were elevated by up to twofold over levels in untreated cells (Figs. 7 B and S5).
Figure 7.

Physiological cell death initiated by Bim does not necessarily coincide with Mcl-1 degradation. (A) Down-regulating Bim attenuates thapsigargin-induced apoptosis. Viability of wild-type (−) or an MCF-7 subclone (+) stably expressing Bim shRNA treated with 1.5 μM thapsigargin for 0–48 h. (B) Treatment of wild-type MCF-7 cells with thapsigargin induced an increase in the levels of Bim (the isoforms include BimEL, BimL, and BimS; middle) that coincided with the onset of apoptosis seen in A. Mcl-1 levels increase upon treatment (top). Only low levels of BimEL were detectable in cells stably expressing Bim shRNA, whereas neither the BimL nor BimS isoforms were apparent. (C) Loss of Bim attenuates ionomycin-induced killing of thymocytes. The viability of wild-type (+/+) or Bim-deficient (−/−) thymocytes determined 0–24 h after treatment with 1 μg/ml ionomycin. Incubation with 50 μM of the pancaspase inhibitor qVD-OPH significantly reduced the killing induced by ionomycin. (D) Treatment of bim+/+ thymocytes with ionomycin increases the levels of BimEL and BimL after 4 h (middle), which coincided with the onset of apoptosis seen in C. There was no significant concomitant loss of Mcl-1 (top). In B and D, immunoblots were reprobed with antibodies to HSP70 for loading control (bottom). In addition, the cells were treated with 50 μM qVD-OPH to avoid confounding effects caused by caspase cleavage of Mcl-1 (Herrant et al., 2004). Data in A and C represent means ± SD (error bars) of three experiments, and B and D are from representative experiments.
Similar results were achieved for primary thymocytes derived from either wild-type or bim −/− mice. Here, treatment with the selective calcium ionophore ionomycin causes significant death to wild-type cells but not to those deficient in Bim (Fig. 7 C). As with thapsigargin treatment of MCF-7 cells, Mcl-1 levels did not significantly change after treatment and, in fact, were slightly elevated at the earlier time point (Fig. 7 D). Together, these results demonstrate that Bim-induced apoptosis does not result in Mcl-1 degradation and can lead to the stabilization of Mcl-1 similar to that seen after the overexpression of BimS (Fig. 3 A; Czabotar et al., 2007). Thus, Mcl-1 degradation is not required for cell death in response to some apoptotic stimuli.
Discussion
Mcl-1 as a target for anticancer therapy
There is great interest in the development of new anticancer drugs that target one or more of the prosurvival Bcl-2 proteins by mimicking BH3 ligands (Fesik, 2005; Zhai et al., 2006). Such therapeutics may be effective in tumors with elevated prosurvival protein levels or with defects in the p53 pathway (Fesik, 2005). Recently, ABT-737 (Oltersdorf et al., 2005) was shown to be a bona fide BH3 mimetic (van Delft et al., 2006). However, despite its high affinity for Bcl-2, Bcl-xL, and Bcl-w, ABT-737 demonstrated single-agent efficacy only in malignancies with low Mcl-1 levels (Oltersdorf et al., 2005; Konopleva et al., 2006; Chauhan et al., 2007; Trudel et al., 2007). This observation suggested that tumors resistant to ABT-737 should succumb when treated in combination with agents that eliminate Mcl-1.
Mcl-1 is believed to be an important survival factor for many cancers (Song et al., 2005; Wuilleme-Toumi et al., 2005; Qin et al., 2006; Sieghart et al., 2006; Cavarretta et al., 2007), and its overexpression contributes to chemoresistance and disease relapse (Kaufmann et al., 1998; Kitada et al., 1998; Saxena et al., 2004; Wuilleme-Toumi et al., 2005). Importantly, strategies that reduce Mcl-1 levels, for example through inhibition of its transcription (such as with the cyclin-dependent kinase inhibitors Seliciclib and Flavopiridol) or translation (with the multikinase inhibitor BAY 43-9006), can induce apoptosis in some cancer cells (Gojo et al., 2002; Rahmani et al., 2005; Raje et al., 2005; Yu et al., 2005). Furthermore, such strategies to eliminate Mcl-1 have been shown to sensitize cells refractory to ABT-737 (van Delft et al., 2006; Chen et al., 2007; Lin et al., 2007). However, these drugs may have global effects within a cell and, thus, might be expected to have side effects when used systemically or be futile in the many tumors that harbor mutations in the p53 pathway. Therefore, a more attractive approach may be to design a small-molecule inhibitor that targets Mcl-1 specifically.
Targeting Mcl-1 does not require its degradation for cell death to proceed
A common view is that antagonizing the prosurvival activity of Mcl-1 requires its elimination from cells to initiate the apoptotic cascade. Indeed, in addition to the aforementioned therapeutic strategies, many death stimuli, including UV irradiation, viral infections, or anoxia, cause a rapid decrease in Mcl-1 levels that correlates with apoptosis (Cuconati et al., 2003; Nijhawan et al., 2003; Brunelle et al., 2007). Therefore, an important question is whether small-molecule antagonists of Mcl-1 would also need to target it for degradation to be effective. This would be a particularly challenging task even though a template already exists in the Noxa BH3 domain that both binds the hydrophobic groove on Mcl-1 with high affinity and appears to form a binding surface to promote its degradation by the proteasome machinery (Willis et al., 2005; Czabotar et al., 2007). Such small-molecule Noxa-like BH3 mimetics would not only need to specifically bind to Mcl-1 with high affinity but would also somehow have to recruit the protein degradation machinery.
Interestingly, although the BimBH3 domain binds to Mcl-1 in a similar fashion to Noxa, it appears to lack the sequence or structural signature required for proteasomal targeting (Czabotar et al., 2007). Importantly, BimS is able to potently kill cells, although its overexpression actually leads to the stabilization of Mcl-1 (Czabotar et al., 2007). Similarly, another BH3-only protein, Puma, which is a potent killer of cells, was also recently shown to stabilize Mcl-1 (Mei et al., 2005). Increasing the pool of Bim or Puma for binding Mcl-1 such as that displaced by Bik from another prosurvival protein (Gillissen et al., 2007) would also be anticipated to stabilize Mcl-1. Here, we have now shown that a physiological stimulus (increase in intracellular calcium) that specifically induces endogenous Bim (Cante-Barrett et al., 2006) leads to (Bim dependent) cell death without the concomitant elimination of Mcl-1. This finding supports the view that strategies to target Mcl-1 do not necessarily need to also induce its degradation.
An Mcl-1–specific ligand
The BimBH3 mutagenesis study demonstrated that the Mcl-1 binding groove is sufficiently different to the other prosurvival proteins to allow ligands highly specific for it to be developed. This is consistent with some of our recent experiments that showed that Mcl-1 is unique in terms of how it engages BH3 peptidyl ligands (Lee et al., 2007). The novel Bim mutant BimS2A binds with high affinity and specificity to Mcl-1. Significantly, BimS2A does not function like Noxa to cause Mcl-1 degradation; instead, it stabilizes Mcl-1 levels like wild-type BimS (possibly by inhibiting interaction with Mule). Indeed, the enforced expression of BimS2A blocks Mcl-1 degradation induced by the overexpression of Noxa. Consistent with its ability to bind only Mcl-1, BimS2A was inert when introduced into cells. Only when combined with strategies to neutralize Bcl-xL, such as by coexpression with Bad, do the cells die. Killing was also observed in the constitutive absence of Bcl-xL because the only restraint left for controlling Bak (Willis et al., 2005), Mcl-1, is now inactivated by BimS2A.
Importantly, fibroblasts or myeloid cells that are refractory to ABT-737 as a single agent (van Delft et al., 2006) become highly sensitive to it when BimS2A is expressed despite the fact that Mcl-1 levels are maintained or are even elevated above those seen in wild-type cells. These cells are as sensitive to ABT-737 as those expressing ligands (such as Noxa) that cause a significant reduction in Mcl-1 levels. Therefore, our study also highlights the limitation of establishing Mcl-1 activity solely by determining its protein level because the increased Mcl-1 protein in BimS2A-overexpressing cells is, in fact, functionally incompetent. Thus, although the absence of Mcl-1 predicts sensitivity to ABT-737 (Konopleva et al., 2006; van Delft et al., 2006; Lin et al., 2007), the presence of detectable protein does not preclude that such a cell could still be readily killed by ABT-737 if the available Mcl-1 is inactivated by associated BH3-only proteins.
We also showed that killing mediated by BimS2A with ABT-737 can occur via either Bax or Bak, suggesting that the elimination of Mcl-1 is not essential for activation of either of these essential cell death pathway components. In addition, our data are consistent with the indirect activation model of apoptosis, as neither direct binding to Bax and/or Bak is required (Fig. 2, E and F) nor are the activator BH3-only proteins (Bim and Bid) required, as cell death readily proceeds in their absence (Fig. 4 D).
Collectively, these data provide proof of principle that ligands specifically targeting Mcl-1 can be developed and do not necessarily need to also target Mcl-1 for degradation to be effective. Such molecules are likely to be useful for extending the range of cancers against which ABT-737 is effective or to treat cancers that are dependent on Mcl-1 for their survival.
Interestingly, long-term neutralization of Mcl-1 through the enforced expression of either BimS2A or Noxa appeared to be poorly tolerated over a (month) long time frame in fibroblasts. Therefore, although we and others have shown the potential of Mcl-1 as a target for pharmacological intervention, especially to potentiate the activity of ABT-737, it will be important to determine the consequences of its long-term neutralization in light of its central role during development (Rinkenberger et al., 2000; Opferman et al., 2003, 2005).
New insights into prosurvival protein–BH3 domain interactions
Structural and limited mutagenesis data suggest that the four conserved hydrophobic residues in BH3 domains all appear to contribute to the binding free energy in interactions with pro-survival proteins (Sattler et al., 1997; Petros et al., 2000; Liu et al., 2003; Czabotar et al., 2007; Kvansakul et al., 2007). One of the key findings that came out of our search for an Mcl-1–specific ligand was that in BimBH3–prosurvival protein complexes, different subsets of these residues were critical depending on the prosurvival protein. These data suggest that different BH3 domain–prosurvival protein interactions are dependent on the receptor–ligand context.
Our structure of the BimBH3 2A peptide in complex with Mcl-1 and accompanying structure-function data illustrate this point. Although the mutation of I65 to alanine on Bim had little effect on Mcl-1 binding, introduction of the same mutation in combination with the L62A mutation was detrimental. Evidently, the very subtle changes observed in orientations of the side chains of residues in the BimBH3 2A–Mcl-1 complex compared with the wild-type complex render the mutant complex highly susceptible to the I65A mutation. This demonstrates the potential challenges associated with the design of BH3 ligands/mimetics given the sensitivity of these interactions to subtle changes in the binding surface of the interacting partners.
Similarly, the design of our Mcl-1–specific ligand, BimBH3 2A, would have been challenging if based solely on rational approaches, as the sequence lacks two of the characteristic features of BH3 domains (a leucine at position h2 and a large hydrophobic residue at position h4). Indeed, although BimBH3 2A is now Noxa-like in its binding profile, this has been achieved in a sequence that is actually less Noxa-like than wild-type BimBH3.
These data demonstrate that the underlying molecular basis that dictates the selectivity of BH3 domains for their prosurvival targets is elusive and does not follow obvious rules of engagement. Thus, the development of small-molecule BH3 mimetic ligands and even peptidic ligands with target specificity will require a combination of both empirical and rational approaches.
Materials and methods
Recombinant proteins and synthetic peptides
The expression and purification of human Bcl-xLΔC25, mouse and human/mouse Mcl-1, or N-terminal His6-tagged human Bcl-2ΔC22 and human Bcl-wΔC29 (C29S and A129E) have been described previously (Chen et al., 2005; Czabotar et al., 2007). Synthetic peptides were synthesized by Mimotopes and purified by reverse-phase HPLC to >90% purity.
Cell lines
The FDC-P1 myelomonocytic cell line and MEFs used in the experiments have been described previously (Willis et al., 2005, 2007) except for the mcl-1−/− MEFs. They were generated from embryonic day 13.5 embryos of targeted mice generated on an inbred C57BL/6 background. All fibroblasts were immortalized with SV40 large T antigen. The MCF-7 cells stably expressing shRNA against Bim were generated as previously described (Puthalakath et al., 2007). Thymocytes derived from bim+/+ and bim−/− mice were prepared as described previously (Bouillet et al., 1999).
Phage display constructs
A 26–amino acid residue peptide encompassing the BH3 domain plus flanking residues of human Bim (Fig. 6 A) was fused via a linker sequence (GGGT) to the N terminus of the M13 phage gene 3 (residues 249–406) in the phagemid vector described previously (Fairlie et al., 2003). To create an N-terminally FLAG-tagged form of the sequence, the FLAG sequence + glycine-serine linker (DYDDDDKGS) was fused to the aforementioned sequence by Kunkel mutagenesis (Kunkel et al., 1991). For the alanine scanning constructs and other point mutations of the BimBH3 sequence, Kunkel mutagenesis on the FLAG-BimBH3 template was performed. Phage particles were isolated from cell supernatants as described previously (Sidhu et al., 2000). All constructs were confirmed by sequencing.
Binding studies
All phage ELISAs were performed as described previously (Fairlie et al., 2003). In each case, either 5 μg/ml of the prosurvival Bcl-2–like family protein or 0.5 μg/ml of the M2 anti-FLAG antibody (Sigma-Aldrich) was coated onto 96-well plates (Maxisorp; Thermo Fisher Scientific) overnight at 4°C. For the competition assays, various concentrations of Bcl-2–like proteins in solution were used to displace a fixed subsaturating dilution of phage-displayed BimBH3 peptide from binding to immobilized Bcl-2–like proteins. Solution competition assays using a biosensor (Biacore 3000; GE Healthcare) were performed as described previously (Chen et al., 2005). The IC50 values of the mutant BH3 domains were divided by the value obtained for its wild-type parent to determine relative binding. ITC studies were performed using a VP-ITC instrument (MicroCal). Proteins were diluted to 4–6.5 μM in TBS, and peptides (prepared in the same buffer from 2-mM stocks) were titrated from a 40–80-μM solution. All experiments were performed at 25°C. Data analysis was performed using Origin software (MicroCal).
Coimmunoprecipitation of prosurvival proteins and Bim
FLAG-tagged mammalian expression vectors for human Bcl-2, mouse Bcl-xL, or mouse Mcl-1 and HA-tagged human BimS or its mutants subcloned into pEF PGKpuro have been previously described (Huang et al., 1997; Moriishi et al., 1999; Chen et al., 2005; Willis et al., 2005). The maintenance, transfection, and metabolic labeling of HEK293T cells with [35S]methionine/cysteine as well as coimmunoprecipitation experiments have been described previously (Huang et al., 1997; O'Connor et al., 1998; Moriishi et al., 1999). Cell lysates were prepared in lysis buffer (20 mM Tris, pH 7.4, 135 mM NaCl, 1.5 mM MgCl2, 1 mM EDTA, and 10% glycerol) containing 1% (vol/vol) Triton X-100 and were supplemented with protease inhibitors (Roche). Immunoprecipitation was performed by incubating lysates with ∼5 μg of antibody (anti-HA HA.11 [Covance Research Products] and anti-FLAG M2 [Sigma-Aldrich]) and control anti–Glu-Glu (anti-EE; Covance Research Products). The proteins were resolved by SDS-PAGE and transferred onto nitrocellulose membranes. 35S-labeled Bim and associated prosurvival proteins were detected by fluorography using Amplify (GE Healthcare).
Coimmunoprecipitation of Bim with Bax/Bak
The transfection of HA-tagged BimS into HEK293T cells was performed using Lipofectamine (Invitrogen). Cell lysates were prepared in lysis buffer as detailed in the previous section and incubated with anti-HA–conjugated agarose resin (Roche). The bound proteins were eluted by boiling in SDS-PAGE sample buffer, resolved by SDS-PAGE, and transferred onto nitrocellulose membranes. Endogenous Bax associated with BimS was detected by Western blotting with the anti-Bax antibody (2D2; Sigma-Aldrich). The HA-tagged BimS/BimS mutants were detected with the anti-HA antibody (3F10; Roche).
For the coimmunoprecipitation of BimS with Bak, FLAG-tagged Bak and HA-tagged BimS/BimS2A/BimS4E or HA-tagged Bcl-xL/Mcl-1 were cotransfected into HEK293T cells. Cell lysates were prepared in lysis buffer as detailed in the previous section and incubated with 5 μg of anti-FLAG antibody (M2; Sigma-Aldrich) for 1 h at 4°C. The resultant complexes were then captured by incubation with protein G–Sepharose (GE Healthcare). Bound proteins were eluted by boiling in SDS-PAGE sample buffer, resolved by SDS-PAGE, and transferred onto nitrocellulose membranes. HA-tagged proteins associated with immunoprecipitated Bak were detected with the anti-HA antibody (3F10; Roche), and FLAG-tagged proteins were detected with the anti-FLAG antibody.
Killing assays
Retroviral expression constructs were made using the pMIG vector (MSCV-IRES-GFP; GFP sequence is that of EGFP) as described previously (Van Parijs et al., 1999; Chen et al., 2005) or the modified vector pMIH, in which the GFP cassette is replaced by one encoding hygromycin B resistance. These plasmids were transiently transfected using Lipofectamine (Invitrogen) into ecotropic packaging cells (Phoenix; Kinsella and Nolan, 1996). Filtered virus-containing supernatants were used to infect SV40 large T-antigen–transformed MEFs or FDC-P1 cells by spin inoculation as described previously (Willis et al., 2005).
For short-term killing assays, cell viability was determined by flow cytometric analyses of infected cells (GFP+ve; FL-1) that excluded 5 μg/ml propidium iodide (PI−ve; FL-3; Sigma-Aldrich) analyzed by using FACScan (Becton Dickinson) either 30 h or at the indicated times after retroviral transduction. For assays performed in combination with ABT-737, cells were incubated with the indicated concentrations of ABT-737, which was added immediately after spin inoculation.
Long-term survival (colony) assays were performed in two ways. Stable cell lines expressing either BimS2A, BimS4E, or Noxa were generated by selection of GFP+ve cells after retrovirus spin inoculation. Assays were performed by plating 150 of the GFP+ve cells in the absence or presence of 1 μM ABT-737 for 6 d. Colonies were then stained with Coomassie blue and counted. Alternatively, stable cell lines expressing either parental pMIH vector, BimS, BimS2A, BimS4E, Noxa, or Bad were first generated by 250 μg/ml hygromycin B (Roche) selection after retrovirus spin inoculation. These stable cell lines were then retrovirally infected with either parental pMIG vector, BimS2A, BimS4E, or BimSBadBH3. Then, 150 GFP+ve cells were plated, and colonies were stained after 6 d and counted.
Alternatively, apoptosis was induced in either MCF-7 cells by treatment with 1.5 μM thapsigargin (Calbiochem) or in primary thymocytes with 1 μg/ml ionomycin (Sigma-Aldrich), and cell survival was quantitated by flow cytometric analysis of cells stained with propidium iodide as described for the short-term killing assays after the indicated times.
The long-term tolerance assay was performed by harvesting stable cell lines expressing BimS4E, BimS2A, Bad, or Noxa every 3 or 4 d over a period of 38 d. At each time point, the proportion of infected cells (GFP+ve; FL-1) that excluded propidium iodide (PI−ve; FL-3) was monitored by flow cytometric analysis.
In vitro cytochrome c release assays
Approximately 107 cells were pelleted, lysed in 0.05% (wt/vol) digitonin (Calbiochem) containing lysis buffer (20 mM Hepes, pH 7.2, 100 mM KCl, 5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, and 250 mM sucrose), and supplemented with protease inhibitors (Roche) for 10 min on ice. The mitochondria-containing crude lysates were incubated with (5 μM) or without ABT-737 at 30°C for 1 h before pelleting. The supernatant was retained as the soluble fraction, whereas the pellet, which contains unpermeabilized mitochondria, was solubilized in radioimmunoprecipitation buffer. Proteins were resolved by SDS-PAGE and transferred onto nitrocellulose membranes. The cytochrome c, Bcl-2, and Apaf-1 proteins were detected with the anti–cytochrome c (7H8.2C12; BD Biosciences), anti–Bcl-2 (3F11; BD Biosciences), and anti–Apaf-1 (18H2; gift from L. O'Reilly, The Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria, Australia) antibodies, respectively.
Mcl-1 degradation
To examine Mcl-1 levels in MCF-7 cells after thapsigargin treatment or in primary thymocytes after ionomycin treatment, cells were cultured in the presence of 50 μM qVD-OPH (MP Biomedicals) to prevent complications caused by caspase cleavage of Mcl-1 (Herrant et al., 2004). To examine basal Mcl-1 levels, bax−/−bak−/ − MEFs were retrovirally infected with either pMIG vector alone or various BimS or Noxa variants as described in Killing assays and GFP+ve cells selected by cell sorting. To determine Mcl-1 levels in Noxa- or BimS4E-expressing cells, wild-type MEFs stably expressing Noxa or BimS4E were reinfected with retroviruses expressing BimS4E, BimS2A, or Noxa. All cells were lysed in buffer containing 1% (vol/vol) Triton X-100, and lysates were analyzed by Western blotting using antibodies against combinations of Mcl-1 (rat monoclonal clone 19C4-15), Bcl-2 (3F11; BD Biosciences), Bim (3C5; Qbiogene), and HSP70 (N6; gift from Drs. W. Welch [University of California, San Francisco, San Francisco, CA] and R. Anderson [Peter MacCallum Cancer Centre, Melbourne, Victoria, Australia]).
Crystallography, data collection, and structure determination
The BimBH3 2A–Mcl-1 complex was produced by combining a 1.3-fold excess of BimBH3 2A peptide with purified human/mouse Mcl-1. The complex was concentrated to 10 mg/ml and set up in hanging drops at 22°C with a reservoir solution consisting of 0.2 M zinc acetate, 0.2 M imidazole, pH 5.75, and 2 mM Tris (2-carbxylethyl) phosphine. Crystals typically took 2–3 d to grow. Crystals were soaked for 30 s in cryoprotectant (0.2 M zinc acetate, 0.2 M imidazole, pH 5.75, and 30% trehalose) and flash frozen in liquid nitrogen. X-ray data were collected at 100 K using an in-house RAXIS-IV++ detector (Rigaku) with a rotating anode x-ray source (Micromax007; Rigaku). Data were integrated and scaled with HKL2000 (Table S1; Otwinowski and Minor, 1997). The structure was determined by molecular replacement with PHASER (Read, 2001; Storoni et al., 2004; McCoy et al., 2005) using the coordinates of Mcl-1 from the wild-type BimBH3–Mcl-1 complex (Czabotar et al., 2007) as a search model. Several rounds of building in COOT (Emsley and Cowtan, 2004) and refinement in REFMAC5 (Murshudov et al., 1997) led to the final model shown in Table S1.
Online supplemental material
Fig. S1 shows BimBH3 alanine scanning data with Bcl-w and hMcl-1. Figs. S2 and S3 show the killing activities of BimS2A and BimSBadBH3, reflecting their complementary prosurvival protein binding profiles either by their coexpression (Fig. S2 A), in the absence of Bcl-xL or Mcl-1 (Fig. S2 B), or in the absence of the putative direct activators Bim and Bid (Fig. S3). Fig. S4 shows that BimS2A can combine with ABT-737 to kill FDC-P1 cells. Fig. S5 shows the quantitation of Bim isoforms after treatment of MCF-7cells with thapsigargin. Table S1 presents crystallographic statistics. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200708096/DC1.
Supplemental Material
Acknowledgments
We thank Abbott Laboratories (S. Fesik, S. Rosenberg, S. Elmore, A. Shoemaker, and colleagues) for providing ABT-737; J. Adams, A. Strasser, L. Chen, S. Cory, and J. Fletcher for discussions and advice; J. Blyth, H. Ierino, T. Pham, K. Knezevic, and M. Evangelista for technical assistance; and A. Strasser, P. Kelly, R. Anderson, M. Baca, G. Dewson, S. Korsmeyer, N. Motoyama, C. Thompson, and W. Welch for reagents.
Our work is supported by grants from the Australian National Health and Medical Research Council (NHMRC; program grant 257502), U.S. National Cancer Institute (grant CA80188), Leukemia and Lymphoma Society (specialized center of research 7015-02), Cancer Council of Victoria (project 461239), Australian Cancer Research Foundation, a Melbourne International Research Scholarship (to M.F. van Delft), fellowships and scholarships from the NHMRC (to D.C.S. Huang, W.D. Fairlie, and P.M. Colman), and the Cancer Council of Victoria (E.F. Lee, S.N. Willis, E.M. Michalak, and M.J. Boyle; Fraser Fellowship to P.M. Colman).
H. Puthalakath's present address is Department of Biochemistry, La Trobe University, Melbourne, Victoria 3086, Australia.
Abbreviations used in this paper: BH3, Bcl-2 homology 3; IRES, internal ribosome entry site; ITC, isothermal titration calorimetry; MEF, mouse embryonic fibroblast; shRNA, short hairpin RNA.
References
- Bouillet, P., D. Metcalf, D.C. Huang, D.M. Tarlinton, T.W. Kay, F. Kontgen, J.M. Adams, and A. Strasser. 1999. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 286:1735–1738. [DOI] [PubMed] [Google Scholar]
- Brunelle, J.K., E.H. Shroff, H. Perlman, A. Strasser, C.T. Moraes, R.A. Flavell, N.N. Danial, B. Keith, C.B. Thompson, and N.S. Chandel. 2007. Loss of Mcl-1 protein and inhibition of electron transport chain together induce anoxic cell death. Mol. Cell. Biol. 27:1222–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cante-Barrett, K., E.M. Gallo, M.M. Winslow, and G.R. Crabtree. 2006. Thymocyte negative selection is mediated by protein kinase C- and Ca2+-dependent transcriptional induction of bim. J. Immunol. 176:2299–2306 (corrected). [DOI] [PubMed] [Google Scholar]
- Cavarretta, I.T., H. Neuwirt, G. Untergasser, P.L. Moser, M.H. Zaki, H. Steiner, H. Rumpold, D. Fuchs, A. Hobisch, J.A. Nemeth, and Z. Culig. 2007. The antiapoptotic effect of IL-6 autocrine loop in a cellular model of advanced prostate cancer is mediated by Mcl-1. Oncogene. 26:2822–2832. [DOI] [PubMed] [Google Scholar]
- Certo, M., V. Del Gaizo Moore, M. Nishino, G. Wei, S. Korsmeyer, S.A. Armstrong, and A. Letai. 2006. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 9:351–365. [DOI] [PubMed] [Google Scholar]
- Chauhan, D., M. Velankar, M. Brahmandam, T. Hideshima, K. Podar, P. Richardson, R. Schlossman, I. Ghobrial, N. Raje, N. Munshi, and K.C. Anderson. 2007. A novel Bcl-2/Bcl-X(L)/Bcl-w inhibitor ABT-737 as therapy in multiple myeloma. Oncogene. 26:2374–2380. [DOI] [PubMed] [Google Scholar]
- Chen, L., S.N. Willis, A. Wei, B.J. Smith, J.I. Fletcher, M.G. Hinds, P.M. Colman, C.L. Day, J.M. Adams, and D.C. Huang. 2005. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell. 17:393–403. [DOI] [PubMed] [Google Scholar]
- Chen, S., Y. Dai, H. Harada, P. Dent, and S. Grant. 2007. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res. 67:782–791. [DOI] [PubMed] [Google Scholar]
- Cheng, E.H., M.C. Wei, S. Weiler, R.A. Flavell, T.W. Mak, T. Lindsten, and S.J. Korsmeyer. 2001. BCL-2, BCL-xL sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell. 8:705–711. [DOI] [PubMed] [Google Scholar]
- Cory, S., D.C.S. Huang, and J.M. Adams. 2003. The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene. 22:8590–8607. [DOI] [PubMed] [Google Scholar]
- Craig, R.W. 2002. MCL1 provides a window on the role of the BCL2 family in cell proliferation, differentiation and tumorigenesis. Leukemia. 16:444–454. [DOI] [PubMed] [Google Scholar]
- Cuconati, A., C. Mukherjee, D. Perez, and E. White. 2003. DNA damage response and MCL-1 destruction initiate apoptosis in adenovirus-infected cells. Genes Dev. 17:2922–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czabotar, P.E., E.F. Lee, M.F. van Delft, C.L. Day, B.J. Smith, D.C. Huang, W.D. Fairlie, M.G. Hinds, and P.M. Colman. 2007. Structural insights into the degradation of Mcl-1 induced by BH3 domains. Proc. Natl. Acad. Sci. USA. 104:6217–6222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley, P., and K. Cowtan. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- Fairlie, W.D., A.D. Uboldi, G.J. Hemmings, B.J. Smith, H.M. Martin, P.O. Morgan, and M. Baca. 2003. A family of leukemia inhibitory factor-binding peptides that can act as antagonists when conjugated to poly(ethylene glycol). Biochemistry. 42:13193–13201. [DOI] [PubMed] [Google Scholar]
- Fesik, S.W. 2005. Promoting apoptosis as a strategy for cancer drug discovery. Nat. Rev. Cancer. 5:876–885. [DOI] [PubMed] [Google Scholar]
- Gillissen, B., F. Essmann, P.G. Hemmati, A. Richter, A. Richter, I. Öztop, G. Chinnadurai, B. Dörken, and P.T. Daniel. 2007. Mcl-1 determines the Bax dependency of Nbk/Bik-induced apoptosis. J. Cell Biol. 179:701–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gojo, I., B. Zhang, and R.G. Fenton. 2002. The cyclin-dependent kinase inhibitor flavopiridol induces apoptosis in multiple myeloma cells through transcriptional repression and down-regulation of Mcl-1. Clin. Cancer Res. 8:3527–3538. [PubMed] [Google Scholar]
- Herrant, M., A. Jacquel, S. Marchetti, N. Belhacene, P. Colosetti, F. Luciano, and P. Auberger. 2004. Cleavage of Mcl-1 by caspases impaired its ability to counteract Bim-induced apoptosis. Oncogene. 23:7863–7873. [DOI] [PubMed] [Google Scholar]
- Huang, D.C.S., L.A. O'Reilly, A. Strasser, and S. Cory. 1997. The anti-apoptosis function of Bcl-2 can be genetically separated from its inhibitory effect on cell cycle entry. EMBO J. 16:4628–4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann, S.H., J.E. Karp, P.A. Svingen, S. Krajewski, P.J. Burke, S.D. Gore, and J.C. Reed. 1998. Elevated expression of the apoptotic regulator Mcl-1 at the time of leukemic relapse. Blood. 91:991–1000. [PubMed] [Google Scholar]
- Kim, H., M. Rafiuddin-Shah, H.C. Tu, J.R. Jeffers, G.P. Zambetti, J.J. Hsieh, and E.H. Cheng. 2006. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat. Cell Biol. 8:1348–1358. [DOI] [PubMed] [Google Scholar]
- Kinsella, T.M., and G.P. Nolan. 1996. Episomal vectors rapidly and stably produce high-titer recombinant retrovirus. Hum. Gene Ther. 7:1405–1413. [DOI] [PubMed] [Google Scholar]
- Kitada, S., J. Andersen, S. Akar, J.M. Zapata, S. Takayama, S. Krajewski, H.G. Wang, X. Zhang, F. Bullrich, C.M. Croce, et al. 1998. Expression of apoptosis-regulating proteins in chronic lymphocytic leukemia: correlations with in vitro and in vivo chemoresponses. Blood. 91:3379–3389. [PubMed] [Google Scholar]
- Konopleva, M., R. Contractor, T. Tsao, I. Samudio, P.P. Ruvolo, S. Kitada, X. Deng, D. Zhai, Y.X. Shi, T. Sneed, et al. 2006. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 10:375–388. [DOI] [PubMed] [Google Scholar]
- Kunkel, T.A., K. Bebenek, and J. McClary. 1991. Efficient site-directed mutagenesis using uracil-containing DNA. Methods Enzymol. 204:125–139. [DOI] [PubMed] [Google Scholar]
- Kuwana, T., L. Bouchier-Hayes, J.E. Chipuk, C. Bonzon, B.A. Sullivan, D.R. Green, and D.D. Newmeyer. 2005. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell. 17:525–535. [DOI] [PubMed] [Google Scholar]
- Kvansakul, M., M.F. van Delft, E.F. Lee, J.M. Gulbis, W.D. Fairlie, D.C. Huang, and P.M. Colman. 2007. A structural viral mimic of prosurvival Bcl-2: a pivotal role for sequestering proapoptotic Bax and Bak. Mol. Cell. 25:933–942. [DOI] [PubMed] [Google Scholar]
- Lee, E.F., P.E. Czabotar, B.J. Smith, K. Deshayes, K. Zobel, P.M. Colman, and W.D. Fairlie. 2007. Crystal structure of ABT-737 complexed with Bcl-x(L): implications for selectivity of antagonists of the Bcl-2 family. Cell Death Differ. 14:1711–1713. [DOI] [PubMed] [Google Scholar]
- Letai, A., M. Bassik, L. Walensky, M. Sorcinelli, S. Weiler, and S. Korsmeyer. 2002. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2:183–192. [DOI] [PubMed] [Google Scholar]
- Lin, X., S. Morgan-Lappe, X. Huang, L. Li, D.M. Zakula, L.A. Vernetti, S.W. Fesik, and Y. Shen. 2007. ‘Seed’ analysis of off-target siRNAs reveals an essential role of Mcl-1 in resistance to the small-molecule Bcl-2/Bcl-XL inhibitor ABT-737. Oncogene. 26:3972–3979. [DOI] [PubMed] [Google Scholar]
- Liu, X., S. Dai, Y. Zhu, P. Marrack, and J.W. Kappler. 2003. The structure of a Bcl-xL/Bim fragment complex: implications for Bim function. Immunity. 19:341–352. [DOI] [PubMed] [Google Scholar]
- McCoy, A.J., R.W. Grosse-Kunstleve, L.C. Storoni, and R.J. Read. 2005. Likelihood-enhanced fast translation functions. Acta Crystallogr. D Biol. Crystallogr. 61:458–464. [DOI] [PubMed] [Google Scholar]
- Mei, Y., W. Du, Y. Yang, and M. Wu. 2005. Puma(*)Mcl-1 interaction is not sufficient to prevent rapid degradation of Mcl-1. Oncogene. 24:7224–7237. [DOI] [PubMed] [Google Scholar]
- Moriishi, K., D.C.S. Huang, S. Cory, and J.M. Adams. 1999. Bcl-2 family members do not inhibit apoptosis by binding the caspase-activator Apaf-1. Proc. Natl. Acad. Sci. USA. 96:9683–9688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov, G.N., A.A. Vagin, and E.J. Dodson. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53:240–255. [DOI] [PubMed] [Google Scholar]
- Nijhawan, D., M. Fang, E. Traer, Q. Zhong, W. Gao, F. Du, and X. Wang. 2003. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 17:1475–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor, L., A. Strasser, L.A. O'Reilly, G. Hausmann, J.M. Adams, S. Cory, and D.C. Huang. 1998. Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 17:384–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltersdorf, T., S.W. Elmore, A.R. Shoemaker, R.C. Armstrong, D.J. Augeri, B.A. Belli, M. Bruncko, T.L. Deckwerth, J. Dinges, P.J. Hajduk, et al. 2005. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 435:677–681. [DOI] [PubMed] [Google Scholar]
- Opferman, J.T. 2006. Unraveling MCL-1 degradation. Cell Death Differ. 13:1260–1262. [DOI] [PubMed] [Google Scholar]
- Opferman, J.T., A. Letai, C. Beard, M.D. Sorcinelli, C.C. Ong, and S.J. Korsmeyer. 2003. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 426:671–676. [DOI] [PubMed] [Google Scholar]
- Opferman, J.T., H. Iwasaki, C.C. Ong, H. Suh, S. Mizuno, K. Akashi, and S.J. Korsmeyer. 2005. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science. 307:1101–1104. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z., and W. Minor. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307–326. [DOI] [PubMed] [Google Scholar]
- Petros, A.M., D.G. Nettseheim, Y. Wang, E.T. Olejniczak, R.P. Meadows, J. Mack, K. Swift, E.D. Matayoshi, H. Zhang, C.B. Thompson, and S.W. Fesik. 2000. Rationale for Bcl-xL/Bad peptide complex formation from structure, mutagenesis, and biophysical studies. Protein Sci. 9:2528–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthalakath, H., L.A. O'Reilly, P. Gunn, L. Lee, P.N. Kelly, N.D. Huntington, P.D. Hughes, E.M. Michalak, J. McKimm-Breschkin, N. Motoyama, et al. 2007. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 129:1337–1349. [DOI] [PubMed] [Google Scholar]
- Qin, J.Z., H. Xin, L.A. Sitailo, M.F. Denning, and B.J. Nickoloff. 2006. Enhanced killing of melanoma cells by simultaneously targeting Mcl-1 and NOXA. Cancer Res. 66:9636–9645. [DOI] [PubMed] [Google Scholar]
- Rahmani, M., E.M. Davis, C. Bauer, P. Dent, and S. Grant. 2005. Apoptosis induced by the kinase inhibitor BAY 43-9006 in human leukemia cells involves down-regulation of Mcl-1 through inhibition of translation. J. Biol. Chem. 280:35217–35227. [DOI] [PubMed] [Google Scholar]
- Raje, N., S. Kumar, T. Hideshima, A. Roccaro, K. Ishitsuka, H. Yasui, N. Shiraishi, D. Chauhan, N.C. Munshi, S.R. Green, and K.C. Anderson. 2005. Seliciclib (CYC202 or R-roscovitine), a small-molecule cyclin-dependent kinase inhibitor, mediates activity via down-regulation of Mcl-1 in multiple myeloma. Blood. 106:1042–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read, R.J. 2001. Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr. D Biol. Crystallogr. 57:1373–1382. [DOI] [PubMed] [Google Scholar]
- Rinkenberger, J.L., S. Horning, B. Klocke, K. Roth, and S.J. Korsmeyer. 2000. Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Dev. 14:23–27. [PMC free article] [PubMed] [Google Scholar]
- Sattler, M., H. Liang, D. Nettesheim, R.P. Meadows, J.E. Harlan, M. Eberstadt, H.S. Yoon, S.B. Shuker, B.S. Chang, A.J. Minn, et al. 1997. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science. 275:983–986. [DOI] [PubMed] [Google Scholar]
- Saxena, A., S. Viswanathan, O. Moshynska, P. Tandon, K. Sankaran, and D.P. Sheridan. 2004. Mcl-1 and Bcl-2/Bax ratio are associated with treatment response but not with Rai stage in B-cell chronic lymphocytic leukemia. Am. J. Hematol. 75:22–33. [DOI] [PubMed] [Google Scholar]
- Sidhu, S.S., H.B. Lowman, B.C. Cunningham, and J.A. Wells. 2000. Phage display for selection of novel binding peptides. Methods Enzymol. 328:333–363. [DOI] [PubMed] [Google Scholar]
- Sieghart, W., D. Losert, S. Strommer, D. Cejka, K. Schmid, S. Rasoul-Rockenschaub, M. Bodingbauer, R. Crevenna, B.P. Monia, M. Peck-Radosavljevic, and V. Wacheck. 2006. Mcl-1 overexpression in hepatocellular carcinoma: a potential target for antisense therapy. J. Hepatol. 44:151–157. [DOI] [PubMed] [Google Scholar]
- Song, L., D. Coppola, S. Livingston, D. Cress, and E.B. Haura. 2005. Mcl-1 regulates survival and sensitivity to diverse apoptotic stimuli in human non-small cell lung cancer cells. Cancer Biol. Ther. 4:267–276. [DOI] [PubMed] [Google Scholar]
- Storoni, L.C., A.J. McCoy, and R.J. Read. 2004. Likelihood-enhanced fast rotation functions. Acta Crystallogr. D Biol. Crystallogr. 60:432–438. [DOI] [PubMed] [Google Scholar]
- Trudel, S., A.K. Stewart, Z. Li, Y. Shu, S.B. Liang, Y. Trieu, D. Reece, J. Paterson, D. Wang, and X.Y. Wen. 2007. The Bcl-2 family protein inhibitor, ABT-737, has substantial antimyeloma activity and shows synergistic effect with dexamethasone and melphalan. Clin. Cancer Res. 13:621–629. [DOI] [PubMed] [Google Scholar]
- van Delft, M.F., A.H. Wei, K.D. Mason, C.J. Vandenberg, L. Chen, P.E. Czabotar, S.N. Willis, C.L. Scott, C.L. Day, S. Cory, et al. 2006. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 10:389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Parijs, L., Y. Refaeli, A.K. Abbas, and D. Baltimore. 1999. Autoimmunity as a consequence of retrovirus-mediated expression of C-FLIP in lymphocytes. Immunity. 11:763–770. [DOI] [PubMed] [Google Scholar]
- Willis, S.N., and J.M. Adams. 2005. Life in the balance: how BH3-only proteins induce apoptosis. Curr. Opin. Cell Biol. 17:617–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis, S.N., L. Chen, G. Dewson, A. Wei, E. Naik, J.I. Fletcher, J.M. Adams, and D.C. Huang. 2005. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 19:1294–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis, S.N., J.I. Fletcher, T. Kaufmann, M.F. van Delft, L. Chen, P.E. Czabotar, H. Ierino, E.F. Lee, W.D. Fairlie, P. Bouillet, et al. 2007. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 315:856–859. [DOI] [PubMed] [Google Scholar]
- Wuilleme-Toumi, S., N. Robillard, P. Gomez, P. Moreau, S. Le Gouill, H. Avet-Loiseau, J.L. Harousseau, M. Amiot, and R. Bataille. 2005. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia. 19:1248–1252. [DOI] [PubMed] [Google Scholar]
- Yu, C., L.M. Bruzek, X.W. Meng, G.J. Gores, C.A. Carter, S.H. Kaufmann, and A.A. Adjei. 2005. The role of Mcl-1 downregulation in the proapoptotic activity of the multikinase inhibitor BAY 43-9006. Oncogene. 24:6861–6869. [DOI] [PubMed] [Google Scholar]
- Zhai, D., C. Jin, A.C. Satterthwait, and J.C. Reed. 2006. Comparison of chemical inhibitors of antiapoptotic Bcl-2-family proteins. Cell Death Differ. 13:1419–1421. [DOI] [PubMed] [Google Scholar]
- Zhong, Q., W. Gao, F. Du, and X. Wang. 2005. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell. 121:1085–1095. [DOI] [PubMed] [Google Scholar]
- Zong, W.X., T. Lindsten, A.J. Ross, G.R. MacGregor, and C.B. Thompson. 2001. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. 15:1481–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}