

Abstract
Voltage-gated Nav channels are required for normal electrical activity in neurons, skeletal muscle, and cardiomyocytes. In the heart, Nav1.5 is the predominant Nav channel, and Nav1.5-dependent activity regulates rapid upstroke of the cardiac action potential. Nav1.5 activity requires precise localization at specialized cardiomyocyte membrane domains. However, the molecular mechanisms underlying Nav channel trafficking in the heart are unknown. In this paper, we demonstrate that ankyrin-G is required for Nav1.5 targeting in the heart. Cardiomyocytes with reduced ankyrin-G display reduced Nav1.5 expression, abnormal Nav1.5 membrane targeting, and reduced Na+ channel current density. We define the structural requirements on ankyrin-G for Nav1.5 interactions and demonstrate that loss of Nav1.5 targeting is caused by the loss of direct Nav1.5–ankyrin-G interaction. These data are the first report of a cellular pathway required for Nav channel trafficking in the heart and suggest that ankyrin-G is critical for cardiac depolarization and Nav channel organization in multiple excitable tissues.
Introduction
Voltage-gated sodium channels (Nav) are responsible for the normal electrical activity of excitable cells, including neurons, skeletal muscle, and cardiomyocytes (Burgess et al., 1995; Cannon, 1996; Escayg et al., 2000; Keating and Sanguinetti, 2001; Lossin et al., 2002; Papadatos et al., 2002; Yu et al., 2005). Nav1.5 (encoded by SCN5A) is the principal Nav channel in vertebrate heart. Nav1.5-dependent activity underlies the rapid upstroke of the cardiac action potential (Keating and Sanguinetti, 2001). Nav1.5 activity depends on precise biophysical properties that regulate inward Na+ current in response to cardiomyocyte membrane potential. Human SCN5A variants that affect biophysical properties of Nav1.5 are associated with several potentially fatal arrhythmias, including type 3 long QT syndrome (LQT3; variants increase inward Na+ current) and Brugada Syndrome (variants reduce inward Na+ current; Priori, 2004; Shah et al., 2005).
In addition to normal biophysical properties, Nav1.5 activity requires proper localization at specialized cardiomyocyte membrane domains. In the vertebrate heart, Nav1.5 is primarily concentrated at the cardiomyocyte intercalated disc (Kucera et al., 2002; Maier et al., 2002; Mohler et al., 2004b). However, Nav1.5 localization at the peripheral sarcolemma and T tubules has also been described previously (Cohen, 1996; Scriven et al., 2000, 2002; Kucera et al., 2002). The molecular mechanisms underlying the targeting and retention of Nav1.5 in cardiac tissue are unknown. Over the past decade, several potential Nav1.5 intermolecular interactions have been hypothesized to regulate Nav1.5 membrane trafficking, stability, and removal (Herfst et al., 2004; Abriel and Kass, 2005). However, the roles for these protein interactions in the context of a cardiomyocyte are unclear.
Ankyrin-G is an adaptor protein required for the targeting of diverse membrane proteins in the central nervous system (Mohler and Bennett, 2005b). Specifically, ankyrin-G is required for the targeting of Nav channel isoforms (Nav1.2 and Nav1.6) to specialized excitable membrane domains (axon initial segments and nodes of Ranvier) in Purkinje and granule cell neurons (Zhou et al., 1998; Jenkins and Bennett, 2001; Garrido et al., 2003; Lemaillet et al., 2003). Mice lacking ankyrin-G expression in the cerebellum display a loss of Nav channel targeting, abnormal neuronal action potentials, and ataxia (Zhou et al., 1998; Jenkins and Bennett, 2001). In 2003, two independent groups identified a small binding motif in the Nav1.2 DII–DIII cytoplasmic loop required for ankyrin-G association (Garrido et al., 2003; Lemaillet et al., 2003).
Our group recently identified 190-kD ankyrin-G expression in the heart (Mohler et al., 2004b). Like Nav1.5, ankyrin-G is expressed at the intercalated disc and transverse tubules and associates with Nav1.5 in coimmunoprecipitation and in vitro binding experiments (Mohler et al., 2004b). The nine–amino acid ankyrin-binding sequence identified in Nav1.2 (Garrido et al., 2003; Lemaillet et al., 2003) is present in Nav1.5 and is required for Nav1.5–ankyrin-G interaction (Mohler et al., 2004b). A human Brugada Syndrome SCN5A variant in the Nav1.5 ankyrin-G–binding motif (E1053K) abolishes ankyrin-G–Nav1.5 interaction, and this Nav channel mutant is not efficiently targeted to cardiomyocyte membranes (Mohler et al., 2004b). Although largely circumstantial, these data support a potential role for an ankyrin-G–dependent pathway in Nav1.5 targeting to excitable membrane domains in the heart.
In this study, we report new data that conclusively links ankyrin-G activity and Nav1.5 membrane expression and localization in cardiomyocytes. Using viral-mediated small hairpin RNA (shRNA) transfer into primary cardiomyocytes, we demonstrate that a full complement of ankyrin-G expression is required for Nav1.5 expression and membrane localization. Specifically, reduced ankyrin-G expression decreases (1) total cellular Nav1.5 expression, (2) efficient membrane localization, and (3) total Na+ membrane current. We also demonstrate that although ankyrin-G is required for normal Nav1.5 membrane expression, reduced ankyrin-G expression does not affect Nav1.5 channel kinetics. Finally, we report the structural requirements for direct ankyrin-G–Nav1.5 interactions and show that direct intermolecular interaction between these two molecules is required for efficient channel membrane localization. Together, these data identify the first clear cellular pathway for Nav channel trafficking in the heart.
Results
Generation of cardiomyocytes lacking ankyrin-G
We developed rat cardiomyocyte primary cultures with reduced ankyrin-G expression using lentiviral-mediated delivery of shRNA. The shRNA was designed to target all identified ankyrin-G mRNAs and was specific for rodent ankyrin-G (versus human; Fig. 1, A and B). We modified the lentiviral construct to encode YFP to enable the identification of transduced cells. A human-specific ankyrin-G shRNA was generated for control experiments (Fig. 1 A).
Figure 1.

Species-specific knockdown of 190-kD ankyrin-G in rat myocytes using lentiviral shRNA. (A) Domain organization of 190-kD ankyrin-G with the site of shRNA target. Rat, mouse, and human shRNA nucleotide target sequences. Note that the human 190-kD ankyrin-G target sequence has three unique nucleotides in wobble base positions (asterisks) that render this target sequence resistant to the rat shRNA. (B) The 21-nucleotide target sequence (sense) is separated by a short loop spacer sequence followed by 21 nucleotides that form the reverse complement of the target sequence. (C) Rat and human species–specific shRNAs reduce 190-kD ankyrin-G expression. Rat myocytes or human HEK293 cells were transduced with control virus, rat ankyrin-G shRNA, or human ankyrin-G shRNA. Equal quantities of protein were analyzed by immunoblotting using affinity-purified ankyrin-G Ig or an antibody to an unrelated protein, NHERF1 (loading control). Note that the rat-specific ankyrin-G shRNA effectively reduces the expression of 190-kD ankyrin-G in rat cardiomyocytes. Moreover, the expression of 190-kD ankyrin-G is reduced in HEK293 cells transduced with human-specific ankyrin-G shRNA. (D) 190-kD ankyrin-G protein levels from whole cell lysates of rat cardiomyocytes transduced with control virus, human-specific ankyrin-G shRNA virus (hAnkG shRNA), or rat-specific ankyrin-G shRNA virus (rAnkG shRNA) were analyzed by immunoblotting and quantitated. Numbers represent the mean ± SD (error bars) from four independent experiments. *, P < 0.05.
Rodent-specific ankyrin-G shRNA is efficient for reducing ankyrin-G expression and does not interfere with human ankyrin-G expression (Fig. 1). Introduction of rodent-specific ankyrin-G shRNA into primary rat cardiomyocytes significantly reduced ankyrin-G protein expression (Fig. 1, C and D). Likewise, human-specific ankyrin-G shRNA reduced ankyrin-G expression in human-derived HEK293 cells (Fig. 1 C). Finally, no significant decrease in ankyrin-G levels was observed in rat cardiomyocytes transduced with human-specific ankyrin-G shRNA (Fig. 1 D). Equivalent viral expression was observed in each cell system (assessed by positive YFP fluorescence).
Ankyrin-G is required for normal Nav1.5 expression and localization
Rat cardiomyocytes lacking a full complement of ankyrin-G expression were used to test the role of ankyrin-G for Nav1.5 expression. As shown in Fig. 1, viral transduction of ankyrin-G shRNA significantly reduces ankyrin-G expression (transduction efficiency of 80–95% based on YFP fluorescence; Fig. 2). Moreover, a striking reduction in Nav1.5 protein levels was observed in the identical cell lysates from rat-specific ankyrin-G shRNA virally transduced myocytes (Fig. 2). This reduction was specific to Nav1.5, as expression differences in cardiomyocyte membrane–associated proteins, including NHERF1, connexin43, ankyrin-B, Cav1.2 (Fig. 2), SERCA2, Na/K ATPase, or Na/Ca exchanger (not depicted), were not observed.
Figure 2.
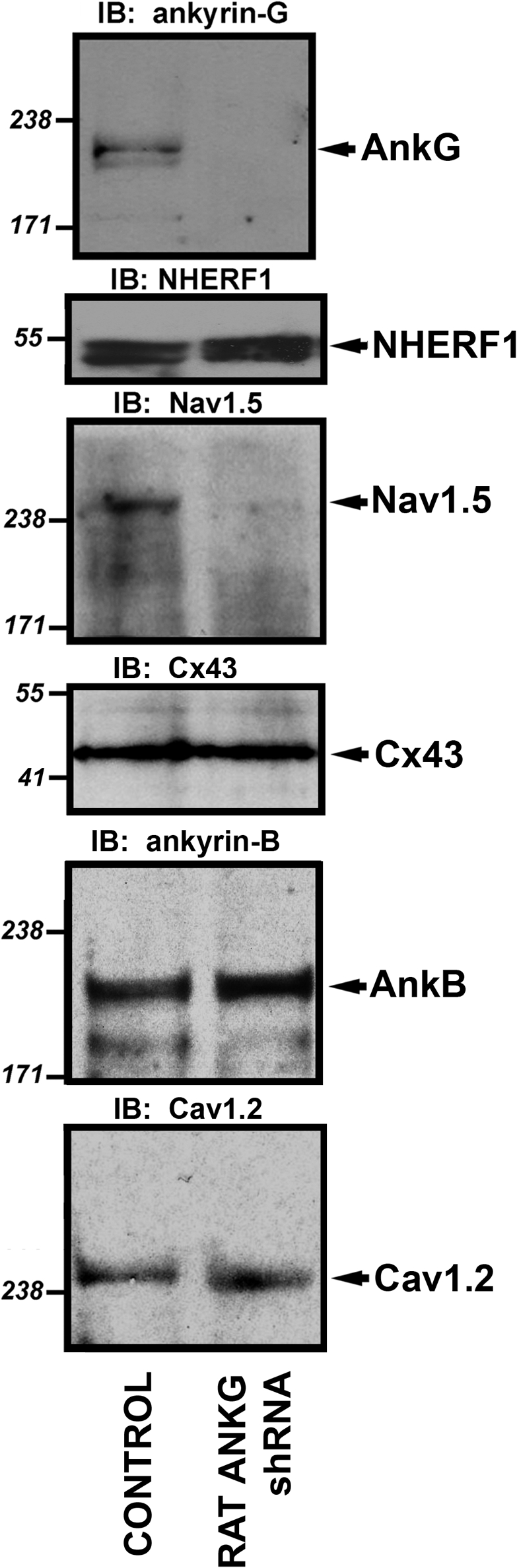
Nav1.5 expression is reduced in rat myocytes with reduced ankyrin-G expression. Neonatal cardiomyocytes were transduced with rat-specific ankyrin-G shRNA virus. After 22 h, myocytes were collected, and whole cell lysates were generated. Equal protein concentrations were analyzed by immunoblot using affinity-purified ankyrin-G Ig and Nav1.5-specific antibody. In parallel, blots were probed with an unrelated Ig to ensure equal protein loading (NHERF1). Note that loss of 190-kD ankyrin-G expression in myocytes transduced with rat-specific ankyrin-G shRNA was paralleled by a significant reduction in Nav1.5 expression. Reduced ankyrin-G expression did not affect the expression of ankyrin-B, connexin43, or Cav1.2. Molecular masses are expressed in kilodaltons.
The role of ankyrin-G on single cardiomyocyte Nav1.5 expression was examined using immunofluorescence and confocal microscopy. In agreement with immunoblot data of cardiomyocyte populations (Fig. 2), reduced ankyrin-G expression resulted in a dramatic loss of Nav1.5 cellular expression (Fig. 3 C) compared with nontransduced (Fig. 3 A), YFP-transduced (Fig. 3 B), and human-specific ankyrin-G shRNA–transduced myocytes (Fig. 3 D). Specifically, Nav1.5 expression appeared to only remain in the perinuclear region of cardiomyocytes lacking ankyrin-G expression (Fig. 3 C). Loss of protein localization was limited to Nav1.5, as cardiomyocytes transduced with ankyrin-G shRNA displayed no difference in the expression or localization of either Cav1.2 or Na/Ca exchanger (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200710107/DC1).
Figure 3.

Nav1.5 expression is reduced in single rat myocytes with reduced ankyrin-G expression. (A–D) Control myocytes (uninfected; A) and cardiomyocytes transduced with control virus (YFP alone; B), rat-specific ankyrin-G shRNA virus (C), or human-specific ankyrin-G shRNA virus (D) were immunolabeled with ankyrin-G and Nav1.5-specific antibodies and imaged using identical confocal settings. Viral transduction was assessed by the presence of YFP fluorescence (pseudocolored in blue in B–D for clarity). Note that only myocytes infected with rat-specific ankyrin-G shRNA virus displayed reduced ankyrin-G expression. These myocytes consistently displayed a decreased expression of Nav1.5. In fact, Nav1.5 expression in myocytes with reduced ankyrin-G expression (arrowheads in C) was limited to the perinuclear region (arrows in C). Bars, 10 μm.
Ankyrin-B is not required for Nav1.5 expression in cardiomyocytes
Ankyrin-B (encoded by ANK2) is required for the membrane targeting of Na/Ca exchanger, InsP3 receptor, and Na/K ATPase in cardiomyocytes (Mohler et al., 2002, 2003, 2005). Ankyrin-B and -G are structurally similar and share binding activity for protein partners, including β-spectrin and Na/K ATPase (Devarajan et al., 1994; Kizhatil and Bennett, 2004; Mohler et al., 2004d, 2005; Kizhatil et al., 2007). We used primary cardiomyocytes from ankyrin-B–null mice to test whether ankyrin-B expression is necessary for Nav1.5 membrane expression. Confocal analyses of wild-type, ankyrin-B+/−, and ankyrin-B–null cardiomyocytes revealed no difference in Nav1.5 immunolocalization (Fig. 4, A–C). Therefore, even in myocytes completely devoid of ankyrin-B expression, Nav1.5 was normally expressed and properly localized. These results demonstrate that ankyrin-G (not ankyrin-B) is the physiological binding partner for Nav1.5 in the heart.
Figure 4.

Nav1.5 is normally expressed in cardiomyocytes with reduced ankyrin-B expression. Immunolocalization of ankyrin-B and Nav1.5 in neonatal myocytes derived from wild-type (A), ankyrin-B+/− (B), and ankyrin-B−/− mice (C). Note that although ankyrin-B levels are reduced ∼50% in ankyrin-B+/− myocytes and nearly 100% in ankyrin-B−/− cardiomyocytes, there is no reduction in Nav1.5 expression/localization. Bars, 10 μm.
Ankyrin-G is required for normal Nav1.5-dependent Na+ current in cardiomyocytes
We made electrophysiological measurements to determine whether reduced ankyrin-G expression significantly affects Nav channel membrane current density in primary cardiomyocytes (Fig. 5). Wild-type rat cardiomyocytes and cardiomyocytes transduced with human- or rat-specific ankyrin-G shRNA virus were analyzed for sodium current using whole-cell patch clamp (see Fig. 5 D [inset] for protocol). Virally transduced cardiomyocytes were selected for electrophysiological measurements on the basis of positive YFP fluorescence. Cardiomyocytes infected with human-specific ankyrin-G shRNA virus displayed no change in peak Na+ current compared with control (nontransduced) myocytes (note the current traces in Fig. 5 [A and B] and the current-voltage relationship in Fig. 5 D). In contrast, myocytes transduced with rat-specific ankyrin-G shRNA virus displayed striking decreases in peak Na+ current density (note the traces in Fig. 5 C; currents normalized for cell capacitance). In fact, several transduced cells that completely lacked detectable Na+ current (data not included in statistical analyses for Figs. 5 and 6; see Materials and methods) were observed. These functional data strongly support a role for ankyrin-G in Nav1.5 membrane surface targeting in cardiomyocytes.
Figure 5.

Reduced sodium current amplitude and current-voltage kinetics in myocytes with reduced ankyrin-G expression. Nontransduced cardiomyocytes (control) and cardiomyocytes transduced with human- (hAnkG) or rat-specific (rAnkG) shRNA virus were analyzed for Nav1.5 current. (A–C) Whole-cell patch clamp sodium current traces elicited control (A; 17 pF), human ankyrin-G–specific shRNA–transduced cells (B; 18 pF), and rat-specific ankyrin-G shRNA–transduced cells (C; 16.8 pF). All cardiomyocytes displayed similar cell capacitance, and all traces are normalized for cell capacitance. (D) Mean and normalized current-voltage relationship for neonatal rat ventricular cardiomyocytes treated with control virus (black squares; n = 10), human ankyrin-G shRNA virus (gray circles; n = 10), and rat ankyrin-G shRNA virus (white squares; n = 10). (E) Normalized maximum sodium current amplitude in myocytes treated with control (black bar) and rat- (white bar) and human (gray bar)-specific ankyrin-G shRNA virus. *, P < 0.05. (F and G) Reduced ankyrin-G expression specifically affects cardiomyocyte sodium current. (F) Amplitude of Na+ and Ca2+ current in nontransduced (control) and rat neonatal cardiomyocytes transduced with ankyrin-G species-specific shRNA viruses. Currents are elicited with the protocol shown in the inset (see Materials and methods for details). Left panels depict Na+ and Ca2+ current in cardiomyocytes treated with control or human ankyrin-G–specific shRNA virus (hAnkG shRNA). Right panels display the significant decrease in INa, but not ICa, in cardiomyocytes transduced with rat-specific ankyrin-G shRNA virus (rAnkG shRNA). All traces are normalized for cell capacitance. (G) Reduced ankyrin-G expression leads to reduced INa but does not affect cardiomyocyte ICa. (E and G) Data are plotted as mean ± SEM (error bars; n = 10). Statistical difference was analyzed by analysis of variance.
Figure 6.

Reduced ankyrin-G expression does not affect Nav1.5 inactivation in primary cardiomyocytes. Superimposed Nav1.5 inactivation curves obtained from neonatal rat cardiomyocytes treated with control (black squares) and species-specific (human, gray circles; rat, white squares) ankyrin-G shRNA viruses. The normalized INa plotted against preconditioning pulse potential was fitted using a Boltzmann equation. Voltage-dependent steady-state inactivation was determined using a paired two-pulse protocol. Each conditioning voltage is paired with a control after 1.5 s. A 500-ms conditioning pulse from −120 to 20 mV in 10-mV increments was followed by a test pulse to −30 mV. The test pulse in each series is separated from the conditioning pulse by a 2-ms interval to −120 mV. Each point (INa) is normalized against the amplitude of corresponding control pulse (INamax). Each point represents mean ± SEM (error bars; n = 5).
To determine the specificity of the ankyrin-G–based cellular pathway for ion channel membrane targeting in cardiomyocytes, we evaluated the effect of reduced ankyrin-G expression on a second critical cardiomyocyte membrane current. Cav1.2-dependent calcium current (ICa) is required for normal cardiac excitability, and human Cav1.2 gene variants are associated with ventricular arrhythmia (Splawski et al., 2004). As expected, myocytes with reduced ankyrin-G expression displayed significant reduction in peak Na+ current amplitude (Fig. 5, F and G). However, these identical myocytes displayed peak calcium current (ICa) measurements that were unchanged from those observed in control (nontransduced) or human-specific ankyrin-G shRNA virally transduced cardiomyocytes (see Materials and methods for protocol details; Fig. 5, F and G). These data (combined with Nav1.5/Cav1.2 immunoblot and immunostaining data in Figs. 2–4 and S1) establish that the cardiac ankyrin-G–targeting pathway is specific for Nav1.5 channels and that loss of Nav1.5 membrane expression in ankyrin-G–deficient cells is not simply the result of the general impairment of default myocyte membrane biogenesis/trafficking pathways.
Nav1.5 channel inactivation is unaffected in cardiomyocytes with reduced ankyrin-G expression
Human Nav1.5 Brugada Syndrome variant E1053K, which lacks ankyrin-G–binding activity, is not efficiently targeted to the cardiomyocyte intercalated disc (Mohler et al., 2004b). However, when introduced into heterologous HEK293 cells, this mutant channel is present at the plasma membrane but displays abnormalities in Nav1.5 inactivation (Mohler et al., 2004b). Furthermore, similar data examining Nav1.2 biophysical properties in TsA201 cells support the concept that ankyrin-G could potentially regulate the biophysical properties of Nav channels in heterologous cells (Shirahata et al., 2006). These studies did not determine whether the inactivation of wild-type Nav1.5 channels requires correct localization through ankyrin-G. Therefore, we tested whether primary cardiomyocytes with reduced ankyrin-G expression displayed abnormalities in Nav channel inactivation. No significant difference in Nav1.5 channel inactivation was observed in wild-type cardiomyocytes versus cardiomyocytes with reduced ankyrin-G expression (Fig. 6). These data demonstrate that in the physiological context of a cardiomyocyte, normal Nav channel inactivation does not require ankyrin-G. Surprisingly, inactivation of wild-type cardiomyocyte Nav1.5 is independent of physiological localization. Thus, localization controls current density but not Nav1.5 inactivation. These data strongly demonstrate the critical nature of studying ankyrin biology in the appropriate physiological context.
Direct interaction between ankyrin-G and Nav1.5 requires ANK repeat 14–15 β-hairpin loop tips
We identified the structural requirements on ankyrin-G for direct Nav1.5 interaction using a series of ankyrin-G ANK repeat mutants (Fig. 7, A–C). These mutants were created based on the identification of exposed, highly accessible residues on the β-hairpin loop tips of the ankyrin-R membrane–binding domain (repeats 13–24) crystal structure (Michaely et al., 2002) as well as the identification of membrane–protein binding sites at these sites in ankyrin-B (Mohler et al., 2004a; Cunha et al., 2007). Specifically, each of the 24 consecutive ANK repeats of an ankyrin membrane–binding domain fold as antiparallel pairs of α helices connected by a β-hairpin loop. The large number of consecutive ANK repeats come together to form a super helix that surrounds a large central cavity (Michaely et al., 2002). Thus, these variable β-hairpin loops are highly accessible to the solvent for potential protein interactions (Fig. 7 C). This strategy has been successfully used to map the binding sites on ankyrin-B for Na/Ca exchanger and InsP3 receptor (Mohler et al., 2004a; Cunha et al., 2007).
Figure 7.

Direct interaction of ankyrin-G with Nav1.5 requires two ANK repeat β-hairpin loop tips on the ankyrin-G membrane–binding domain. (A) 190-kD ankyrin-G includes a membrane-binding domain comprised of 24 consecutive ANK repeats (green), a spectrin-binding domain (black), a death domain (blue), and a C-terminal domain (red). (B) ANK repeat mutants were engineered in the context of full-length GFP 190-kD ankyrin-G and display alanine substitutions for the two residues located at the tip of each ANK repeat β-hairpin loop (red arrowheads in B and purple sites in C). (C) Crystal structure diagram of membrane-binding domain ANK repeats 13–24 (Michaely et al., 2002). Exposed charged residues on β-hairpin loop tips (sites of alanine mutagenesis) are colored in purple. (D) Relative binding (compared with wild-type GFP 190-kD ankyrin-G) of GFP 190-kD ankyrin-G ANK repeat mutants with purified Nav1.5 DII–DIII cytoplasmic domain (n = 3; *, P < 0.05). Binding levels are corrected for the relative expression of each GFP–ankyrin-G mutant. Error bars represent SEM. (E) Nav1.5 binding sites (arrows) superimposed on the deduced crystal structure of the ankyrin-G membrane–binding domain (ANK repeats 13–24). Ankyrin-G membrane–binding domain structure is based on the crystal structure of ANK repeats 13–24 of ankyrin-R (Michaely et al., 2002).
24 individual ankyrin-G ANK repeat mutants were generated for binding analyses. Each ankyrin-G mutant harbored two alanine substitutions in the variable β-hairpin loop tips connecting each pair of ANK repeats within the membrane-binding domain (ANK repeats 1–24; Fig. 7, B and C). Mutagenesis was performed in the context of GFP-tagged ankyrin-G (Mohler et al., 2002). Mutant plasmids were completely sequenced to verify that no additional mutations were introduced during PCR amplification. Wild-type ankyrin-G and each GFP ankyrin-G mutant were expressed in HEK293 cells. Expressed proteins were immunopurified from the detergent-soluble fraction of the cells using immobilized affinity-purified GFP Ig and were incubated with purified His-tagged Nav1.5 DII–DIII cytoplasmic loop (site for ankyrin-G association on Nav1.5; Mohler et al., 2004b). The quantity of pure Nav1.5 DII–DIII bound to each mutant was determined by immunoblotting using a His tag Ig. Using similar methods, the relative level of GFP–ankyrin-G mutant expression for each data point was determined. The amount of Nav1.5 His-tagged DII–DIII bound to each ankyrin-G mutant was corrected for relative GFP–ankyrin-G expression in each experimental sample.
Although the majority of ankyrin-G mutants associated with Nav1.5 His-tagged DII–DIII at levels similar to wild-type GFP–ankyrin-G, there was a significant reduction in DII–DIII binding to ankyrin-G mutants R13, R14, and R15 (Fig. 7, D and E). In fact, GFP–ankyrin-G mutants R14 and R15 displayed a near complete loss of Nav1.5 DII–DIII binding activity in these assays. Together, our findings demonstrate that ankyrin-G–Nav1.5 binding is exclusively dependent on three critical elements (two β-hairpin loops on ankyrin-G and a nine–amino acid motif on Nav1.5; Mohler et al., 2004b).
Human ankyrin-G expression rescues abnormal Nav1.5 localization
We performed rescue assays to determine whether the loss of Nav1.5 localization in cardiomyocytes with reduced ankyrin-G expression was specifically caused by the monogenic loss of ankyrin-G. Rat neonatal cardiomyocytes with reduced levels of ankyrin-G and Nav1.5 (as a result of the presence of rat-specific ankyrin-G shRNA virus) were transfected with cDNA encoding human GFP–ankyrin-G. This GFP–ankyrin-G cDNA is resistant to the rat ankyrin-G shRNA (Fig. 1). As expected, shRNA-transduced cardiomyocytes displayed decreased expression and abnormal localization of Nav1.5 (Fig. 8 B). In contrast, shRNA-transduced cardiomyocytes expressing human GFP–ankyrin-G cDNA displayed a Nav1.5 distribution similar to wild-type cardiomyocytes (Fig. 8 C). These data demonstrate that abnormal Nav1.5 localization in ankyrin-G–null cardiomyocytes can be rescued by the exogenous expression of human ankyrin-G and reinforce the critical role for ankyrin-G in the subcellular localization of Nav1.5 in cardiomyocytes.
Figure 8.

Nav1.5 targeting requires direct interaction with 190-kD ankyrin-G. (A and B) Immunolocalization of ankyrin-G and Nav1.5 in control (nontransduced) and rat ankyrin-G shRNA virally transduced neonatal cardiomyocytes. Note the localization of Nav1.5 in the perinuclear region of shRNA-transduced myocytes (white arrows). Yellow arrows denote remaining ankyrin-G staining in transduced myocytes, and asterisks mark sites of complete knockdown. Virally transduced myocytes in the figure are denoted by positive YFP fluorescence (pseudocolored in blue). (C) Cardiomyocytes expressing rat-specific ankyrin-G shRNA (note the blue color denoting YFP expression) were transfected with GFP-labeled human ankyrin-G cDNA and immunolabeled with Nav1.5 and ankyrin-G antibodies. Note that human GFP–ankyrin-G restores the localization of Nav1.5 to normal (compare Nav1.5 in B and C). (D and E) GFP-human ankyrin-G mutants (R14 and R15) that lack binding activity for Nav1.5 (see Fig. 6) are unable to rescue to normal the aberrant localization of Nav1.5 (note perinuclear distribution; arrows) in myocytes stably transduced with rat ankyrin-G shRNA (note positive YFP fluorescence). Bars, 10 μm.
Direct ankyrin-G–Nav1.5 interaction is required for Nav1.5 localization
We used the aforementioned ankyrin-G rescue assay with ankyrin-G mutants that lack Nav1.5-binding activity (Fig. 7) to determine whether direct ankyrin-G–Nav1.5 interactions are required for Nav1.5 localization in cardiomyocytes. Ankyrin-G mutants lacking Nav1.5-binding activity were introduced into full-length human GFP–ankyrin-G cDNA to create human GFP–ankyrin-G R14 and R15 (both resistant to rat ankyrin-G shRNA). Cardiomyocytes with reduced ankyrin-G expression (transduced with rat ankyrin-G YFP virus) were transfected with human GFP–ankyrin-G or human ankyrin-G R14 or R15 mutant cDNAs. YFP-positive cardiomyocytes (express shRNA) were fixed and immunostained. GFP–ankyrin-G is properly targeted in neonatal cardiomyocytes and rescues the normal expression of Nav1.5 (Fig. 8 C). In contrast, although both GFP–ankyrin-G R14 and GFP–ankyrin-G R15 were properly localized in transfected cardiomyocytes, there was no detectable rescue of Nav1.5 localization as observed with wild-type human GFP–ankyrin-G (Fig. 8 C). In fact, the localization of Nav1.5 in myocytes transfected with GFP–ankyrin-G R14–15 was similar to nontransfected myocytes lacking endogenous ankyrin-G expression (Fig. 8, B, D, and E). Therefore, direct association of ankyrin-G with Nav1.5 is required for the membrane expression of Nav1.5 in ventricular cardiomyocytes.
Ankyrin-G is required for normal Nav1.5 expression in adult cardiomyocytes
To test whether our findings in neonatal myocytes can also be applied to adult cardiomyocytes, we performed ankyrin-G shRNA viral transduction of freshly isolated adult rat cardiomyocytes. Nav1.5 expression is most pronounced at the intercalated disc of adult cardiomyocytes (see Nav1.5 localization in the control myocyte; Fig. S2 A, available at http://www.jcb.org/cgi/content/full/jcb.200710107/DC1). Therefore, we tested whether ankyrin-G expression is required for Nav1.5 localization at the mature cardiomyocyte intercalated disc. Reduced ankyrin-G expression in adult myocytes results in the reduced membrane expression of Nav1.5 (note the loss of Nav1.5 intercalated disc staining in Fig. S2 B), which is consistent with results in neonatal cardiomyocytes. In fact, identical with our findings in neonatal cells, we observed that Nav1.5 localized to the perinuclear region (Fig. S2 B) in adult cardiomyocytes with reduced ankyrin-G expression. These data from isolated adult cardiomyocytes further confirm the role of the ankyrin-G–based protein-targeting pathway for Nav1.5 membrane expression in the heart.
Discussion
In this study, we present the first report of a cellular pathway required for Nav channel trafficking in cardiomyocytes. We demonstrate that direct interaction of the ankyrin-G membrane–binding domain and Nav1.5 DII–DIII loop is necessary for Nav1.5 expression and localization at the cardiomyocyte membrane surface. Our new data provide compelling evidence that ankyrin-G–dependent targeting of Nav1.5 is a fundamental requirement for cardiomyocytes and likely other excitable cells.
Ankyrin polypeptides have likely coordinately evolved to regulate electrical activity in the heart by targeting key ion channels/transporters involved in controlling the cardiac action potential. Ankyrin-G directly associates with and targets Nav1.5 to the membrane surface (Fig. 9 A) to regulate inward Na+ current and, thus, action potential initiation and cardiomyocyte depolarization. Therefore, it is not surprising that human SCN5A variants that disrupt ankyrin-G interactions are associated with the Brugada Syndrome (Priori et al., 2002; Mohler et al., 2004b), a cardiac syndrome associated with reduced inward INa (for review see Napolitano and Priori, 2006). Clinical features of the Brugada Syndrome include fast polymorphic ventricular tachycardia typically occurring at rest or during sleep (Wilde and Priori, 2000). In contrast, ankyrin-B targets distinct ion channels and transporters with central roles in cytosolic calcium extrusion during cardiac repolarization (Mohler et al., 2002, 2003, 2004c, 2005, 2007; Cunha et al., 2007). Ankyrin-B (product of the ANK2 gene) is required for targeting Na/Ca exchanger, Na/K ATPase, and InsP3 receptor to the transverse tubule and sarcoplasmic reticulum in cardiomyocytes (Fig. 9 B; Mohler et al., 2003, 2005).
Figure 9.

Ankyrin-G and ankyrin-B ion channel/transporter complexes in the heart. (A) Ankyrin-G is required for the targeting of Nav1.5 to the cardiomyocyte intercalated disc. Although other Nav1.5-interacting proteins have been identified, it is not yet clear whether these are present in the ankyrin-G–dependent protein complex. (B) Ankyrin-B is required for the targeting of Na/Ca exchanger and Na/K ATPase to transverse tubule membranes in the heart. InsP3 receptor targeting to the sarcoplasmic reticulum membrane requires direct interaction with ankyrin-B. Cardiac ankyrin-B protein partners also include PP2A and β2-spectrin.
Dysfunction in ankyrin-B–based cellular pathways as a result of human ANK2 variants are associated with ankyrin-B syndrome, a cardiac arrhythmia with a host of phenotypes, including sinoatrial node dysfunction, atrial fibrillation, conduction defects, and ventricular tachycardia and/or sudden cardiac death (Mohler et al., 2003, 2004c, 2007). In contrast to Nav1.5-associated Brugada Syndrome, ventricular tachycardia and syncopal events in the ankyrin-B syndrome are most often associated with adrenergic stimulation (e.g., emotional stress and/or exercise, similar to catecholaminergic polymorphic arrhythmia; Mohler et al., 2003, 2004c, 2007; Liu et al., 2007).
In addition to targeting ion channels in cardiomyocytes, ankyrin-B was recently shown to be required for targeting of the regulatory subunit of the PP2A complex in primary cardiomyocytes (Fig. 9 B; Bhasin et al., 2007). This broad targeting role of ankyrin-B for both integral membrane and signaling proteins in the heart suggests that the ankyrin-G–targeting pathway may similarly facilitate the localization of additional myocyte proteins. Moreover, our new findings for ankyrin-G in Nav1.5 targeting identify an attractive, unconventional candidate disease gene for cardiac (arrhythmia and myopathy) and other excitable cell diseases.
Based on the role of ankyrin-R in the erythrocyte (Bennett and Stenbuck, 1979), cardiac ankyrin-G may simply act as a membrane scaffold to link integral membrane proteins (such as Nav1.5) with the underlying actin- and spectrin-based cytoskeleton. In support of this role, we observe a significant level of Nav1.5 clustering on the plasma membrane surface with ankyrin-G as assessed by immunoelectron microscopy of cardiomyocyte membrane sheets (Fig. 10). Alternatively, ankyrin-G may play an active role in the cellular trafficking of Nav1.5 to specific membrane domains. In theory, ankyrin-G could have multiple roles in both trafficking and stabilization/retention of Nav1.5 channels in the heart. Although these findings are beyond the scope of this study, elucidating the specific cellular roles of ankyrin polypeptides in excitable cells is an obvious future goal for the field.
Figure 10.

Nav1.5 is clustered at the cardiomyocyte membrane surface with ankyrin-G. Immunogold electron microscopy of adult rat ventricular cardiomyocyte plasma membrane sheets. Anti–ankyrin-G Ig particles (10 nm gold) and anti-Nav1.5 Ig particles (5 nm gold; arrows) are found in clusters at the plasma membrane. The majority of Nav1.5-positive gold particles were clustered in proximity (<10 nm) to ankyrin-G–positive gold particles (see A, B, and D–F). However, we also observed a small fraction of Nav1.5-positive particles that were physically isolated from ankyrin-G (C). Membrane sheets labeled with gold-conjugated secondary antibodies (negative control) were clear of any gold particles (not depicted). Bar, 50 nm.
Reduction of ankyrin-G in cardiomyocytes significantly affects the expression (>75%; Fig. 2) and cellular localization of Nav1.5 (Fig. 3). Although functional experiments on ankyrin-G shRNA myocytes reveal a striking reduction in INa (50–65% reduction), residual INa current was observed in these cells (Fig. 5). This current may represent residual Nav1.5 activity. Alternatively, this remaining current may reflect the combined activity of other Nav isoforms (Rogart et al., 1989; Baruscotti et al., 1997; Dhar Malhotra et al., 2001; Maier et al., 2002, 2003, 2004; Haufe et al., 2005). In theory, these Nav channel isoforms may be targeted to the membrane via an ankyrin-G–independent cellular pathway and, therefore, may be resistant to ankyrin-G knockdown. These interesting data form the foundation for future experiments to dissect the specificity of the ankyrin-G–based cellular pathway for Nav channel isoform trafficking in the heart.
Our electrophysiological measurements illustrate the importance of studying ankyrin–membrane protein interactions in the physiological context of a native cell. Our previous work suggested that Nav1.5 E1053K lacking ankyrin-G–binding activity displayed minor yet significant abnormalities in activation and inactivation when expressed in HEK293 cells (Mohler et al., 2004b). However, when expressed in native cardiomyocytes, these biophysical abnormalities were inconsequential, as the majority of the channel lacked sufficient targeting information to even reach the plasma membrane (Mohler et al., 2004b). Our new findings demonstrate that in myocytes lacking ankyrin-G, the small number of residual Nav1.5 channels that reach the plasma membrane have effectively normal biophysical characteristics. These findings are in contrast to a recent publication that demonstrated abnormal inactivation gating of Nav1.2 in TsA201 cells (modified HEK293 cell) lacking ankyrin-G (Shirahata et al., 2006). Our new data suggest that these measurements should be reevaluated in the context of a neuron.
The critical role of ion channels and transporters for normal cardiac function has been highlighted by the linkage of gene mutations in cardiac ion channels and associated subunits with human arrhythmia (Lehnart et al., 2007). Specifically, human gene variants in KCNQ1 (KvLQT1), KCNH2 (HERG), SCN5A (Nav1.5), KCNE1 (minK), KCNE2 (MiRP), KCNJ2 (Kir2.1), and CACNA1C (Cav1.2) have been associated with potentially fatal long QT arrhythmias (Lehnart et al., 2007). Most channel variants affect channel biophysics (Lehnart et al., 2007). However, recent findings suggest that dysfunction in channel trafficking mechanisms may explain a significant number of long QT variant cellular phenotypes (Zhou et al., 1999; Mohler et al., 2003; Ye et al., 2003; Gouas et al., 2004; Krumerman et al., 2004; Liu et al., 2005; Anderson et al., 2006; Ballester et al., 2006; Schmitt et al., 2007).
Cardiomyocytes have clearly evolved unique channel trafficking pathways for the precise localization of specific ion channels to unique cardiomyocyte membrane domains. For example, although Nav1.5, Kv4.2, and connexin 43 are concentrated at the intercalated disc (Kanter et al., 1992; Barry et al., 1995; Maier et al., 2002; Mohler et al., 2004b), Kir2.1, Kir2.3, and Cav1.2 are primarily localized to transverse tubule membranes (Carl et al., 1995; Sun et al., 1995;Clark et al., 2001; Melnyk et al., 2002). Moreover, significant populations of KvLQT1, ERG, minK, MiRP, Na/Ca exchanger, and Na/K ATPase are found at transverse tubule and peripheral sarcolemma membranes (Frank et al., 1992; Kieval et al., 1992; McDonough et al., 1996; Pond et al., 2000; Rasmussen et al., 2004; Wu et al., 2006). Finally, high resolution imaging techniques have revealed unique transverse tubule membrane domains that further segregate cardiac ion channel signaling complexes (Scriven et al., 2000). Clearly, the molecular and structural characteristics of cardiac membrane domains represent a central feature for the regulation of local ion channel pathways and represent a relatively unexplored field in cell biology.
Ankyrin-based ion channel trafficking pathways have unique roles in specific cells. Recently, Pan et al. (2006) demonstrated the requirement of ankyrin-G association for the targeting of KCNQ2 and KCNQ3 (encode axonal M currents responsible for stabilizing neuronal resting potential) to axon initial segments. Specifically, ankyrin-G cerebellar-specific null mice display loss of KCNQ2/3 clustering at axonal initial segments (Pan et al., 2006). Moreover, the ankyrin-G–binding motif originally described in Nav1.2 and Nav1.5 (Garrido et al., 2003; Lemaillet et al., 2003; Mohler et al., 2004b) is present in the C-terminal domain of KCNQ2/3 and is required for neuronal KCNQ2/3 targeting (Chung et al., 2006; Pan et al., 2006; Rasmussen et al., 2007). Interestingly, although nearly structurally identical, KCNQ1 (cardiac KvLQT1) lacks the C-terminal ankyrin-G–binding motif (Pan et al., 2006). Consistent with this data, ankyrin-G and KCNQ1 are differentially targeted in ventricular cardiomyocytes (Mohler et al., 2004b; Rasmussen et al., 2004). Therefore, these data strongly suggest that ankyrin-based pathways operate in a cell type–specific manner.
In summary, our new findings show that ankyrin-G is the physiological binding partner for Nav1.5 in cardiomyocytes. Loss of direct interaction between ankyrin-G and Nav1.5 results in abnormal Nav1.5 channel localization in primary cardiomyocytes. Moreover, the loss of ankyrin-G expression affects Nav1.5 expression, membrane localization, and, therefore, whole cell Nav1.5 activity. Based on recent ankyrin findings in the brain (Dubreuil, 2006; Pan et al., 2006; Yang et al., 2007), heart (Lencesova et al., 2004; Mohler and Bennett, 2005a), and skeletal muscle (Bagnato et al., 2003; Kontrogianni-Konstantopoulos et al., 2004, 2006), our new findings likely represent only the first component of a larger ankyrin-G macromolecular signaling complex at cardiomyocyte excitable membrane domains.
Materials and methods
shRNA targets/cDNA constructs
Ankyrin-G–specific shRNA targets were selected based on their unique sequence presence of flanking 5′ AA and 3′ TT nucleotides (Brummelkamp et al., 2002). Human- and rat-specific targets were conferred by three unique nucleotides located at wobble positions. Primers were created and ligated into a modified pFIV lentiviral vector (System Biosciences, Inc.). The vector was modified to contain YFP driven by an independent H1PGK promoter to track transduced cells. Feline immunodeficiency viruses were titered before transduction experiments. Ankyrin-G constructs were generated using 190-kD ankyrin-G cDNA. GFP 190-kD ankyrin-G mutant constructs were made using QuikChange Mutagenesis (Stratagene). Loop mutations were designed to replace ANK repeat β-hairpin loop tip residues with two alanines. Rescue constructs were made using GFP 190-kD ankyrin-G and ANK repeat mutants R14 and R15. All constructs were completely sequenced and confirmed to express protein in HEK293 cells.
Immunoblots
After viral and/or rescue treatment, cells were collected into PBS, pH 7.4. Cells were lysed using radioimmunoprecipitation assay buffer containing protease inhibitor cocktail. Detergent-soluble fractions were collected after high speed centrifugation, and protein concentrations were determined using a bicinchoninic acid protein assay kit (Thermo Fisher Scientific). Equal quantities of protein (20 μg) were analyzed by SDS-PAGE and immunoblotting. Equal loading was assessed by transfer to Ponceau S and blotting with NHERF1.
Neonatal rat cardiomyocytes
Hearts were dissected from postnatal day 1 rats and cultured as described previously (Bhasin et al., 2007).
Adult cardiomyocytes
Myocytes were isolated from 250–300 g of Sprague-Dawley rats (Grueter et al., 2006). Acutely isolated cardiomyocytes were then plated at high density (∼2 × 106 per well), infected with rat-specific ankyrin-G shRNA virus or control virus for 9 h, and maintained for 22 h at 37°C before experiments.
Binding experiments
GFP 190-kD ankyrin-G and mutants were expressed in HEK293 cells and purified using affinity-purified GFP Ig coupled to protein A agarose beads. Cells were lysed in homogenization buffer plus 1.0% Triton X-100 and 0.5% deoxycholate (Mohler et al., 2005). The extract was centrifuged at 100,000 g, and the supernatant was incubated with GFP Ig coupled to protein A–Sepharose. Nav1.5 DII–DIII cytoplasmic loop was subcloned into pET15b for expression and purification as a hexahistidine fusion protein. Purified proteins were incubated with GFP or control Ig coupled to protein A–Sepharose. Protein bound to each mutant GFP 190-kD ankyrin-G was eluted, analyzed by immunoblotting, normalized for relative GFP ankyrin-G expression, and compared with wild-type GFP 190-kD ankyrin-G binding.
Statistics
When appropriate, data were analyzed using a two-tailed t test, and P < 0.05 was considered significant. Values are expressed as the mean ± SD.
Virus generation
Ankyrin-G shRNAs were engineered into the pFIV lentiviral vector and packaged into viral pseudoparticles (System Biosciences, Inc.). Constructs were cotransfected with packaging plasmids into HEK293 cells using Effectene. The pseudoparticle-containing supernatant was concentrated using Centriplus YM-30 columns. The concentrate was stored at −80°C.
Immunostaining/confocal microscopy
Cardiomyocytes were isolated, cultured, and processed for immunofluorescence as described previously (Mohler et al., 2003; Cunha et al., 2007). Secondary antibodies included anti–rabbit and anti–mouse Igs conjugated to AlexaFluor488 or 568 (Invitrogen). Phalloidin-conjugated AlexaFluor633 was used for double-labeling experiments in Fig. S1. After secondary antibody treatment, cells were extensively washed, covered with Vectashield imaging medium (Vector Laboratories), and coverslips (#1) were applied. Images were collected on a confocal microscope (510 Meta; Carl Zeiss, Inc.) with a 63× oil 1.40 NA or 40× oil 1.30 NA lens (pinhole equals 1.0 airy disc; Carl Zeiss, Inc.) using imaging software (release version 4.0 SP1; Carl Zeiss, Inc.). Images were collected using similar confocal protocols at room temperature. In experiments in which both AlexaFluor488 and YFP were analyzed, the emission signal from AlexaFluor488 was collected from 500 to 515 nm, and the emission from YFP was collected from 530 to 565 nm. Because YFP was only used to identify virus-positive cells (versus identifying the localization of a YFP fusion protein), we used minimal laser power and detector gain (thus the minimal YFP image resolution) to collect YFP images to prevent potential signal bleed-through to the AlexaFluor488 image, in which protein immunolocalization was crucial. For images in Figs. 3, Figs.8, and S1, the YFP image was pseudocolored. Images were imported into Photoshop CS (Adobe) for cropping and linear contrast adjustment.
Plasma membrane sheet preparation/immunoelectron microscopy
Ventricular cardiac myocytes from adult Sprague-Dawley rats were obtained as described previously (Shibata et al., 2006). Immunolabeling of the membrane sheets was performed by placing the grids on drops of primary antibody solution for 1 h on ice and rinsing six times for 5 min in PBS containing BSA. Grids were then incubated on drops of gold-conjugated secondary antibody solution (5 nm of goat anti–rabbit diluted 1:50 or 10 nm of goat anti–mouse diluted 1:50 in PBS containing BSA; Electron Microscopy Sciences) for 1 h on ice. Grids were then rinsed three times for 5 min in PBS containing BSA followed by a 2-min fixation in 2.5% glutaraldehyde. Finally, the grids were washed for 5 min in deionized water before being allowed to air dry. Negative controls included grids labeled with secondary antibody alone. Grids were visualized on a transmission electron microscope (H-70000; Hitachi).
Antibodies
Antibodies used include anti-Nav1.5 (Alomone Laboratories), anti-NCX1 (Swant), anti–connexin43 (Invitrogen), anti-Cav1.2 (Affinity BioReagents), anti-NHERF1 (Sigma-Aldrich), affinity-purified Igs against ankyrin-B, ankyrin-G, and GFP (monoclonal and polyclonal), and goat anti–rabbit AlexaFluor568 (Invitrogen).
Electrophysiology experiments
Voltage-dependent Na+ and Ca+2 currents were measured using standard patch clamp techniques. Whole-cell currents were recorded with an amplifier (Axopatch 200B; MDS Analytical Technologies), and the analogue signal was filtered using an eight-pole filter (Bessel) with a bandwidth of 5 kHz and was digitized at a sampling rate of 50 kHz. Borosilicate glass capillaries (VWR Scientific) were used to fabricate patch pipettes. Electrode resistances ranged from 1 to 1.5 MΩ, and seal resistances were 1–5 GΩ. Pipette seal resistances were compensated to >85% of the uncompensated value. The whole-cell bath solution contained 10 mM NaCl, 130 mM choline chloride, 4.5 mM KCl, 1.8 mM CaCl2, 2.0 mM MgCl2, 10.0 mM Hepes, and 5.5 mM glucose, pH 7.35, titrated with KOH. The pipette solution contained 130 mM CsCl, 0.5 mM CaCl2, 2 mM MgCl2, 5 mM Na2ATP, 0.5 mM GTP, 5 mM EGTA, and 10 mM Hepes, pH 7.3, titrated with CsOH. All electrophysiology experiments were performed at room temperature (21–23°C).
Whole-cell voltage clamp Na+ current data were elicited from a holding potential of −120 mV to membrane potentials ranging from −110 to 30 mV in the presence of 2.0 mM CoCl2. Voltage-dependent steady-state inactivation was determined using a paired two-pulse protocol. Each conditioning voltage was paired with a control after 1.5 s. A 500-ms conditioning pulse from −120 to 20 mV in 10-mV increments was followed by a test pulse to −30 mV. The test pulse in each series was separated form the conditioning pulse by a 2-ms interval to −120 mV. The steady-state inactivation curves were constructed by normalizing currents to the maximal Na+ current elicited from a holding potential of −120 to −30 mV for a duration of 20 ms. The resulting curve was fitted using a Boltzmann distribution equation of the form INa = INa, max/(1 + exp([Vm − V1/2]/k)), where Vm is the conditioning pulse voltage, V1/2 is the voltage at half-inactivation, and k is the slope factor. The whole-cell voltage clamp protocol showing INa and ICa in Fig. 5 F was elicited using the two-pulse paradigm shown in Fig. 5 G. From a holding potential of −100 mV, INa was elicited by a 30-ms pulse to −30 mV. The membrane potential was then hyperpolarized to −70 mV for 10 ms. ICa was then elicited by a 50-ms pulse to 0 mV. This protocol allowed the simple differentiation of INa from ICa as indicated by the unique ICa and INa current signatures in Fig. 5. All presented currents were normalized for cell capacitance. Data were collected and analyzed using pCLAMP 9.0 software (MDS Analytical Technologies) and OriginPro 7.5 (OriginLab Corp.). Analysis of variance was used to compare the nominal change in current density among the means. Statistical significance is defined as P < 0.05.
Online supplemental material
Fig. S1 demonstrates that cardiomyocytes with reduced ankyrin-G expression display normal localization and distribution of Cav1.2 and Na/Ca exchanger. Fig. S2 demonstrates that a full complement of ankyrin-G expression is required for normal Nav1.5 expression in adult rat cardiomyocytes. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200710107/DC1.
Supplemental Material
Acknowledgments
We acknowledge financial support from the National Institutes of Health (grants HL084583 and HL083422 to P.J. Mohler, grant HL 075541 to E. Shibata, and grants HL079031, HL62494, and HL70250 to M.E. Anderson) and the Pew Scholars Trust (grant to P.J. Mohler). J.S. Lowe is a member of the Vanderbilt University Graduate Program in Cellular and Molecular Pathology.
Abbreviation used in this paper: shRNA, small hairpin RNA.
References
- Abriel, H., and R.S. Kass. 2005. Regulation of the voltage-gated cardiac sodium channel Nav1.5 by interacting proteins. Trends Cardiovasc. Med. 15:35–40. [DOI] [PubMed] [Google Scholar]
- Anderson, C.L., B.P. Delisle, B.D. Anson, J.A. Kilby, M.L. Will, D.J. Tester, Q. Gong, Z. Zhou, M.J. Ackerman, and C.T. January. 2006. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation. 113:365–373. [DOI] [PubMed] [Google Scholar]
- Bagnato, P., V. Barone, E. Giacomello, D. Rossi, and V. Sorrentino. 2003. Binding of an ankyrin-1 isoform to obscurin suggests a molecular link between the sarcoplasmic reticulum and myofibrils in striated muscles. J. Cell Biol. 160:245–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballester, L.Y., D.W. Benson, B. Wong, I.H. Law, K.D. Mathews, C.G. Vanoye, and A.L. George Jr. 2006. Trafficking-competent and trafficking-defective KCNJ2 mutations in Andersen syndrome. Hum. Mutat. 27:388. [DOI] [PubMed] [Google Scholar]
- Barry, D.M., J.S. Trimmer, J.P. Merlie, and J.M. Nerbonne. 1995. Differential expression of voltage-gated K+ channel subunits in adult rat heart. Relation to functional K+ channels? Circ. Res. 77:361–369. [DOI] [PubMed] [Google Scholar]
- Baruscotti, M., R. Westenbroek, W.A. Catterall, D. DiFrancesco, and R.B. Robinson. 1997. The newborn rabbit sino-atrial node expresses a neuronal type I-like Na+ channel. J. Physiol. 498:641–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, V., and P.J. Stenbuck. 1979. The membrane attachment protein for spectrin is associated with band 3 in human erythrocyte membranes. Nature. 280:468–473. [DOI] [PubMed] [Google Scholar]
- Bhasin, N., S.R. Cunha, M. Mudannayake, M.S. Gigena, T.B. Rogers, and P.J. Mohler. 2007. Molecular basis for PP2A regulatory subunit B56{alpha } targeting in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 293:H109–H119. [DOI] [PubMed] [Google Scholar]
- Brummelkamp, T.R., R. Bernards, and R. Agami. 2002. A system for stable expression of short interfering RNAs in mammalian cells. Science. 296:550–553. [DOI] [PubMed] [Google Scholar]
- Burgess, D.L., D.C. Kohrman, J. Galt, N.W. Plummer, J.M. Jones, B. Spear, and M.H. Meisler. 1995. Mutation of a new sodium channel gene, Scn8a, in the mouse mutant ‘motor endplate disease’. Nat. Genet. 10:461–465. [DOI] [PubMed] [Google Scholar]
- Cannon, S.C. 1996. Sodium channel defects in myotonia and periodic paralysis. Annu. Rev. Neurosci. 19:141–164. [DOI] [PubMed] [Google Scholar]
- Carl, S.L., K. Felix, A.H. Caswell, N.R. Brandt, W.J. Ball Jr., P.L. Vaghy, G. Meissner, and D.G. Ferguson. 1995. Immunolocalization of sarcolemmal dihydropyridine receptor and sarcoplasmic reticular triadin and ryanodine receptor in rabbit ventricle and atrium. J. Cell Biol. 129:672–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, H.J., Y.N. Jan, and L.Y. Jan. 2006. Polarized axonal surface expression of neuronal KCNQ channels is mediated by multiple signals in the KCNQ2 and KCNQ3 C-terminal domains. Proc. Natl. Acad. Sci. USA. 103:8870–8875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, R.B., A. Tremblay, P. Melnyk, B.G. Allen, W.R. Giles, and C. Fiset. 2001. T-tubule localization of the inward-rectifier K(+) channel in mouse ventricular myocytes: a role in K(+) accumulation. J. Physiol. 537:979–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, S.A. 1996. Immunocytochemical localization of rH1 sodium channel in adult rat heart atria and ventricle. Presence in terminal intercalated disks. Circulation. 94:3083–3086. [DOI] [PubMed] [Google Scholar]
- Cunha, S.R., N. Bhasin, and P.J. Mohler. 2007. Targeting and stability of na/ca exchanger 1 in cardiomyocytes requires direct interaction with the membrane adaptor ankyrin-B. J. Biol. Chem. 282:4875–4883. [DOI] [PubMed] [Google Scholar]
- Devarajan, P., D.A. Scaramuzzino, and J.S. Morrow. 1994. Ankyrin binds to two distinct cytoplasmic domains of Na,K-ATPase alpha subunit. Proc. Natl. Acad. Sci. USA. 91:2965–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar Malhotra, J., C. Chen, I. Rivolta, H. Abriel, R. Malhotra, L.N. Mattei, F.C. Brosius, R.S. Kass, and L.L. Isom. 2001. Characterization of sodium channel alpha- and beta-subunits in rat and mouse cardiac myocytes. Circulation. 103:1303–1310. [DOI] [PubMed] [Google Scholar]
- Dubreuil, R.R. 2006. Functional links between membrane transport and the spectrin cytoskeleton. J. Membr. Biol. 211:151–161. [DOI] [PubMed] [Google Scholar]
- Escayg, A., B.T. MacDonald, M.H. Meisler, S. Baulac, G. Huberfeld, I. An-Gourfinkel, A. Brice, E. LeGuern, B. Moulard, D. Chaigne, et al. 2000. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat. Genet. 24:343–345. [DOI] [PubMed] [Google Scholar]
- Frank, J.S., G. Mottino, D. Reid, R.S. Molday, and K.D. Philipson. 1992. Distribution of the Na(+)-Ca2+ exchange protein in mammalian cardiac myocytes: an immunofluorescence and immunocolloidal gold-labeling study. J. Cell Biol. 117:337–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido, J.J., P. Giraud, E. Carlier, F. Fernandes, A. Moussif, M.P. Fache, D. Debanne, and B. Dargent. 2003. A targeting motif involved in sodium channel clustering at the axonal initial segment. Science. 300:2091–2094. [DOI] [PubMed] [Google Scholar]
- Gouas, L., C. Bellocq, M. Berthet, F. Potet, S. Demolombe, A. Forhan, R. Lescasse, F. Simon, B. Balkau, I. Denjoy, et al. 2004. New KCNQ1 mutations leading to haploinsufficiency in a general population; defective trafficking of a KvLQT1 mutant. Cardiovasc. Res. 63:60–68. [DOI] [PubMed] [Google Scholar]
- Grueter, C.E., S.A. Abiria, I. Dzhura, Y. Wu, A.J. Ham, P.J. Mohler, M.E. Anderson, and R.J. Colbran. 2006. L-type Ca(2+) channel facilitation mediated by phosphorylation of the beta subunit by CaMKII. Mol. Cell. 23:641–650. [DOI] [PubMed] [Google Scholar]
- Haufe, V., J.A. Camacho, R. Dumaine, B. Gunther, C. Bollensdorff, G.S. von Banchet, K. Benndorf, and T. Zimmer. 2005. Expression pattern of neuronal and skeletal muscle voltage-gated Na+ channels in the developing mouse heart. J. Physiol. 564:683–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herfst, L.J., M.B. Rook, and H.J. Jongsma. 2004. Trafficking and functional expression of cardiac Na+ channels. J. Mol. Cell. Cardiol. 36:185–193. [DOI] [PubMed] [Google Scholar]
- Jenkins, S.M., and V. Bennett. 2001. Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J. Cell Biol. 155:739–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanter, H.L., J.E. Saffitz, and E.C. Beyer. 1992. Cardiac myocytes express multiple gap junction proteins. Circ. Res. 70:438–444. [DOI] [PubMed] [Google Scholar]
- Keating, M.T., and M.C. Sanguinetti. 2001. Molecular and cellular mechanisms of cardiac arrhythmias. Cell. 104:569–580. [DOI] [PubMed] [Google Scholar]
- Kieval, R.S., R.J. Bloch, G.E. Lindenmayer, A. Ambesi, and W.J. Lederer. 1992. Immunofluorescence localization of the Na-Ca exchanger in heart cells. Am. J. Physiol. 263:C545–C550. [DOI] [PubMed] [Google Scholar]
- Kizhatil, K., and V. Bennett. 2004. Lateral membrane biogenesis in human bronchial epithelial cells requires 190-kDa ankyrin-G. J. Biol. Chem. 279:16706–16714. [DOI] [PubMed] [Google Scholar]
- Kizhatil, K., W. Yoon, P.J. Mohler, L.H. Davis, J.A. Hoffman, and V. Bennett. 2007. Ankyrin-G and beta2-spectrin collaborate in biogenesis of lateral membrane of human bronchial epithelial cells. J. Biol. Chem. 282:2029–2037. [DOI] [PubMed] [Google Scholar]
- Kontrogianni-Konstantopoulos, A., D.H. Catino, J.C. Strong, W.R. Randall, and R.J. Bloch. 2004. Obscurin regulates the organization of myosin into A bands. Am. J. Physiol. Cell Physiol. 287:C209–C217. [DOI] [PubMed] [Google Scholar]
- Kontrogianni-Konstantopoulos, A., D.H. Catino, J.C. Strong, S. Sutter, A.B. Borisov, D.W. Pumplin, M.W. Russell, and R.J. Bloch. 2006. Obscurin modulates the assembly and organization of sarcomeres and the sarcoplasmic reticulum. FASEB J. 20:2102–2111. [DOI] [PubMed] [Google Scholar]
- Krumerman, A., X. Gao, J.S. Bian, Y.F. Melman, A. Kagan, and T.V. McDonald. 2004. An LQT mutant minK alters KvLQT1 trafficking. Am. J. Physiol. Cell Physiol. 286:C1453–C1463. [DOI] [PubMed] [Google Scholar]
- Kucera, J.P., S. Rohr, and Y. Rudy. 2002. Localization of sodium channels in intercalated disks modulates cardiac conduction. Circ. Res. 91:1176–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnart, S.E., M.J. Ackerman, D.W. Benson Jr., R. Brugada, C.E. Clancy, J.K. Donahue, A.L. George Jr., A.O. Grant, S.C. Groft, C.T. January, et al. 2007. Inherited arrhythmias: a National Heart, Lung, and Blood Institute and Office of Rare Diseases workshop consensus report about the diagnosis, phenotyping, molecular mechanisms, and therapeutic approaches for primary cardiomyopathies of gene mutations affecting ion channel function. Circulation. 116:2325–2345. [DOI] [PubMed] [Google Scholar]
- Lemaillet, G., B. Walker, and S. Lambert. 2003. Identification of a conserved ankyrin-binding motif in the family of sodium channel alpha subunits. J. Biol. Chem. 278:27333–27339. [DOI] [PubMed] [Google Scholar]
- Lencesova, L., A. O'Neill, W.G. Resneck, R.J. Bloch, and M.P. Blaustein. 2004. Plasma membrane-cytoskeleton-endoplasmic reticulum complexes in neurons and astrocytes. J. Biol. Chem. 279:2885–2893. [DOI] [PubMed] [Google Scholar]
- Liu, K., T. Yang, P.C. Viswanathan, and D.M. Roden. 2005. New mechanism contributing to drug-induced arrhythmia: rescue of a misprocessed LQT3 mutant. Circulation. 112:3239–3246. [DOI] [PubMed] [Google Scholar]
- Liu, N., B. Colombi, E.V. Raytcheva-Buono, R. Bloise, and S.G. Priori.2007. Catecholaminergic polymorphic ventricular tachycardia. Herz. 32:212–217. [DOI] [PubMed] [Google Scholar]
- Lossin, C., D.W. Wang, T.H. Rhodes, C.G. Vanoye, and A.L. George Jr. 2002. Molecular basis of an inherited epilepsy. Neuron. 34:877–884. [DOI] [PubMed] [Google Scholar]
- Maier, S.K., R.E. Westenbroek, K.A. Schenkman, E.O. Feigl, T. Scheuer, and W.A. Catterall. 2002. An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc. Natl. Acad. Sci. USA. 99:4073–4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier, S.K., R.E. Westenbroek, T.T. Yamanushi, H. Dobrzynski, M.R. Boyett, W.A. Catterall, and T. Scheuer. 2003. An unexpected requirement for brain-type sodium channels for control of heart rate in the mouse sinoatrial node. Proc. Natl. Acad. Sci. USA. 100:3507–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier, S.K., R.E. Westenbroek, K.A. McCormick, R. Curtis, T. Scheuer, and W.A. Catterall. 2004. Distinct subcellular localization of different sodium channel alpha and beta subunits in single ventricular myocytes from mouse heart. Circulation. 109:1421–1427. [DOI] [PubMed] [Google Scholar]
- McDonough, A.A., Y. Zhang, V. Shin, and J.S. Frank. 1996. Subcellular distribution of sodium pump isoform subunits in mammalian cardiac myocytes. Am. J. Physiol. 270:C1221–C1227. [DOI] [PubMed] [Google Scholar]
- Melnyk, P., L. Zhang, A. Shrier, and S. Nattel. 2002. Differential distribution of Kir2.1 and Kir2.3 subunits in canine atrium and ventricle. Am. J. Physiol. Heart Circ. Physiol. 283:H1123–H1133. [DOI] [PubMed] [Google Scholar]
- Michaely, P., D.R. Tomchick, M. Machius, and R.G. Anderson. 2002. Crystal structure of a 12 ANK repeat stack from human ankyrinR. EMBO J. 21:6387–6396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohler, P.J., and V. Bennett. 2005. a. Ankyrin-based cardiac arrhythmias: a new class of channelopathies due to loss of cellular targeting. Curr. Opin. Cardiol. 20:189–193. [DOI] [PubMed] [Google Scholar]
- Mohler, P.J., and V. Bennett. 2005. b. Defects in ankyrin-based cellular pathways in metazoan physiology. Front. Biosci. 10:2832–2840. [DOI] [PubMed] [Google Scholar]
- Mohler, P.J., A.O. Gramolini, and V. Bennett. 2002. The ankyrin-B C-terminal domain determines activity of ankyrin-B/G chimeras in rescue of abnormal inositol 1,4,5-trisphosphate and ryanodine receptor distribution in ankyrin-B (−/−) neonatal cardiomyocytes. J. Biol. Chem. 277:10599–10607. [DOI] [PubMed] [Google Scholar]
- Mohler, P.J., J.J. Schott, A.O. Gramolini, K.W. Dilly, S. Guatimosim, W.H. duBell, L.S. Song, K. Haurogne, F. Kyndt, M.E. Ali, et al. 2003. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 421:634–639. [DOI] [PubMed] [Google Scholar]
- Mohler, P.J., J.Q. Davis, L.H. Davis, J.A. Hoffman, P. Michaely, and V. Bennett. 2004. a. Inositol 1,4,5-trisphosphate receptor localization and stability in neonatal cardiomyocytes requires interaction with ankyrin-B. J. Biol. Chem. 279:12980–12987. [DOI] [PubMed] [Google Scholar]
- Mohler, P.J., I. Rivolta, C. Napolitano, G. Lemaillet, S. Lambert, S.G. Priori, and V. Bennett. 2004. b. Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proc. Natl. Acad. Sci. USA. 101:17533–17538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohler, P.J., I. Splawski, C. Napolitano, G. Bottelli, L. Sharpe, K. Timothy, S.G. Priori, M.T. Keating, and V. Bennett. 2004. c. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc. Natl. Acad. Sci. USA. 101:9137–9142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohler, P.J., W. Yoon, and V. Bennett. 2004. d. Ankyrin-B targets {beta }2-spectrin to an intracellular compartment in neonatal cardiomyocytes. J. Biol. Chem. 279:40185–40193. [DOI] [PubMed] [Google Scholar]
- Mohler, P.J., J.Q. Davis, and V. Bennett. 2005. Ankyrin-B coordinates the Na/K ATPase, Na/Ca exchanger, and InsP(3) receptor in a cardiac T-tubule/SR microdomain. PLoS Biol. 3:e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohler, P.J., S. Le Scouarnec, I. Denjoy, J.S. Lowe, P. Guicheney, L. Caron, I.M. Driskell, J.J. Schott, K. Norris, A. Leenhardt, et al. 2007. Defining the cellular phenotype of “ankyrin-B syndrome” variants: human ANK2 variants associated with clinical phenotypes display a spectrum of activities in cardiomyocytes. Circulation. 115:432–441. [DOI] [PubMed] [Google Scholar]
- Napolitano, C., and S.G. Priori. 2006. Brugada syndrome. Orphanet J. Rare Dis. 1:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, Z., T. Kao, Z. Horvath, J. Lemos, J.Y. Sul, S.D. Cranstoun, V. Bennett, S.S. Scherer, and E.C. Cooper. 2006. A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J. Neurosci. 26:2599–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadatos, G.A., P.M. Wallerstein, C.E. Head, R. Ratcliff, P.A. Brady, K. Benndorf, R.C. Saumarez, A.E. Trezise, C.L. Huang, J.I. Vandenberg, et al. 2002. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc. Natl. Acad. Sci. USA. 99:6210–6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pond, A.L., B.K. Scheve, A.T. Benedict, K. Petrecca, D.R. Van Wagoner, A. Shrier, and J.M. Nerbonne. 2000. Expression of distinct ERG proteins in rat, mouse, and human heart. Relation to functional I(Kr) channels. J. Biol. Chem. 275:5997–6006. [DOI] [PubMed] [Google Scholar]
- Priori, S.G. 2004. Inherited arrhythmogenic diseases: the complexity beyond monogenic disorders. Circ. Res. 94:140–145. [DOI] [PubMed] [Google Scholar]
- Priori, S.G., C. Napolitano, M. Gasparini, C. Pappone, P. Della Bella, U. Giordano, R. Bloise, C. Giustetto, R. De Nardis, M. Grillo, et al. 2002. Natural history of Brugada syndrome: insights for risk stratification and management. Circulation. 105:1342–1347. [DOI] [PubMed] [Google Scholar]
- Rasmussen, H.B., M. Moller, H.G. Knaus, B.S. Jensen, S.P. Olesen, and N.K. Jorgensen. 2004. Subcellular localization of the delayed rectifier K(+) channels KCNQ1 and ERG1 in the rat heart. Am. J. Physiol. Heart Circ. Physiol. 286:H1300–H1309. [DOI] [PubMed] [Google Scholar]
- Rasmussen, H.B., C. Frokjaer-Jensen, C.S. Jensen, H.S. Jensen, N.K. Jorgensen, H. Misonou, J.S. Trimmer, S.P. Olesen, and N. Schmitt. 2007. Requirement of subunit co-assembly and ankyrin-G for M-channel localization at the axon initial segment. J. Cell Sci. 120:953–963. [DOI] [PubMed] [Google Scholar]
- Rogart, R.B., L.L. Cribbs, L.K. Muglia, D.D. Kephart, and M.W. Kaiser. 1989. Molecular cloning of a putative tetrodotoxin-resistant rat heart Na+ channel isoform. Proc. Natl. Acad. Sci. USA. 86:8170–8174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt, N., K. Calloe, N.H. Nielsen, M. Buschmann, E.J. Speckmann, E. Schulze-Bahr, and M. Schwarz. 2007. The novel C-terminal KCNQ1 mutation M520R alters protein trafficking. Biochem. Biophys. Res. Commun. 358:304–310. [DOI] [PubMed] [Google Scholar]
- Scriven, D.R., P. Dan, and E.D. Moore. 2000. Distribution of proteins implicated in excitation-contraction coupling in rat ventricular myocytes. Biophys. J. 79:2682–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scriven, D.R., A. Klimek, K.L. Lee, and E.D. Moore. 2002. The molecular architecture of calcium microdomains in rat cardiomyocytes. Ann. NY Acad. Sci. 976:488–499. [DOI] [PubMed] [Google Scholar]
- Shah, M., F.G. Akar, and G.F. Tomaselli. 2005. Molecular basis of arrhythmias. Circulation. 112:2517–2529. [DOI] [PubMed] [Google Scholar]
- Shibata, E.F., T.L. Brown, Z.W. Washburn, J. Bai, T.J. Revak, and C.A. Butters. 2006. Autonomic regulation of voltage-gated cardiac ion channels. J. Cardiovasc. Electrophysiol. 17:S34–S42. [DOI] [PubMed] [Google Scholar]
- Shirahata, E., H. Iwasaki, M. Takagi, C. Lin, V. Bennett, Y.Y. Okamura, and K. Hayasaka. 2006. Ankyrin-G regulates inactivation gating of the neuronal sodium channel, Nav1.6. J. Neurophysiol. 96:1347–1357. [DOI] [PubMed] [Google Scholar]
- Splawski, I., K.W. Timothy, L.M. Sharpe, N. Decher, P. Kumar, R. Bloise, C. Napolitano, P.J. Schwartz, R.M. Joseph, K. Condouris, et al. 2004. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 119:19–31. [DOI] [PubMed] [Google Scholar]
- Sun, X.H., F. Protasi, M. Takahashi, H. Takeshima, D.G. Ferguson, and C. Franzini-Armstrong. 1995. Molecular architecture of membranes involved in excitation-contraction coupling of cardiac muscle. J. Cell Biol. 129:659–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilde, A.A., and S.G. Priori. 2000. Brugada syndrome and sudden death. Eur. Heart J. 21:1483–1484. [DOI] [PubMed] [Google Scholar]
- Wu, D.M., M. Jiang, M. Zhang, X.S. Liu, Y.V. Korolkova, and G.N. Tseng. 2006. KCNE2 is colocalized with KCNQ1 and KCNE1 in cardiac myocytes and may function as a negative modulator of I(Ks) current amplitude in the heart. Heart Rhythm. 3:1469–1480. [DOI] [PubMed] [Google Scholar]
- Yang, Y., Y. Ogawa, K.L. Hedstrom, and M.N. Rasband. 2007. βIV spectrin is recruited to axon initial segments and nodes of Ranvier by ankyrinG. J. Cell Biol. 176:509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, B., C.R. Valdivia, M.J. Ackerman, and J.C. Makielski. 2003. A common human SCN5A polymorphism modifies expression of an arrhythmia causing mutation. Physiol. Genomics. 12:187–193. [DOI] [PubMed] [Google Scholar]
- Yu, F.H., V. Yarov-Yarovoy, G.A. Gutman, and W.A. Catterall. 2005. Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacol. Rev. 57:387–395. [DOI] [PubMed] [Google Scholar]
- Zhou, D., S. Lambert, P.L. Malen, S. Carpenter, L.M. Boland, and V. Bennett. 1998. AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J. Cell Biol. 143:1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Z., Q. Gong, and C.T. January. 1999. Correction of defective protein trafficking of a mutant HERG potassium channel in human long QT syndrome. Pharmacological and temperature effects. J. Biol. Chem. 274:31123–31126. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}