

Abstract
Integrin binding to matrix proteins such as fibronectin (FN) leads to formation of focal adhesion (FA) cellular contact sites that regulate migration. RhoA GTPases facilitate FA formation, yet FA-associated RhoA-specific guanine nucleotide exchange factors (GEFs) remain unknown. Here, we show that proline-rich kinase-2 (Pyk2) levels increase upon loss of focal adhesion kinase (FAK) in mouse embryonic fibroblasts (MEFs). Additionally, we demonstrate that Pyk2 facilitates deregulated RhoA activation, elevated FA formation, and enhanced cell proliferation by promoting p190RhoGEF expression. In normal MEFs, p190RhoGEF knockdown inhibits FN-associated RhoA activation, FA formation, and cell migration. Knockdown of p190RhoGEF-related GEFH1 does not affect FA formation in FAK−/− or normal MEFs. p190RhoGEF overexpression enhances RhoA activation and FA formation in MEFs dependent on FAK binding and associated with p190RhoGEF FA recruitment and tyrosine phosphorylation. These studies elucidate a compensatory function for Pyk2 upon FAK loss and identify the FAK–p190RhoGEF complex as an important integrin-proximal regulator of FA formation during FN-stimulated cell motility.
Introduction
Cell migration is a highly regulated process that involves the continuous formation and turnover of cell–substratum contact sites termed focal adhesions (FAs), which serve as points of traction and as signaling centers (Ridley et al., 2003; Romer et al., 2006). FAs, which link integrins to the actin cytoskeleton, control the migratory potential of cells (Geiger and Bershadsky, 2001; Webb et al., 2003). Despite the importance of regulated FA formation and turnover in cell migration, the molecular mechanisms controlling these events remain loosely defined (Vicente-Manzanares et al., 2005; Moissoglu and Schwartz, 2006).
Rho family GTPases are molecular switches involved in the regulation of many cellular processes. The RhoGTPase members Rho, Rac, and Cdc42 control signaling pathways regulating actin and FA assembly or disassembly (Hall, 2005). RhoA promotes stress fiber and FA formation, in part through Rho kinase–mediated cell contractility. Temporal regulation of RhoA is important, as constitutively active RhoA impedes cell motility, in part through enhanced FA formation. Integrin binding to fibronectin (FN) generates intracellular signals, leading to the transient inhibition of RhoA followed by extended RhoA reactivation (Ren et al., 1999). Rho GTPases are activated by guanine nucleotide exchange factors (GEFs), which catalyze the exchange of GDP for GTP (Rossman et al., 2005; Bos et al., 2007). Rho GTPases return to an inactive state upon hydrolysis of GTP to GDP, a reaction enhanced by GTPase-activating proteins (GAPs). Transient inhibition of RhoA during FN adhesion is mediated in part by the Src family protein tyrosine kinase (PTK) phosphorylation of p190RhoGAP, which leads to elevated RhoGAP activity (Arthur et al., 2000). The GEFs important in facilitating RhoA reactivation and FA formation upon FN adhesion remain unknown.
Many PTKs facilitate Rac- and Cdc42-specific GEF activation, whereas Rho-specific GEF activation by PTKs is less common (Schiller, 2006). FAK activation by integrins facilitates the recruitment of Src family PTKs into a signaling complex localized to FAs (Mitra et al., 2005; Mitra and Schlaepfer, 2006). FAK- or Src-mediated tyrosine phosphorylation of various RhoGEFs is associated with RhoA activation (Chikumi et al., 2002; Medley et al., 2003; Zhai et al., 2003), but it remains unclear whether these events are linked to FA formation or the regulation of cell motility. An unexplained phenomenon is that FAK-null (FAK−/−) mouse embryonic fibroblasts (MEFs) exhibit constitutively high RhoA activity, enhanced FA formation, and refractory cell motility responses (Ilic et al., 1995; Owen et al., 1999; Sieg et al., 1999; Ren et al., 2000). Inhibition of RhoA (Ren et al., 2000) or Rho kinase (Chen et al., 2002) in FAK−/− MEFs decreases FA formation. Although FAK can suppress RhoA activity via p190RhoGAP tyrosine phosphorylation (Holinstat et al., 2006), it is unclear whether loss of FAK accounts for constitutive RhoA activation. Alternatively, expression of the FAK-related proline-rich kinase 2 (Pyk2) PTK is elevated in FAK−/− MEFs (Sieg et al., 1998), but the role of Pyk2 in FAK−/− MEFs remains undefined.
In this paper, we show that Pyk2 promotes FAK−/− MEF proliferation and aberrant FA formation through the regulation of p190RhoGEF expression affecting RhoA activation. p190RhoGEF is a ubiquitously expressed RhoA-specific GEF that can bind microtubules and also associates with FAK (Gebbink et al., 1997; van Horck et al., 2001; Zhai et al., 2003). In FAK−/− MEFs, Pyk2 associates with p190RhoGEF, and knockdown of Pyk2 or p190RhoGEF results in FAK−/− MEFs with normal FN-associated RhoA regulation but extreme motility and trailing-edge retraction defects. These findings show that some FAK-null phenotypes are caused by compensatory Pyk2 signaling effects. In normal MEFs with FAK and little Pyk2, p190RhoGEF knockdown limits RhoA activation, FA formation, and cell motility on FN. Knockdown of p190RhoGEF-related GEFH1 (Ren et al., 1998; Rossman et al., 2005) was without effects on FA formation in FAK−/− and normal MEFs. As deletion of the p190RhoGEF FAK-binding region prevents p190RhoGEF localization to FAs, inhibits p190RhoGEF-associated RhoA activation and FA formation, and blocks integrin-stimulated p190RhoGEF tyrosine phosphorylation, our studies have identified the FAK–p190RhoGEF signaling complex as an important integrin-proximal regulator of FA formation during cell migration.
Results
Compensatory Pyk2 expression upon FAK loss in mouse and human fibroblasts
FAK−/− MEFs exhibit elevated Pyk2 expression (Sieg et al., 1998), and it has been proposed that this may reflect genetic instability associated with p53 coinactivation (Tilghman et al., 2005). To determine whether Pyk2 levels change upon FAK inactivation in culture, primary FAK(loxP/loxP) MEFs were transduced with adenoviral (Ad) Cre-recombinase and FAK-Pyk2 levels were monitored by immunoblotting (Fig. 1 A). High Cre levels were observed within 48 h of transduction with slight reduction of FAK but no changes in Pyk2 expression. Loss of FAK was complete by day 7 after Ad-Cre transduction at which time Pyk2 levels were greatly elevated (Fig. 1 A). To determine whether this compensatory change in Pyk2 occurs in other cells, human diploid fibroblasts were infected with scrambled (Scr) or anti-FAK short hairpin RNA (shRNA) lentivirus (Fig. 1 B). After 7 d, FAK expression was selectively inhibited by FAK shRNA expression and Pyk2 levels were greatly elevated relative to actin and control Scr shRNA–expressing cells. Although stable FAK shRNA–mediated knockdown in various tumor cell lines did not result in alterations in Pyk2 expression (Mitra et al., 2006a,b), compensatory Pyk2 expression occurs upon loss of FAK in MEFs and human fibroblasts.
Figure 1.

Pyk2 knockdown affects FAK−/− MEF morphology, FA formation, and paxillin tyrosine phosphorylation. (A and B) Compensatory increase in Pyk2 levels upon inactivation of FAK expression. Lysates from FAK+/+, FAK−/−, and FAK+/+(loxP/loxP) MEFs were analyzed by anti-FAK, Pyk2, Cre recombinase, and paxilllin blotting. Lysates from Ad-Cre–infected FAK+/+(loxP/loxP) MEFs were analyzed after 48 h or 7 d. Paxillin levels were used as a loading control. (B) Human diploid fibroblasts were treated with lentiviral Scr or FAK shRNA and protein lysates were evaluated by anti-FAK, Pyk2, and actin blotting after 7 d. (C) Stable Pyk2 reduction in FAK−/− MEFs by lentiviral anti-Pyk2 shRNA expression. Lysates from the indicated MEFs were analyzed by antiphosphotyrosine (pY), Pyk2, actin, and GFP blotting. (D) Phase-contrast images show that FAK−/− Pyk2 shRNA MEFs exhibit more of a fibroblast phenotype than FAK−/− Scr shRNA control MEFs. Bars, 30 μm. (E) F-actin was reduced and fewer FAs formed in FN-replated (60 min) FAK−/− Pyk2 shRNA compared with FAK−/− Scr shRNA MEFs as determined by phalloidin and anti-paxillin costaining, respectively. Alexa Fluor 350 phalloidin stain is pseudocolored green. Bar, 20 μm. (F) Increased FAK−/− FA size upon reduced Pyk2 expression. Box-and-whisker plots of paxillin-stained area within FAK−/− Scr shRNA or FAK−/− Pyk2 shRNA MEFs plated on FN for 60 min (n = 15 cells per point). Box-and-whisker diagrams show the distribution of the data: square, mean; bottom line, 25th percentile; middle line, median; top line, 75th percentile; and whiskers, 5th or 95th percentiles. This representation applies for all box-and-whisker plots in this paper. (G) Decreased FAK−/− FA formation upon reduced Pyk2 expression. Box-and-whisker plots of paxillin-positive–stained points within FAK−/− Scr shRNA or FAK−/− Pyk2 shRNA MEFs plated on FN for 60 min (n = 15 cells per point). (H) Decreased FAK−/− F-actin accumulation upon reduced Pyk2 expression. Relative Alexa Fluor phalloidin staining per FAK−/− Scr shRNA MEF (set to 100) and FAK−/− Pyk2 shRNA MEF upon FN plating (60 min; n = 15 cells per point). Error bars represent the SD. (I) Replating assays with FAK−/− Scr or FAK−/− Pyk2 shRNA MEFs show differences in FN-stimulated paxillin Y118 phosphorylation. Lysates from serum-starved cells, cells held in suspension (45 min), or cells replated with FN for the indicated times were analyzed by antiphosphotyrosine blotting. Paxillin immunoprecipitations from the same lysates were sequentially analyzed by anti-paxillin phosphotyrosine 118 phosphospecific and paxillin blotting.
Pyk2 knockdown in FAK−/− MEFs reduces FA formation and paxillin tyrosine phosphorylation
To elucidate the role of Pyk2 in FAK−/− MEFs, Pyk2 expression was inhibited by lentiviral-mediated Pyk2-specific shRNA (Fig. 1 C). The shRNA lentivirus coexpresses GFP, and FAK−/− vector-only, Scr, or Pyk2 shRNA MEF pooled cell populations were enriched by FACS. Stable Pyk2 knockdown of ∼90% in FAK−/− Pyk2 shRNA MEFs resulted in cells with a normal fibroblast-like phenotype compared with the more rounded FAK−/− parental (not depicted) or control FAK−/− Scr shRNA MEFs (Fig. 1 D). When plated onto FN for 60 min, Scr shRNA and Pyk2 shRNA FAK−/− MEFs were equally spread (Fig. 1 E), but cells with less Pyk2 formed fewer FAs of larger size (Fig. 1, F and G) and contained less polymerized F-actin compared with control FAK−/− Scr shRNA MEFs (Fig. 1 H). The number and size of FAs in FAK−/− Pyk2 shRNA MEFs was equivalent to normal MEFs (unpublished data). Tyrosine phosphorylation of the FA-associated protein paxillin is elevated in FAK−/− compared with normal MEFs (Ilic et al., 1995), and this was reduced by Pyk2 shRNA expression in growing (Fig. 1 C) and FN-plated FAK−/− MEFs (Fig. 1 I). As these changes in FAK−/− Pyk2 shRNA MEFs were not associated with alterations in α5, β1, or αv integrin subunit surface expression (unpublished data), our data support the notion that Pyk2 facilitates enhanced FA formation and paxillin tyrosine phosphorylation in FAK−/− MEFs.
Pyk2 promotes FAK−/− MEF proliferation, but not motility, at a level equal to FAK
FAK signaling can promote cell cycle progression (Zhao et al., 2003), but FAK−/− MEFs do not exhibit proliferation defects in culture compared with FAK-reconstituted (clone DP3) and normal FAK+/+ MEFs (Fig. 2 A). Analyses of FAK−/− Scr shRNA MEFs showed no differences in growth compared with control FAK−/− MEFs (Fig. 2, A and B), whereas FAK−/− Pyk2 shRNA MEFs grow slower than FAK−/− Scr and vector shRNA controls (Fig. 2 B). Importantly, transient Ad-FAK or human Ad-Pyk2 expression in FAK−/− Pyk2 shRNA MEFs enhanced cell proliferation at a level equivalent to FAK−/− controls (Fig. 2, C and D). These results support the conclusion that compensatory Pyk2 expression within FAK−/− MEFs promotes cell growth.
Figure 2.

Pyk2 facilitates FAK−/− MEFs' proliferation and motility. (A) FAK−/−, FAK+/+, and FAK-reconstituted (DP3) MEF proliferation was evaluated over 5 d and shows that FAK−/− MEFs do not exhibit growth defects. (B) FAK−/− vector, FAK−/− Scr shRNA, and FAK−/− Pyk2 shRNA MEF proliferation was evaluated over 5 d and shows that Pyk2 shRNA expression is associated with reduced growth. (C) Transient Ad-FAK or Ad-Pyk2 expression enhances FAK−/− Pyk2 shRNA MEF proliferation. Cells were infected for 24 h with the indicated control Ad-TA, Ad-FAK, or Ad-Pyk2 constructs before initiating growth analyses. Values in A–C are means ± SD of three experiments. (D) Lysates from FAK−/− vector and Pyk2 shRNA MEFs infected with Ad-FAK or Ad-Pyk2 were resolved by SDS-PAGE and analyzed by HA-tag, Pyk2, and actin blotting. (E) FAK promotes FAK−/− MEF motility better than Pyk2. The indicated MEFs were analyzed for 10-μg/ml-FN–stimulated Boyden chamber motility over 4 h and values are expressed as a percentage of FAK−/− vector control (set to 100). As indicated, FAK−/− vector shRNA or FAK−/− Pyk2 shRNA MEFs were infected with Ad-FAK or Ad-Pyk2 for 48 h and analyzed for motility responses. Values ± SD are from three separate experiments.
In addition to growing slower, FAK−/− Pyk2 shRNA MEFs exhibit severe motility defects in wound-healing assays under conditions where control FAK−/− MEFs achieve wound closure (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200708194/DC1). In FN-stimulated Boyden chamber motility assays, FAK−/− Pyk2 shRNA MEFs exhibited virtually no motility (Fig. 2 E). Importantly, transient Ad-FAK expression could rescue the motility defect of FAK−/− Pyk2 shRNA MEFs to ∼75% of the level of FAK-reconstituted (DP3) or FAK+/+ MEFs and twice the level of FAK−/− controls (Fig. 2 E). Notably, Ad-Pyk2 overexpression only weakly promoted FAK−/− Pyk2 shRNA MEF motility to ∼70% of the level of FAK−/− controls (Fig. 2 E). Time-lapse microscopy revealed that FAK−/− Pyk2 shRNA MEFs spread on FN but exhibited limited random movement compared with FAK-reconstituted MEFs (Fig. 3, A and B; and Fig. S2). Normal and FAK-reconstituted MEF movement is characterized by leading lamellipodia protrusions with coordinated cell contraction associated with trailing-tail region release (Fig. 3 A and Video 1). FAK−/− MEFs exhibit membrane ruffling in all directions and limited directional movement (Video 2). Although reduced Pyk2 expression prevents excessive FA formation (Fig. 1), when FAK−/− Pyk2 shRNA MEFs moved in one direction, cells became very stretched, pieces broke off, and movement occurred in a snapping fashion (Fig. 3 B and Video 3). This snapping phenotype was rescued by FAK reexpression (unpublished data). These results are consistent with the inability of FAK−/− Pyk2 shRNA MEFs to facilitate trailing-edge FA release as FAK−/− MEFs exhibit decreased FA turnover dynamics (Ren et al., 2000; Webb et al., 2004). Moreover, these results show that compensatory Pyk2 expression is not causal for FAK−/− MEF motility defects.
Figure 3.

FAK−/− Pyk2 shRNA MEFs exhibit tail-retraction motility defects. (A) FAK-reconstituted DP3 MEFs were plated on 1 μg/ml FN and monitored for random cell motility in 10% FBS over the indicated time (images obtained from Video1, available at http://www.jcb.org/cgi/content/full/jcb.200708194/DC1). Cells show normal protrusive activity at the leading edge and coordinated tail retraction (arrows) as the cell moves forward. Bar, 30 μm. (B) FAK−/− Pyk2 shRNA MEFs on 1 μg/ml FN exhibit membrane-ruffling activity in the presence of 10% FBS (images obtained from Video 3). Efficient motility does not occur as cells exhibit defects in tail-associated FA release (arrows). A cell-snapping phenotype is common with pieces of cell material left behind. Bar, 30 μm.
The connection between Pyk2 and RhoA activation
The regulation of FA formation is, in part, associated with RhoA GTPase activation. MEF binding and spreading on FN results in the transient inhibition of RhoA at 15–30 min, followed by an extended phase of RhoA activation associated with FA formation and maturation (Ren et al., 1999; Vicente-Manzanares et al., 2005). An affinity binding assay can be used to determine the amount of activated RhoA-GTP as a function of total RhoA (Ren et al., 1999). Both FAK−/− and FAK+/+ MEFs exhibit elevated RhoA-GTP levels in suspension (Fig. 4 A). In FAK−/− MEFs, RhoA-GTP levels remain high upon initial cell binding to FN (15 and 30 min), whereas RhoA-GTP levels are low when FAK+/+ MEFs bind to FN (Fig. 4, A and B). By 90 min, RhoA-GTP levels are equally elevated in FAK−/− and FAK+/+ MEFs. Plating FAK−/− Pyk2 shRNA MEFs on FN revealed the transient inhibition of RhoA-GTP levels, which is similar to RhoA regulation in FAK+/+ MEFs (Fig. 4, A and B). This result supports the notion that Pyk2 is facilitating aberrant RhoA activation in FAK−/− MEFs.
Figure 4.

Pyk2 signaling activates RhoA and promotes p190RhoGEF expression. (A) Pyk2 knockdown restores normal RhoA regulation during FN adhesion of FAK−/− MEFs. RhoA activation analyses were performed using the indicated serum-starved MEFs held in suspension for 30 min and then replated on 10 μg/ml FN for the indicated times. GTP-bound RhoA was determined by GST-RBD pulldown and blotting for RhoA levels in lysates. (B) Quantitation of FN-associated RhoA-GTP binding. RhoA values are means ± SD of three independent experiments and are plotted relative to RhoA-GTP levels in suspended FAK−/− vector shRNA MEFs. (C) Pyk2 kinase activity and Y402 phosphorylation are required to promote Rho activation. FAK−/− Pyk2 shRNA MEFs were preinfected with Ad-TA control, Myc-Pyk2 WT, Myc-Pyk2 F402, or Myc-Pyk2 A457 (kinase inactive), subjected to FN replating, and analyzed for Rho-GTP binding as in A. (D) Quantitation of FN-stimulated RhoA-GTP binding from C. Values are means ± SD of two independent experiments and are plotted relative to RhoA-GTP levels in suspended FAK−/− Pyk2 shRNA MEFs. (E) Elevated p190RhoGEF mRNA levels in FAK−/− MEFs. RT-PCR analyses using p190RhoGEF- and β-actin–specific primers. Shown is an inverted ethidium bromide–stained gel. (F) p190RhoGEF protein expression is regulated by Pyk2. FAK−/− Pyk2 shRNA MEFs were infected with Ad-FAK or Ad-Pyk2 for 48 h. p190RhoGEF or actin protein expression were analyzed by blotting and compared with lysates from FAK+/+ and FAK−/− MEFs. (G) Kinase- and Y402-dependent Pyk2 signaling promotes Pyk2 expression. FAK−/− Pyk2 shRNA MEFs were infected with Ad-TA control, Ad-Pyk2 WT, Ad-Pyk2 F402, or Ad-Pyk2 A457 for 48 h. p190RhoGEF, Pyk2, and actin expression were analyzed by blotting.
To determine whether Pyk2 signaling promotes RhoA activation, FAK−/− Pyk2 shRNA MEFs were transduced with either human Ad-Pyk2 wild type (WT), Ad-Pyk2 A457 (kinase dead), or Ad-Pyk2 F402 or Ad–tetracycline transactivator (TA; control), and RhoA activity was evaluated after FN replating (Fig. 4 C). Ad-Pyk2 WT promoted constitutively activated RhoA, which is similar to what occurs in FAK−/− MEFs (Fig. 4, A and C). Expression of Pyk2 kinase dead or Pyk2 F402 had slight inhibitory effects on RhoA regulation upon FN plating (Fig. 4, C and D). Collectively, these results show that Pyk2 kinase activity and Pyk2 Y402 phosphorylation are needed to facilitate RhoA activation in FAK−/− MEFs.
Pyk2 regulates p190RhoGEF expression
Integrin-induced RhoA regulation is compromised in Pyk2−/− macrophages (Okigaki et al., 2003) but the signaling linkage between Pyk2 and RhoA remains undefined. GEFs function as RhoGTPase activators (Rossman et al., 2005), and we previously identified p190RhoGEF as a FAK-binding partner involved in laminin-stimulated RhoA activation in neuronal cells (Zhai et al., 2003). p190RhoGEF is a ubiquitously expressed RhoA-specific GEF of unclear biological function (Gebbink et al., 1997; van Horck et al., 2001). By semiquantitative RT-PCR analyses, low levels of p190RhoGEF mRNA were detected in FAK+/+ MEFs, whereas more than fivefold higher p190RhoGEF mRNA levels were present in FAK−/− MEFs (Fig. 4 E). By protein blotting, p190RhoGEF expression was three- to fourfold higher in FAK−/− compared with FAK+/+ MEFs, and Pyk2 shRNA expression was associated with reduced p190RhoGEF levels in FAK−/− MEFs (Fig. 4 F). Transient Ad-Pyk2, but not Ad-FAK, expression increased p190RhoGEF mRNA (not depicted) and protein levels about twofold by 48 h in FAK−/− Pyk2 shRNA MEFs (Fig. 4 F). This increase in p190RhoGEF protein was dependent on intrinsic Pyk2 kinase activity and Y402 phosphorylation (Fig. 4 G). These results show that Pyk2 signaling controls p190RhoGEF expression in FAK−/− MEFs, and this linkage is also conserved in rat pheochromocytoma PC12 cells (unpublished data).
p190RhoGEF promotes deregulated RhoA activity and growth of FAK−/− MEFs
To determine the function of p190RhoGEF in FAK−/− MEFs, lentiviral-mediated shRNA was used to stably knock down p190RhoGEF expression >85% (Fig. 5 A). Strikingly, FAK−/− p190RhoGEF shRNA MEFs exhibited a conversion from an epithelial-like to a fibroblast morphology and formed fewer FAs than FAK−/− Scr shRNA MEFs upon FN plating (Fig. 5, B and C). Reduction in p190RhoGEF was also associated with the cessation of deregulated RhoA activity in FAK−/− MEFs (Fig. 5 D). Despite the relatively normal appearance of FAK−/− p190RhoGEF shRNA MEFs, Boyden chamber FN motility assays revealed severe motility defects (Fig. 5 E). Time-lapse microscopy showed that FAK−/− p190RhoGEF shRNA MEFs spread on FN and possessed leading-edge membrane–ruffling activity but exhibited limited directional movement upon serum stimulation (Fig. S2 and Video 4, available at http://www.jcb.org/cgi/content/full/jcb.200708194/DC1). These results support a role for p190RhoGEF in MEF motility. Additionally, as FAK−/− p190RhoGEF shRNA MEFs form FAs within 60 min on FN (Fig. 5 B) under conditions of low RhoA-GTP binding (Fig. 5 D), our results support the notion that FA formation can also occur through alternative pathways (Zaidel-Bar et al., 2004) and that low levels of RhoA activity in FAK−/− p190RhoGEF shRNA MEFs likely represent contributions of other GEFs downstream of integrins.
Figure 5.

p190RhoGEF contributes to aberrant FAK−/− MEF morphology, RhoA activity, and FA formation. (A) Stable p190RhoGEF knockdown. FAK−/− MEFs were infected with lentiviral Scr or p190RhoGEF shRNA and sorted for GFP expression, and lysates were analyzed by p190RhoGEF, Pyk2, actin, and GFP blotting. (B) Phase-contrast images show that FAK−/− p190RhoGEF shRNA MEFs exhibit more of a fibroblast phenotype than FAK−/− Scr shRNA control MEFs. Reduced numbers of FAs connected to F-actin after FN replating (60 min) upon p190RhoGEF knockdown as determined by anti-paxillin and Alexa Fluor 350 phalloidin costaining. Bars, 30 μm. (C) Decreased FAK−/− FA formation upon p190RhoGEF knockdown. Box-and-whisker plots of paxillin-positive–stained points within FAK−/− Scr shRNA or FAK−/− p190RhoGEF shRNA MEFs plated on FN for 60 min (n = 15 cells per point). (D) p190RhoGEF knockdown prevents deregulated FAK−/− MEF RhoA activation. GTP-bound RhoA was determined by GST-RBD pulldown assays from lysates of suspended and FN-replated FAK−/− Scr shRNA and FAK−/− p190RhoGEF shRNA MEFs followed by blotting for total RhoA levels. (E) Loss of p190RhoGEF inhibits motility. 10-μg/ml-FN–stimulated FAK+/+, FAK−/− Scr, and FAK−/− p190RhoGEF shRNA MEF Boyden chamber motility over 4 h. Means ± SD were determined from three experiments and are a percentage of FAK−/− control (set to 100). (F) p190RhoGEF contributes to FAK−/− MEF proliferation. Cell growth assays over 5 d show that FAK−/− p190RhoGEF shRNA MEFs grow slower than FAK−/− Scr shRNA MEF controls and that Ad-Pyk2 overexpression does not function to enhance FAK−/− p190RhoGEF shRNA MEF cell proliferation. Cells were infected 24 h before initiating growth analyses. Values are means ± SD from two experiments. (G) Pyk2 overexpression weakly activates RhoA in FAK−/− p190RhoGEF shRNA MEFs. Cells were preinfected with Ad-Pyk2 WT, subjected to FN replating, and analyzed for Rho-GTP binding followed by blotting for total RhoA and Myc-Pyk2 levels.
To determine whether p190RhoGEF contributes to FAK−/− growth, cell proliferation assays were performed. FAK−/− p190RhoGEF shRNA MEFs grew significantly (P < 0.01 at day 3) slower than FAK−/− Scr shRNA MEFs (Fig. 5 F). Rescue assays were inconclusive because of shRNA inhibition of mouse p190RhoGEF reexpression (unpublished data). Interestingly, Ad-Pyk2 overexpression did not significantly promote FAK−/− p190RhoGEF shRNA MEF growth (Fig. 5 F; P > 0.05) and only weakly facilitated RhoA-GTP binding upon FN plating (Fig. 5 G). This result contrasts with strong RhoA activation (Fig. 4 C) and increased cell proliferation (Fig. 2 C) upon Pyk2 reexpression within FAK−/− Pyk2 shRNA MEFs. This result supports the conclusion that a Pyk2-p190RhoGEF signaling linkage promotes FAK−/− MEF proliferation and that p190RhoGEF is an important target for Pyk2-mediated RhoA activation.
p190RhoGEF functions downstream of integrins in normal MEFs
It is well established that Rho GTPase activation and cell contractility contribute to FA formation (Wehrle-Haller and Imhof, 2002; Zaidel-Bar et al., 2003). However, it remains unclear which GEFs downstream of integrins participate in this process. Lentiviral p190RhoGEF shRNA was used to stably reduce p190RhoGEF levels in normal (FAK+/+) MEFs (Fig. 6 A), and this resulted in the inhibition of RhoA activation at 90 and 120 min after FN plating compared with parental and Scr shRNA–expressing MEFs (Fig. 6, B and C). FAK+/+ MEFs with reduced p190RhoGEF did not show differences in binding to or spreading on FN. At 60 min on FN, FAK+/+ p190RhoGEF shRNA MEFs were fully spread but contained greater than twofold fewer FAs per cell than control FAK+/+ Scr MEFs (Fig. 6, D and E). This difference in FA formation upon p190RhoGEF reduction was also associated with the inhibition of FN-stimulated motility (Fig. 6 F). These results support the notion that p190RhoGEF functions as an important regulator of RhoA activity and FA formation downstream of integrins.
Figure 6.

p190RhoGEF facilitates integrin-associated RhoA regulation, FA formation, and motility of normal MEFs. (A) Stable p190RhoGEF knockdown. FAK+/+ MEFs were infected with lentiviral Scr or p190RhoGEF shRNA and sorted for GFP expression and lysates were analyzed by p190RhoGEF, FAK, actin, and GFP blotting. (B) p190RhoGEF knockdown limits RhoA activation upon FN adhesion. GTP-bound RhoA was determined by GST-RBD pulldown assays from lysates of suspended and FN-replated FAK+/+ parental, FAK+/+ Scr shRNA, and FAK+/+ p190RhoGEF shRNA MEFs, followed by blotting for total RhoA levels. (C) Quantitation of FN-stimulated RhoA-GTP binding from assays shown in B. Values are means ± SD of two independent experiments and are plotted relative to RhoA-GTP levels in suspended FAK+/+ MEFs. (D) p190RhoGEF knockdown limits FA formation but not MEF spreading on FN. FAK+/+ Scr and FAK+/+ p190RhoGEF shRNA MEFs were plated onto 1-μg/ml-FN–coated coverslips for 60 min and costained with antibodies to paxillin and Alexa Fluor 350 phalloidin for F-actin. Bar, 30 μm. (E) Decreased FA formation upon p190RhoGEF knockdown. Box-and-whisker plots of paxillin-positive–stained points within FAK+/+ Scr shRNA or FAK+/+ p190RhoGEF shRNA MEFs plated on FN for 60 min (n = 15 cells per point). (F) 10-μg/ml-FN–stimulated FAK+/+ (parental), FAK+/+ Scr, and FAK+/+ p190RhoGEF shRNA MEF Boyden chamber motility over 4 h. Means ± SD were determined from three experiments and are a percentage of FAK+/+ control (set to 100).
To evaluate the specificity of p190RhoGEF action, transient siRNA knockdowns were performed with a closely related (Rossman et al., 2005) RhoA-specific GEF termed GEFH1 (Ren et al., 1998). Efficient GEFH1 knockdown was accomplished in both FAK−/− and FAK+/+ MEFs, but there were no differences in paxillin-stained FAs or actin structures upon loss of GEFH1 (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200708194/DC1). Alternatively, transient GFP-GEFH1 expression revealed a circular microtubule distribution consistent with GEFH1 binding to microtubules (Ren et al., 1998), whereas expression of GFP-p190RhoGEF was cytoplasmic distributed and selectively enriched within the tips of cell projections in normal MEFs (Fig. S4). Both GEFs enhanced F-actin generation and this was inhibited by GEF-inactivating point mutations in the catalytic domains of GEFFH1 (Y393A) and p190RhoGEF (Y1003A; unpublished data).
To explore the similarities and differences of these GEFs, GFP fusions of GEFH1 WT (or catalytically inactive Y393A GEFH1) and p190RhoGEF (or catalytically inactive Y1003A or Δ1292-1301, a FAK-binding mutant) were stably expressed in normal MEFs via lentiviral transduction (Fig. 7 A). GFP–p190RhoGEF WT and Y1003A were expressed at levels equivalent to endogenous p190RhoGEF (not depicted), whereas GFP–GEFH1 WT expression was low (Fig. 7 A) and associated with a rounded and multinucleated phenotype, which is consistent with a role for GEFH1 in cytokinesis (Birkenfeld et al., 2007). GFP-GEFH1 and GFP-p190RhoGEF facilitated elevated RhoA activation upon FN plating and these effects were inactivated by Y393A or by Y1003A or Δ1292-1301 mutations in GEFH1 and p190RhoGEF, respectively (Fig. 7 B and not depicted). Interestingly, GFP–p190RhoGEF WT–expressing MEFs exhibited a rounded phenotype identical to FAK−/− MEFs (Fig. 7 C), with an approximately threefold increase in FA formation (Fig. 7 D) and inhibited motility on FN (Fig. 7 E) compared with normal GFP-expressing MEFs. As changes in p190RhoGEF expression either prevent or stimulate FA formation with corresponding negative effects on cell motility, these results point to the importance of p190RhoGEF in FA regulation for cell movement.
Figure 7.

p190RhoGEF-stimulated RhoA activity and FA formation are dependent on FAK binding. (A) GFP-p190RhoGEF or GFP-GEFH1 overexpression in normal MEFs. (A) FAK+/+ MEFs were infected with a lentiviral expression vector for GFP or GFP fusions of p190RhoGEF WT, p190RhoGEF Y1003A (catalytically inactive), p190RhoGEF Δ1292-1301 (FAK-binding mutant), GEFH1 WT, or GEFH1 Y393A (catalytically inactive) and sorted for GFP expression. Shown is the flow cytometry profile of GFP expression (open peaks) compared with FAK+/+ autofluorescence (shaded peaks). (B) GFP-p190RhoGEF–mediated RhoA activation is dependent on FAK binding. GTP-bound RhoA was determined by GST-RBD pulldown assays from lysates of suspended and FN-replated FAK+/+-expressing GFP or the indicated GFP-190RhoGEF constructs followed by blotting for RhoA expression. (C) FAK+/+ p190RhoGEF WT MEFs show enhanced FA formation and actin fibers similar to FAK−/− MEFs. Cells were plated onto 1-μg/ml-FN–coated coverslips for 60 min and costained with antibodies to vinculin and Alexa Fluor 350 phalloidin for F-actin. Bar, 30 μm. (D) p190RhoGEF and GEFH1 promote FA formation in MEFs. Box-and-whisker plots of paxillin-positive–stained points within MEFs stably overexpressing the indicated GFP-p190RhoGEF or GFP-GEFH1 constructs upon FN plating for 60 min (n = 15 cells per point). (E) GEF-mediated increased FA formation is associated with decreased cell motility. MEFs overexpressing the indicated GFP-p190RhoGEF or GFP-GEFH1 constructs were evaluated for 10-μg/ml-FN–stimulated Boyden chamber motility over 4 h. Means ± SD were determined from two experiments and are a percentage of FAK+/+ GFP MEFs (set to 100). Asterisks indicate that the difference between FAK+/+ GFP and FAK+/+ GFP–p190RhoGEF Δ1292-1301 or FAK+/+ GFP–p190RhoGEF Y1003A is significant (P < 0.05).
FAK regulation of p190RhoGEF FA localization and tyrosine phosphorylation
Many regulatory and signaling proteins that participate in integrin signaling events are transiently recruited to FAs upon FN plating (Vicente-Manzanares et al., 2005). Although localization studies for endogenous p190RhoGEF distribution were inconclusive because of high nonspecific staining (not depicted), GFP–p190RhoGEF WT partially colocalized with paxillin at FAs upon FN plating of MEFs (Fig. 8). Interestingly, GFP–p190RhoGEF Δ1292-1301 with a deletion in the FAK-binding region or catalytically inactive GFP–p190RhoGEF Y1003A did not exhibit strong FA localization (Fig. 8) and did not function to facilitate RhoA activation (Fig. 7 B) or alter FA formation (Fig. 7 D). This differs from GFP-GEFH1, which was primarily cytoplasmic distributed (Fig. 8) and was actively promoting FA formation (Fig. 7 D). Although overexpression of GFP–p190RhoGEF Δ1292-1301 or GFP–p190RhoGEF Y1003A had weak but significant (P < 0.05) inhibitory effects on FN-stimulated motility (Fig. 7 E), this was not associated with alterations in cell morphology (Fig. 8) or FA formation (Fig. 7 D) and may reflect alterations in endogenous p190RhoGEF function. Collectively, these results show that although GEFH1 was active but not FA localized, p190RhoGEF activity required FAK binding and FA association in normal MEFs.
Figure 8.

FAK binding directs p190RhoGEF localization to FAs upon FN plating. FAK+/+ MEFs stably expressing GFP or the indicated GFP-p190RhoGEF or GFP-GEFH1 constructs were plated onto 1-μg/ml-FN–coated coverslips for 1 h, fixed, and then stained with antibodies to paxillin. Shown is the cellular GFP (left) or paxillin (middle) distribution and the merged images (right). Bar, 30 μm.
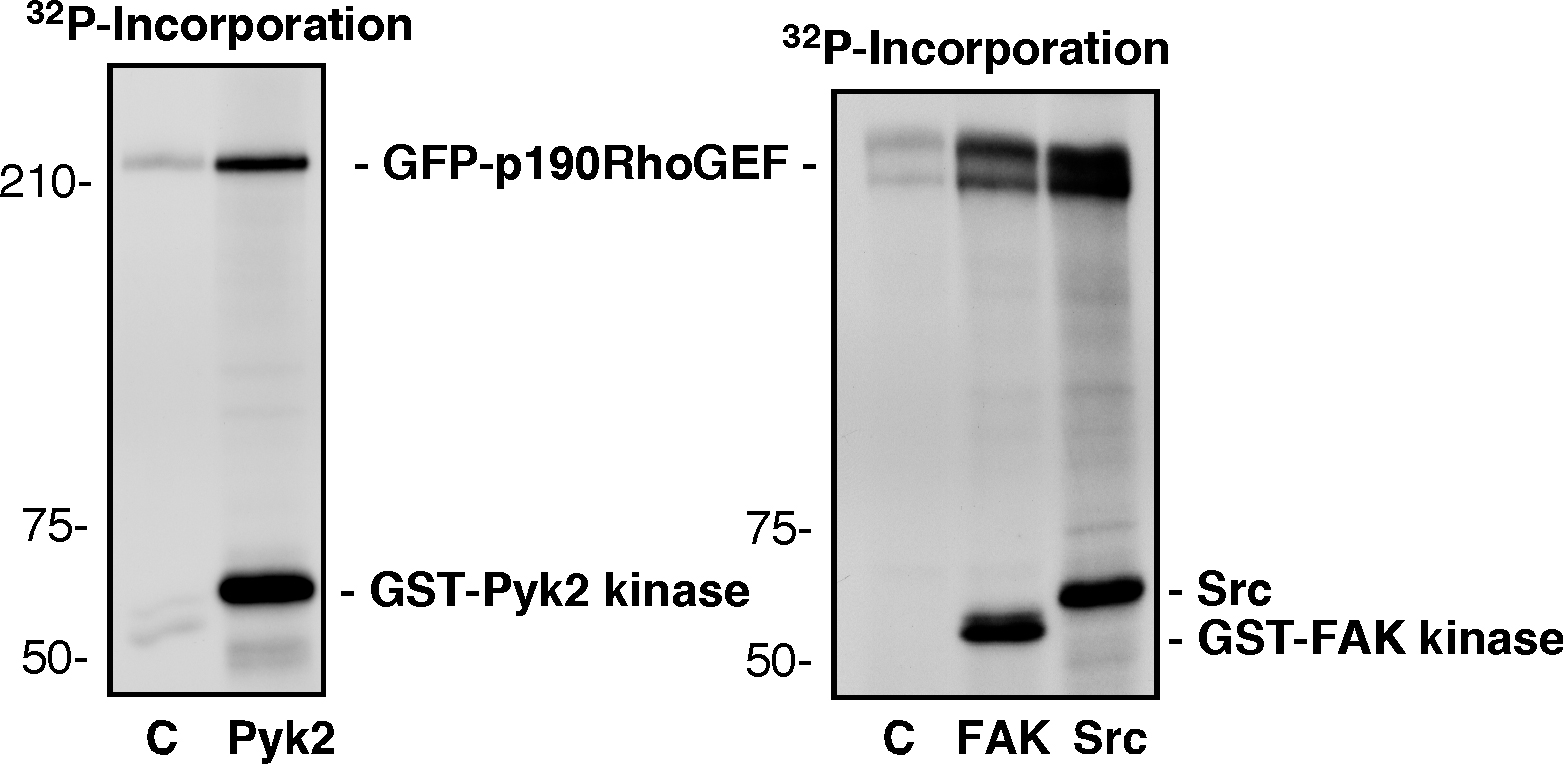
To determine if FA localization of p190RhoGEF is essential for its effects on FA formation, GFP-p190RhoGEF was transiently expressed in FAK−/− MEFs. Surprisingly, GFP-p190RhoGEF was not FA localized but exhibited potent effects on enhancing FA formation (Fig. 9 A). By coimmunoprecipitation analyses, endogenous p190RhoGEF forms a complex with Pyk2 in FAK−/− MEFs (Fig. 9 B) and is tyrosine phosphorylated upon FN plating (Fig. 9 C). Importantly, Pyk2 also does not localize to FAs in FAK−/− MEFs (Sieg et al., 1998). However, as GFP-p190RhoGEF did not function to promote FA formation or localize to FAs in FAK−/− Pyk2 shRNA MEFs (unpublished data), these results are consistent with the importance of FAK in mediating p190RhoGEF FA localization and with potentially overlapping roles for Pyk2 or FAK in promoting p190RhoGEF tyrosine phosphorylation and activation. In vitro kinase assays using purified Pyk2 or FAK kinase domains or full-length Src showed that each of these PTKs could directly phosphorylate p190RhoGEF (Fig. S5, available at http://www.jcb.org/cgi/content/full/jcb.200708194/DC1). Analysis of GFP-p190RhoGEF–expressing FAK+/+ MEFs revealed adhesion-dependent p190RhoGEF WT tyrosine phosphorylation and the formation of a FAK–paxillin–p190RhoGEF signaling complex (Fig. 9 D). FAK association and p190RhoGEF tyrosine phosphorylation upon FN plating were inhibited by p190RhoGEF catalytically inactivating (Y1003A) mutation or deletion of the FAK-binding region (Δ1292-1301). Together, these results support the notion that p190RhoGEF activity is regulated in part by FAK and or Pyk2 binding leading to enhanced p190RhoGEF tyrosine phosphorylation and the formation of an adhesion-dependent signaling complex.
Figure 9.

Adhesion-dependent regulation of p190RhoGEF tyrosine phosphorylation. (A) GFP-p190RhoGEF is cytoplasmic distributed in FAK−/− MEFs. Cells were transiently transfected with GFP–p190RhoGEF WT, plated on FN for 1 h, and fixed and coanalyzed for GFP distribution and paxillin staining as an FA marker. Bar, 30 μm. (B) p190RhoGEF associates with Pyk2. Control rabbit IgG or anti-Pyk2 immunoprecipitations from FAK−/− MEFs were sequentially analyzed by p190RhoGEF and Pyk2 blotting. (C) p190RhoGEF is tyrosine phosphorylated upon FAK−/− MEF adhesion to FN. Antiphosphotyrosine (pY) immunoprecipitates were made from lysates of FAK−/− MEFs held in suspension or FN replated for 1 h and were analyzed by anti-p190RhoGEF blotting. The amount of p190RhoGEF in suspended or FN-replated whole cell lysates (WCLs) was determined by blotting. (D) The FAK-binding region is important for FN-stimulated p190RhoGEF tyrosine phosphorylation. Lysates were prepared from FAK+/+ MEFs expressing GFP or the indicated GFP-p190RhoGEF constructs after cell suspension or FN replating for 1 h, and anti-GFP immunoprecipitates were sequentially blotted for phosphotyrosine and GFP. In a second experiment, anti-GFP immunoprecipitates were sequentially blotted for FAK and paxillin.
Discussion
Many investigators have compared FAK+/+ and FAK−/− MEFs to define the role of FAK in various signaling pathways (Mitra et al., 2005). One complication of these analyses is that expression of the FAK-related PTK Pyk2 is elevated in FAK−/− MEFs. We show that increased Pyk2 expression occurs after conditional FAK inactivation within FAK(loxP/loxP) MEFs and upon FAK knockdown by shRNA expression within human diploid fibroblasts. The same shRNA was used to knock down FAK within human MCF-7 and MDA-MB-231 breast carcinoma, PC3 prostate carcinoma, or NB8 neuroblastoma cells without detectable changes in Pyk2 expression (Mitra et al., 2006a,b). Thus, compensatory changes in Pyk2 levels are independent of the means to inactivate FAK and may reflect differences in cell origin or transformation.
We found that Pyk2 shRNA knockdown within FAK−/− MEFs slows cell growth that could be rescued by either transient FAK or Pyk2 reexpression. Previous studies have linked FAK signaling to MAPK activation and increased cell cycle progression (Zhao et al., 2003). Although Pyk2 overexpression in FAK−/− MEFs enhances MAPK activation (Sieg et al., 1998), we found that Pyk2-enhanced FAK−/− MEF proliferation was dependent on p190RhoGEF expression and was correlated with the ability of Pyk2 to stimulate RhoA activation. This result is consistent with the finding that Rho kinase inhibition restored adhesion-dependent control of FAK−/− MEF proliferation (Pirone et al., 2006) and that a Pyk2-p190RhoGEF signaling linkage promotes FAK−/− MEF proliferation.
Although stable Pyk2 knockdown in FAK−/− MEFs decreased paxillin tyrosine phosphorylation to normal levels, the most striking finding of our study was that aberrant FAK−/− MEF phenotypes of elevated FA formation, altered F-actin distribution, and a rounded epithelial-like morphology were linked to Pyk2-enhanced p190RhoGEF expression promoting constitutive RhoA activation (Fig. 10). Interestingly, FAK-null keratinocytes also display altered actin and FA formation. Pyk2 is expressed and activated in these cells but it remains unclear whether elevated Rho activity in the absence of FAK within keratinocytes is associated with a Pyk2-p190RhoGEF signaling linkage (Schober et al., 2007). p190RhoGEF is a ubiquitously expressed RhoA-specific GEF (Gebbink et al., 1997; van Horck et al., 2001). p190RhoGEF contains a central tandem Dbl-pleckstrin homology domain, an N-terminal leucine-rich region, a cysteine-rich zinc finger domain, a large C-terminal domain with a potential coiled-coil region shown to bind microtubules (van Horck et al., 2001), the 3′ untranslated region of neurofilament mRNA (Canete-Soler et al., 2001), and FAK (Zhai et al., 2003). In neuronal cells, p190RhoGEF possesses antiapoptosis activity (Wu et al., 2003), is implicated in the pathogenesis of motor neuron degeneration (Lin et al., 2005), and plays a negative role in axonal branching and synapse formation downstream of FAK (Rico et al., 2004). Through combined knockdown or transient overexpression studies, we found that kinase- and Y402-dependent signals from Pyk2, but not FAK, regulate p190RhoGEF expression in FAK−/− MEFs and in PC12 cells. Although studies have identified p190RhoGEF as a protein induced by CD40 stimulation of B cells (Lee et al., 2003), the important signaling pathways controlling either Pyk2- or CD40-induced p190RhoGEF expression remain to be defined.
Figure 10.

Model of p190RhoGEF function in FAK−/− and FAK+/+ MEFs. Elevated Pyk2 levels and signaling in FAK−/− MEFs promote p190RhoGEF mRNA and protein expression. Knockdown of Pyk2 reduces FAK−/− proliferation, whereas Pyk2 reexpression enhances cell growth that is dependent on p190RhoGEF expression. Pyk2 phosphorylates p190RhoGEF, Pyk2 forms a complex with p190RhoGEF that is cytoplasmic distributed, and Pyk2-p190RhoGEF acts to promote aberrant RhoA activation connected to enhanced FAK−/− MEF FA formation. Knockdown of Pyk2 or p190RhoGEF in FAK−/− MEFs restores normal integrin-regulated RhoA activity and FA formation but do not rescue FAK−/− MEF motility defects that involve lack of FA release. In normal MEFs, transient RhoA inhibition after FN plating is mediated in part through Src phosphorylation and activation of p190RhoGAP (Arthur et al., 2000). p190RhoGEF knockdown limits RhoA activation and FA formation upon FN plating, whereas p190RhoGEF overexpression overrides p190RhoGAP-mediated RhoA regulation to promote RhoA activation and enhanced FA formation and recapitulates a partial FAK−/− cell phenotype. Both p190RhoGEF overexpression and knockdown prevent cell movement as the formation of too many or too few FAs limits motility. p190RhoGEF transiently localizes to FAs upon FN replating. This is dependent on FAK binding and is associated with p190RhoGEF tyrosine phosphorylation. Unlike other cytoplasmic GEFs, such as GEFH1, that can activate RhoA, the ability of p190RhoGEF to stimulate RhoA is associated with FAK-Pyk2 binding, tyrosine phosphorylation, or FA localization. This paper has elucidated a novel role for p190RhoGEF as an integrin-proximal regulator of FA formation and cell motility.
In FAK−/− MEFs, stable Pyk2 or p190RhoGEF knockdown restored normal FN-associated regulation of RhoA activity and FA formation but these reversals did not enhance cell motility. Therefore, deregulated RhoA activity likely contributes to but is not causal for FAK-null motility defects (Ren et al., 2000). Importantly, knockdown of GEFH1 that is closely related to p190RhoGEF did not reduce FA formation FAK−/− MEFs, and this further supports the specificity of our findings. Importantly, Pyk2 or p190RhoGEF knockdown results in FAK−/− MEFs with normal ruffling and lamellipodia formation but these cells exhibit tail retraction and severe motility defects. We speculate that either cell polarization (Tilghman et al., 2005) or rear FA-release defects (Ren et al., 2000) act to limit FAK−/− Pyk2 shRNA MEF motility. As transient FAK reexpression can rescue the motility defects of FAK−/− Pyk2 shRNA MEFs, stable reconstitution with WT FAK and various mutants will yield a powerful cell system to elucidate the molecular connections of FAK-mediated cell motility.
In normal MEFs, reduction in p190RhoGEF expression did not affect binding to or spreading on FN, but these cells formed fewer FAs under conditions of low RhoA activity. FAK+/+ p190RhoGEF shRNA MEFs exhibited reduced motility on FN, supporting the notion that p190RhoGEF functions as an important regulator of RhoA activity and FA formation downstream of integrins and is needed for efficient cell movement. As FN-stimulated FAK and tyrosine phosphorylation was also reduced in FAK+/+ p190RhoGEF shRNA MEFs (unpublished data) and contractility-associated mechanisms within cells can facilitate FAK recruitment to and activation at growing FAs (Kirchner et al., 2003), our results support the existence of a FAK–p190RhoGEF regulatory circuit controlling FA formation and cell motility (Fig. 10). Additionally, as FAK activation and paxillin phosphorylation can influence the distribution of these proteins at FAs (Zaidel-Bar et al., 2007), there may be other feedback mechanisms limiting FA formation upon p190RhoGEF knockdown in normal MEFs.
GEFs are multidomain proteins regulated in a complex fashion (Bos et al., 2007). This can include protein–protein, protein–lipid, and second messenger binding and posttranslational modification of GEFs. These interactions or modifications can induce GEF translocation to sites of small G protein localization, autoinhibition release, or allosteric conformational activation of the catalytic region. The importance of FAK in p190RhoGEF regulation was supported by comparisons of MEFs overexpressing WT or the FAK-binding mutant (Δ1292-1301) of p190RhoGEF. Although p190RhoGEF WT partially localized to FAs, promoted RhoA activation, and enhanced FA formation, p190RhoGEF Δ1292-1301 overexpression was cytoplasmic distributed and did not affect FN-associated RhoA activity or FA formation. Although it is also possible that the Δ1292-1301 deletion has global effects on catalytic domain folding, overexpressed GEFH1 was not FA localized but still functioned similar to p190RhoGEF WT in activating RhoA and promoting FA formation. These results support a model whereby p190RhoGEF binding to FAK facilitates its localization to and activation at FAs, whereas the related GEFH1 does not share this mode of regulation. Additionally, p190RhoGEF catalytic activity was also important in promoting FA localization through undefined mechanisms. As catalytically inactive (Y1003A) p190RhoGEF could weakly colocalize with FA structures (unpublished data), we speculate that increased numbers of FAs formed by WT, but not p190RhoGEF Y1003A overexpression influences the equilibrium and distribution of both endogenous and exogenous p190RhoGEF localized to FAs in MEFs.
One additional level of p190RhoGEF regulation is that adhesion-dependent p190RhoGEF tyrosine phosphorylation was inhibited by both Δ1292-1301 and Y1003A mutations. Although it remains unclear how phosphorylation may affect p190RhoGEF function, this modification, as well as FAK binding, correlated with p190RhoGEF-mediated RhoA activation. Interestingly, p190RhoGEF WT did not localize to the many FAs in FAK−/− MEFs yet still functioned to enhance FAK−/− FA formation. As we find that p190RhoGEF binds to cytoplasmic-distributed Pyk2 within FAK−/− MEFs and is tyrosine phosphorylated upon FAK−/− MEF adhesion to FN, we hypothesize that p190RhoGEF activity is regulated in part by FAK and/or Pyk2 binding leading to enhanced p190RhoGEF tyrosine phosphorylation.
In summary, our studies have uncovered a novel role of p190RhoGEF as a mediator of aberrant FAK−/− MEF phenotypes and as an important regulator of FN-associated RhoA activation and FA formation in normal MEFs. As the formation of too many or too few FAs limit cell movement, future studies on the regulation of p190RhoGEF activity and the FAK–p190RhoGEF complex will yield important insights into the temporal balance of RhoA activity needed for cell migration.
Materials and methods
Antibodies and reagents
mAbs to phosphotyrosine (clone 4G10), vinculin (clone V284), and FAK (clone 4.47) were obtained from Millipore. mAbs to p190RhoGAP (clone 30), Pyk2 (clone 11), paxillin (clone M107), and phosphospecific paxillin pTyr-118 (clone 30) were obtained from BD Biosciences. Rabbit polyclonal anti-RhoA (sc-179) was obtained from Santa Cruz Biotechnology, Inc. Texas red–phalloidin and Alexa fluor 350 phalloidin were obtained from Invitrogen. HA tag (clone 16B12), Myc tag (clone 9E10), GFP (clone B34), and Cre recombinase (clone 7.23) mAbs were obtained from Covance Research. Anti–β-actin (clone AC-17) mAb was obtained from Sigma-Aldrich. Rabbit polyclonal antibodies (7801/7802) were generated to mouse p190RhoGEF residues 1457–1582 expressed as a GST fusion protein (University of California, San Diego). Antibodies were affinity purified using GST-p190RhoGEF (1457–1582) covalently coupled to glutathione agarose (Sigma-Aldrich) using dimethylpimelimidate as described previously (Sieg et al., 1998). Antibody specificity was confirmed by Western blot analysis combined with soluble GST-p190RhoGEF (1457–1582) as a competitive inhibitor. pCDNA3-Y1003A p190RhoGEF was provided by J. Lee (Ewha Womans University, Seoul, Korea). Purified bovine plasma FN was obtained from EMD.
Lentiviral constructs
shRNA constructs were created using pLentiLox3.7 as previously described (Rubinson et al., 2003). Human FAK shRNA was created and used as previously described (Mitra et al., 2006a). For mouse Pyk2 and p190RhoGEF, sense oligos used were as follows: Pyk2, 5′-tGA AGT AGT TCT TAA CCG CAT tca aga gAT GCG GTT AAG AAC TAC TTC ttt ttt c-3′; p190RhoGEF-1, 5′-tGA ACA GAG ATC ATA TAG TTT tca aga gAA ACT ATA TGA TCT CTG TTC ttt ttt c-3′; p190RhoGEF-2, 5′-tGA ACC AGA CTG TAA TAT CTT tca aga gAA GAT ATT ACA GTA TGG TTC TTT TTT C-3′; and Scr, 5′-tGT CTC CGA ACG TGT CAC GTT tca aga gAA CGT GAT ACG TTC GGA GAC ttt ttt c-3′. The 5′t belongs to the U6 promoter and lowercase letters represent components of the shRNA loop. The 3′c was added to generate an XhoI site. Antisense oligos used were as follows: Pyk2, 5′-tcg aga aaa aaG AAG TAG TTC TTA ACC GCA Tct ctt gaA TGC GGT TAA GAA CTA CTT Ca-3′; p190RhoGEF-1, 5′-tcg aga aaa aaG AAC AGA GAT CAT ATA GTT Tct ctt gaA AAC TAT ATG ATC TCT GTT Ca-3′; p190RhoGEF-2, 5′-tcg aga aaa aaG AAC CAG ACT GTA ATA TCT Tct ctt gaA AGA TAT TAC AGT CTG GTT Ca-3′; and Scr, 5′-tcg aga aaa aaG TCT CCG AAC GTG TCA CGT Tct ctt gaA ACG TGA CAC GTT CGG AGA Ca-3′. An additional four nucleotides at the 5′ end were added to generate XhoI site. pLentiLox3.7 expresses the shRNA and GFP from distinct promoters, and virus production was achieved by 293T cell transfection as previously described (Mitra et al., 2006a). Media containing viruses was used to infect target cells after 48 h. To create N-terminal GFP fusions of p190RhoGEF WT, p190RhoGEF Y1003A and Δ1292-1301 RhoGEF (Zhai et al., 2003) were digested with KpnI and XbaI and cloned into the same sites within pEGFPC2. For lentiviral expression, GFP-p190RhoGEF was removed by NheI–XbaI digestion and cloned into NheI–SwaI–digested pCDH1-MCS1 (System Biosciences). GFP–GEFH1 WT and catalytically inactive Y393A (Krendel et al., 2002) were removed from pCMV5 by EcoR1–ClaI digestion and blunt-end cloned into pCDH1-MSC1. Viruses were produced by 293T cell transfection, concentrated by polyethylene glycol precipitation, and used to infect target cells.
RT-PCR
Total RNA was extracted with Trizol reagent (Invitrogen) and cDNA was synthesized from 5 μg using SuperScript first-strand synthesis system for RT-PCR (Invitrogen). A 1-μl aliquot of the reverse-transcription reaction was then used for subsequent PCR amplifications. Primers used were the following: p190RhoGEF forward, 5′-GAGCAAGGAGGTAACAGAACT-3′; p190RhoGEF reverse, 5′-CGCCATCGGAAGACTTAGTT-3′; β-actin forward, 5′-TGTGATGGTGGGAATGGGTCAG-3′; and β-actin reverse, 5′-TTTGATGTCACGCACGATTTCC-3′. p190RhoGEF was amplified for 30 cycles and primers for actin were added after cycle 10.
Cell culture and viruses
FAK(loxP/loxP) mice (Beggs et al., 2003) were obtained from H. Beggs (University of California, San Francisco, San Francisco, CA) and were crossed with p53-null mice (Ilic et al., 1995). Embryonic day–8.5 mouse embryos were isolated, placed within a drop of Matrigel (BD BioSciences), and cultured for 10 d. MEFs that grew out of embryos were disaggregated, plated onto 10-μg/ml-FN–coated dishes, and expanded. At passage two, 1/3 of MEFs were used for genomic DNA isolation genotype confirmation, 1/3 were expanded, and 1/3 were transduced with Ad-Cre recombinase at 25 MOI. Cre protein expression was detected at 48 h of infection but was undetectable after 7 d and subsequent passages. FAK+/+, FAK−/−, and FAK-reconstituted (DP3) MEFs are p53-null and were maintained as described previously (Sieg et al., 1999). Human diploid fibroblasts and PC12 cells were obtained from American Type Culture Collection. Human fibroblasts were grown in the same media as mouse MEFs (Sieg et al., 1999). PC12 cells were grown in DME with 5% horse serum, 10% FBS, 50 U/ml penicillin, and 50 μg/ml streptomycin. Recombinant adenovirus for HA-tagged FAK, Myc-tagged Pyk2, Myc-tagged Pyk2 F402, and Myc-tagged Pyk2 A457 in pADtet7 contain Tet-responsive enhancer sequences within a minimal cytomegalovirus promoter, and expression was driven by coinfection with Ad-Tet transactivator as previously described (Hsia et al., 2005). Cells were infected at 5 MOI per cell for Ad-TA and at 50 MOI for Ad-FAK or Ad-Pyk2 constructs. Lentiviral-infected fibroblasts expressing GFP, GFP-p190RhoGEF, or GFP-GEFH1 were obtained by FACS and maintained as pool populations.
Cell lysis, immunoprecipitation, blotting, and pulldown assays
Cells were washed in ice-cold PBS and solubilized in modified RIPA lysis buffer containing 1% Triton X-100, 0.5% sodium deoxycholate, and 0.1% SDS (Sieg et al., 1998). For immunoprecipitations, 2.5 μg of antibodies were incubated with lysates (each sample containing 500–1,000 μg of total protein) for 3 h at 4°C and collected by binding to protein G plus or protein A–agarose beads. SDS-PAGE, antibody blotting, and sequential membrane reprobing was performed as previously described (Sieg et al., 1998). Rho activity was measured by pulldown assays using GST–Rhotekin Rho binding domain (GST-RBD) (Ren et al., 2000). Clarified RIPA cell lysates were incubated with 20 μg GST-RBD beads at 4°C for 45 min and beads were washed four times with buffer B (Tris buffer containing 1% Triton X-100, 150 mM NaCl, 10 mM MgCl2, 10 μg/ml leupeptin, 10 μg/ml aprotinin, and 0.1 mM PMSF). Bound Rho proteins were detected by polyclonal RhoA blotting. Image J version1.37 (National Institutes of Health) was used to obtain densitometric values from blots, and Rho activity was determined as the amount of RBD-bound Rho versus total Rho in the lysate.
Cell migration and proliferation assays
For cell migration assays, cells were used 48 h after Ad infection and serum starved in 0.5% FBS overnight. Millicell chambers with 8-μm pores (Millipore) were coated on the both sides with 10 μg/ml FN for 2 h at room temperature. Membranes were washed with PBS and air dried for 2 h. Cells were suspended by limited EDTA-trypsin treatment, washed by 0.25 mg/ml soybean trypsin inhibitor in DME, suspended in Migration medium (0.5% BSA in DME), and counted. 5 × 104 cells in 0.3 ml (held in suspension for 1 h) were added to each MilliCell chamber and incubated for 4 h at 37°C. Cells were fixed and stained with crystal violet. Each data point represents three individual chambers enumerated as previously described (Hsia et al., 2005). For scratch-wound assays, cells were seeded in six-well plates and starved for 24 h. Cells were wounded by scratching with pipette tip, washed with PBS, photographed (0-h point), and placed into growth media containing 0.5 μg/ml mitomycin-C to block cell division. After 16 h, matched-pair wound regions were photographed. For phase-contrast time-lapse video microscopy, cells were plated onto 1-μg/ml-FN–coated glass coverslips in growth media. After 3 h, images were collected with a charge coupled device camera (Orca 285; Hamamatsu) at 5-min intervals over 4 h with a 20× lens on an automated (Metamorph 6.3; Universal Imaging Corporation) microscope (Nikon TE300) with a heated and motorized stage. For each experiment, 12 individual cell fields were recorded. Cell trajectories were measured by tracking the position of the cell nucleus over time. Cells that did not spread or remain within the field of view during the experimental period were not evaluated. For cell proliferation assays, 5 × 105 cells were plated into 6-well plates precoated with 0.5% gelatin and grown in DME with 10% FBS. At 24-h intervals, two wells were trypsinized and individually enumerated using an automated hemocytometer. Each experiment was repeated three times. One-way analysis of variance was used to determine statistical significance for proliferation and motility assays.
Immunofluorescent staining
Cells were plated onto 10-μg/ml-FN–coated glass coverslips in the absence of serum for the indicated times. Cells were fixed in 3.7% paraformaldehyde for 15 min and permeabilized with 0.1% Triton X-100 for 10 min. Blocking was performed with 100 μg/ml ChromPure donkey IgG in PBS (Jackson ImmunoResearch Laboratories) at room temperature for 1 h. Vinculin (1:50) or paxillin (1:50) antibodies were diluted in PBS and incubated overnight at 4°C. After three washes in PBS, coverslips were incubated for 45 min in PBS containing rhodamine-conjugated donkey anti–mouse IgG and fluorescein-conjugated phalloidin to visualize actin stress fibers. In cells expressing GFP or GFP fusion proteins, actin was visualized with Alexa Fluor 350 phalloidin. After washing, coverslips were mounted in Vectashield (Vector Laboratories). Images from wide UV (excitation, 330–85 nm; dichroic mirror, 400 nm; emission, 420 nm), wide blue (excitation, 450–80 nm; dichroic mirror, 500 nm; emission, 515 nm), and wide green (excitation, 510–550 nm; dichroic mirror, 570 nm; emission, 590 nm) were sequentially captured at 60× (UPLFL objective, 1.25 NA; Olympus) using a controller (MAC5000) and shutter (LEP; Ludl), a monochrome charge-coupled device camera (ORCA ER; Hamamatsu), an inverted microscope (IX51; Olympus), and Openlab software (Improvision). Images were pseudocolored using Image J 1.38 and overlayed and merged using Photoshop CS3 (Adobe). Integrated morphometry analysis was performed using Image J 1.38 on thresholded images to select classified objects of a size range of 0.1–20 pixels as FAs, based on anti-paxillin staining. The analyze particles command was used to measure both FA size and number. F-actin intensity was measured on spread cells stained with FITC-conjugated or Alexa Flour 350 phalloidin using Image J analysis of mean pixel intensity within a defined region of interest.
Mouse GEFH1 (Arhgef2), Scr siRNA, and siGLO green transfection indicator were obtained from Dharmacon. 100 pmol siRNA + 100 pmol siGLO was used to transfect (Lipofectamine 2000) FAK−/− or FAK+/+ MEFs. After 48 h, GEFH1 knockdown was confirmed by blotting (Birkenfeld et al., 2007). Cells were starved overnight, trypsinized, suspended for 1 h, and plated onto coverslips coated with 10 μg/ml FN for 1 h. Cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and costained for actin and paxillin using standard methods.
Online supplemental material
Fig. S1 shows scratch-wound healing motility defects of FAK−/− Pyk2 shRNA MEFs. Fig. S2 shows time-lapse analyses of random MEF motility. Fig. S3 shows that GEFH1 knockdown does not affect FN-associated FA formation. Fig. S4 shows the differential localization of GEFH1 and p190RhoGEF in MEFs. Fig. S5 shows that FAK, Pyk2, and Src can directly phosphorylate p190RhoGEF in vitro. Videos 1–4 show random motility of the indicated MEFs plated on 1-μg/ml-FN–coated glass slides in the presence of 10% FBS at 37°C. Phase-contrast images were obtained every 5 min for 10 h. Files were sharpened and contrast-adjusted using Photoshop. Videos were created using QuickTime Pro (Apple), Cinepak compression, at 450 × 375 pixel frame size. Each file represents 105 sequential images (8 h and 45 min) at 15 frames per second. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200708194/DC1.
Supplemental Material
Acknowledgments
This project was supported by National Institutes of Health grant CA87038 to D. Schlaepfer. Y. Lim was supported in part by Korea Research Foundation grant M01-2005-000-10071-0. M. Gardel was supported by a Jane Coffin Childs fellowship. W. Schlaepfer is supported by grant NS515722, C. Waterman-Storer by grant GM67230, and D. Schlaepfer by grants CA75240 and CA102310 from the National Institutes of Health. C. Waterman-Storer and D. Schlaepfer are Established Investigators of the American Heart Association.
Y. Lim, S.-T. Lim, and A. Tomar contributed equally to this paper.
Y. Lim's present address is Mogam Biotechnology Research Institute, Yong-In, Korea 449-799.
Abbreviations used in this paper: Ad, adenoviral; FA, focal adhesion; FN, fibronectin; GAP, GTPase-activating protein; GEF, guanine nucleotide exchange factor; GST-RBD, GST–Rhotekin Rho binding domain; MEF, mouse embryonic fibroblast; PTK, protein tyrosine kinase; Pyk2, proline-rich kinase 2; Scr, scrambled; shRNA, short hairpin RNA; TA, tetracycline transactivator; WT, wild type.
References
- Arthur, W.T., L.A. Petch, and K. Burridge. 2000. Integrin engagement suppresses RhoA activity via a c-Src-dependent mechanism. Curr. Biol. 10:719–722. [DOI] [PubMed] [Google Scholar]
- Beggs, H.E., D. Schahin-Reed, K. Zang, S. Goebbels, K.A. Nave, J. Gorski, K.R. Jones, D. Sretavan, and L.F. Reichardt. 2003. FAK deficiency in cells contributing to the basal lamina results in cortical abnormalities resembling congenital muscular dystrophies. Neuron. 40:501–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkenfeld, J., P. Nalbant, B.P. Bohl, O. Pertz, K.M. Hahn, and G.M. Bokoch. 2007. GEF-H1 modulates localized RhoA activation during cytokinesis under the control of mitotic kinases. Dev. Cell. 12:699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos, J.L., H. Rehmann, and A. Wittinghofer. 2007. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 129:865–877. [DOI] [PubMed] [Google Scholar]
- Canete-Soler, R., J. Wu, J. Zhai, M. Shamim, and W.W. Schlaepfer. 2001. p190RhoGEF Binds to a destabilizing element in the 3′ untranslated region of light neurofilament subunit mRNA and alters the stability of the transcript. J. Biol. Chem. 276:32046–32050. [DOI] [PubMed] [Google Scholar]
- Chen, B.H., J.T. Tzen, A.R. Bresnick, and H.C. Chen. 2002. Roles of Rho-associated kinase and myosin light chain kinase in morphological and migratory defects of focal adhesion kinase-null cells. J. Biol. Chem. 277:33857–33863. [DOI] [PubMed] [Google Scholar]
- Chikumi, H., S. Fukuhara, and J.S. Gutkind. 2002. Regulation of G protein-linked guanine nucleotide exchange factors for Rho, PDZ-RhoGEF, and LARG by tyrosine phosphorylation: evidence of a role for focal adhesion kinase. J. Biol. Chem. 277:12463–12473. [DOI] [PubMed] [Google Scholar]
- Gebbink, M.F., O. Kranenburg, M. Poland, F.P. van Horck, B. Houssa, and W.H. Moolenaar. 1997. Identification of a novel, putative Rho-specific GDP/GTP exchange factor and a RhoA-binding protein: control of neuronal morphology. J. Cell Biol. 137:1603–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger, B., and A. Bershadsky. 2001. Assembly and mechanosensory function of focal contacts. Curr. Opin. Cell Biol. 13:584–592. [DOI] [PubMed] [Google Scholar]
- Hall, A. 2005. Rho GTPases and the control of cell behaviour. Biochem. Soc. Trans. 33:891–895. [DOI] [PubMed] [Google Scholar]
- Holinstat, M., N. Knezevic, M. Broman, A.M. Samarel, A.B. Malik, and D. Mehta. 2006. Suppression of RhoA activity by focal adhesion kinase-induced activation of p190RhoGAP: role in regulation of endothelial permeability. J. Biol. Chem. 281:2296–2305. [DOI] [PubMed] [Google Scholar]
- Hsia, D.A., S.T. Lim, J.A. Bernard-Trifilo, S.K. Mitra, S. Tanaka, J. den Hertog, D.N. Streblow, D. Ilic, M.H. Ginsberg, and D.D. Schlaepfer. 2005. Integrin {alpha }4{beta }1 promotes focal adhesion kinase-independent cell motility via {alpha }4 cytoplasmic domain-specific activation of c-Src. Mol. Cell. Biol. 25:9700–9712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilic, D., Y. Furuta, S. Kanazawa, N. Takeda, K. Sobue, N. Nakatsuji, S. Nomura, J. Fujimoto, M. Okada, T. Yamamoto, and S. Aizawa. 1995. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 377:539–544. [DOI] [PubMed] [Google Scholar]
- Kirchner, J., Z. Kam, G. Tzur, A.D. Bershadsky, and B. Geiger. 2003. Live-cell monitoring of tyrosine phosphorylation in focal adhesions following microtubule disruption. J. Cell Sci. 116:975–986. [DOI] [PubMed] [Google Scholar]
- Krendel, M., F.T. Zenke, and G.M. Bokoch. 2002. Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat. Cell Biol. 4:294–301. [DOI] [PubMed] [Google Scholar]
- Lee, J.R., Y.J. Ha, and H.J. Kim. 2003. Cutting edge: induced expression of a RhoA-specific guanine nucleotide exchange factor, p190RhoGEF, following CD40 stimulation and WEHI 231 B cell activation. J. Immunol. 170:19–23. [DOI] [PubMed] [Google Scholar]
- Lin, H., J. Zhai, and W.W. Schlaepfer. 2005. RNA-binding protein is involved in aggregation of light neurofilament protein and is implicated in the pathogenesis of motor neuron degeneration. Hum. Mol. Genet. 14:3643–3659. [DOI] [PubMed] [Google Scholar]
- Medley, Q.G., E.G. Buchbinder, K. Tachibana, H. Ngo, C. Serra-Pages, and M. Streuli. 2003. Signaling between focal adhesion kinase and trio. J. Biol. Chem. 278:13265–13270. [DOI] [PubMed] [Google Scholar]
- Mitra, S.K., and D.D. Schlaepfer. 2006. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 18:516–523. [DOI] [PubMed] [Google Scholar]
- Mitra, S.K., D.A. Hanson, and D.D. Schlaepfer. 2005. Focal adhesion kinase: in command and control of cell motility. Nat. Rev. Mol. Cell Biol. 6:56–68. [DOI] [PubMed] [Google Scholar]
- Mitra, S.K., S.T. Lim, A. Chi, and D.D. Schlaepfer. 2006. a. Intrinsic focal adhesion kinase activity controls orthotopic breast carcinoma metastasis via the regulation of urokinase plasminogen activator expression in a syngeneic tumor model. Oncogene. 25:4429–4440. [DOI] [PubMed] [Google Scholar]
- Mitra, S.K., D. Mikolon, J.E. Molina, D.A. Hsia, D.A. Hanson, A. Chi, S.T. Lim, J.A. Bernard-Trifilo, D. Ilic, D.G. Stupack, et al. 2006. b. Intrinsic FAK activity and Y925 phosphorylation facilitate an angiogenic switch in tumors. Oncogene. 25:5969–5984. [DOI] [PubMed] [Google Scholar]
- Moissoglu, K., and M.A. Schwartz. 2006. Integrin signalling in directed cell migration. Biol. Cell. 98:547–555. [DOI] [PubMed] [Google Scholar]
- Okigaki, M., C. Davis, M. Falasca, S. Harroch, D.P. Felsenfeld, M.P. Sheetz, and J. Schlessinger. 2003. Pyk2 regulates multiple signaling events crucial for macrophage morphology and migration. Proc. Natl. Acad. Sci. USA. 100:10740–10745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen, J.D., P.J. Ruest, D.W. Fry, and S.K. Hanks. 1999. Induced focal adhesion kinase (FAK) expression in FAK-null cells enhances cell spreading and migration requiring both auto- and activation loop phosphorylation sites and inhibits adhesion-dependent tyrosine phosphorylation of Pyk2. Mol. Cell. Biol. 19:4806–4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirone, D.M., W.F. Liu, S.A. Ruiz, L. Gao, S. Raghavan, C.A. Lemmon, L.H. Romer, and C.S. Chen. 2006. An inhibitory role for FAK in regulating proliferation: a link between limited adhesion and RhoA-ROCK signaling. J. Cell Biol. 174:277–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, X.D., W.B. Kiosses, and M.A. Schwartz. 1999. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 18:578–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, X.D., W.B. Kiosses, D.J. Sieg, C.A. Otey, D.D. Schlaepfer, and M.A. Schwartz. 2000. Focal adhesion kinase suppresses Rho activity to promote focal adhesion turnover. J. Cell Sci. 113:3673–3678. [DOI] [PubMed] [Google Scholar]
- Ren, Y., R. Li, Y. Zheng, and H. Busch. 1998. Cloning and characterization of GEF-H1, a microtubule-associated guanine nucleotide exchange factor for Rac and Rho GTPases. J. Biol. Chem. 273:34954–34960. [DOI] [PubMed] [Google Scholar]
- Rico, B., H.E. Beggs, D. Schahin-Reed, N. Kimes, A. Schmidt, and L.F. Reichardt. 2004. Control of axonal branching and synapse formation by focal adhesion kinase. Nat. Neurosci. 7:1059–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley, A.J., M.A. Schwartz, K. Burridge, R.A. Firtel, M.H. Ginsberg, G. Borisy, J.T. Parsons, and A.R. Horwitz. 2003. Cell migration: integrating signals from front to back. Science. 302:1704–1709. [DOI] [PubMed] [Google Scholar]
- Romer, L.H., K.G. Birukov, and J.G. Garcia. 2006. Focal adhesions: paradigm for a signaling nexus. Circ. Res. 98:606–616. [DOI] [PubMed] [Google Scholar]
- Rossman, K.L., C.J. Der, and J. Sondek. 2005. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 6:167–180. [DOI] [PubMed] [Google Scholar]
- Rubinson, D.A., C.P. Dillon, A.V. Kwiatkowski, C. Sievers, L. Yang, J. Kopinja, D.L. Rooney, M.M. Ihrig, M.T. McManus, F.B. Gertler, et al. 2003. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat. Genet. 33:401–406. [DOI] [PubMed] [Google Scholar]
- Schiller, M.R. 2006. Coupling receptor tyrosine kinases to Rho GTPases–GEFs what's the link. Cell. Signal. 18:1834–1843. [DOI] [PubMed] [Google Scholar]
- Schober, M., S. Raghavan, M. Nikolova, L. Polak, H.A. Pasolli, H.E. Beggs, L.F. Reichardt, and E. Fuchs. 2007. Focal adhesion kinase modulates tension signaling to control actin and focal adhesion dynamics. J. Cell Biol. 176:667–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieg, D.J., D. Ilic, K.C. Jones, C.H. Damsky, T. Hunter, and D.D. Schlaepfer. 1998. Pyk2 and Src-family protein-tyrosine kinases compensate for the loss of FAK in fibronectin-stimulated signaling events but Pyk2 does not fully function to enhance FAK− cell migration. EMBO J. 17:5933–5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieg, D.J., C.R. Hauck, and D.D. Schlaepfer. 1999. Required role of focal adhesion kinase (FAK) for integrin-stimulated cell migration. J. Cell Sci. 112:2677–2691. [DOI] [PubMed] [Google Scholar]
- Tilghman, R.W., J.K. Slack-Davis, N. Sergina, K.H. Martin, M. Iwanicki, E.D. Hershey, H.E. Beggs, L.F. Reichardt, and J.T. Parsons. 2005. Focal adhesion kinase is required for the spatial organization of the leading edge in migrating cells. J. Cell Sci. 118:2613–2623. [DOI] [PubMed] [Google Scholar]
- van Horck, F.P., M.R. Ahmadian, L.C. Haeusler, W.H. Moolenaar, and O. Kranenburg. 2001. Characterization of p190RhoGEF, a RhoA-specific guanine nucleotide exchange factor that interacts with microtubules. J. Biol. Chem. 276:4948–4956. [DOI] [PubMed] [Google Scholar]
- Vicente-Manzanares, M., D.J. Webb, and A.R. Horwitz. 2005. Cell migration at a glance. J. Cell Sci. 118:4917–4919. [DOI] [PubMed] [Google Scholar]
- Webb, D.J., C.M. Brown, and A.F. Horwitz. 2003. Illuminating adhesion complexes in migrating cells: moving toward a bright future. Curr. Opin. Cell Biol. 15:614–620. [DOI] [PubMed] [Google Scholar]
- Webb, D.J., K. Donais, L.A. Whitmore, S.M. Thomas, C.E. Turner, J.T. Parsons, and A.F. Horwitz. 2004. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 6:154–161. [DOI] [PubMed] [Google Scholar]
- Wehrle-Haller, B., and B. Imhof. 2002. The inner lives of focal adhesions. Trends Cell Biol. 12:382–389. [DOI] [PubMed] [Google Scholar]
- Wu, J., J. Zhai, H. Lin, Z. Nie, W.W. Ge, L. Garcia-Bermejo, R.J. Muschel, W.W. Schlaepfer, and R. Canete-Soler. 2003. Cytoplasmic retention sites in p190RhoGEF confer anti-apoptotic activity to an EGFP-tagged protein. Brain Res. Mol. Brain Res. 117:27–38. [DOI] [PubMed] [Google Scholar]
- Zaidel-Bar, R., C. Ballestrem, Z. Kam, and B. Geiger. 2003. Early molecular events in the assembly of matrix adhesions at the leading edge of migrating cells. J. Cell Sci. 116:4605–4613. [DOI] [PubMed] [Google Scholar]
- Zaidel-Bar, R., M. Cohen, L. Addadi, and B. Geiger. 2004. Hierarchical assembly of cell-matrix adhesion complexes. Biochem. Soc. Trans. 32:416–420. [DOI] [PubMed] [Google Scholar]
- Zaidel-Bar, R., R. Milo, Z. Kam, and B. Geiger. 2007. A paxillin tyrosine phosphorylation switch regulates the assembly and form of cell-matrix adhesions. J. Cell Sci. 120:137–148. [DOI] [PubMed] [Google Scholar]
- Zhai, J., H. Lin, Z. Nie, J. Wu, R. Canete-Soler, W.W. Schlaepfer, and D.D. Schlaepfer. 2003. Direct interaction of focal adhesion kinase with p190RhoGEF. J. Biol. Chem. 278:24865–24873. [DOI] [PubMed] [Google Scholar]
- Zhao, J., Z.C. Bian, K. Yee, B.P. Chen, S. Chien, and J.L. Guan. 2003. Identification of transcription factor KLF8 as a downstream target of focal adhesion kinase in its regulation of cyclin D1 and cell cycle progression. Mol. Cell. 11:1503–1515. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}