

Abstract
The exocyst is an octameric protein complex implicated in tethering post-Golgi secretory vesicles at the plasma membrane in preparation for fusion. However, it is not clear how the exocyst is targeted to and physically associates with specific domains of the plasma membrane and how its functions are regulated at those regions. We demonstrate that the N terminus of the exocyst component Sec3 directly interacts with phosphatidylinositol 4,5-bisphosphate. In addition, we have identified key residues in Sec3 that are critical for its binding to the guanosine triphosphate–bound form of Cdc42. Genetic analyses indicate that the dual interactions of Sec3 with phospholipids and Cdc42 control its function in yeast cells. Disrupting these interactions not only blocks exocytosis and affects exocyst polarization but also leads to defects in cell morphogenesis. We propose that the interactions of Sec3 with phospholipids and Cdc42 play important roles in exocytosis and polarized cell growth.
Introduction
Polarized exocytosis is important for many biological processes ranging from the secretion of neurotransmitters to the establishment of epithelia membrane asymmetry. The budding yeast Saccharomyces cerevisiae undergoes polarized growth (budding), which requires exocytosis of newly synthesized materials at the daughter cell membrane for cell wall remodeling and surface expansion. This property, combined with its facile genetics, makes the budding yeast an excellent model system to study the molecular mechanisms of polarized exocytosis.
Exocytosis is accomplished when the exocytic vesicles are docked to and fused with the plasma membrane. The initial contact of the vesicles with the plasma membrane (also known as tethering) is believed to be mediated by the exocyst complex, which consists of Sec3, 5, 6, 8, 10, and 15 and Exo70 and 84 (for reviews see Pfeffer, 1999; Guo et al., 2000; Whyte and Munro, 2002; Hsu et al., 2004). In yeast, defects in exocyst proteins lead to accumulation of secretory vesicles in cells (Novick et al., 1980; Guo et al., 1999a, Zhang et al., 2005a,b). Members of the exocyst are localized to the bud tip or mother–daughter junction, where active exocytosis and cell surface expansion take place (TerBush and Novick, 1995; Finger et al., 1998; Guo et al., 1999a, 2001; Zajac et al., 2005). The exocyst component Sec15 directly interacts with the Rab protein Sec4, a master regulator of post-Golgi secretion (Guo et al., 1999b). Sec3 and Exo70 interact with the Rho family of small GTPases, which are key regulators of polarized cell growth in yeast (Adamo et al., 1999; Robinson et al., 1999; Guo et al., 2001; Zhang et al., 2001). Recent crystallographic studies suggest that many of the exocyst components are composed of contiguous helical bundles with an overall rodlike extended conformation, and the exocyst complex assembly involves orderly packing of these rodlike structures (for review see Munson and Novick, 2006).
In this paper, we find that Sec3 directly interacts with phosphatidylinositol 4,5-bisphosphate (PIP2) in the plasma membrane. In addition, we have identified key residues on Sec3 that are critical for its binding to Cdc42. We demonstrate that both PIP2 and Cdc42 binding are required for the polarization and function of Sec3 at the bud tip membrane. Disruption of the interaction of Sec3 with Cdc42 or PIP2 not only blocks exocytosis but also causes defects in cell morphology. Our studies shed light on the molecular basis of vesicle targeting to the plasma membrane and reveal the critical role the exocyst plays as a downstream effector of PIP2 and Cdc42 during cell polarization.
Results
sec3ΔN is synthetic lethal with the exo70-38 mutant
We have previously shown that the N terminus of Sec3 (Sec3N, aa 1–320) interacts with the Rho GTPases (Guo et al., 2001; Zhang et al., 2001). Surprisingly, cells expressing N terminus–deleted Sec3 (sec3ΔN) as the only copy of Sec3 grew normally at all temperatures tested, and these cells did not have any defects in secretion (Guo et al., 2001). A recent study showed that Exo70, like Sec3, resides on the bud tip membrane and that the other subunits, arriving via secretory vesicles, interact with Sec3 and Exo70 at the bud tip for exocyst assembly (Boyd et al., 2004). Furthermore, it was shown that Exo70 binds directly to PIP2 (He et al., 2007b; Liu et al., 2007). It is thus possible that Sec3 and Exo70 function together in exocyst targeting in yeast. In that case, mutations in Sec3 or Exo70 that alone cause little or no phenotype may have a synthetic defect when combined. We recently obtained Exo70 mutants (He et al., 2007a,b) that allowed us to test this hypothesis. Both sec3ΔN and exo70-38 were expressed under their endogenous promoters in CEN plasmids. sec3ΔN has no growth defect. exo70-38 can survive at 25 but not 37°C (He et al., 2007a). sec3ΔN is synthetic lethal with exo70-38, as at all temperatures tested the sec3ΔN and exo70-38 double mutant sec3ΔN exo70-38 cannot survive on 5–fluoroorotic acid (5-FOA) plates on which the URA3-based wild-type SEC3 balancer is eliminated (Fig. 1 A). The genetic interaction between sec3ΔN and exo70-38 suggests that the N terminus of Sec3 becomes indispensable in the exo70 mutant background. Besides exo70-38, sec3ΔN also has synthetic defects with other exocyst mutants (Roumanie et al., 2005; unpublished data).
Figure 1.

The gic2-sec3 chimera is able to rescue synthetic lethality between sec3ΔN and exo70-38. (A) sec3ΔN is synthetic lethal with exo70-38. sec3ΔN and exo70-38 were expressed under SEC3 and EXO70 promoters in CEN plasmids. The sec3ΔN exo70-38 double mutant supplemented with a CEN, URA3, SEC3 balancer was streaked out on the plates with (right) or without (left) 5-FOA and incubated for 5 d at 25°C. The sec3ΔN and exo70-38 single mutants were used as controls. exo70-38 sec3ΔN could not survive when losing the SEC3 balancer on the 5-FOA plate (right). (B) Diagram of Sec3 and gic2-sec3 chimera in which the N terminus of Sec3 (aa 1–307) was replaced with the N terminus of Gic2 (aa 1–155; gray). The interaction with Cdc42 is indicated by the arrows. (C) The chimera gic2-sec3 is able to rescue the synthetic lethality between sec3ΔN and exo70-38. gic2-sec3 was expressed under the endogenous SEC3 promoter in a CEN plasmid supplemented with a CEN, URA, SEC3 balancer. Although sec3ΔN and exo70-38 were synthetic lethal, the exo70-38 gic2-sec3 grew well at 25°C in the presence of 5-FOA. (D) The CRIB domain and polybasic region at the N terminus of Gic2 are essential for the functional replacement of wild-type SEC3 with gic2-sec3 chimera in yeast. Chimeras, including gic2-sec3 and gic2 (CRIB mutation)-sec3, in which the key residues of the CRIB domain are replaced by alanine (I134A, S135A, and P137A), and gic2 (CRIBΔ)-sec3, gic2 (CRIBΔ & polybasicΔ)-sec3, and gic2-sec3 (K109A, K110A, K119A, K120A, and K121A), in which the polybasic region of Gic2 was mutated, were expressed under the SEC3 promoter in CEN plasmids and tested for their synthetic lethality with exo70-38. All chimeras except gic2-sec3 are synthetic lethal with exo70-38.
Functional replacement of the Sec3ΔN terminus with Gic2 N terminus
We have previously shown that the N terminus of Sec3 directly interacts with Cdc42, but we were unable to assess the functional importance of this interaction (Zhang et al., 2001). Now, taking advantage of the synthetic lethality assay, we tested whether the lethality of the sec3ΔN exo70-38 double mutant could be rescued by adding the N terminus of Gic2 (Gic2N, aa 1–155) to sec3ΔN (Fig. 1 B). Gic2 is a well-characterized effector of Cdc42 (Brown et al., 1997, Chen et al., 1997). Gic2N contains a Cdc42/Rac interactive binding (CRIB) domain that interacts with the GTP-bound form of Cdc42. We tested whether this gic2-sec3 chimera, when expressed under the SEC3 promoter, can function as well as the wild-type SEC3 in the exo70-38 background. We found that although exo70-38 and sec3ΔN were synthetic lethal, growth of the exo70-38 gic2-sec3 strain was similar to that of the exo70-38 SEC3 strain on plates at 25°C (Fig. 1 C) and all other temperatures tested (not depicted).
Next, we performed experiments to identify the sequences of Gic2N that are crucial for its functional replacement of the Sec3 N terminus. First, we mutated or deleted the CRIB domain in Gic2N and tested the synthetic effects of these mutants with exo70-38. As shown in Fig. 1 D, the gic2-sec3 chimeras with the CRIB domain mutated or deleted were synthetic lethal with exo70-38, whereas the mutant proteins were expressed at similar levels to the wild-type protein (not depicted). Gic2 also contains a cluster of positively charged residues (aa 109–121) adjacent to the CRIB domain that are implicated in membrane association. As shown in Fig. 1 D, replacing these basic residues with alanine (K109A, K110A, K119A, K120A, and K121A) led to synthetic lethality between gic2-sec3 and exo70-38. These genetic analyses indicate that both the CRIB domain and the polybasic region of Gic2 are essential for the ability of the gic2-sec3 chimera to functionally replace SEC3 in yeast cells.
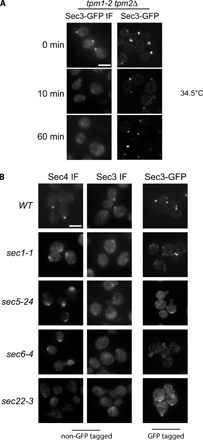
Localization of Sec3 in tropomyosin mutants and cells treated with latrunculin
It was previously reported by Finger et al. (1998) that polarized localization of Sec3-GFP to the bud tip is independent of actin. In addition, fluorescence recovery after photobleaching (FRAP) of Sec3-GFP and Exo70-GFP at the bud was found to take place properly in the presence of latrunculin, which disrupted actin cables in cells (Boyd et al., 2004). Consistent with these observations, Sec3-GFP was found to remain polarized in the tropomyosin mutant, tpm1-2 tpm2Δ (Zajac et al., 2005), in which actin cables were disrupted upon shifting to the restrictive temperature (Pruyne et al., 1998). However, using an anti-Sec3 antibody, Roumanie et al. (2005) reported that Sec3 immunofluorescence signals at the bud were lost in the tropomyosin mutant. In this study, we performed the immunostaining experiment using a Sec3 antibody (generated against a fusion protein containing aa 445–711 of Sec3) in parallel with GFP-tagged Sec3. This antibody detects Sec3 in wild-type (SEC3) but not sec3-null (sec3Δ) cells by Western blotting or immunofluorescence (Fig. 2 A). The endogenous Sec3 was polarized in the TPM1 tpm2Δ cells shifted to 34.5°C for 10 min (72% of the cells; n = 300) or 60 min (58% of the cells; n = 300; Fig. 2 B). Sec3 remained polarized in many of the tpm1-2 tpm2Δ cells shifted to 34.5°C for 10 min (53% of the cells; n = 300) or 60 min (36% of the cells; n = 300). However, in many cases, the polarized signals in these mutant cells were not concentrated as tight patches like those observed in the control cells. We also observed flat disc-shaped signals near the mother–daughter connections in the cells after zymolyase treatment (not counted as polarized signals in the experiments). In contrast, the Rab protein Sec4 was completely depolarized in the mutant cells. Sec3-GFP was well polarized in the tpm1-2 tpm2Δ mutant cells. We noticed that some of the tpm1-2 tpm2Δ mutant cells were easily lysed during the immunofluorescence procedure. The zymolyase and SDS treatments during yeast cell wall removal and membrane permeabilization steps may have led to the partial dispersion of the Sec3 signals in tpm mutant cells. This is supported by a side-by-side comparison of the procedures for direct GFP observation and immunofluorescence in cells (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200704128/DC1). In addition, the antibodies used in Roumanie et al. (2005) and in this study were generated against different regions of Sec3, which may also contribute to the discrepancy. In fact, different antibodies and staining methods have been successfully used to reveal different pools of exocyst in mammalian cells (Yeaman et al., 2001; Oztan et al., 2007).
Figure 2.

Localization of Sec3 in the tropomyosin mutant and cells treated with latrunculin. (A) The affinity-purified anti-Sec3 antibody recognizes Sec3 in wild-type (SEC3) but not sec3 deletion (sec3Δ) cells by Western blotting (left; molecular masses indicated to the left) and immunofluorescence microscopy (right). (B) Sec3 remains polarized in tpm1-2 tpm2Δ mutant cells. The TPM1 tpm2Δ (left) and tpm1-2 tpm2Δ (right) cells were grown at 25°C and then shifted to 34.5°C for 10 and 60 min before immunostaining with anti-Sec3 or anti-Sec4 antibodies. Both Sec4 and Sec3 were polarized in TPM1 tpm2Δ cells (left). Sec4 was completely depolarized in the tpm1-2 tpm2Δ cells after the temperature shift, whereas Sec3 was mostly polarized, albeit less concentrated than the control cells. Sec3 in these mutants monitored by GFP tagging (Sec3-GFP) was also polarized. (C) Yeast cells were arrested at G0 phase and then released into fresh medium at 25°C for 90 min in the presence of 100 μM Lat B (left) or DMSO (right). Cells were fixed and then processed for immunostaining with the anti-Sec3 antibody. In cells treated with Lat B, Sec3 still formed a patch in the presumptive budding site. Actin cables were disrupted by this treatment. As controls, both Sec3 and actin were polarized in cells treated with DMSO (right). Bars, 5 μm.
To avoid the cell lysis problems, we took another approach commonly used to study the role of actin cables in the localization of proteins involved in yeast cell polarity (Ayscough et al., 1997). Cells were first arrested at G0 phase and then released to fresh medium for growth in the presence of latrunculin B (Lat B), which disrupts actin in yeast cells. Immunostaining was then performed in these cells to detect Sec3 localization. We found that although actin cables were clearly disrupted in the Lat B–treated cells, Sec3 remained polarized (66%; n = 300; Fig. 2 C).
Polarization of sec3ΔN-GFP is sensitive to latrunculin
The sec3ΔN protein, when expressed as the only copy of Sec3 in the cell, was well polarized to the bud tip (Guo et al., 2001). We compared the targeting of sec3ΔN-GFP and Sec3-GFP to the emerging bud after G0 release in the presence of latrunculin. As shown in Fig. 3 A, in cells treated with Lat B, sec3ΔN-GFP was dispersed throughout the cells. However, the full-length Sec3 still formed a patch in the presumptive buds. As a control, both Sec3-GFP and sec3ΔN-GFP were well polarized in cells treated with DMSO. These results suggest that, unlike full-length Sec3, the delivery of sec3ΔN to the bud requires intact actin cables in a fashion similar to the other exocyst components that associate with secretory vesicles (Boyd et al., 2004). Because the gic2-sec3 chimera is able to functionally replace SEC3 in yeast, we tested whether the initial targeting of gic2-sec3 to the bud is, as with the full-length Sec3, independent of actin cables. As shown in Fig. 3 B, similar to Sec3-GFP, gic2-sec3–GFP was localized to the presumptive bud sites in cells treated with Lat B. We conclude that the N terminus of Sec3 confers its actin-independent localization at the bud tip and that the interaction of Cdc42 with Sec3 is important for Sec3 targeting.
Figure 3.

The actin-independent localization of Sec3 is conferred by its N terminus. (A) The targeting of sec3ΔN to the bud tip is dependent on actin. Yeast cells expressing Sec3-GFP or sec3ΔN-GFP under the SEC3 promoter as the sole copy of Sec3 were arrested at G0. Cells were then released into fresh medium at 25°C for 90 min in the presence of 100 μM Lat B or DMSO. Cells were then fixed for fluorescence microscopy. In cells treated with Lat B (left), the full-length Sec3 formed a patch in the presumptive budding site, whereas sec3ΔN-GFP was dispersed throughout the cells (left). As controls, both Sec3-GFP and sec3ΔN-GFP were well polarized in cells treated with DMSO (right). (B) The targeting of gic2-sec3 to the bud tip is independent of actin. gic2-sec3–GFP was expressed under the SEC3 promoter in sec3Δ background. Cells were arrested at G0 phase and released into fresh medium in the presence of 100 μM Lat B. gic2-sec3–GFP was polarized at the presumptive buds when actin was disrupted. Bars, 5 μm.
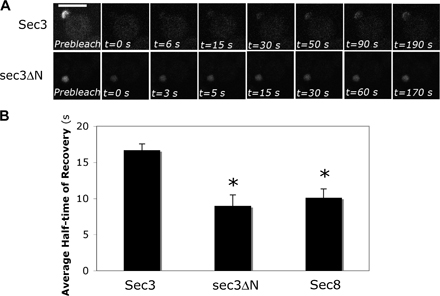
Previous FRAP analyses demonstrated that Sec3-GFP fluorescence recovery at the bud can take place even in the presence of latrunculin (Boyd et al., 2004). We asked whether sec3ΔN-GFP fluorescence could recover after photobleaching if actin is disrupted. Cells expressing Sec3-GFP and sec3ΔN-GFP under the SEC3 promoter were grown at 25°C. Small buds of these cells were bleached with laser, and pre-and postbleach images were recorded over time. As shown in Fig. 4, the full-length Sec3-GFP was able to recover its fluorescence at the bud tip in the presence of latrunculin, which is consistent with the previous observation (Boyd et al., 2004). On the contrary, sec3ΔN-GFP could not recover. As controls, both Sec3-GFP and sec3ΔN-GFP recovered in the presence of DMSO. We also compared the recovery time of Sec3-GFP and sec3ΔN-GFP (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200704128/DC1). sec3ΔN-GFP, like Sec8-GFP, reached nearly full recovery by 30 s, whereas Sec3-GFP did not fully recover until ∼90 s. The data suggest that sec3ΔN may localize to the bud tip through its association with the other exocyst components.
Figure 4.

FRAP of sec3ΔN-GFP at the bud relies on actin cables. (A) Recovery of Sec3-GFP or sec3ΔN-GFP fluorescence in cells treated with DMSO or 100 μM Lat B. Bar, 5 μm. (B) Fluorescence recovery graphs of cells expressing either Sec3-GFP (top) or sec3ΔN-GFP (bottom) treated with DMSO or Lat B. Each data point represents the mean ± SEM (n = 3; P < 0.05 for every time point after the initial two for sec3ΔN-GFP ± Lat B).
The N terminus of Sec3 directly interacts with PIP2
Sequence analysis reveals that Sec3, like Gic2, also has a cluster of basic residues, including K134, K135, K136, and R137, located at its N terminus. In many cases, clusters of basic residues are implicated in direct interaction with the negatively charged phospholipids, including PIP2 and phosphatidylserine (PS), distributed in the inner leaflet of the plasma membrane. Because Sec3 is stably localized to the bud tip membrane, it is likely that Sec3 directly interacts with phospholipids via its N-terminal basic residues. To test this, we examined the binding of recombinant Sec3N to large unilamellar vesicles (LUVs) containing various phospholipids. As shown in Fig. 5 A, Sec3N bound to LUVs containing PIP2 but not to LUVs containing phosphatidylcholine (PC). Sec3N also bound to PS; however, the binding was indistinct unless the molar ratio of PS in the LUVs was raised to 60%. Noticeably, although Sec3N only bound diminutively to 20% PS LUVs, combining PIP2 and PS (5% PIP2 + 20% PS) significantly increased the affinity of Sec3N for the LUVs. As a control, GST did not bind to LUVs of any lipid composition. To measure the affinity of these interactions, we examined the binding of Sec3N to LUVs with increasing lipid concentrations. As shown in Fig. 5 B, although Sec3N barely bound to 20% PS, the affinity of Sec3N for LUVs containing 5% PIP2 was greatly enhanced when 20% PS was added (dissociation constant [Kd] = 14.4 ± 4 μM). This result suggests that membrane association of Sec3 involves multiple Sec3–phospholipid interactions. It is possible that PS, though binding to Sec3 with low affinity, synergistically contributes to Sec3 binding to PIP2 in physiological membranes.
Figure 5.

The N terminus of Sec3 directly binds to phospholipids. (A) 0.3 μm GST-Sec3N (aa 1–320) purified from bacteria was incubated with liposomes containing 100% PC, 5% PIP2, 20% PS, 60% PS, or a combination of 5% PIP2 and 20% PS. After ultracentrifugation, proteins in supernatant (S) and pellet (P) were subjected to SDS-PAGE and visualized by SYPRO red staining (top). Sec3N bound to vesicles containing 5% PIP2 and more strongly to vesicles containing both 5% PIP2 and 20% PS. GST did not bind to LUVs with any lipid compositions (bottom). (B) 0.3 μM Sec3N was incubated with increasing concentrations of LUVs composed of 5% PIP2, 20% PS, or 5% PIP2 + 20% PS for the binding reaction. The percentage of lipid-bound Sec3N was plotted with increasing LUV concentration with a single rectangular hyperbola equation (B = BmaxX/[Kd + X]) using SigmaPlot. Each point is the mean of three measurements. Error bars represent SD. (C) Changing the polybasic region to alanine impaired Sec3N binding to phospholipids. Wild-type and mutant Sec3N (sec3N-mt) fusion proteins were incubated with 30 μM of phospholipids containing 2% PIP2 and 20% PS. Proteins in supernatant and pellet were analyzed by SDS-PAGE.
We next tested whether mutating the polybasic region of Sec3 affects its binding to phospholipids. Mutagenesis was performed to change these residues to alanine (K134A, K135A, K136A, and R137A). As shown in Fig. 5 C, mutating theseresidues impaired the ability of Sec3 to bind to LUVs containing PIP2.
Identification of residues on Sec3 that are crucial for Cdc42 binding
After finding the interaction between Sec3 and Cdc42 (Zhang et al., 2001), we performed several domain-mapping experiments to narrow down the region in Sec3 that mediates its interaction with Cdc42. We found that aa 140–155 were important for the binding (Fig. 6 A). We mutated four residues (I140A, L141A, S142A, and P145A) in this region and tested the mutant (named sec3-201) for Cdc42 binding. The wild-type and mutant forms of the Sec3 N-terminal sequence (Sec3N and sec3-201N; aa 1–320) were expressed as GST fusion proteins, and Cdc42 was expressed as a Hisx6-tagged fusion protein. These recombinant proteins were purified from bacteria and used in an in vitro binding experiment. As shown in Fig. 6 B, the wild-type Sec3N bound strongly to Cdc42 in the presence of GTPγS, which is consistent with our previous observation (Zhang et al., 2001). However, the sec3-201 mutant had almost no detectable binding to Cdc42. We also mutated four residues in the polybasic region (K134, K135, K136, and R137) into alanine (sec3-202) or negatively charged glutamic acid (sec3-203). The sec3-202 mutant (K/R→A) was able to bind to Cdc42 at a level comparable to wild-type Sec3. The sec3-203 mutant (K/R→E) had reduced binding to Cdc42. The dramatic charge reversion probably led to a certain degree of perturbation of the Sec3N structure. The sec3-201 mutant that failed in Cdc42 binding remains capable of binding to PIP2-containing lipids in the LUV sedimentation assay (Fig. 6 C).
Figure 6.

The Cdc42 binding domain and the polybasic region of Sec3 are important for Sec3 function. (A) Diagram of the Sec3 sequence containing the potential Cdc42 binding site and the polybasic region. Residues before R137 may be involved in lipid binding and residues after I140 may be involved in Cdc42 binding. (B) The sec3-201 mutant was not able to bind to Cdc42 in vitro. GST fusion proteins containing the N terminus (aa 1–320) of Sec3, sec3-201 (I140A, L141A, S142A, and P145A), sec3-202 (K134A, K135A, K136A, and R137A), and sec3-203 (K134E, K135E, K136E, and R137E) were purified and conjugated to glutathione sepharose. Cdc42 was expressed as a Hisx6 fusion and purified from bacteria. The in vitro binding assay was performed using GST-Sec3N and Hisx6-Cdc42 in the presence of GTPγS. The Hisx6-Cdc42 fusion protein bound to the GST-Sec3N sepharose was detected by Western blotting with anti-Hisx6 antibody (top). Equal amounts of wild-type and mutant Sec3 fusion proteins were used in the binding assay (bottom; Ponceau S staining). Cdc42 bound to GST-Sec3N and GST–sec3-202N but not to GST–sec3-201N. GST–sec3-203N has reduced binding to Cdc42. (C) Mutations at the Cdc42 binding domain do not impair Sec3 binding to phospholipids. GST fusion proteins of wild-type Sec3N and sec3-201N mutant were incubated with 30 μM LUV containing 2% PIP2 and 20% PS. Proteins in supernatant and pellet were analyzed by SDS-PAGE. + and −, with or without phospholipids in the protein-lipid binding assays, respectively. (D) Synthetic growth defects of sec3 mutants with exo70-38. sec3-201 has mutations within the Cdc42 binding domain. sec3-202 and sec3-203 have mutations at the polybasic region (K/R→A or K/R→E, respectively). exo70-38, exo70-38 sec3-201, exo70-38 sec3-202, and exo70-38 sec3-203 were serially diluted and spotted onto SC medium plates. Cells were incubated at 25 or 32°C for 5 d. All sec3 mutants showed clear synthetic growth defects with exo70-38 at 32°C, whereas sec3-201 had the greatest defects and was unable to survive with exo70-38 over 32°C. (E) sec3 mutants with combined mutations at both the Cdc42 binding domain and polybasic region are synthetic lethal with exo70-38. Various sec3 mutants expressed under the SEC3 promoter in CEN plasmids were introduced into exo70-38 supplemented with a CEN, URA3, SEC3 balancer. sec3-204 has combined mutations of sec3-201 and sec3-202. sec3-205 combines the mutations of sec3-201 and sec3-203. The cells were serially diluted onto SC plates with or without 5-FOA and incubated for 5 d at 25°C. The sec3-204 and sec3-205 mutants were synthetic lethal with exo70-38 at 25°C.
The polybasic region and the Cdc42-binding region of Sec3 are critical for Sec3 function
Taking advantage of the synthetic lethality assay (Fig. 1), we assessed the functional implications of the Sec3 interaction with phospholipids and Cdc42. First, SEC3 was replaced with the Cdc42-binding–deficient mutant sec3-201 in exo70-38 cells. As shown in Fig. 6 D, the double mutant exo70-38 sec3-201 was inviable at 32°C. Next, we examined the synthetic genetic interaction between exo70-38 and sec3-202 or sec3-203. Both sec3-202 and sec3-203 had clear synthetic growth defects with exo70-38 at 32°C. The K/R→E mutation in sec3-203 led to a more severe growth defect than the K/R→A mutation in sec3-202. When mutations in the Cdc42 binding domain (sec3-201) were combined with the mutations in the polybasic region (sec3-202 and sec3-203), the resulting sec3 mutants sec3-204 and sec3-205 became synthetic lethal with exo70-38, even at 25°C (Fig. 6 E). Combining these results, we conclude that the dual interactions of Sec3 with Cdc42 and phospholipids are important for Sec3 function in cells.
Cdc42 and PIP2 interactions are important for Sec3 targeting to the bud tip
Using these sec3 mutants, we asked whether the interactions of Sec3 with Cdc42 and phospholipids are important for its polarization to the bud tip. The sec3 mutants were integrated into the SEC3 locus in the yeast chromosome to replace the endogenous SEC3. These sec3 mutants were then C-terminally tagged with GFP by chromosomal integration. The cells were arrested in G0 phase and then released into fresh medium in the presence of Lat B. Polarization of sec3-201, sec3-202, and sec3-203 to the presumptive bud emergence site were all affected to various extents in the presence of Lat B (Fig. 7 A). The sec3-204 and sec3-205 mutants that combine the Cdc42-binding and phospholipid-binding mutations failed to polarize to the bud tip. Quantification of the cells that polarized, partially polarized, or depolarized Sec3-GFP is presented in Fig. 7 B. These results suggest that the Cdc42 and phospholipid interactions synergistically control the actin-independent targeting of Sec3 to the bud tip during budding.
Figure 7.

Mutations in the Cdc42 binding region or the polybasic region of Sec3 affect its targeting to the bud tip in the presence of Lat B. (A) Yeast cells expressing SEC3 or various sec3 mutants under the SEC3 promoter as the only copy of Sec3 were GFP tagged and arrested at G0 phase. Cells were then released into fresh medium with 100 μM Lat B (left) or DMSO (right) at 25°C for 90 min. Cells were fixed for fluorescence microscopy. Although the wild-type Sec3 formed a clear patch in the presumptive buds when treated with Lat B, the mutant sec3 proteins were dispersed to different extents. (B) Quantification of the percentages of Sec3-GFP cells that were polarized, partially polarized, or depolarized after treatment with Lat B (n = 300). Partially polarized was scored when the GFP signals were localized at one end of the cell but appeared as multiple patches. (C) Polarization of exo70-38 protein depends on actin. Cells expressing Exo70-GFP or exo70-38–GFP were arrested at G0 phase. These cells were released into fresh medium at 25°C for 90 min in the presence of 100 μM Lat B or DMSO and fixed for fluorescence microscopy. In cells treated with Lat B (left), the wild-type Exo70 still formed a patch in the presumptive budding site. exo70-38–GFP was, however, dispersed throughout the cells. As controls, both Exo70-GFP and exo70-38–GFP were well polarized in cells treated with DMSO. Bars, 5 μm.
Using the same method, we also examined the polarization of the exo70-38 protein in cells treated with Lat B. In cells treated with Lat B, the wild-type Exo70 still formed a patch in the presumptive budding site; however, exo70-38–GFP was dispersed throughout the cells. As controls, both Exo70-GFP and exo70-38–GFP were well polarized in cells treated with DMSO (Fig. 7 C).
The sec3 exo70-38 double mutants display severe secretion defects
We examined the secretion of the periplasmic enzyme invertase in the exo70-38 sec3-201 and exo70-38 sec3-203 double mutants. As shown in Fig. 8 A, after a 2-h shift to 35°C, the exo70-38 or sec3ΔN single mutant cells secreted >90% of the total invertase, whereas the exo70-38 sec3-201 cells and the exo70-38 sec3-203 cells only secreted 34.1 and 38.8% of the total invertase, respectively. We also examined the secretion of the cell wall modification enzyme Bgl2. As shown in Fig. 8 B, no Bgl2 accumulation was detected in the exo70-38 or exo70-38 sec3-201 cells at 25°C, and only a small amount of Bgl2 was accumulated in the exo70-38 sec3-203 cells. After a 2-h shift to 35°C, although the exo70-38 cells accumulated only a moderate amount of Bgl2, the exo70-38 sec3-201 and exo70-38 sec3-203 cells showed a much more pronounced amount of Bgl2 accumulation. As a control, the sec3ΔN cells did not accumulate Bgl2 at either 25 or 35°C. These results indicate that combining sec3-201 or sec3-203 with exo70-38 greatly aggravates the Bgl2 secretion defects. Overall, these results suggest that disruption of the Cdc42 or lipid interaction of Sec3 in the exo70-38 background blocks secretion.
Figure 8.

The exo70-38 sec3 double mutants display severe secretion defect. (A) The exo70-38 sec3 double mutants are defective in invertase secretion. The exo70-38 sec3-201 and exo70-38 sec3-203 mutants were tested for the secretion of the invertase after being shifted to the restrictive temperature of 35°C for 2 h. sec3ΔN and exo70-38 mutant strains were used as controls in the assay (n = 3). The percentage of external invertase (secreted) versus total invertase was measured. (B) The exo70-38 sec3 double mutants display aggravated defects in Bgl2 secretion. Western blot analysis of the internal and external pools of Bgl2 in sec3ΔN, exo70-38, exo70-38 sec3-201, and exo70-38 sec3-203 cells. Cells were either grown at 25°C or shifted to 35°C for 2 h. Alcohol dehydrogenase (ADH) was used as a control to show that equal amounts of proteins were loaded. (C) exo70-38 sec3 double mutants accumulate a large amount of secretory vesicles at the restrictive temperature. The sec3ΔN, exo70-38, exo70-38 sec3-201, and exo70-38 sec3-203 cells were grown to early log phase at 25°C (top), and then shifted to 35°C for 2 h and processed for thin-section EM. Bars, 500 nm. (D) Quantification of the number of secretory vesicles per section in the single and double mutant cells (n = 30; P < 0.01). Error bars represent SD.
We also performed thin-section electron microscopy on these mutants. As shown in Fig. 8 (C and D), the exo70-38 cells began to accumulate vesicles (100 ± 38 vesicles/section) at 35°C, whereas the exo70-38 sec3-201 and exo70-38 sec3-203 double mutants accumulated much larger amounts of vesicles (366 ± 84 and 330 ± 69 vesicles/section, respectively). The double mutants accumulated vesicles even at 25°C. The sec3ΔN cells did not accumulate vesicles at any temperature tested. These results clearly show that the secretion defects are aggravated when the Cdc42–Sec3 or lipid–Sec3 interaction is disrupted in exo70-38 cells.
Exocyst localization and morphologies of the exo70-38 sec3 double mutants
To examine the localization of exocyst components in the mutant cells, we GFP tagged the exocyst components Sec3, 5, and 8 and Exo84 by chromosomal integration in the single or double mutants of exo70 and sec3. As shown in Fig. 9, at 35°C, in the sec3 and exo70 single mutants, the exocyst components were polarized to the bud tip. However, in the exo70-38 sec3-201 and exo70-38 sec3-203 double mutants, the exocyst proteins were either completely depolarized or diffused inside or in the vicinity of the daughter cells. The assembly of the exocyst complex was mostly unaffected in the exo70-38 sec3ΔN double mutants (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200704128/DC1). In addition, Sec4 and actin are mostly polarized in these mutants, whereas Sec4 is less concentrated in the bud tip (Fig. S4).
Figure 9.

Localization of the exocyst components in the exo70-38 sec3 double mutants. The exocyst components in exo70-38, sec3-201, sec3-203, and exo70-38 sec3 double mutant cells were GFP tagged by chromosomal integration. The cells were shifted from 25 to 35°C for 2 h before fluorescence microscopy. GFP-tagged Sec3, 5, and 8 and Exo84 remained polarized to the bud tip in all single mutants but were no longer polarized in double mutants exo70-38 sec3-201 (top) and exo70-38 sec3-203 (bottom) after the temperature shift. Bars, 5 μm.
Because the correct targeting of the exocytic vesicles to the bud tip is required for polarized growth of the yeast cells, disrupting the interaction of Sec3 with Cdc42 or phospholipids in the exo70 mutant background would affect cell morphogenesis. As shown in Fig. 10 A, at 25°C, in contrast to the ellipsoidal shapes of the single mutants, the exo70-38 sec3 double mutants are significantly rounder. The double mutants have smaller axial ratios (length/width) compared with the single mutants (Fig. 10 B). The rounder morphology in the mutant cells suggest that although some vesicles are delivered to the daughter cells, their tethering is not restricted to the bud tip. Instead, vesicles are diffusely tethered to the daughter cell plasma membrane resulting in isotropic daughter cell expansion. When these round daughter cells reach the mother stage, the mother cells also appear round in shape. In addition to the rounder shape, the double mutant cells are also clearly larger in size (approximately twice as large as either the exo70-38 or the sec3 single mutants). This morphology suggests that some of the secretion occurs even in mother cells, leading to isotropic mother cell surface expansion. The cell biological characterization of the various sec3 mutant alleles generated in this study is summarized in Table I.
Figure 10.

Morphological defects of the exo70-38 sec3 double mutants. (A) Morphology of sec3ΔN, exo70-38, exo70-38 sec3-201, and exo70-38 sec3-203. The exo70 sec3 double mutants were significantly larger and rounder than each single mutant, even at 25°C. Bar, 5 μm. (B) Quantification of the mean axial ratios (length/width) of mother cells from each indicated strains. Error bars represent SD among measured samples. Asterisks represent significant difference between mutants and control cells (n = 30; P < 0.05).
Table I. Summary of the phenotypic characterization of the sec3 mutants.
|
sec3 single mutants
|
sec3 exo70-38 double mutants
|
|||||
|---|---|---|---|---|---|---|
| Mutation sites |
Exocyst localization |
Lat B treatment |
Synthetic effects |
Exocyst localization |
Vesicle accumulation |
|
| SEC3 | N/A | Polarized | 66% polarized | None | Polarized | 100 ± 38 (35°C) (in exo70-38 single mutant) |
| sec3-201 | I140A, L141A, S142A, P145A |
Polarized | 25.7% polarized | Inviable >32°C | Depolarized | 366 ± 80 (35°C) |
| sec3-202 | K134A, K135A, K136A, R137A |
Polarized | 31.9% polarized | Inviable >35°C | Depolarized | ND |
| sec3-203 | K134E, K135E, K136E, R137E |
Polarized | 27.9% polarized | Inviable >33°C | Depolarized | 330 ± 69 (35°C) |
| sec3-204 | K134A, K135A, K136A, R137A, I140A, L141A, S142A, P145A. |
Polarized | 11.7% polarized | Synthetic lethal | ND | ND |
| sec3-205 | K134E, K135E, K136E, R137E, I140A, L141A, S142A, P145A |
Polarized | 6.8% polarized | Synthetic lethal | ND | ND |
Discussion
The exocyst mediates the tethering of post-Golgi secretory vesicles at specific areas of the plasma membrane (Pfeffer, 1999; Guo et al., 2000; Whyte and Munro, 2002; Hsu et al., 2004). As vesicle tethering takes place at a step before vesicle docking and fusion, regulation of the exocyst is crucial to the spatial and temporal control of exocytosis in the cell. Cdc42 is a master regulator of yeast cell polarity. It not only interacts with cytoskeleton regulators for polarized actin organization (for review see Pruyne et al., 2004) but also regulates exocytosis (Adamo et al., 2001; Zhang et al., 2001; Roumanie et al., 2005). However, the molecular mechanism by which Cdc42 regulates exocytosis is unclear. We have previously found that the GTP-bound form of Cdc42 directly interacts with Sec3 through its N terminus (Zhang et al., 2001). However, as deletion of this region (sec3ΔN) does not cause any detectable secretion or growth defects, we were unable to assess the functional implication of this interaction. In this study, we found that sec3ΔN was synthetic lethal with the exo70-38 mutant, and this synthetic lethality provided us with an effective assay to study the function of the Cdc42–Sec3 interaction in yeast. We demonstrate that Cdc42 spatially and functionally regulates Sec3 in exocytosis and polarized cell growth. This study for the first time establishes the functional significance of the Sec3–Cdc42 interaction in the cell.
These genetic analyses, together with the sequence comparison of the N terminus of Sec3 with that of Gic2, have also led to the identification of a polybasic region in Sec3 that is crucial for its function. Using the vesicle sedimentation assay, we found that the polybasic region interacts with PIP2. Disruption of the Sec3–PIP2 interaction (sec3-202 and sec3-203) affects Sec3 function. What is the functional implication of this interaction? One possibility is that this interaction may serve to make Sec3 more accessible as a downstream effector of Cdc42 at the plasma membrane. However, blocking Cdc42 binding in the sec3-201 mutant affected, but did not totally abolish, the function of Sec3N. Only when PIP2 and Cdc42 binding were both disrupted did the sec3 mutants (sec3-204 and sec3-205) become synthetic lethal with exo70-38. Thus, both Cdc42 and PIP2 are needed for the complete function of Sec3N. Another possibility is that PIP2 acts in concert with Cdc42 to coactivate Sec3. The synergy in which PIP2 acts together with the GTP-binding proteins to activate downstream effectors has been observed in other biological situations. One well-characterized example is neural Wiskott-Aldrich syndrome protein (N-WASP), which binds to both PIP2 and Cdc42. Both interactions are required for the full activation of N-WASP for subsequent Arp2/3 activation and actin polymerization (Prehoda et al., 2000; Rohatgi et al., 2000). Unlike N-WASP, there is no in vitro enzymatic approach to assay Sec3 function at present. However, assays for proper secretion and the polarized localization of Sec3 in yeast provided readouts that strongly support the role of Cdc42 and PIP2 in controlling Sec3 function in exocytosis. Finally, it is also possible that the interaction of Sec3 with Cdc42, a signaling GTPase, may not be sufficient for the stable association of Sec3 with the plasma membrane. PIP2 and Cdc42 may act together to control the localization and function of Sec3 at the bud tip membrane. Our localization studies demonstrate that the N terminus of Sec3 is important for its polarization when actin is disrupted. Our detailed mutagenesis analyses of the polybasic or the Cdc42 binding region of full-length Sec3 further indicate that PIP2 binding and Cdc42 interaction control the localization of Sec3 to the bud tip membrane. Overall, we found that the interaction of Sec3 with Cdc42 and PIP2 is important for its function in exocytosis and cell polarity. Besides Cdc42, Sec3 also binds to Rho1 (Guo et al., 2001). Cdc42 and Rho1 compete for their binding to Sec3 in vitro (Zhang et al., 2001), suggesting that their controls over Sec3 are executed at different times or under different physiological conditions. It is known that the major role of Rho1 is to coordinate cellular processes during yeast cell wall stress response and remodeling (Levin, 2005). Experiments are being performed to analyze the role of Rho1 and exocytosis during these processes.
Analysis of the mammalian Sec3 N-terminal sequence (aa 1–125) suggests that it forms a structure consisting of α strands interspersed with β sheets, which is similar to that of yeast Sec3 aa 75–240, which contains the Cdc42 and PIP2-binding regions (Xu, Z., personal communication). Future functional and structural analyses of the mammalian and yeast Sec3 will help us understand the conservation of exocyst function in vesicle tethering across species.
Materials and methods
Plasmids and yeast strains
All mutagenesis was performed using the QuikChange site-directed mutagenesis kit (Stratagene) and verified by sequencing. sec3ΔN (nt 922–4011) was placed behind the native SEC3 promoter in a CEN plasmid (LEU2). The resulting plasmid (pG1215) was used as a backbone for later mutagenesis and gic2-sec3 chimera construction. Various gic2 N-terminal (gic2NT) mutants were generated from a GIC2 2μ plasmid by PCR using different mutagenesis primers. PCR fragments were placed in front of sec3ΔN in frame to make the final gic2-sec3 chimera constructs. We also mutated key residues in the CRIB domain that have been previously shown to be essential for Cdc42 interaction (Burbelo et al., 1995) and used these in the chimera construction. Various sec3 mutants were generated based on a SEC3 plasmid with the native SEC3 promoter in a CEN plasmid (LEU2). The sec3 constructs harboring various mutations were summarized in Table I and Table S1 (available at http://www.jcb.org/cgi/content/full/jcb.200704128/DC1).
Standard methods were used for yeast media and genetic manipulations. To make various exo70 sec3 double mutants, a SEC3 URA3 CEN plasmid (pG1216) was transformed into the exo70-38 cells as a balancer, and then the endogenous SEC3 was deleted with a PCR product containing the SEC3 promoter and terminator flanking the Kanamycin gene (sec3∷KanR). The resulting strain (GY2650) was transformed with various sec3 mutants and gic2-sec3 chimera constructs, and the transformants were tested for growth at different temperatures upon losing the SEC3 balancer on 5-FOA plates. For GFP tagging of the sec3 mutants, the sec3 mutants were first integrated into the SEC3 locus in the yeast chromosome to replace the endogenous SEC3 using genetic methods as previously described (Zhang et al., 2001). The GFP sequence was then introduced to the C terminus of sec3 by integration (Finger et al., 1998). All of the major strains used in this study are listed in Table S1.
Secretion assays
To measure the total and external invertase activities, cells were grown overnight at 25°C in yeast extract/peptone/dextrose medium to early log phase. For each sample, part of the cells was immediately pelleted, resuspended in ice-cold buffer containing 10 mM NaN3 and 10 mM Tris-HCl (pH 7.5), and stored on ice as the 0-h control. The remaining cells were incubated in YP plus 0.1% glucose for 2 h at 25 or 35°C for invertase induction. After the temperature shift, the 0 and 2-h samples were collected and diluted into 0.6 OD600. Total invertase level was determined by subjecting equal volumes of samples to a beads beater to release the extract from the cells. Intact cells not subject to bead beating were used to measure external invertase. Total and external invertase activities were measured at the beginning and end of the shift. The percentage of invertase secretion was calculated using the following equation: % secretion = Δexternal/Δtotal. The Bgl2 secretion assay was performed as described in He et al. (2007a).
Microscopy
Chromosomal tagging of the exocyst components by GFP was performed as previously described (Finger et al., 1998; Guo et al., 1999a). Cells were grown to early log phase (0.6 OD600) in synthetic complete (SC) media and fixed by methanol/acetone before microscopy (Zajac et al., 2005). The signals were scored as mislocalized in small-budded cells when they appeared diffused or in multiple patches in the mother cells. For immunofluorescence staining, cells were fixed with 4.4% formaldehyde for 1 h and then spheroplasted with 1 mg/ml zymolyase for 30–45 min. The cells were permeabilized with 0.3% SDS and subsequently incubated with primary antibodies at 4°C overnight. Affinity-purified anti-Sec3 polyclonal antibody (a gift from D. TerBush, Uniformed Services University of Health Sciences, Bethesda, MD) was used at 1:100 dilution and anti-Sec4 polyclonal antibody was used at 1:1,000 dilution. The Alexa Fluor 488–conjugated goat anti–rabbit IgG antibody was used as the secondary antibody. For actin staining, cells were stained with Alexa Fluor 594 phalloidin after fixation and permeabilization. Latrunculin treatment and protein localization in yeast cells released from G0 was examined as previously described (Ayscough et al., 1997; Zajac et al., 2005). All images were captured by a microscope (DM IRB; Leica) using a 100× oil immersion objective and a high resolution charge-coupled device camera (ORCA-ER; Hamamatsu Photonics). Immunofluorescence signals were quantified as pixels using OpenLab 5.0.2 software (Improvision).
The dynamic localization of GFP-tagged Sec3, sec3ΔN, and Sec8 in yeast bud was analyzed by FRAP. Cells were grown overnight in SC media at 25°C, diluted to 0.6 OD600 the next morning, and grown for an additional 1.5–2 h. 1 or 2 ml of culture was pelleted and resuspended in 20 μl of fresh SC media. Slides containing agar pads were prepared according to the general protocol from Tran et al. (2004), with 2% agar dissolved in SC medium containing the appropriate amino acids. 2 μl of the suspension was dropped onto the agar pads, coverslips were placed on top of the agar pads, and a mixture of Vaseline, lanolin, and candle wax (a modified form of VALAP) was used to seal in the cells. Bleaching and imaging was performed using a laser scanning confocal microscope (TCS SL; Leica) with an oil immersion 63× objective. For each cell, three prebleach images were taken, and then small buds were photobleached using an argon laser at 488 nm. Bleaching was done at 100% intensity and cells underwent three bleaches. To observe both fast and slow recoveries, postbleach images were collected every 3 s for 30 s and then every 20 s for ∼3 min. For each time point, the background intensity was subtracted, and in some cases the data were normalized by dividing each data point by the mean prebleached value. The intensity versus time was plotted, and the graph was used to measure the time when the cells reach half of the final intensity (half-life).
Yeast cell electron microscopy was performed as previously described (Zhang et al. 2005b). All of the statistical analyses were performed using the Student's t test.
In vitro binding between the N terminus of Sec3 and Cdc42
Wild-type and mutant forms of Sec3 N terminus (Sec3N; aa 1–320) were expressed as GST fusion proteins. Cdc42 was expressed as Hisx6-tagged fusion protein (Hisx6-Cdc42). 0.3 μM each of the purified recombinant fusion proteins was used in the in vitro binding experiment, as previously described (Zhang et al., 2001).
LUV sedimentation assay
LUV sedimentation assay was performed as previously described (Hokanson and Ostap, 2006; He et al., 2007b). The percentages of PS and PIP2 indicated in the text are the molar percentages of total PS and PIP2, with the remainder being PC. Lipid concentrations are given as total lipid. The binding of Sec3N and Sec3N mutants to LUVs was determined by sedimentation assays conducted in 200 μl of total volume in an ultracentrifuge rotor (TLA-100; Beckman Coulter). The tubes were preincubated for 1 h in a 50-μM solution of PC in the HNa100 buffer to prevent nonspecific binding of Sec3 to polycarbonate centrifuge tubes. Sucrose-loaded LUVs were precipitated at 150,000 g for 30 min at 25°C. The supernatants and pellets were subjected to 12% SDS-PAGE and stained with SYPRO-red (Invitrogen) for quantification of free and bound materials with Image Quant software (Molecular Dynamics, Inc.).
Online supplemental material
Fig. S1 shows side-by-side comparison of Sec3 localization in cells as detected by immunofluorescence imaging and GFP imaging. Fig. S2 shows the different recovery of Sec3-GFP and sec3ΔN-GFP fluorescence after photobleaching in yeast cells. Fig. S3 shows the composition of the exocyst complex in the exo70-38 sec3 double mutants. Fig. S4 shows the localization of Sec4 and F-actin in the exo70-38 sec3 double mutants. Table S1 shows the major yeast stains and their genotypes used in this study. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200704128/DC1.
Supplemental Material
Acknowledgments
We are grateful to Drs. Michael Ostap and David Hokanson for their advice on lipid binding experiments, Drs. John Murray and Lee Peachy for their advice with the FRAP experiments, and Dr. Dan TerBush for sharing anti-Sec3 antibodies and comparing immunofluorescence results before publication.
This work is supported by National Institutes of Health (RO1-GM64690) and Pew Scholars Program grants to W. Guo. X. Zhang is partially supported by American Heart Association.
Abbreviations used in this paper: 5-FOA, 5–fluoroorotic acid; CRIB, Cdc42/Rac interactive binding; FRAP, fluorescence recovery after photobleaching; Lat B, latrunculin B; LUV, large unilamellar vesicle; N-WASP, neural Wiskott-Aldrich syndrome protein; PC, phosphatidylcholine; PIP2, phosphatidylinositol 4,5-bisphosphate; PS, phosphatidylserine; SC, synthetic complete.
References
- Adamo, J.E., G. Rossi, and P. Brennwald. 1999. The Rho GTPase Rho3 has a direct role in exocytosis that is distinct from its role in actin polarity. Mol. Biol. Cell. 10:4121–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamo, J.E., J.J. Moskow, A.S. Gladfelter, D. Viterbo, D.J. Lew, and P.J. Brennwald. 2001. Yeast Cdc42 functions at a late step in exocytosis, specifically during polarized growth of the emerging bud. J. Cell Biol. 155:581–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayscough, K.R., J. Stryker, N. Pokala, M. Sanders, P. Crews, and D.G. Drubin. 1997. High rates of actin filament turnover in budding yeast and roles for actin in establishment and maintenance of cell polarity revealed using the actin inhibitor latrunculin-A. J. Cell Biol. 137:399–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd, C., T. Hughes, M. Pypaert, and P. Novick. 2004. Vesicles carry most exocyst subunits to exocytic sites marked by the remaining two subunits, Sec3p and Exo70p. J. Cell Biol. 167:889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, J.L., M. Jaquenoud, M.P. Gulli, J. Chant, and M. Peter. 1997. Novel Cdc42-binding proteins Gic1 and Gic2 control cell polarity in yeast. Genes Dev. 11:2972–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbelo, P.D., D. Drechsel, and A. Hall. 1995. A conserved binding motif defines numerous candidate target proteins for both Cdc42 and Rac GTPases. J. Biol. Chem. 270:29071–29074. [DOI] [PubMed] [Google Scholar]
- Chen, G.C., Y.J. Kim, and C.S. Chan. 1997. The Cdc42 GTPase-associated proteins Gic1 and Gic2 are required for polarized cell growth in Saccharomyces cerevisiae. Genes Dev. 11:2958–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger, F.P., T.E. Hughes, and P. Novick. 1998. Sec3p is a spatial landmark for polarized secretion in budding yeast. Cell. 92:559–571. [DOI] [PubMed] [Google Scholar]
- Guo, W., A. Grant, and P. Novick. 1999. a. Exo84p is an exocyst protein essential for secretion. J. Biol. Chem. 274:23558–23564. [DOI] [PubMed] [Google Scholar]
- Guo, W., D. Roth, C. Walch-Solimena, and P. Novick. 1999. b. The exocyst is an effector for Sec4p, targeting secretory vesicles to sites of exocytosis. EMBO J. 18:1071–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, W., M. Sacher, J. Barrowman, S. Ferro-Novick, and P. Novick. 2000. Protein complexes in transport vesicle targeting. Trends Cell Biol. 10:251–255. [DOI] [PubMed] [Google Scholar]
- Guo, W., F. Tamanoi, and P. Novick. 2001. Spatial regulation of the exocyst complex by Rho1 GTPase. Nat. Cell Biol. 3:353–360. [DOI] [PubMed] [Google Scholar]
- He, B., F. Xi, J. Zhang, D. TerBush, X. Zhang, and W. Guo. 2007. a. Exo70p mediates the secretion of specific exocytic vesicles at early stages of the cell cycle for polarized cell growth. J. Cell Biol. 176:771–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, B., F. Xi, X. Zhang, J. Zhang, and W. Guo. 2007. b. Exo70 interacts with phospholipids and mediates the targeting of the exocyst to the plasma membrane. EMBO J. 26:4053–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hokanson, D.E., and M. Ostap. 2006. Myo1c binds tightly and specifically to phosphatidylinositol 4,5-bisphosphate and inositol 1,4,5-trisphosphate. Proc. Natl. Acad. Sci. USA. 103:3118–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, S.C., D. TerBush, M. Abraham, and W. Guo. 2004. The exocyst complex in polarized exocytosis. Int. Rev. Cytol. 233:243–265. [DOI] [PubMed] [Google Scholar]
- Levin, D.E. 2005. Cell wall integrity signaling in Saccharomyces cerevisiae.Microbiol. Mol. Biol. Rev. 69:262–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J. X. Zuo,P. Yue,and W. Guo,2007. Phosphatidylinositol 4,5-biphosphate mediates the targeting of the exocyst to the plasma membrane for exocytosis in mammalian cells. Mol. Biol. Cell. 18:4483–4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munson, M., and P. Novick. 2006. The exocyst defrocked, a framework of rods revealed. Nat. Struct. Mol. Biol. 13:577–581. [DOI] [PubMed] [Google Scholar]
- Novick, P., C. Field, and R. Schekman. 1980. Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell. 21:205–215. [DOI] [PubMed] [Google Scholar]
- Oztan, A., M. Silvis, O.A. Weisz, N.A. Bradbury, S.C. Hsu, J.R. Goldenring, C. Yeaman, and G. Apodaca. 2007. Exocyst requirement for endocytic traffic directed toward the apical and basolateral poles of polarized MDCK cells. Mol. Biol. Cell. 18:3978–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer, S.R. 1999. Transport-vesicle targeting: tethers before SNAREs. Nat. Cell Biol. 1:E17–E22. [DOI] [PubMed] [Google Scholar]
- Prehoda, K.E., J.A. Scott, R.D. Mullins, and W.A. Lim. 2000. Integration of multiple signals through cooperative regulation of the N-WASP-Arp2/3 complex. Science. 290:801–806. [DOI] [PubMed] [Google Scholar]
- Pruyne, D.W., D.H. Schott, and A. Bretscher. 1998. Tropomyosin-containing actin cables direct the Myo2p-dependent polarized delivery of secretory vesicles in budding yeast. J. Cell Biol. 143:1931–1945. [DOI] [PubMed] [Google Scholar]
- Pruyne, D., A. Legesse-Miller, L. Gao, Y. Dong, and A. Bretscher. 2004. Mechanisms of polarized growth and organelle segregation in yeast. Annu. Rev. Cell Dev. Biol. 20:559–591. [DOI] [PubMed] [Google Scholar]
- Robinson, N.G., L. Guo, J. Imai, E.A. Toh, Y. Matsui, and F. Tamanoi. 1999. Rho3 of Saccharomyces cerevisiae, which regulates the actin cytoskeleton and exocytosis, is a GTPase which interacts with Myo2 and Exo70. Mol. Cell. Biol. 19:3580–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi, R., H.Y. Ho, and M.W. Kirschner. 2000. Mechanism of N-WASP activation by CDC42 and phosphatidylinositol 4, 5-bisphosphate. J. Cell Biol. 150:1299–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roumanie, O., H. Wu, J.N. Molk, G. Rossi, K. Bloom, and P. Brennwald. 2005. Rho GTPase regulation of exocytosis in yeast is independent of GTP hydrolysis and polarization of the exocyst complex. J. Cell Biol. 170:583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TerBush, D.R., and P. Novick. 1995. Sec6, Sec8, and Sec15 are components of a multisubunit complex which localizes to small bud tips in Saccharomyces cerevisiae. J. Cell Biol. 130:299–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran, P.T., A. Paoletti, and F. Chang. 2004. Imaging green fluorescent protein fusions in living fission yeast cells. Methods. 33:220–225. [DOI] [PubMed] [Google Scholar]
- Whyte, J.R., and S. Munro. 2002. Vesicle tethering complexes in membrane traffic. J. Cell Sci. 115:2627–2637. [DOI] [PubMed] [Google Scholar]
- Yeaman, C., K.K. Grindstaff, J.R. Wright, and W.J. Nelson. 2001. Sec6/8 complexes on trans-Golgi network and plasma membrane regulate late stages of exocytosis in mammalian cells. J. Cell Biol. 155:593–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zajac, A., X. Sun, J. Zhang, and W. Guo. 2005. Cyclical regulation of the exocyst and cell polarity determinants for polarized cell growth. Mol. Biol. Cell. 16:1500–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X., E. Bi, P. Novick, L. Du, K.G. Kozminski, J. Lipschultz, and W. Guo. 2001. Cdc42 interacts with the exocyst and regulates polarized secretion. J. Biol. Chem. 276:46745–46750. [DOI] [PubMed] [Google Scholar]
- Zhang, X., A. Zajac, J. Zhang, P. Wang, M. Li, J. Murray, D. Terbush, and W. Guo. 2005. a. The critical role of Exo84p in the organization and polarized localization of the exocyst complex. J. Biol. Chem. 280:20356–20364. [DOI] [PubMed] [Google Scholar]
- Zhang, X., P. Wang, A. Gangar, J. Zhang, P. Brennwald, D. Terbush, and W. Guo. 2005. b. Lethal giant larvae proteins interact with the exocyst complex and are involved in polarized exocytosis. J. Cell Biol. 170:273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}