

Abstract
Prohibitin 1 (PHB1) is a highly conserved protein that is mainly localized to the inner mitochondrial membrane and has been implicated in regulating mitochondrial function in yeast. Because mitochondria are emerging as an important regulator of vascular homeostasis, we examined PHB1 function in endothelial cells. PHB1 is highly expressed in the vascular system and knockdown of PHB1 in endothelial cells increases mitochondrial production of reactive oxygen species via inhibition of complex I, which results in cellular senescence. As a direct consequence, both Akt and Rac1 are hyperactivated, leading to cytoskeletal rearrangements and decreased endothelial cell motility, e.g., migration and tube formation. This is also reflected in an in vivo angiogenesis assay, where silencing of PHB1 blocks the formation of functional blood vessels. Collectively, our results provide evidence that PHB1 is important for mitochondrial function and prevents reactive oxygen species–induced senescence and thereby maintains the angiogenic capacity of endothelial cells.
Introduction
In the vascular system, mitochondria play an important role, not only as the powerhouse of the cell, but also as an important regulator of vascular function (Moncada and Erusalimsky, 2002; Quintero et al., 2006; Madamanchi and Runge, 2007). Functional mitochondria consume oxygen to form ATP but also produce a certain amount of superoxide (O2−) during respiration. Within mitochondria, complex I and III have been shown to be the sites of superoxide production (Boveris et al., 1976; Takeshige and Minakami, 1979). Under normal physiological conditions, the amount of superoxide produced is estimated to be ∼1% of oxygen uptake (Du et al., 1998). O2− can be further converted to hydrogen peroxide (H2O2) or react with nitric oxide (NO) to form peroxynitrite (NOO−; Cai, 2005). All these reactive oxygen species (ROS) have been proven to act as signaling molecules impacting many basic cellular functions such as proliferation and apoptosis. For example, H2O2 has been shown to activate various pathways in endothelial cells (ECs) and smooth muscle cells (SMCs) including PKC, MAPK, phosphatidyl inositol 3-kinase (PI3-K)/protein kinase B (Akt), p38 MAPK as well as Ca2+ signaling, thereby modulating vascular homeostasis (Yang et al., 1998; Connor et al., 2005; Lucchesi et al., 2005; Oeckler et al., 2005). When mitochondrial function is affected, mitochondrial ROS production can be increased and hence influence the described pathways to trigger cellular dysfunction (Gutierrez et al., 2006). For example, ROS produced from mitochondria has been shown to contribute to EC dysfunction and the progression of atherosclerosis (Madamanchi and Runge, 2007). Reducing mitochondrial ROS levels by overexpressing thioredoxin in the mitochondria of ECs has been shown to improve EC function and reduce atherosclerosis (Zhang et al., 2007). Thus, there is compelling evidence that mitochondria are essential for vascular homeostasis.
A protein that has been shown to be important for maintaining mitochondrial function in lower organisms is prohibitin-1 (PHB1). PHB1 forms a high molecular complex with PHB2 in the inner mitochondrial membrane (Tatsuta et al., 2005; Ahn et al., 2006); this complex exerts a chaperonelike function for newly synthesized mitochondrial proteins and thereby regulates mitochondrial function and biogenesis in yeast (Steglich et al., 1999). Additionally, its deficiency is associated with defective mitochondrial biogenesis in Caenorhabditis elegans (Artal-Sanz et al., 2003), yet the importance of PHB1 in mammalian mitochondria remains elusive. However, PHB1 expression could be linked to cellular senescence, a stage of low metabolic turnover caused either by telomere shortening or DNA-damaging stress factors such as ROS (Passos and Von Zglinicki, 2006). PHB1 is down-regulated in aging animal cells as well as human fibroblasts (Coates et al., 2001) and deletion mutants or knockdown of prohibitins in yeast, bacteria, and plants display a senescence-like phenotype (Roskams et al., 1993; Coates et al., 1997; Piper et al., 2002; Chen et al., 2005). Additionally, the expression of PHB1 is also down-regulated after induction of oxidative stress in epithelial cells in vitro as well as in diseases linked to enhanced ROS such as ulcerative colitis and Crohn's disease (Theiss et al., 2007). Cellular senescence, increased ROS, and mitochondrial dysfunction are associated with vascular disease (Minamino and Komuro, 2007), however, it is not known if PHB1 is able to collectively control these events or have an impact on vascular homeostasis.
Here, we show that PHB1 is amply expressed in mitochondria of ECs and, by regulating mitochondrial integrity, ROS production via complex I, and delaying senescence in EC, controls signaling pathways that govern diverse functions of the endothelium such as cell proliferation, migration, tube formation, and angiogenesis in vivo.
Results
PHB1 is expressed in the vascular system and localizes to mitochondria
Initially, the expression of PHB1 in the vascular system was examined in vivo and in vitro. The descending aorta of C57B6 mice was isolated and sections analyzed by indirect immunofluorescence for PHB1. As seen in Fig. 1 A, PHB1 was found in SMCs (Fig. 1 A, arrows) as well as ECs (arrowheads) in the vessel wall labeled with platelet EC adhesion molecule 1 (PECAM-1), a marker protein for ECs (Fig. 1 B). Prohibitins have been identified in several intracellular locations including mitochondria, nuclei, lipid rafts, and the cytoplasm, depending on the cell type analyzed (Ikonen et al., 1995; Wang et al., 2002a; Morrow and Parton, 2005; Rastogi et al., 2006b). To examine the localization of PHB1 in EC, immunofluorescence microscopy of PHB1 in bovine aortic ECs (BAECs) was performed. A small fraction of PHB1 was localized to nuclear regions, however, the main fraction of PHB1 colocalized with the vital mitochondrial marker MitoTracker (Fig. 1 B) throughout the cells. PHB1 was also detectable by Western blotting in BAECs and mouse aortic and carotid arterial lysates (Fig. 1 C). PHB1 was also present in a human EC line (EA.hy926), human umbilical vein ECs (HUVECs), and mouse lung ECs (unpublished data).
Figure 1.

Expression of PHB1 in the vascular system. (A) Mouse aortas were paraffin embedded and sectioned and PHB1 protein expression was analyzed by immunofluorescence (red) microscopy compared with PECAM-1 (as an EC marker, green) with a 20× objective. Arrows and arrowheads depict PHB1 in SMCs and ECs, respectively. Bars, 10 μm. (B) Localization of PHB1 (green) was analyzed in ECs by indirect immunofluorescence compared with MitoTracker (red) and colocalization is shown (orange; blue, DAPI). Bars, 10 μm.(C) Western blot analysis of PHB1 in lysates from BAECs and mouse aorta and carotid arteries.
Knockdown of PHB1 causes a senescence-like phenotype in ECs
Because mitochondrial localized PHB1 is involved in regulating mitochondrial function in yeast (Roskams et al., 1993; Steglich et al., 1999; Piper et al., 2002) and loss of PHB1 homologues delays cellular senescence in yeast, bacteria, and plants (Roskams et al., 1993; Piper et al., 2002; Chen et al., 2005), we sought to examine the function of mitochondrial PHB1 in ECs using an RNAi approach to reduce PHB1 protein levels. Transfection of ECs with control RNAi had no effect on the levels of PHB1, whereas sequence-specific RNAi to PHB1 markedly reduced PHB1 levels (Fig. 2 A). Knockdown of PHB1 in ECs changed their morphology to a more flat but densely packed morphology that is a characteristic of senescent cells (Fig. 2 B). To examine if the loss of PHB1 induces cellular senescence in ECs, we tested for several hallmarks for senescence upon knockdown of PHB1. As one functional hallmark of cellular senescence, knockdown of PHB1 resulted in a decrease in cellular proliferation (Fig. 2 C) that was caused by an increase in both G1 and sub-G1 populations of the cell cycle with slightly enhanced apoptosis (Table I). The loss of PHB1 also increased the expression of the negative regulators of cell cycle progression p16 and p21, which are biochemical markers of cell senescence (Fig. 2 D; Alcorta et al., 1996). Finally as a well-accepted biomarker of cellular senescence, we tested for senescence-associated β-galactosidase expression at pH 6 in ECs (Dimri et al., 1995). Although hardly any positive staining was detectable under control conditions, the cells depleted of PHB1 showed a significant increase in β-galactosidase expression (Fig. 2, E and F).
Figure 2.

RNAi-mediated knockdown of PHB1 causes a senescence-like phenotype in ECs. (A) Western blot analysis of PHB1 and β-actin in EC lysates from control and PHB1 knockdown cells. (B) Cell morphology was analyzed by live imaging phase contrast microscopy. Bars, 50 μm. (C) Proliferation assays were performed after knockdown of PHB1 for the indicated time points and compared with control cells. n = 6; *, P < 0.05. (D) Expression of p21, p16, and β-actin in PHB1 knockdown and control HUVECs was determined by Western blotting. (E) BAECs transfected with control or PHB1-specific siRNA were stained for β-galactosidase with x-gal at pH 6 and images were taken using a 10× phase contrast objective. Bars, 100 μm. (F) β-galactosidase–positive cells were quantified by counting positive and negative cells in three random vision fields and averaging the results. Error bars show the calculated standard deviation. n = 3; *, P < 0.05.
Table I. Cell cycle analysis of PHB1 knockdown cells.
| 24 h | 48 h | 72 h | |
|---|---|---|---|
| Control RNAi | |||
| G1 | 29.94 ± 3.50 | 52.47 ± 3.20 | 61.99 ± 3.93 |
| S | 48.86 ± 2.36 | 32.52 ± 2.51 | 24.74 ± 2.09 |
| G2 | 19.32 ± 0.67 | 11.98 ± 1.29 | 9.36 ± 1.39 |
| Sub-G1 | 1.89 ± 0.49 | 3.03 ± 0.72 | 3.90 ± 0.61 |
| PHB RNAi | |||
| G1 | 38.52 ± 1.16 | 76.71 ± 2.72 | 78.48 ± 1.84 |
| S | 41.71 ± 1.73 | 8.80 ± 1.40 | 7.35 ± 0.99 |
| G2 | 17.24 ± 0.97 | 7.56 ± 0.74 | 5.09 ± 0.60 |
| Sub-G1 | 2.53 ± 0.40 | 6.93 ± 0.83 | 9.08 ± 1.56 |
ECs were transfected with control or PHB1 RNAi, cells were stained with propidium iodide, and the cell cycle was analyzed via FACS analysis after the indicated time points. n = 3.
Loss of PHB1 alters mitochondrial function
Mitochondrial localized PHB1 is involved in mitochondrial function in yeast (Roskams et al., 1993; Steglich et al., 1999; Piper et al., 2002) and mitochondrial dysfunction, which is characterized by a drop in mitochondrial membrane potential and increased ROS production, is often involved in cellular senescence. Therefore, we examined if PHB1 was regulating these two additional indices of senescence.
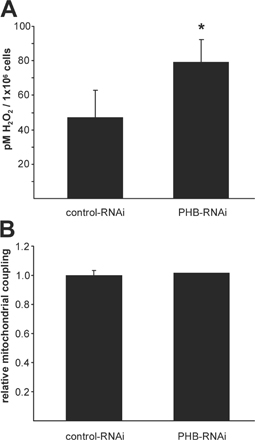
ROS levels were measured by FACS analysis of control and PHB1-depleted ECs upon labeling with CM-H2-DCFDA (Fig. 3 A). ROS levels were significantly increased in cells lacking PHB1 (Fig. 3 A, red) compared with control (solid gray) and could be reversed by preincubation with the H2O2 scavenger polyethylene glycol (PEG)–catalase (green). This result was confirmed by measuring H2O2 levels in cell lysates with the fluorometric probe Amplex red, also showing a significant accumulation of H2O2 after knockdown of PHB1 (Fig. S1 A, available at http://www.jcb.org/cgi/content/full/jcb.200706072/DC1).
Figure 3.

PHB1 regulates mitochondrial function. (A) ROS production was analyzed by FACS using the dye CM-H2-DCFDH. Control RNAi cells are depicted in gray, PHB1 RNAi cells are depicted in red, and PHB1 RNAi cells preincubated with PEG-catalase are depicted in green. Positive control cells were preincubated with H2O2 (blue). (B) Respirating mitochondria were stained with reduced MitoTracker CM-H2 XRos (red) in BAECs (top) and rho0 ECs (bottom) and immunofluorescence analyses were performed (blue, DAPI). Bars, 10 μm. (C) ROS production of rho0 ECs transfected with control (red) or PHB1 siRNA (blue) was analyzed by FACS using CM-H2-DCFDH. (D) Control (top) and PHB1-depleted BAECs (bottom) were stained with the radiometric dye JC-1 to monitor depolarization of the mitochondrial membrane and intensity of red (left) and green (right) fluorescence was analyzed by indirect immunofluorescence. Bar, 10 μm. (E) Control cells (top left) and PHB1 knockdown cells pretreated with (bottom right) or without (bottom left) PEG-catalase were stained with JC-1 and both FL-1 and FL-2 fluorescence was analyzed by FACS. Valinomycin-treated cells served as a positive control for depolarization of the mitochondrial membrane (top right). As a mathematical indicator for mitochondrial membrane depolarization, the ratio of means between red and green fluorescence was calculated. (F and G) Complex I and III activity of PHB1-depleted cells was calculated and normalized to control cells by measuring oxygen consumption in the presence and absence of rotenone, antimycin-A, and azide. Error bars show the calculated standard deviation. n = 3; *, P < 0.05.
Because there are several possible sources of ROS in ECs, e.g., mitochondria, the NADPH oxidase, xanthine oxidase, and endothelial NO synthase, we generated mitochondrial DNA–depleted BAEC, e.g., rho° ECs, and measured ROS levels after knockdown of PHB1. Immunofluorescence analysis of rho° ECs upon staining with reduced MitoTracker confirmed that no respiring mitochondria were present in these cells (Fig. 3 B). Also, in these cells, the increase of ROS production upon loss of PHB1 was eliminated (Fig. 3 C), clearly indicating mitochondrial respiration as the source of elevated ROS levels upon loss of PHB1.
Mitochondrial membrane potential (ΔΨ) was assessed using the radiometric dye JC-1, which localizes as a red monomer at the mitochondrial membrane under normal conditions and forms a green dimer when the mitochondrial membrane depolarizes. Compared with the control, both indirect immunofluorescence (Fig. 3 D) and FACS analysis (Fig. 3 E) revealed a drop in red fluorescence and an increase in green fluorescence after knockdown of PHB1, indicating a depolarization of the mitochondrial membrane. The potassium ionophore valinomycin served as a positive control in Fig. 3 E.
The general idea of the interplay between depolarization of the mitochondrial membrane and ROS production is that, upon mitochondrial damage or dysfunction, mitochondrial pores open and allowing the influx of potassium and calcium cations, thereby depolarizing the mitochondrial membrane, which in turn induces ROS production and release (Boveris and Chance, 1973). Because the damage of the mitochondrial membrane can also be caused by initial doses of ROS triggering ROS-induced ROS release (Zorov et al., 2000), it remained unclear if in our system the initial trigger is an increase in ROS causing depolarization or vice versa. However, scavenging of intracellular superoxide with PEG-catalase upon knockdown of PHB1 did not reverse the depolarization of the mitochondrial membrane (Fig. 3 E, bottom right), indicating that the drop in membrane potential results in increased ROS production and not vice versa.
Mitochondrial ROS are produced by complex I and/or III (Boveris et al., 1976; Takeshige and Minakami, 1979) and ROS production can be elevated upon inhibition of complex I and/or III activity (Boveris and Chance, 1973). Therefore, we investigated the relative contribution of complex I and III activity to overall mitochondrial respiration by measuring oxygen consumption before and after inhibition of these complexes with rotenone and antimycin-A, respectively. We found that the contribution of complex I activity in PHB1-depleted ECs was 30% lower than in control cells (Fig. 3 F), whereas the contribution of complex III activity was virtually identical (Fig. 3 G). Additionally, the degree of mitochondrial coupling was analyzed by measuring oxygen consumption in the absence and presence of oligomycin, which inhibits ATP synthase and is predicted to reduce oxygen consumption in efficiently coupled mitochondria. Oligomycin inhibited respiration to the same degree in control and PHB-inhibited ECs, which suggests that a similar degree of coupling was maintained despite the decreased complex I contribution to respiration in response to PHB knockdown (Fig. S1 B). Collectively, these results indicate that mitochondrial electron transport is partially blocked at complex I and that this is likely the source of ROS production in the PHB1-inhibited cells. However, the total mitochondrial oxygen consumption was maintained apparently by compensatory mechanisms that allow efficient electron flow to continue through complexes II and III despite the complex I inhibition.
PHB1 regulates Rac1 activity via the PI3-K–Akt pathway
Because ROS are known to participate in signal transduction pathways (Yang et al., 1998; Connor et al., 2005; Lucchesi et al., 2005; Oeckler et al., 2005), we checked for alterations in several redox-sensitive pathways upon knockdown of PHB1. Compared with ECs treated with control RNAi, knockdown of PHB1 increased VEGF-simulated Akt phosphorylation on serine 473 (S473). The increased phosphorylation of Akt was prevented by preincubation of cells with the H2O2 scavenger PEG-catalase (Fig. 4 A), which suggests that H2O2 was responsible for Akt activation. Furthermore, the addition of exogenous H2O2 to ECs activated Akt phosphorylation on S473 (Fig. 4 B), underlining the concept of ROS-dependent Akt activation.
Figure 4.

PHB1 regulates Akt and Rac1 activity. (A) The level of phosphorylated (P473) and total Akt (after 100-ng/ml VEGF stimulation for different time points) was examined via Western blotting after PHB1 knockdown in ECs (middle) in comparison to control cells (left) and PHB1 knockdown cells preincubated with PEG-catalase (right). (B) H2O2 activates Akt phosphorylation, as shown by Western blotting. (C) Rac1 activity assays were performed using control and PHB1 knockdown cells, and active Rac1 pulldowns as well as total cell lysates were Western blotted against Rac1 and PHB1. Cells were treated with 100 ng/ml VEGF and/or PI3-K inhibitors wortmannin (50 nM) and LY294002 (30 μM) as indicated. (D) Phospho-Akt (P473) and active Rac1 were monitored in control and PHB1 knockdown cells upon 100-ng/ml VEGF stimulation for the indicated time points. (E) Quantification of Akt phosphorylation (top) and active Rac1 (bottom) normalized to total Akt and Rac, respectively, from three independent experiments. *, P < 0.05. (F) Rac1 activity in ECs pretreated with VEGF and/or a Rac1 inhibitor as indicated by Western blot analysis of total cell lysates for p-Akt, total Rac1, PHB1, and active Rac1. Error bars show the calculated standard deviation.
Next, we examined pathways downstream of Akt, i.e., eNOS and Rac1, in ECs deficient in PHB1. A reduction in PHB1 increased the phosphorylation of eNOS on its Akt-sensitive site S1179 and also promoted NO release from cells (Fig. S2, A and B, available at http://www.jcb.org/cgi/content/full/jcb.200706072/DC1). Another downstream partner of Akt that is involved in cell motility and thus could be responsible for the observed change in cell morphology (Fig. 2 B) is the small GTPase Rac1 (Burridge and Wennerberg, 2004). VEGF-stimulated Rac1 activity was clearly elevated in cells lacking PHB1 and Rac1 activity was dependent on the PI3-K pathway because preincubation with PI3-K inhibitors wortmannin or LY294002 blocked Rac1 activation (Fig. 4 C).
Persistent hyperactivation of Akt could be the explanation for the observed senescent phenotype. Therefore, we checked the long-term activation status of Akt and its downstream target Rac1. In PHB1-depleted cells, both Akt and Rac1 were persistently hyperactivated throughout a time range of 120 min upon VEGF stimulation (Fig. 4 D and E).
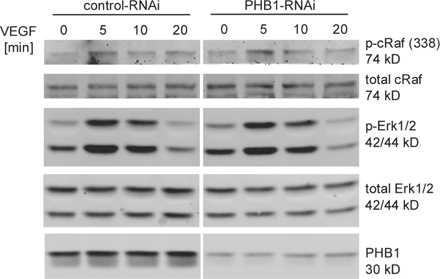
Akt may lie downstream of Rac1 (Gonzalez et al., 2006) but this was not the case in our experimental system because a Rac1 inhibitor had no impact on Akt S473 phosphorylation in PHB1 knockdown cells (Fig. 4 F) but did block VEGF-stimulated Rac activation. PHB1 has recently been described to regulate the Ras–Raf–Erk pathways in epithelial cells (Rajalingam et al., 2005). However, we could not detect any differences in c-Raf or Erk-1/2 phosphorylation upon knockdown of PHB1 in ECs (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200706072/DC1). Similar results were obtained in human ECs using different RNAi constructs (unpublished data).
Loss of PHB1 provokes an antiangiogenic phenotype by influencing the cytoskeletal balance in vitro
Because a deficiency of PHB1 increases ROS production, which activates Akt and Rac1, we examined changes in cytoskeletal architecture. Rac1 and its downstream partners are involved in balancing the dynamics of the actin cytoskeleton, which in turn impacts cell motility. As seen in Fig. 5 A, in control RNAi-treated ECs, VEGF induced the formation of F-actin–mediated stress fibers as expected (Soga et al., 2001), whereas in PHB1 knockdown cells, VEGF-induced stress fiber formation was attenuated and demonstrated a higher abundance of cortical actin at the periphery of the cells. This impairment in VEGF-induced stress fiber formation was rescued by preincubation of cells with PEG-catalase and was therefore ROS dependent (Fig. 5 A). Furthermore, the observed change in senescent cell morphology upon PHB1 knockdown (Fig. 2 B) could be rescued by scavenging H2O2 with PEG-catalase in live cell imaging experiments (Fig. 5 B) comparing PHB1 knockdown cells before (left) and after (right) addition of PEG-catalase for 30 min. Because dynamic changes in the cytoskeleton control cell motility, which is highly important for the motile phenotype of ECs, we examined VEGF-stimulated migration and 3D tube formation. Knockdown of PHB1 caused a decrease in VEGF-stimulated directional cell migration that was also rescued by scavenging H2O2 with PEG-catalase (Fig. 5 C). Furthermore, VEGF-induced 3D tube formation was almost completely blunted in cells lacking PHB1 (Fig. 5 D). Both migration and tube formation experiments were performed at early time points (5 h for both migration and tube formation) to obviate the antiproliferative phenotype of the PHB knockdown. Thus, upon loss of PHB1, increased ROS production leads to increased Akt and Rac1 activity that, via cytoskeletal rearrangements, blunts the angiogenic capacity of ECs in vitro. Experiments in Fig. 5 (C and D) were also performed in EA.hy926 cells using different RNAi constructs and showed similar results (not depicted).
Figure 5.

Loss of PHB provokes an antiangiogenic phenotype by influencing the cytoskeletal balance in vitro. (A) Actin stress fiber formation (stained with phalloidin) was examined via indirect immunofluorescence analysis using control (upper left) and PHB1 knockdown (top right and bottom) cells stimulated with VEGF (all) and PEG-catalase (bottom) and stained with rhodamin-phalloidin (yellow) and DAPI (blue). Bars, 10 μm. (B) EC morphology after knockdown of PHB1 (left) and incubation with PEG-catalase (right) using life imaging phase contrast microscopy. Insets show a higher magnification. Bars, 100 μm. (C) Migration of control and PHB1 knockdown cells pretreated with and without PEG-catalase was examined via migration assay in the presence and absence of VEGF. n = 4; *, P < 0.05. (D) Tube formation assays on matrigel plugs were performed and quantified using control and PHB1 knockdown cells. Representative examples of tubes are shown on the right. Error bars show the calculated standard deviation. n = 3; *, P < 0.05. Bars, 100 μm.
PHB1 knockdown reduces blood vessel formation in vivo
Next, we wanted to examine if the antiangiogenic phenotype in vitro was also applicable in vivo. Because knockout animals for PHB1 were not available, we performed siRNA-based matrigel angiogenesis assays in vivo. This method has been shown to successfully knock down sphingosine-1-phosphate expression in vivo (Chae et al., 2004). To reduce immune driven angiogenesis, growth factor–reduced matrigel and immunodeficient severe combined immunodeficiency mice were used. The matrigel was mixed with basic FGF, VEGF, and RNAi constructs and injected subcutaneously. 12 d after injection, gels were harvested, fixed, and paraffin embedded for histochemical analysis. Immunostaining for PHB1 in the gel showed successful knockdown of PHB1 in cells invading the gel compared with the control (Fig. 6 A). Interestingly, total cell counting revealed less overall cellular invasion into the PHB1 knockdown gels compared with control (Fig. 6 B). To quantify the number of ECs invading the gel, sections were stained for the EC marker PECAM-1. EC invasion was inhibited to a greater extent than overall cell invasion (Fig. 6 C), which could be an effect of reduced motility of ECs with reduced PHB1 expression. Furthermore, the reduced number of PECAM-1–positive cells was corroborated by a marked reduction in red blood cell–positive structures in the PHB1 knockdown gels (Fig. 6 D).
Figure 6.

Loss of PHB reduces angiogenesis in vivo. Matrigel angiogenesis assays in vivo were performed using gels mixed with control or PHB1 knockdown constructs. Gels were dissected, sectioned, and analyzed immunohistochemically (n = 5). (A) Both control and PHB1 knockdown gel slides were stained for PHB1 to show successful knockdown and images were taken using a 20× objective. Bars, 10 μm. (B) After hematoxylin and eosin staining, images were taken using a 4× objective to quantify total cells invading the gels. Representative images of control and a PHB1 knockdown gel are shown on the bottom. n = 5; *, P < 0.05. Bars, 100 μm. (C) Angiogenic ECs in both control and PHB1 knockdown gels were quantified by staining the slides for PECAM-1 after taking images with a 10× objective. An example of PECAM-1–positive structures (arrows) in a control gel is shown on the right. n = 5; *, P < 0.05. Bar, 50 μm. (D) Red blood cell–positive vascular structures (indicated by arrows in the representative example from a control gel shown on the right) were quantified in both control and PHB1 knockdown gels after staining with hematoxylin and eosin and taking images with a 10× objective. Error bars show the calculated standard deviation. n = 5; *, P < 0.05. Bar, 50 μm.
Discussion
The data provided herein is the first characterization of PHB1 in the vascular system and provides an interesting connection between mitochondrial function, senescence, and vascular homeostasis. We show that PHB1 is expressed in ECs and that the loss of PHB1 results in a block in electron transport at complex I, increased mitochondrial ROS generation, and cellular senescence that culminates in the loss of several endothelial functions including cell migration, cell proliferation, and blood vessel formation. These results stand in contrast to the putative nuclear roles of PHB1 as a tumor suppressor gene and regulator of transcription (Wang et al., 2002b; Fusaro et al., 2003; Rastogi et al., 2006a) and suggests that the main function of PHB1 in primary mammalian cells is to regulate mitochondrial integrity.
PHB1 is a highly conserved and broadly expressed protein defining the heterologous protein family of prohibitins. PHB1 has been shown to be localized to different cell compartments including mitochondria and the nucleus as well as lipid rafts in the plasma membrane (Morrow and Parton, 2005). We show that PHB1 is expressed in both ECs and SMCs in the murine aorta as well as in a variety of primary ECs in vitro. Our colocalization experiments show that PHB1 is primarily localized in the mitochondria of ECs (Fig. 1) despite evidence supporting its localization in the nucleus (Fig. 1) and in lipid rafts (not depicted). To examine the function of PHB1 in the vascular system, we chose a loss-of-function strategy in primary ECs using RNAi because overexpression and knockdown studies in cancer cell lines have suggested its role as a tumor suppressor (Wang et al., 2002b; Fusaro et al., 2003; Rastogi et al., 2006a), vertebrate knockout models were not available, and the knockout of the PHB1 homologue in Drosophila melanogaster revealed a lethal phenotype (Eveleth and Marsh, 1986).
The knockdown of PHB1 induced a cellular senescent phenotype characterized by a change in morphology, cell cycle block, decreased proliferation, increased expression of the cell cycle inhibitors p16 and p21, and senescence-associated β-galcatosidase expression. This is in contrast to a recent study documenting that RNAi knockdown of PHB1 in the breast carcinoma cell line MCF-7 prevents cellular senescence and increases cell growth (Rastogi et al., 2006a). Perhaps PHB1 exerts a different function in primary cells (i.e., ECs) versus cancer cells with multiple genetic mutations in known oncogenic pathways. Senescence usually occurs by telomere shortening but can also be telomere independent, thus maintaining cells in a quiescent state of low metabolism. Telomere-independent cellular senescence is believed to be triggered by stress factors such as DNA-damaging agents, radiation, oxidative stress, retinoids, oncogenes, or hypermitogenic stimuli such as overexpression of Raf (Sewing et al., 1997) or constitutive active Akt (Miyauchi et al., 2004). In the case of decreased PHB1 expression, an alteration of mitochondrial function resulted in increased O2− production. Increased mitochondrial ROS production was induced by depolarization of the mitochondrial membrane and a block of electron flow at complex I, which together with complex III is the accepted source of mitochondrial ROS (Boveris et al., 1976; Takeshige and Minakami, 1979). Because the contribution of complex III activity and the degree of mitochondrial coupling was similar in PHB1 knockdown cells, we hypothesize that a substantial amount of the electrons within the inhibited complex I are used for superoxide production when PHB levels are reduced. However, we propose that, under these circumstances, there are compensatory mechanisms at play that maintain cytochrome oxidase (complex IV) activity and overall ATP production. Such mechanisms likely involve an increase in electron flow via complex II and/or III. Other sources of ROS, e.g., NADPH oxidase, NO synthase, or xanthine oxidase, could be ruled out because BAECs depleted of respiring mitochondria (rho° ECs) did not demonstrate ROS production after knockdown of PHB1.
Increased ROS production resulted in subsequent hyperactivation of Akt, triggering the senescent phenotype. Accordingly, ROS production, cellular morphology, hyperactivated Akt, stress fiber formation, and the loss of migrative capacity could all be rescued by preincubation of cells with the ROS scavenger PEG-catalase. Indeed, elevated ROS as well as overexpression of constitutively active Akt are able to promote senescence in the vascular system (Miyauchi et al., 2004; Madamanchi and Runge, 2007). Also, the idea that ROS modulates the actin cytoskeleton, thereby inducing senescence, is a well-accepted concept (Gourlay and Ayscough, 2005). Additionally, expression of PHB1 itself has been shown to prevent senescence in both plants and yeast (Piper et al., 2002; Chen et al., 2005). Hyperactivation of other mitogenic pathways as a trigger of senescence could be excluded because we were not able to observe changes in p38 MAPK (unpublished data) or c-Raf and Erk1/2 phosphorylation after PHB1 knockdown, although PHB1 has been previously shown to influence Ras/c-Raf signaling in epithelial cells (Rajalingam et al., 2005). The loss of the nuclear functions of PHB1, e.g., interaction with E2F and p53 transcription factors, as a further trigger of senescence is possible but not likely in primary ECs because these interactions would result in increased cell growth in the absence of PHB1 as is observed in cancer cells (Wang et al., 2002b; Fusaro et al., 2003).
Downstream of Akt, the activity of the small GTPase Rac1 plays an important role in regulating cytoskeletal dynamics (Burridge and Wennerberg, 2004). The signaling pathways downstream of Rac1 are complex because various pathways are affected, dependent on cell system, stimulus, and the duration of the stimulus. Two common and accepted pathways through which Rac1 influences actin polymerization are via p21-activated kinase (PAK) through LIM-kinase to cofilin and via PAK through myosin heavy chain and light chain kinases (Edwards et al., 1999; Kiosses et al., 1999; Sells et al., 1999; van Leeuwen et al., 1999). Because Rac1 activity assays were determined using the PAK-binding domain for active Rac1, it is very likely that the observed hyperactivation of Rac1 signals through either of the described pathways to the actin cytoskeleton. In fact, a dramatic change in actin stress fiber formation in ECs occurred upon PHB1 knockdown, which could be rescued by scavenging ROS with PEG-catalase. Most importantly, these observations are reliable functional assays that are known to be dependent on cytoskeletal dynamics and properly balanced Rac1 activity (Fukata et al., 2003) because both migration and tube formation were clearly reduced in ECs lacking PHB1. These observations cannot be explained by reduced proliferation because each assay was designed to last 5 h or less. Furthermore, reduced migration could be rescued by scavenging H2O2, supporting the concept that ROS- and Akt-mediated senescence leads to cytoskeletal rearrangements and decreased cell mobility.
Because angiogenesis is a multistep process involving cell proliferation, migration, and morphogenesis, we examined the in vivo function of PHB1 using RNAi constructs injected subcutaneously into mice and monitored cell invasion and blood vessel formation (Fig. 6). This approach has been successful in showing the proangiogenic actions of sphingosine-1-phosphate in vivo (Chae et al., 2004) and is a relatively easy approach when knockout mice are nonviable or not available. RNAi-mediated knockdown of PHB1 reduced overall cellular infiltration and specifically reduced EC-positive structures, demonstrating that PHB1 function is required for normal angiogenesis in vivo.
In summary, PHB1 expression in ECs is mandatory for maintaining both mitochondrial and vascular function by regulating ROS production and cellular senescence. These findings underscore the emerging concept of mitochondria as an important signaling organelle and ROS as important signaling molecules in vascular biology (Quintero et al., 2006; Madamanchi and Runge, 2007). Because both senescence as well as mitochondrial function have been linked to pathological events in the vascular system such as atherosclerosis (Madamanchi and Runge, 2007), PHB1 could become an interesting candidate for further studies in this field.
Materials and methods
Materials
JC-1, CM-H2-DCFDA, valinomycin, rhodamine-phalloidin, rotenone, antimycin-A, and both reduced and nonreduced MitoTracker were obtained from Invitrogen. Growth factor–reduced matrigel was obtained from BD Biosciences and PEG-catalase, ethidium bromide, and uridine were obtained from Sigma-Aldrich. Human VEGF165 was a gift of Genentech. Antibodies against Akt, phospho-Akt, and the senescence β-galactosidase staining kit were obtained from Cell Signaling Technology. Antibodies against PECAM-1 (clone M-20), p16 (clone C-20), and p21 (clone C-19) were obtained from Santa Cruz Biotechnology, Inc. The Rac1 antibody was obtained from BD Biosciences. Antibodies against β-actin and PHB1 were obtained from Sigma-Aldrich and Thermo Fisher Scientific, respectively. The antibody against phospho-eNOS was obtained from Invitrogen and antibodies against eNOS and c-Raf were obtained from BD Biosciences. phospho-c-Raf (clone 56A6), Erk1/2, phosphor-Erk1/2 (clone E10) antibodies were obtained from Cell Signaling Technology. The Amplex red hydrogen peroxide kit was obtained from Invitrogen.
siRNA contructs
Nonmodified and high performance purified siRNA duplexes were obtained from QIAGEN. The constructs were directed against the following sequences: 5′-AATGTGGATGCTGGGCACAGA-3′ for knocking down PHB1 in BAECs and the in vivo angiogenesis assay and 5′-AAAGGCTGAGCAACAGAAAAA-3′ for knocking down PHB1 in HUVECs and EA.hy926. A negative control (QIAGEN) and a scrambled sequence of the first construct 5′-AAGAGTGTCGGATGCAGGATC-3′ were used as controls.
Cell culture and transfection with RNAi constructs
BAECs and HUVECs were isolated from freshly harvested aorta or umbilical cords, respectively, and cultured and characterized as described previously (Bernatchez et al., 2001). BAECs and HUVECs were not passaged for more than eight and five passages, respectively. For knockdown of PHB1, ECs were transfected with 25 nM of the corresponding RNAi constructs using Oligofectamine (Invitrogen) according to the manufacturer's instructions. The cells were maintained in OptiMem (Invitrogen) for 8 h and then incubated in full medium for a total of 48 h if not stated otherwise.
Generation of rho0 ECs
Rho0 ECs were generated by depleting BAECs of respiring mitochondria. Therefore, BAECs were incubated in full medium also containing 250 ng/ml ethidium bromide, 50 μg/ml uridine, and 1 mM pyruvate for 2 wk. The medium was changed every other day.
Western blotting
Protein samples were dissolved in sample buffer (200 mM Tris-HCl, pH 6.8, 6% SDS, 20% glycerol, 10% DTT, and 0.1 mg/ml bromophenol blue), separated by SDS-PAGE, and transferred to nitrocellulose membranes. The membranes were blocked in TBS containing 0.1% casein or 5% BSA. Detection was performed with primary antibodies as indicated in combination with Alexa Fluor 680 (Invitrogen)– or IRDye 800 (Rockland)–conjugated antibodies to rabbit or mouse IgG, respectively, and the Odyssey detection system (Li-Cor).
Quantification of Western blots
Western blot bands of phosphorylated Akt and active Rac1 were quantified and normalized against total Akt and Rac1, respectively, using ImageJ software. For calculating relative intensities, Western blots from three independent experiments were quantified and averaged.
Rac1 activity assays
Rac1 activity was analyzed using the Rac1 assay kit (Millipore) according to the manufacturer's instructions. Confluent BAECs were starved for 6–8 h. After stimulation with 100 ng/ml VEGF for 15 min, cells were lysed, insoluble material was spun down, and protein concentration was assessed. 500 μg of total protein per sample was used to pull down active Rac1. Both active Rac1 and total cell lysates were analyzed for Rac1 by Western blotting using the Rac1 antibody (BD Biosciences).
Indirect immunofluorescence and live imaging
For indirect immunofluorescence, cells were seeded on coverslips fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and blocked with PBS containing 0.1% Tween 20 (PBST), 5% BSA, and 5% serum. Afterward, cells were incubated with primary antibodies followed by incubation with Alexa Fluor 488– or 568–coupled secondary antibodies (Invitrogen) and 1 μg/ml DAPI. For staining of F-actin stress fibers, cells were incubated with rhodamine-phalloidin. Pictures were taken using a 63× oil immersion objective (Carl Zeiss, Inc.) at room temperature unless stated otherwise. For live imaging, pictures were taken using a 20× phase contrast objective (Carl Zeiss, Inc.) together with a 37°C incubation chamber. Images were acquired using a microscope (Axiovert 200; Carl Zeiss, Inc.) in combination with OpenLab software (Improvision).
Immunohistochemistry
Specimens were fixed in 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, embedded in paraffin, and sectioned (5 μm). Upon antigen retrieval with 20 mM sodium citrate, pH 6, and incubation with a primary antibody, sections were stained using biotin-coupled secondary antibodies (Jackson ImmunoResearch Laboratories) together with the Vectastain ABC kit (Vector Laboratories) and Nova red (Vector Laboratories) as a substrate. Pictures were taken and acquired using 4, 10, or 20× objectives with a microscope (Axioskop2 plus; Carl Zeiss, Inc.) in combination with a camera (AxioCam MRc) and MRGrab software (both from Carl Zeiss, Inc.) at room temperature.
β-Galactosidase assay for senescent cells
BAECs were seeded on glass coverslips and transfected with siRNA constructs, and the expression of β-galactosidase was monitored using the senescence β-galactosidase staining kit (Cell Signaling Technology) according to the manufacturer's instructions. Pictures were taken with a 10× phase contrast objective (Carl Zeiss, Inc.) on a microscope (Eclipse 80i) with a camera (DXM 1200C; both from Nikon) at room temperature using NIS-Elements D software (Nikon).
Cell cycle analysis
Cell cycle of control and PHB1 knockdown transfected cells via propidium iodide staining and FACS analysis was performed as described previously (Schleicher et al., 2005).
Proliferation assay
104 cells were seeded on 24-well dishes, transfected with RNAi constructs upon attachment, and cultured in full medium. Cells were trypsinized, resuspended, and counted at indicated time points using a Coulter counter (Z-1; Beckman Coulter).
FACS analysis of mitochondrial membrane potential and ROS production
Cells were trypsinized, washed, and resuspended in full medium. For analysis of the mitochondrial membrane potential, resuspended cells were incubated with 1 μg/ml JC-1 for 10 min and 5 μg/ml propidium iodide for 30 min at 37°C. As a positive control, transfected cells were preincubated with 50 μM valinomycin for 2 h. For analysis of ROS production, cells were starved for 2 h in DME containing 0.2% FCS and resuspended cells were incubated with 5 μM CM-H2-DCDA and 5 μg/ml propidium iodide for 30 min at 37°C. As a positive control, cells were preincubated with 100 μM H2O2 for 20 min. To ensure that DCDA was detecting H2O2, PHB1 knockdown transfected cells were preincubated with 10 μg/ml PEG-catalase for 2 h. Living cells were gated according to propidium iodide staining and FL-1 and FL-2 fluorescence was measured using a FACScalibur cell analyzer and CellQuest software (Beckton Dickinson). Data were analyzed using WinMDI software (provided by J. Trotter, Scripps Research Institute, La Jolla, CA).
Measurement of H2O2 in cell lysates
ECs transfected with control or PHB1 knockdown constructs on a 10-cm dish were scraped in 200-μl assay buffer and frozen/thawed in liquid nitrogen three times. 50 μl of the supernatants were used to measure H2O2 concentrations by fluorometric analysis with the Amplex red kit according to the manufacturer's protocol on a 96-well scale.
Measurement of mitochondrial complex activity
siRNA transfected, exponentially growing BAECs were trypsinized and resuspended in full medium (106 cells/ml). 5 ml of cell suspension was used for measuring oxygen consumption with an oxygen probe in the absence and presence of inhibitors for complex I (10 μM rotenone), complex III (10 μM antimycin-A), or mitochondrial respiration (0.05% azide). Complex I and III activities were calculated with the following formula: activity = 1 − [(Slopeinhibitor cons − Slopeazide cons)/(Slopetotal cons − Slopeazide cons)]. Afterward, complex activities of PHB1 knockdown cells were normalized to control cells.
Measurement of mitochondrial coupling
siRNA transfected, exponentially growing BAECs were trypsinized and resuspended in full medium (106 cells/ml). 5 ml of cell suspension was used for measuring oxygen consumption with an oxygen probe in the absence and presence of an uncoupler (0.5 μg/ml oligomycine) or an inhibitor of mitochondrial respiration (0.05% azide).
The degree of coupling was calculated with the following formula: coupling = 1 − [(Slopeuncoupler cons − Slopeazide cons)/(Slopetotal cons − Slopeazide cons)]. Afterward, the degree of coupling of PHB1 knockdown cells was normalized to control cells.
Tube formation assay
3 × 104 cells previously transfected with RNAi constructs and cultured for 48 h were trypsinized, washed, and seeded on top of growth factor–reduced matrigel plugs in DME containing 0.2% FCS with or without 100 ng/ml VEGF. Images of formed tubes were taken using a 5× phase contrast objective mounted on an Axiovert 200 microscope at room temperature after 5 h and tube lengths were measured using OpenLab software. Only closed tubes were taken into the measurement.
Cell migration assay
Cell migration was evaluated using a microchamber (Boyden). Near-confluent BAECs were washed with DME, trypsinized, and resuspended in DME/0.2% FCS, and then 4 × 104 cells in 50 μl were seeded in the upper chamber and the lower chamber was filled with DME containing 0.2% FCS with or without 100 ng/ml VEGF and separated by an 8-μm gelatin-coated polycarbonate filter. 5 h after incubation at 37°C, the nonmigrated cells were scraped with a plastic policeman and the migrated cells were stained using Quick-Diff (Shandon, Inc.) solutions and counted.
Matrigel angiogenesis assay in vivo
The assay was performed as described previously (Chae et al., 2004) with minor modifications. In brief, female severe combined immunodeficiency mice were injected with 0.5 ml of growth factor–reduced Matrigel premixed with 150 ng/ml VEGF, 50 ng/ml FGF, and 2 μM of the siRNA constructs. After 10 d, matrigel plugs were harvested, fixed in 4% paraformaldehyde, and embedded in paraffin. Sections were stained with hematoxylin and eosin, anti-PECAM-1, or anti-PHB1.
NO release experiments
BAECs were grown to confluence in 6-well tissue culture plates (Becton Dickinson). The medium was removed and serum-free DME containing various concentrations of peptides or vehicle (0.1% DMSO) was added and incubated for 6 h. After pretreatment, the medium was removed and cells were stimulated with DME containing peptides and/or VEGF for 30 min. After a 30-min incubation, the medium was collected and cells were trypsinized and counted with a Coulter counter. NO release from confluent BAECs was assessed in the medium by measuring nitrite levels using a NO-specific chemiluminescence analyzer. Data are reported as picomole of nitrite per 106 cells.
Statistical significance
Experiments were performed with at least n = 3. Error bars show the calculated standard deviation. Significance was calculated by a t test.
Online supplemental material
Fig. S1 shows measurement of hydrogen peroxide in cell lysates as an Amplex red complex and analysis of relative mitochondria coupling in control versus knockdown ECs. Fig. S2 shows Western blot analysis of phospho-eNOS and NO production upon stimulation with VEGF in control versus knockdown ECs. Fig. S3 shows Western blot analysis of phospho-Raf and phosphor Erk1/2 upon stimulation with VEGF in control versus knockdown cells. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200706072/DC1.
Supplemental Material
Acknowledgments
We thank Drs. Michelle Lin and Pascal Bernatchez for helpful discussions as well as Roger Babbitt for excellent technical assistance.
This work was supported by grants from the National Institutes of Health (RO1 HL64793, RO1 HL 61371, RO1 HL 57665, and PO1 HL 70295) and a National Heart, Lung, and Blood Institute Yale Proteomics Contract (NO1-HV-28186) to W.C. Sessa. M. Schleicher was supported by a research fellowship from the Deutsche Forschungsgemeinschaft (SCHL1818/1-1). B.R. Shepherd was supported by a National Institutes of Health grant (1-RO1 HL 085416). C. Fernandez-Hernando was supported by a fellowship from Ministerio de Educacion y Ciencia and a Philip Morris External Research Postdoctoral Fellowship. Y. Suarez was supported by a Program 3 + 3 Fellowship from the Centro Nacional de Investigaciones Cardivasculares. G. Shadel was supported by a National Institutes of Health grant (RO1 NIH-NINDS #NS056206). Y. Pan was supported as a Medical Research Scholar of the Yale Cell Biology graduate program.
Abbreviations used in this paper: BAEC, bovine aortic endothelial cell; EC, endothelial cell; HUVEC, human umbilical vein EC; PAK, p21-activated kinase; PECAM-1, platelet EC adhesion molecule 1; PEG, polyethylene glycol; PHB1, prohibitin-1; PI3-K, phosphatidyl inositol 3-kinase; ROS, reactive oxygen species; SMC, smooth muscle cell.
References
- Ahn, C.S., J.H. Lee, A. Reum Hwang, W.T. Kim, and H.S. Pai. 2006. Prohibitin is involved in mitochondrial biogenesis in plants. Plant J. 46:658–667. [DOI] [PubMed] [Google Scholar]
- Alcorta, D.A., Y. Xiong, D. Phelps, G. Hannon, D. Beach, and J.C. Barrett. 1996. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc. Natl. Acad. Sci. USA.. 93:13742–13747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artal-Sanz, M., W.Y. Tsang, E.M. Willems, L.A. Grivell, B.D. Lemire, H. van der Spek, and L.G. Nijtmans. 2003. The mitochondrial prohibitin complex is essential for embryonic viability and germline function in Caenorhabditis elegans. J. Biol. Chem. 278:32091–32099. [DOI] [PubMed] [Google Scholar]
- Bernatchez, P.N., M.V. Winstead, E.A. Dennis, and M.G. Sirois. 2001. VEGF stimulation of endothelial cell PAF synthesis is mediated by group V 14 kDa secretory phospholipase A2. Br. J. Pharmacol. 134:197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boveris, A., and B. Chance. 1973. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem. J. 134:707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boveris, A., E. Cadenas, and A.O.M. Stoppani. 1976. Role of ubiquinone in the mitochondrial generation of hydrogen peroxide. Biochem. J. 156:435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge, K., and K. Wennerberg. 2004. Rho and Rac take center stage. Cell. 116:167–179. [DOI] [PubMed] [Google Scholar]
- Cai, H. 2005. Hydrogen peroxide regulation of endothelial function: origins, mechanisms, and consequences. Cardiovasc. Res. 68:26–36. [DOI] [PubMed] [Google Scholar]
- Chae, S.S., J.H. Paik, H. Furneaux, and T. Hla. 2004. Requirement for sphingosine 1-phosphate receptor-1 in tumor angiogenesis demonstrated by in vivo RNA interference. J. Clin. Invest. 114:1082–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J.C., C.Z. Jiang, and M.S. Reid. 2005. Silencing a prohibitin alters plant development and senescence. Plant J. 44:16–24. [DOI] [PubMed] [Google Scholar]
- Coates, P.J., D.J. Jamieson, K. Smart, A.R. Prescott, and P.A. Hall. 1997. The prohibitin family of mitochondrial proteins regulate replicative lifespan. Curr. Biol. 7:607–610. [DOI] [PubMed] [Google Scholar]
- Coates, P.J., R. Nenutil, A. McGregor, S.M. Picksley, D.H. Crouch, P.A. Hall, and E.G. Wright. 2001. Mammalian prohibitin proteins respond to mitochondrial stress and decrease during cellular senescence. Exp. Cell Res. 265:262–273. [DOI] [PubMed] [Google Scholar]
- Connor, K.M., S. Subbaram, K.J. Regan, K.K. Nelson, J.E. Mazurkiewicz, P.J. Bartholomew, A.E. Aplin, Y.T. Tai, J. Aguirre-Ghiso, S.C. Flores, and J.A. Melendez. 2005. Mitochondrial H2O2 regulates the angiogenic phenotype via PTEN oxidation. J. Biol. Chem. 280:16916–16924. [DOI] [PubMed] [Google Scholar]
- Dimri, G.P., X. Lee, G. Basile, M. Acosta, G. Scott, C. Roskelley, E.E. Medrano, M. Linskens, I. Rubelj, O. Pereira-Smith, et al. 1995. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA.. 92:9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, G., A. Mouithys-Mickalad, and F.E. Sluse. 1998. Generation of superoxide anion by mitochondria and impairment of their functions during anoxia and reoxygenation in vitro. Free Radic. Biol. Med. 25:1066–1074. [DOI] [PubMed] [Google Scholar]
- Edwards, D.C., L.C. Sanders, G.M. Bokoch, and G.N. Gill. 1999. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat. Cell Biol. 1:253–259. [DOI] [PubMed] [Google Scholar]
- Eveleth, D.D. Jr., and J.L. Marsh. 1986. Sequence and expression of the Cc gene, a member of the dopa decarboxylase gene cluster of Drosophila: possible translational regulation. Nucleic Acids Res. 14:6169–6183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata, M., M. Nakagawa, and K. Kaibuchi. 2003. Roles of Rho-family GTPases in cell polarisation and directional migration. Curr. Opin. Cell Biol. 15:590–597. [DOI] [PubMed] [Google Scholar]
- Fusaro, G., P. Dasgupta, S. Rastogi, B. Joshi, and S. Chellappan. 2003. Prohibitin induces the transcriptional activity of p53 and is exported from the nucleus upon apoptotic signaling. J. Biol. Chem. 278:47853–47861. [DOI] [PubMed] [Google Scholar]
- Gonzalez, E., R. Kou, and T. Michel. 2006. Rac1 modulates sphingosine 1-phosphate-mediated activation of phosphoinositide 3-kinase/Akt signaling pathways in vascular endothelial cells. J. Biol. Chem. 281:3210–3216. [DOI] [PubMed] [Google Scholar]
- Gourlay, C.W., and K.R. Ayscough. 2005. The actin cytoskeleton: a key regulator of apoptosis and ageing? Nat. Rev. Mol. Cell Biol. 6:583–589. [DOI] [PubMed] [Google Scholar]
- Gutierrez, J., S.W. Ballinger, V.M. Darley-Usmar, and A. Landar. 2006. Free radicals, mitochondria, and oxidized lipids: the emerging role in signal transduction in vascular cells. Circ. Res. 99:924–932. [DOI] [PubMed] [Google Scholar]
- Ikonen, E., K. Fiedler, R.G. Parton, and K. Simons. 1995. Prohibitin, an antiproliferative protein, is localized to mitochondria. FEBS Lett. 358:273–277. [DOI] [PubMed] [Google Scholar]
- Kiosses, W.B., R.H. Daniels, C. Otey, G.M. Bokoch, and M.A. Schwartz. 1999. A role for p21-activated kinase in endothelial cell migration. J. Cell Biol. 147:831–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchesi, P.A., S. Belmadani, and K. Matrougui. 2005. Hydrogen peroxide acts as both vasodilator and vasoconstrictor in the control of perfused mouse mesenteric resistance arteries. J. Hypertens. 23:571–579. [DOI] [PubMed] [Google Scholar]
- Madamanchi, N.R., and M.S. Runge. 2007. Mitochondrial dysfunction in atherosclerosis. Circ. Res. 100:460–473. [DOI] [PubMed] [Google Scholar]
- Minamino, T., and I. Komuro. 2007. Vascular cell senescence: contribution to atherosclerosis. Circ. Res. 100:15–26. [DOI] [PubMed] [Google Scholar]
- Miyauchi, H., T. Minamino, K. Tateno, T. Kunieda, H. Toko, and I. Komuro. 2004. Akt negatively regulates the in vitro lifespan of human endothelial cells via a p53/p21-dependent pathway. EMBO J. 23:212–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncada, S., and J.D. Erusalimsky. 2002. Does nitric oxide modulate mitochondrial energy generation and apoptosis? Nat. Rev. Mol. Cell Biol. 3:214–220. [DOI] [PubMed] [Google Scholar]
- Morrow, I.C., and R.G. Parton. 2005. Flotillins and the PHB domain protein family: rafts, worms and anaesthetics. Traffic. 6:725–740. [DOI] [PubMed] [Google Scholar]
- Oeckler, R.A., E. Arcuino, M. Ahmad, S.C. Olson, and M.S. Wolin. 2005. Cytosolic NADH redox and thiol oxidation regulate pulmonary arterial force through ERK MAP kinase. Am. J. Physiol. Lung Cell. Mol. Physiol. 288:L1017–L1025. [DOI] [PubMed] [Google Scholar]
- Passos, J.F., and T. Von Zglinicki. 2006. Oxygen free radicals in cell senescence: are they signal transducers? Free Radic. Res. 40:1277–1283. [DOI] [PubMed] [Google Scholar]
- Piper, P.W., G.W. Jones, D. Bringloe, N. Harris, M. MacLean, and M. Mollapour. 2002. The shortened replicative life span of prohibitin mutants of yeast appears to be due to defective mitochondrial segregation in old mother cells. Aging Cell. 1:149–157. [DOI] [PubMed] [Google Scholar]
- Quintero, M., S.L. Colombo, A. Godfrey, and S. Moncada. 2006. Mitochondria as signaling organelles in the vascular endothelium. Proc. Natl. Acad. Sci. USA.. 103:5379–5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajalingam, K., C. Wunder, V. Brinkmann, Y. Churin, M. Hekman, C. Sievers, U.R. Rapp, and T. Rudel. 2005. Prohibitin is required for Ras-induced Raf-MEK-ERK activation and epithelial cell migration. Nat. Cell Biol. 7:837–843. [DOI] [PubMed] [Google Scholar]
- Rastogi, S., B. Joshi, P. Dasgupta, M. Morris, K. Wright, and S. Chellappan. 2006. a. Prohibitin facilitates cellular senescence by recruiting specific corepressors to inhibit E2F target genes. Mol. Cell. Biol. 26:4161–4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rastogi, S., B. Joshi, G. Fusaro, and S. Chellappan. 2006. b. Camptothecin induces nuclear export of prohibitin preferentially in transformed cells through a CRM-1-dependent mechanism. J. Biol. Chem. 281:2951–2959. [DOI] [PubMed] [Google Scholar]
- Roskams, A.J., V. Friedman, C.M. Wood, L. Walker, G.A. Owens, D.A. Stewart, M.S. Altus, D.B. Danner, X.T. Liu, and J.K. McClung. 1993. Cell cycle activity and expression of prohibitin mRNA. J. Cell. Physiol. 157:289–295. [DOI] [PubMed] [Google Scholar]
- Schleicher, M., F. Brundin, S. Gross, W. Muller-Esterl, and S. Oess. 2005. Cell cycle-regulated inactivation of endothelial NO synthase through NOSIP-dependent targeting to the cytoskeleton. Mol. Cell. Biol. 25:8251–8258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sells, M.A., J.T. Boyd, and J. Chernoff. 1999. p21-activated kinase 1 (Pak1) regulates cell motility in mammalian fibroblasts. J. Cell Biol. 145:837–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sewing, A., B. Wiseman, A.C. Lloyd, and H. Land. 1997. High-intensity Raf signal causes cell cycle arrest mediated by p21Cip1. Mol. Cell. Biol. 17:5588–5597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soga, N., N. Namba, S. McAllister, L. Cornelius, S.L. Teitelbaum, S.F. Dowdy, J. Kawamura, and K.A. Hruska. 2001. Rho family GTPases regulate VEGF-stimulated endothelial cell motility. Exp. Cell Res. 269:73–87. [DOI] [PubMed] [Google Scholar]
- Steglich, G., W. Neupert, and T. Langer. 1999. Prohibitins regulate membrane protein degradation by the m-AAA protease in mitochondria. Mol. Cell. Biol. 19:3435–3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshige, K., and S. Minakami. 1979. NADH- and NADPH-dependent formation of superoxide anions by bovine heart submitochondrial particles and NAD-ubiquinone-reductase preparation. Biochem. J. 180:129–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsuta, T., K. Model, and T. Langer. 2005. Formation of membrane-bound ring complexes by prohibitins in mitochondria. Mol. Biol. Cell. 16:248–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theiss, A.L., R.D. Idell, S. Srinivasan, J.M. Klapproth, D.P. Jones, D. Merlin, and S.V. Sitaraman. 2007. Prohibitin protects against oxidative stress in intestinal epithelial cells. FASEB J. 21:197–206. [DOI] [PubMed] [Google Scholar]
- van Leeuwen, F.N., S. van Delft, H.E. Kain, R.A. van der Kammen, and J.G. Collard. 1999. Rac regulates phosphorylation of the myosin-II heavy chain, actinomyosin disassembly and cell spreading. Nat. Cell Biol. 1:242–248. [DOI] [PubMed] [Google Scholar]
- Wang, S., G. Fusaro, J. Padmanabhan, and S.P. Chellappan. 2002. a. Prohibitin co-localizes with Rb in the nucleus and recruits N-CoR and HDAC1 for transcriptional repression. Oncogene. 21:8388–8396. [DOI] [PubMed] [Google Scholar]
- Wang, S., B. Zhang, and D.V. Faller. 2002. b. Prohibitin requires Brg-1 and Brm for the repression of E2F and cell growth. EMBO J. 21:3019–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z.W., T. Zheng, A. Zhang, B.T. Altura, and B.M. Altura. 1998. Mechanisms of hydrogen peroxide-induced contraction of rat aorta. Eur. J. Pharmacol. 344:169–181. [DOI] [PubMed] [Google Scholar]
- Zhang, H., Y. Luo, W. Zhang, Y. He, S. Dai, R. Zhang, Y. Huang, P. Bernatchez, F.J. Giordano, G. Shadel, et al. 2007. Endothelial-specific expression of mitochondrial thioredoxin improves endothelial cell function and reduces atherosclerotic lesions. Am. J. Pathol. 170:1108–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov, D.B., C.R. Filburn, L.O. Klotz, J.L. Zweier, and S.J. Sollott. 2000. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon on accompanying induction of mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 192:1001–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}