

Abstract
In the absence of activation signals, circulating human neutrophils and eosinophils undergo spontaneous apoptosis. The glucocorticoid Dexamethasone (Dex) accelerates apoptosis in inflammatory cells such as eosinophils, but uniquely delays neutrophil apoptosis. Corresponding to the opposite effects of Dex on granulocyte apoptosis, we demonstrate that in neutrophils and eosinophils Dex oppositely affects expression of the anti-apoptotic Bcl-2 family protein Mcl-1L. Mcl-1L expression declines over time in vitro; however, Dex maintains Mcl-1L expression in neutrophils. In contrast, Dex accelerates Mcl-1L protein loss in eosinophils. Neither Mcl-1S, a pro-apoptotic splice variant, nor Bax were affected. Dex treatment in the presence of a translation inhibitor stabilized existing Mcl-1L protein in neutrophils, while Mcl-1L stability in eosinophils was unaffected. Accordingly, delay of neutrophil apoptosis by Dex was prevented by antisense Mcl-1L siRNA. Our findings suggest that regulation of Mcl-1L degradation plays an important role in the opposite effects of Dex on granulocyte apoptosis.
Keywords: Neutrophils, Eosinophils, Apoptosis, Mcl-1, Dexamethasone, Bax, Inflammation
1. Introduction
Neutrophils and eosinophils are closely related granulocytes, which are non-dividing, fully differentiated cells. These granulocytes produce an arsenal of antimicrobial defenses that are contained in granules within the cell, which include reactive oxygen species and potent cytotoxic and degradative enzymes [1,2]. Upon stimulation, granulocytes are activated to kill invading pathogens by releasing these degradative enzymes in the local millieu. However, if activated at inappropriate times, the release of anti-microbials from granulocytes can cause local tissue damage and chronic inflammation.
Glucocorticoids are used to treat a broad range of inflammatory and autoimmune diseases. In asthmatic patients, glucocorticoids suppress airway inflammation, reduce eosinophil numbers in bronchial mucosa and peripheral blood, and decrease neutrophil migration into tissues [3]. A portion of the anti-inflammatory effects of Dex can be attributed to their ability to induce apoptosis in a number of pro-inflammatory cell types. In contrast to their pro-apoptotic effects on inflammatory cells, Dex inhibits apoptosis in tissue resident cells such as lung epithelium [4]. Paradoxically, glucocorticoids oppositely affect eosinophil and neutrophil survival. Circulating granulocytes undergo spontaneous apoptosis in the absence of activation signals; this apoptosis triggers their clearance by macrophages [5–8]. In the absence of cytokines, neutrophils undergo spontaneous apoptosis in vitro after 6–24 h [9]. Eosinophils have a longer life span; they undergo apoptosis in vitro within 2–3 days [10]. The glucocorticoid Dexamethasone (Dex) dramatically induces eosinophil apoptosis; but Dex actually delays neutrophil apoptosis [11–13]. The goal of the experiments presented here was to determine the basis for these opposite apoptotic responses of granulocytes to Dex.
According to previous studies, neutrophils and eosinophils contain few mitochondria. These mitochondria are not a significant source of respiration; however, they are vital in regulating apoptosis [14,15]. The release of apoptosis mediators from the mitochondria is primarily regulated by Bcl-2 family members [16,17]. Pro-apoptotic members of the Bcl-2 family (such as Bax, Bad, Bcl-XS, Bak, and Bid) trigger the release of apoptosis mediators, such as cytochrome c (Cyt c). Anti-apoptotic members (such as Bcl-2, Bcl-XL, Mcl-1L, and A1) stabilize the mitochondrial membrane and neutralize pro-apoptotic proteins [18]. The balance of pro-apoptotic to anti-apoptotic Bcl-2 family proteins is vital in determining the fate of the cell [19]. This led us to investigate the role of Bcl-2 family proteins in the opposite effects of Dex on granulocyte apoptosis.
In both neutrophils and eosinophils, the predominant pro-apoptotic Bcl-2 family member expressed is Bax [20]. The constitutively high level of Bax expressed in granulocytes is suspected to play a part in their relatively short life spans. An anti-apoptotic Bcl-2 family protein, Mcl-1L was also of particular interest due to its half-life of only 2 to 3 h [21]. The short half-life of Mcl-1L suggests the protein is rapidly expressed in response to a survival signal; and then it is rapidly degraded once the signal is absent. Additionally, Mcl-1S, a splice variant of Mcl-1L, has recently been identified [22,23]. Unlike Mcl-1L, Mcl-1S is pro-apoptotic, but still retains the protein sequence motif thought to target the protein for rapid degradation. The relative balance of expression of Mcl-1L, Mcl-1S, and Bax proteins is likely important in the regulation of granulocyte apoptosis by Dex. Therefore, we hypothesized that Dex has an opposite effect on neutrophil and eosinophil apoptosis through alternate effects on expression of Bcl-2 family members such as Mcl-1L, Mcl-1S, and Bax.
2. Materials and Methods
2.1 Neutrophil and Eosinophil Isolation
Healthy human donors with absolute peripheral blood eosinophil counts <350/mm3 were used in a protocol approved by our Institutional Review Board. Written, informed consent was obtained from each blood donor according to Internal Review Board regulation. Granulocytes were isolated from whole blood by gravity sedimentation over Ficoll-Hypaque as described previously [24,25]. Eosinophils were separated by negative magnetic selection from neutrophils tagged by adherence of anti-FcRγIII coupled to magnetic beads [14,26] (anti- FcRγIII [3G8] was a gift from Jay Unkeless, Mt. Sinai School of Medicine, NY). Isolated eosinophils were >97% pure. Neutrophils and eosinophils were cultured at 37°C in RPMI-1640 media with 100 U/ml penicillin, 100 mg/ml streptomycin, 10% FBS, and L-glutamine at 1 × 106 cells/ml (Invitrogen, Carlsbad, CA).
2.2 Assessment of Apoptotic Morphology
Granulocytes were cytocentrifuged and stained with Diff-Quik (Baxter, McGraw Park, IL) and observed under light microscopy for morphological changes, including condensation of the nucleus and decreased cell volume. At least two hundred cells were examined per condition.
2.3 Immunohistochemistry
Cell samples of 1 × 106 were resuspended in 200 μl PBS and spun onto poly-L-lysine coated slides (Cytospin, Thermo-Shandon). Slides were fixed with 10% Formalin for 15 min, washed in PBS, and then placed in 1%BSA/0.1%Saponin at 4°C until processed. Rabbit anti-Bax (PharMingen) was added at 1:100 in 1% BSA/0.1% Saponin and incubated for 45 min. Non-specific flourochrome binding in eosinophils was blocked with 0.2% chromotrope 2R between primary and secondary antibody incubations. Alexa 488 goat anti-rabbit IgG was added at 1:800 in 1% BSA/0.1% Saponin and incubated for 45 min. Cells were mounted with gel mount (Biomeda, Foster City, CA) and examined by fluorescence microscopy.
2.4 Generation of Mcl-1S Polyclonal Antibody
The frame shift resulting from the skipping of exon 2 produces a unique C-terminal peptide sequence in Mcl-1S that was used to generate a specific antibody. A 15 amino acid peptide (RGPRRWHQECAAGFC, corresponding to amino acids 239-253) was used to immunize rabbits (Genemed Synthesis, San Francisco, CA). Anti-serum was subjected to ammonium sulfate precipitation and then peptide-specific affinity purification using the SulfoLink Kit from Pierce (Rockford, IL) following the manufacturer’s protocol. Purified antibody recognized a single band when used in a western blot. The single band was eliminated when the antibody was pre-incubated with the Mcl-1S specific peptide used to immunize the rabbit (Fig. 4E), confirming that the antibody binding is specific for the generated peptide sequence. The anti-Mcl-1S antibody was designated as anti-Mcl-1S 239.
Fig 4.

Mcl-1S protein expression remained constant during Dex treatment in neutrophils and eosinophils, despite changes in mRNA. A) Total RNA was isolated from neutrophils incubated in media alone or with Dex (20 μM) at 0, 3, 5, 7, and 12 h after incubation. B) RNA was isolated from untreated and Dex-treated eosinophils at 0, 6, and 12 h after incubation. Mcl-1S mRNA expression was analyzed by real time PCR and was normalized against GAPDH mRNA expression. The ratio of expression of Mcl-1S mRNA in Dex-treated cells versus control cells was calculated at each time point. * denotes p<0.05 for Dex-treated cells versus control cells at each time point. 1 × 107 neutrophils (C) and 1 × 106 eosinophils (D) were processed and examined for Mcl-1S protein expression by Western blot. E) The specificity of the Mcl-1S 239 antibody was tested by probing blots that contained boiled cell lysates of either 1 × 106 or 1 × 107 total neutrophils. The two sets of samples were run on the same gel, transferred, and then cut and blotted with either the Mcl-1S 239 antibody or the Mcl-1S 239 antibody pre-incubated with the peptide used to raise it (RGPRRWHQECAAGFC, corresponding to the unique amino acids 239-253 of Mcl-1S). The detection of a single band with the antibody alone, and the disappearance of the band upon addition of the peptide confirmed the specificity of the Mcl-1S 239 antibody (representative experiment shown, n=2).
2.5 Western blot analysis
Neutrophil samples of 1 × 107 cells/condition for Mcl-1 blots and 1 × 106 cells/condition for Bax blots were used. Eosinophil samples of 1 × 106 cells/condition were used for both protein blots. For Mcl-1 stability experiments, cells were treated with CHX at 10 μg/ml. At each time point, cells were counted and equal cell numbers were lysed in SDS sample buffer (125 mM Tris pH 6.8, 2% SDS, 10% Glycerol, and 10 mM DTT), boiled for 5 min, and then frozen at −80°C until samples were processed. Proteins were separated on Criterion Tris-HCl SDS-PAGE gels (10% for Mcl-1 Westerns and 18% for Bax Westerns, BioRad, Hercules, CA) and then transferred onto PVDF (Immobilon, Millipore, Bedford, MA). Membranes were blocked with 5% nonfat dry milk in PBS for 1 h at room temperature. Mcl-1 antibody (2 μg/ml) (BD PharMingen, San Diego, CA), anti-Mcl-1S 239 antibody (1 μg/ml), Bax antibody (1 μg/ml) (BD PharMingen), or GAPDH antibody (10 μg/ml) (Chemicon, Temecula, CA) was added and incubated for 1 h. Membranes were then washed in PBS + 0.05% Tween. A goat anti-mouse horseradish peroxidase secondary antibody (ICN/Cappel, Aurora, OH) was added at a dilution of 1:2000 in 1% milk and incubated at room temperature for 1 h. Protein bands were then detected using SuperSignal West Fempto Maximum Sensitivity Substrate and CL-Xposure film (Pierce, Rockford, IL). BioRad Quantity One software was used to compare protein expression between samples. Equal loading was confirmed by GAPDH western blots or by analyzing actin levels by Ponceau S staining.
2.6 Real Time PCR
Total RNA was extracted using RNA STAT-60 (Tel-Test, Inc., Friendswood, TX) according to the Tel-Test protocol. RNA was reverse transcribed using the GeneAmp Gold RNA PCR Core Kit (PE Biosystems, Foster City, CA) with Oligo dT according to manufacturer’s instructions. cDNA samples were then analyzed by semi-quantitative, real-time PCR using an ABI 7000 Sequence Detection System (Applied Biosystems, Foster City, CA). Primers and Taqman probes were designed using ABI Primer Express Software and obtained from Applied Biosystems. The Mcl-1L forward primer was 5′-CGC AAC CAC GAG ACG G-3′ and the reverse primer was 5′-TCA CAT CGT CTT CGT TTT TGA T-3′. The sequence of the Taqman Mcl-1L probe was 5′-FAM-TCC AAG GCA TGC TTC GGA AAC TG-TAMRA-3′. The Mcl-1S forward primer was 5′-GCA ACC ACG AGA CGG CC-3′ and the reverse primer was 5′-GAT GCC ACC TTC TAG GTC CTC TAC-3′. The sequence of the Taqman Mcl-S probe was 5′-FAM-TGG AAG AAC TCC ACA AAC CCA TCC TTG G-TAMRA-3′. Primers were used at 300 nM and probe was used at 200 nM. Real-time reactions were performed using Taqman Universal PCR Master Mix (Applied Biosystems) according to manufacturer’s instructions. The amount of Mcl-1 mRNA was normalized against levels of GAPDH mRNA using Taqman GAPDH Control Reagents (Applied Biosystems). All data was analyzed using the Comparative CT Method [27].
2.7 Mcl-1 Antisense Oligonucleotides
Antisense Mcl-1 (5′-GGGGCTTCCATCTCCTCAA-3′) and sense Mcl-1 (5′-CCCCGAAGGTAGAGGAGTT-3′) oligos designed by Leuenroth et al were obtained from Oligos Etc. (Wilsonville, OR) [28]. Oligos were complexed with the cationic lipid DOTAP (Sigma). Five μg DOTAP was added to every 1μg oligos and allowed to complex for 15 minutes at room temperature prior to adding to culture. Granulocytes were cultured as described above with a final concentration of 2 μM oligos. Cells were harvested at shown times, cytospin slides were made, and cells were assessed for apoptotic morphology. Mcl-1 protein knockdown was confirmed by western blot after 6 h of treatment using 2.5 × 106 treated neutrophils/condition.
2.8 Statistical Analysis
All experiments were conducted a minimum of 3 times, unless stated. Results are displayed as mean (± standard error of the mean). Significance was determined by two-tailed, paired t-test.
3. Results
3.1 Clustering of Bax protein occurs concurrently with the appearance of apoptotic morphology in neutrophils and eosinophils
It is unknown at what point in the apoptotic pathway at which Dex has its divergent effects on neutrophil and eosinophil apoptosis. One key step in the mitochondrial apoptosis pathway is release of Cyt c from the mitochondria. According to immunofluorescence studies, organellar localization of Cyt c in neutrophils was lost after 20 h of incubation in media alone. This resulted in a diffuse labeling throughout the cytoplasm. However, in neutrophils incubated with Dex for 20 h, Cyt c remained in an organellar localization. In neutrophils, the ability of Dex to maintain organellar Cyt c localization corresponds with the ability of Dex to delay apoptosis. In eosinophils, Cyt c was still localized to discrete organelles after 20 h in culture. The persistence of Cyt c organellar localization is consistent with the longer life span of eosinophils. In contrast, most eosinophils treated with Dex showed diffuse cytoplasmic labeling of Cyt c after 20 h (data not shown). Since Dex maintained Cyt c mitochondrial localization in neutrophils while Dex induced Cyt c release in eosinophils, we concluded that the opposite effect of Dex on apoptosis occurs upstream of Cyt c release from mitochondria.
In the cytoplasm of healthy cells, pro-apoptotic Bax is maintained in a monomeric state. Once apoptosis is induced, Bax is activated which triggers it to homodimerize and translocate to mitochondria. However, this translocation of Bax is prevented if it is bound and retained in the cytoplasm by anti-apoptotic Bcl-2 proteins [29]. The movement of Bax from the cytoplasm to the mitochondria in response to Dex was examined by immunofluoresence. As expected, freshly isolated neutrophils and eosinophils showed diffuse labeling throughout the cell (not shown). Isolated neutrophils cultured in vitro for 12 h showed a clustered pattern of Bax labeling consistent with Bax translocation to organelles such as mitochondria. In Dex-treated neutrophils, in which apoptosis is delayed, Bax clustering was also delayed (Fig. 1A). Isolated eosinophils retained diffuse, cytoplasmic labeling of Bax; but Dex treatment induced Bax clustering (Fig. 1A). In both granulocytes, the appearance of apoptotic morphology (loss of cell volume and condensed nuclei) and clustered labeling of Bax occurred with similar time courses. This data further supports a role for Bax translocation in granulocyte apoptosis (Fig. 1B and C).
Fig 1.

Organellar, clustered localization of Bax closely corresponds with the induction of apoptosis in neutrophils and eosinophils. (A) Neutrophils and eosinophils were incubated in media alone or with Dex (20 μM). At designated time points, cells were fixed, permeabilized, probed with an anti-Bax antibody, and labeled with an Alexa Fluor 488-conjugated secondary antibody. To compare the movement of Bax with the induction of apoptosis, the percent of neutrophils (B) and eosinophils (C) demonstrating apoptotic morphology was graphed along with the percentage of cells showing clustered labeling of Bax. To examine apoptotic morphology, 1 × 105 cells were collected at each time point, cytocentrifuged, stained with Diff-Quik, and analyzed for apoptotic features including decreased cell volume and condensed nuclei. At least 200 cells were examined per condition. * denotes p<0.05 for Dex-treated cells versus control cells at each time point. At the time points indicated, 1 × 106 neutrophils (D) or eosinophils (E) were harvested and examined for Bax protein expression by Western blot.
Over a 24 h culture of untreated and Dex-treated granulocytes, Bax protein expression remained constant (Fig 1D and E). Although Bax protein levels in eosinophils declined to approximately 50% of 0 h control levels by 36 h, there was no difference in Bax protein levels between untreated and Dex-treated eosinophils (Fig. 1E). Thus, Dex does not regulate Bax at the level of protein expression; and the divergent effects of Dex on neutrophil and eosinophil apoptosis appear to occur upstream of Bax activation.
3.2 Treatment with Dex maintained Mcl-1L protein expression in neutrophils and decreased Mcl-1L protein expression in eosinophils
In response to agents such as GM-CSF, interleukin-1β, and lipopolysaccharide, Mcl-1L protein levels have been shown to correlate with changes in the survival rate of neutrophils [30]. To determine the effect of Dex on Mcl-1L protein expression in neutrophils and eosinophils, granulocytes were cultured in media alone or with Dex and examined by Western blot for Mcl-1L. When neutrophils were cultured in media alone, Mcl-1L expression decreased rapidly. In contrast, even 12 h after treatment, Mcl-1L expression remained high in Dex-treated neutrophils (Fig. 2A and C). Mcl-1L protein in untreated neutrophils declined to 50% of 0 h levels by approximately 6.5 h. In Dex-treated neutrophils, Mcl-1L levels did not decline by half until 10.5 h. The level of apoptosis was then determined by observing changes in morphology indicative of apoptosis, such as loss of cell volume and condensed nuclei. Fifty percent of untreated neutrophils had entered apoptosis by 15 h in culture. Fitting with the known Dex effects on neutrophils, 50% of Dex-treated neutrophils did not enter apoptosis until after 25 h in culture (Fig. 2E). Therefore, corresponding to the ability of Dex to delay apoptosis in neutrophils, the loss of Mcl-1L in neutrophils precedes changes in apoptotic morphology and is delayed following Dex treatment.
Fig 2.

Dex maintains Mcl-1L expression in neutrophils and accelerates the loss of Mcl-1L in eosinophils. Neutrophils (A) and eosinophils (B) were incubated in the absence or presence of Dex (20 μM). Neutrophils (1 × 107cells) and eosinophils (1 × 106 cells) were processed and examined for Mcl-1L expression by Western blot at each of the designated time points. Densitometry of Mcl-1L Western blots shows the expression of Mcl-1L in neutrophils (C) and eosinophils (D) as a percent of 0 h control. At each time point, 1 × 105 neutrophils (E) and eosinophils (F) were cytocentrifuged, stained with Diff-Quik, and analyzed for apoptotic morphology. At least 200 cells were examined per condition. * denotes p<0.05 for Dex-treated cells versus control cells at each time point.
Due to the dramatic differences in neutrophil and eosinophil life spans in vitro, later time points were investigated in the eosinophil in order to examine similar stages in the apoptotic process of both granulocytes. Mcl-1L expression in untreated eosinophils declined by approximately 50% of the initial levels by 36 h in culture; and Mcl-1L protein was still present at 48 h. However, Mcl-1L protein expression in eosinophils treated with Dex was dramatically lower than in untreated eosinophils, requiring only 18 h to decline by 50% from 0 h levels (Fig. 2B and D). This acceleration of Mcl-1L decline is consistent with the enhanced apoptosis induced by Dex. After 48 h, only 21% of eosinophils cultured in media alone had entered apoptosis. In contrast, 50% of eosinophils treated with Dex entered apoptosis by 27 h in culture (Fig. 2F).
3.3 Dex has a similar effect on Mcl-1L mRNA expression in neutrophils and eosinophils, in contrast to the opposite effect of Dex on Mcl-1L protein expression
Dex exerts a large portion of its effects through binding to the intracellular glucocorticoid receptor. This steroid-receptor complex translocates to the nucleus where it interacts with glucocorticoid response elements in the DNA and thereby, alters gene transcription [31]. Mcl-1L mRNA expression was quantified by real time PCR to determine if the opposite effect of Dex on Mcl-1L protein in granulocytes occurs at the transcriptional level. In Dex-treated neutrophils, Mcl-1L mRNA levels peaked around 3 h at which time Mcl-1L mRNA was 3.5 (± 0.5) fold greater than in control neutrophils and Mcl-1L mRNA returned to control levels by 12h post-treatment (Fig. 3A). Dex also increased levels of Mcl-1L mRNA in eosinophils. Mcl-1L mRNA was 4.2 (± 0.3) fold higher at 6 h post-treatment and at 12 h Mcl-1L mRNA expression was at or below control levels (Fig. 3B). Fewer time points were examined in eosinophils due to limiting amounts of eosinophils and RNA that can be recovered from healthy normal donors. Although Dex maintained Mcl-1L mRNA expression above that expressed in untreated cells in both granulocytes, this does not account for the opposite effect of Dex on Mcl-1L at the protein level. Therefore, the differing effect of Dex on Mcl-1L protein occurs downstream of early transcriptional regulation of Mcl-1L.
Fig 3.
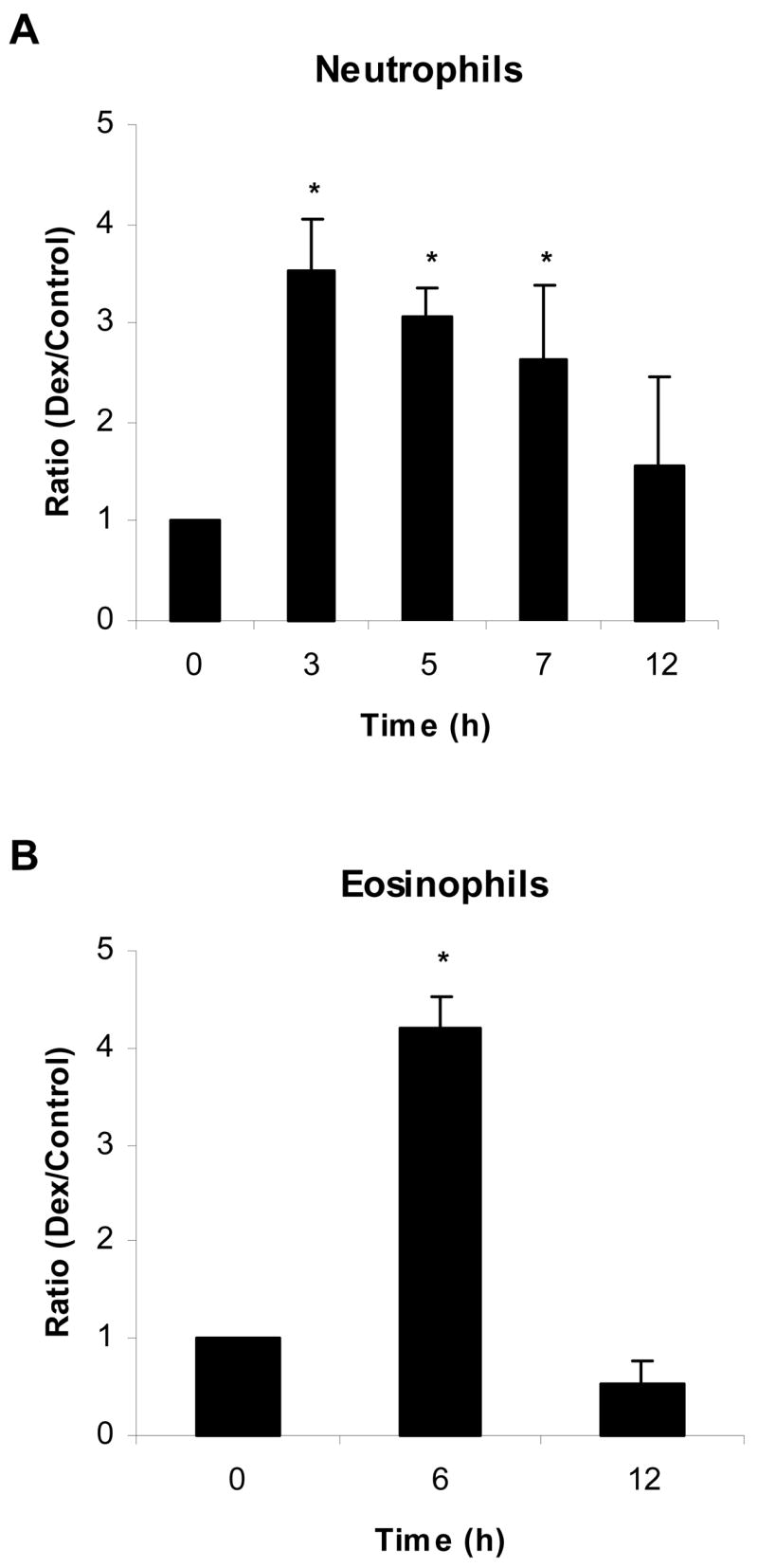
Dex up-regulates Mcl-1L mRNA expression in both neutrophils and eosinophils. Neutrophils and eosinophils were cultured in media alone or with Dex (20 μM). A) Total RNA was isolated from neutrophils at times 0, 3, 5, 7, and 12 h. B) Total RNA was isolated from eosinophils at 0, 6, and 12 h after incubation. Real time PCR was used to examine Mcl-1L mRNA levels over these time courses. Mcl-1L mRNA levels were normalized against GAPDH mRNA expression. At each time point, we determined the ratio of expression of Mcl-1L mRNA expression in Dex-treated cells versus control cells. * denotes p<0.05 for each time point versus 0 h.
3.4 In contrast to Mcl-1L, Dex does not significantly alter Mcl-1S protein expression
A splice variant of Mcl-1 mRNA encodes Mcl-1S, which is pro-apoptotic in contrast to the anti-apoptotic activity of Mcl-1L. To determine the effect of Dex on Mcl-1S mRNA expression, a real time PCR probe, specific for Mcl-1S, was designed to span the gap created by the skipped exon in this transcript. In neutrophils, Mcl-1S mRNA levels were unchanged in the presence of Dex (Fig. 4A). However, after 6 h there was about a 3 fold increase in Mcl-1S mRNA in Dex-treated eosinophils (Fig. 4B).
A polyclonal anti-peptide antibody, anti-Mcl-1S 239 was generated to allow for a specific examination of the effect of Dex on Mcl-1S protein expression (Fig. 4E). Despite changes in Mcl-1S mRNA in eosinophils, Western blot analysis detected no change in the amount of Mcl-1S protein expressed in neutrophils or eosinophils after treatment with Dex (Fig. 4C, D). Therefore, while Dex alters Mcl-1L protein expression in granulocytes, expression of the splice variant Mcl-1S remains constant.
3.5 Dex increases Mcl-1L stability in neutrophils, but does not alter Mcl-1L stability in eosinophils
As demonstrated by Derouet et al, the treatment of neutrophils with the proteasome inhibitor MG-132 slows the loss of Mcl-1L [32]. Those studies show the short half-life of Mcl-1L is due in part to its rapid turnover via the proteasome. Therefore, the effect of Dex on Mcl-1L protein stability in granulocytes was determined by incubating the cells for 6 h with the translation inhibitor cycloheximide (CHX) (Fig. 5). A portion of these cells were treated with Dex in addition to CHX to determine if Dex affected the stability of existing Mcl-1L in the absence of de novo protein synthesis. After treatment with CHX for 2 h, levels of Mcl-1L protein in neutrophils were dramatically reduced; and after 4 h of CHX treatment, Mcl-1L was barely detectable. However, in Dex-treated neutrophils, Mcl-1L protein levels remained high at 2 h; and Mcl-1L was still evident at 4 h despite the lack of de novo protein synthesis (Fig. 5A). Therefore, Dex maintains Mcl-1L expression in neutrophils in part by slowing the turnover rate of the protein. This was not the case in eosinophils. Mcl-1L also showed decay in eosinophils within 6 h in the presence of CHX; but unlike the rapid Dex effect in neutrophils, Dex did not exert a significant effect on the rate of Mcl-1L turnover in eosinophils (Fig. 5B).
Fig 5.
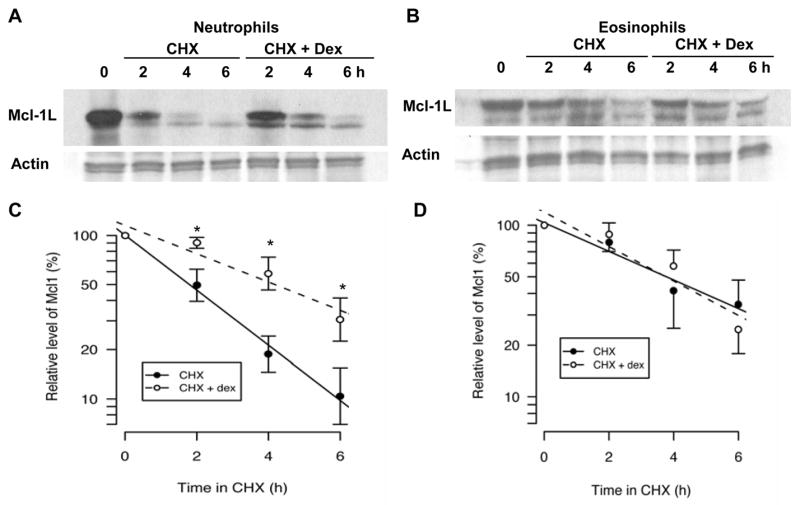
Dex enhances Mcl-1L protein stability in neutrophils, but not in eosinophils. Neutrophils (A and C) and eosinophils (B and D) were cultured with CHX (10 μg/ml) to eliminate de novo protein synthesis. Cells were either cultured in the absence or presence of Dex (20 μM) to determine the effect of Dex on the stability of existing Mcl-1L protein. Neutrophils and eosinophils were analyzed by Western blot, as in figure 2, at 0, 2, 4, and 6 h to determine how much of the initial Mcl-1L was still present. Densitometry of western blots is graphed on a log scale as a percent of the 0 h control value for neutrophils (C) and eosinophils (D) treated with CHX alone (closed circles) or treated with CHX and Dex (open circles). The solid lines represent the best fit exponential decay for CHX-treated samples and the dashed lines represent the best fit exponential decay for CHX and Dex-treated samples. * denotes p<0.05 for Dex-treated cells versus control cells at each time point.
These data allowed us to calculate the half-life of Mcl-1L in neutrophils and eosinophils in the presence of CHX alone or with CHX and Dex. In eosinophils, Mcl-1L had a half-life of approximately 3.6 h when treated with CHX. Addition of Dex resulted in a Mcl-1L half-life of 3 h, but this difference was not statistically significant (Fig. 5D). In CHX-treated neutrophils, Mcl-1L displayed a shorter half-life of only 1.8 h. In contrast to Dex effects on Mcl-1L in eosinophils, Dex actually increased the half-life of Mcl-1L in neutrophils to approximately 3.5 h (Fig. 5C). Therefore, Dex treatment extends the half-life of Mcl-1L in neutrophils to the half-life that was observed in untreated eosinophils. This data demonstrates that in neutrophils Dex decreases Mcl-1L degradation; while in eosinophils Dex exerts a negligible effect on Mcl-1L degradation. Therefore, an important factor in the opposite effects of Dex on neutrophil and eosinophil apoptosis is the early alteration of Mcl-1L protein stability in neutrophils.
3.6 Anti-apoptotic Effect of Dex on Neutrophils is Prevented by Mcl-1 Antisense Oligonucleotides
Mcl-1 antisense oligonucleotides, previously designed by Leuenroth, were used to determine the importance of Mcl-1 in the Dex-induced delay of neutrophil apoptosis [28]. Within 12 h, Mcl-1 antisense oligos significantly induced neutrophil apoptosis above control levels (Fig 6A, p < 0.005, n=6); whereas Dex alone decreased apoptosis significantly (less than 5% apoptosis, p < 0.02). Similar results were seen out to 20 h (not shown). When neutrophils were treated with Dex following uptake of Mcl-1 antisense oligos, the ability of Dex to delay apoptosis was significantly inhibited (p < 0.001, n=6 for Dex plus antisense versus Dex alone). Therefore, we conclude that Mcl-1L protein expression is essential in Dex inhibition of neutrophil apoptosis. As end-differentiated phagocytic cells, uptake of liposomes alone or liposomes with sense constructs appeared to activate the neutrophils and tended to inhibit apoptosis. However, Mcl-1 antisense oligos overcame this possible protective effect and significantly induced neutrophil apoptosis. Additionally, only Mcl-1 antisense oligos abrogated the protective effect of Dex, sense oligos did not, further supporting the importance of Mcl-1L expression in preventing neutrophils from entering apoptosis. Western blotting confirmed antisense oligos knocked down Mcl-1 protein expression (Fig 6B), as shown by Leuenroth [28].
Fig 6.
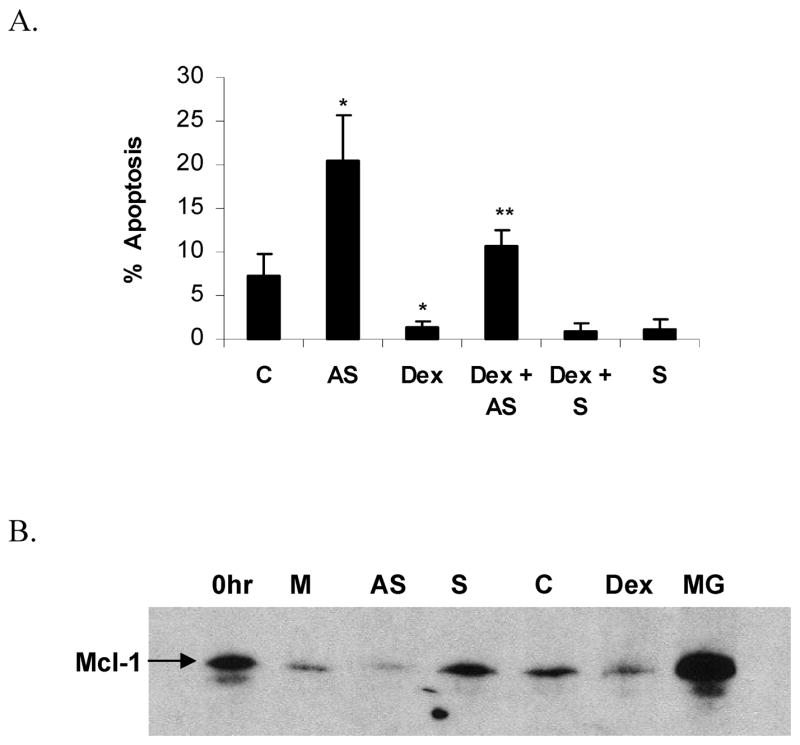
Mcl-1 antisense oligos induce neutrophil apoptosis and inhibit the anti-apoptotic effect of Dex. Neutrophils were cultured in media alone (C), or with the addition of Mcl-1 antisense oligos (AS), Dex, Dex plus Mcl-1 antisense (Dex + AS), Dex plus Mcl-1 sense (Dex + S), or Mcl-1 sense oligos (S), for 12 h. A) Neutrophils were cytocentrifuged onto slides, and stained with Diff Quik in order to determine the percentage of cells demonstrating apoptotic morphology. * denotes p< 0.02 for Dex versus control and for antisense versus control, ** denotes p< 0.02 for Dex + AS versus Dex alone, n=6. B) Western blot of neutrophil protein confirmed that Mcl-1 antisense oligos (AS) knocked down Mcl-1 protein after 6 h of treatment relative to media alone (M), sense oligos (S), vehicle control (C), Dex, or proteasome protection by MG-132 (MG) (representative experiment shown, n=2).
4. Discussion
As suggested by our results, spontaneous granulocyte apoptosis proceeds through the mitochondrial pathway. Cyt c release from mitochondria is affected in an opposite fashion by Dex; this opposite effect parallels the opposite effect of Dex on granulocyte apoptosis. This led us to hypothesize that Dex was exerting these opposite effects on granulocytes by differentially regulating a factor involved in the control of apoptosis mediator release from mitochondria.
Mitochondrial membrane potential and release of apoptosis mediators from mitochondria into the cytoplasm are regulated by Bcl-2 related proteins [17,33]. According to our studies, clustering of the pro-apoptotic Bcl-2 related protein Bax around mitochondria closely corresponds to induction of granulocyte apoptosis. The delay of neutrophil apoptosis by Dex is accompanied by a delay in Bax movement to the mitochondria. Similarly, Dex-induced apoptosis in eosinophils corresponds to a mitochondria-like organellar localization of Bax. As suggested by recent studies, pro-apoptotic Bcl-2 related proteins that contain Bcl-2 homology (BH) regions 1-3, such as Bax, are directly responsible for executing the induction of Cyt c release [34]. In both granulocytes, this is consistent with our data that show the features of apoptotic morphology and organellar clustering of Bax appear concurrently. Bax protein levels were not altered by Dex in either granulocyte. This suggests that Dex does not affect Bax at the level of expression but rather alters activation or localization of Bax at the post-translational level.
As neutrophils undergo spontaneous apoptosis, the anti-apoptotic Bcl-2 protein Mcl-1L decreases; in addition, Mcl-1L protein levels appear to be closely linked to cytokine-mediated neutrophil survival [28,30,35]. These previous findings prompted our investigation of Mcl-1L expression in relation to Dex treatment. Our data make the novel observation that Dex has an opposite effect on Mcl-1L protein expression in neutrophils and eosinophils. This opposite effect of Dex on Mcl-1L corresponds to the opposite effect of Dex on neutrophil and eosinophil apoptosis. As compared to untreated neutrophils, Mcl-1L expression following Dex treatment was sustained. The ability of Dex to maintain expression of anti-apoptotic Mcl-1L in neutrophils is consistent with the ability of Dex to delay neutrophil apoptosis.
Once again in contrast to the Dex-treated neutrophils, the decline of Mcl-1L protein in Dex-treated eosinophils was accelerated. In untreated eosinophils, Mcl-1L did not decline to 50% of its initial level until 36 h. Dex treatment cut that time in half. As demonstrated in neutrophils, Mcl-1L protein levels also corresponded to levels of eosinophil apoptosis. Therefore, Dex maintained expression of Mcl-1L in neutrophils; while Dex increased Mcl-1L loss in eosinophils. Since real time PCR demonstrated that Dex had similar effects on Mcl-1L mRNA expression in both granulocytes, this divergent effect of Dex on Mcl-1L protein expression is likely to occur at the post-transcriptional level.
CHX treatment of granulocytes allowed us to examine the effect of Dex on Mcl-1L protein stability in the absence of de novo protein synthesis. The addition of Dex to eosinophils in the presence of CHX demonstrated no significant effects on Mcl-1L decay. However, Dex treatment of neutrophils in the absence of de novo protein synthesis prolonged the life of existing Mcl-1L protein. A critical step in the divergent effects of Dex on granulocyte apoptosis, as suggested by our data, is the delay in Mcl-1L protein degradation in neutrophils; however, this is not the case in eosinophils. In eosinophils, examination of the half-life of Mcl-1L protein in the CHX experiments demonstrated that Mcl-1L has a half-life around 3 h. As previously noted, Dex treatment did not alter the half-life of Mcl-1L protein in eosinophils. In neutrophils, Mcl-1L demonstrated a shorter half-life of only 1.8 h. In contrast to the negligible effect of Dex on Mcl-1L stability in eosinophils, Dex treatment increased the half-life of Mcl-1L to approximately 3.5 h in neutrophils. This delay of Mcl-1L degradation is consistent with the pattern we observe with Dex-treated neutrophils in which Dex induces both delayed loss of Mcl-1L and delayed apoptosis. Our observation that Mcl-1 activity can be regulated through control of its rate of degradation has been recently confirmed. As shown by Kato et al, inhibition of neutrophil apoptosis by cyclic AMP agonists prevents Mcl-1 degradation [36]. It has also been shown that treatment of neutrophils with the proteasome inhibitor MG-132 prevents loss of Mcl-1L [32]. Mcl-1L degradation is controlled in part by polyubiquination. This polyubiquination is believed to be a result of the action of a enzyme homologous to the BH3-only Bcl-2 related proteins, recently identified by Zhong et al, termed Mcl-1 ubquitin ligase E3 (Mule) [37]. Therefore, we speculate that Mcl-1L expression is regulated in part through proteasomal degradation. This may indicate another point of divergent regulation between neutrophils and eosinophils that remains to be delineated.
In contrast to the opposite effect of Dex on Mcl-1L protein expression and stability, Dex exhibits similar effects on Mcl-1L mRNA expression in both cell types. This does not account for the decrease in Mcl-1L protein expression in eosinophils that is detected following 18–24 h of Dex treatment. In the absence of transcription and translation, Dex treatment of eosinophils does not induce a loss of Mcl-1L protein. Therefore, the data suggests that de novo protein synthesis is necessary in eosinophils for either the eventual increase in Mcl-1L degradation or decrease in Mcl-1L translation. It is also worth noting that the protein stability of Mcl-1L is inherently greater in eosinophils than in neutrophils. Since recent findings have detected a specific regulator of Mcl-1L processing and degradation, it will be of future interest to determine the expression and role of Mule in Dex-induced or Dex-delayed apoptosis in these two granulocytes.
Results from Mcl-1 antisense oligonucleotide experiments confirmed the importance of Mcl-1L protein regulation in the ability of Dex to delay neutrophil apoptosis. Within 12 h in culture, inhibition of Mcl-1L protein production in neutrophils increased the rate of apoptosis over neutrophils incubated in media alone. When Dex was added following Mcl-1 antisense oligo uptake, a large portion of its anti-apoptotic effect was lost. These results demonstrated that the full effect of Dex on neutrophil apoptosis is dependent on its ability to regulate Mcl-1L protein stability within the cell.
A splice variant of Mcl-1L produces the pro-apoptotic BH3-only protein Mcl-1S [22,23]. Mcl-1S, as shown by previous yeast two-hybrid studies, can bind Mcl-1L [22]. This binding could allow for another level of Mcl-1L regulation. Mcl-1S could displace Mcl-1L from its associated proteins (such as Bax), therefore, inhibiting the anti-apoptotic effects of Mcl-1L. However, our analysis of Mcl-1S protein showed that Dex had no significant effect on expression of the splice variant in either granulocyte. Despite the fact that Mcl-1S also contains the PEST motifs thought to target Mcl-1L for rapid degradation, a decrease in Mcl-1S protein was not observed at early time points when apoptotic changes were seen. Therefore, we conclude that there are unique mechanisms regulating turnover of Mcl-1L versus Mcl-1S.
Data presented here imply that glucocorticoids affect regulators of Mcl-1L turnover in neutrophils through a mechanism not entirely dependent on protein synthesis, since we observe these effects even in the presence of CHX. Along with the ability of activated glucocorticoid receptors to directly alter transcription by binding to DNA, the glucocorticoid receptor has also been shown to bind to transcription factors such as NF-κB and AP-1 [38,39]. Studies suggest that most DNA binding and protein-protein interactions of the activated GR work to alter transcription. However, since our CHX data show that protein synthesis is not necessary for alterations in Mcl-1L decay, we conclude that some effects of Dex on granulocyte apoptosis are mediated through other mechanisms, potentially including protein-protein interactions that are not dependent on the ability of Dex to alter transcription.
In our granulocyte model, the anti-apoptotic effects of Mcl-1L are believed to antagonize the pro-apoptotic effect of Bax. As shown by yeast two-hybrid and co-immunoprecipitation experiments, Mcl-1L can bind Bax [40,41]. This suggests the apoptosis inhibiting ability of Mcl-1L could be due in part to its ability to directly bind Bax, therefore, preventing movement of Bax to mitochondria. As demonstrated by Gardai et al, Bax is phosphorylated in its inactive state and this phosphorylated form of Bax shows enhanced heterodimerization with Mcl-1L in neutrophils. Since Mcl-1S can bind Mcl-1L, Mcl-1S could function by displacing Bax from Mcl-1L. This competition for binding to Mcl-1L would free Bax to induce apoptosis. Bax and Mcl-1S levels remain constant even in the presence of Dex, while Mcl-1L levels changed in response to Dex treatment. Though interactions with other Bcl-2 family members are undoubtedly involved, our data indicate Mcl-1L expression is a key regulatory step in granulocyte apoptosis.
Mcl-1L protein loss occurs more rapidly in Dex-treated eosinophils than in eosinophils incubated in media alone; and Mcl-1L loss parallels the enhanced rate of apoptosis in Dex-treated eosinophils. Potentially, this decline in Mcl-1L would leave a larger proportion of Bax free to induce apoptosis. In contrast, Dex slows the loss of Mcl-1L protein in neutrophils. Consistent with our model, maintenance of Mcl-1L protein by Dex, due in part to delayed degradation in neutrophils, would maintain Mcl-1L in a surplus over Mcl-1S. This would allow excess Mcl-1L to inhibit the apoptotic effect of Bax. At early time points, we do not observe changes in Mcl-1L half-life in response to Dex in eosinophils. Therefore, another mechanism may be involved in the eosinophil that causes the increased rate of decline in Mcl-1L protein and accelerated induction of apoptosis by 18 h. While the reduction of Mcl-1 degradation by Dex appears to be transcription-independent in neutrophils, the induction of Mcl-1 degradation in eosinophils may be a result of the well characterized transcriptional effects of Dex. Therefore, the increase in Mcl-1 degradation at later time points may be the result of a build up of pro-apoptotic or degradative proteins produced upon initial transcriptional activation by Dex. It will be of future interest to determine the mechanism which regulates eosinophil Mcl-1 degradation in response to Dex.
Recent findings of Madsen-Bouterse et al in bovine blood neutrophils confirm the importance of Bcl-2 family expression in response to Dex [42]. These studies found that Dex induced an increase in mRNA and protein expression of another anti-apoptotic Bcl-2 family member, A1. Unlike Mcl-1L, A1 protein levels do not decrease as neutrophils undergo spontaneous apoptosis [43]. Therefore, it will be necessary to examine knockdown or over-expression studies with A1 to determine its role in Dex-delayed apoptosis. Our findings demonstrated that lowering the levels of Mcl-1L protein greatly inhibited the anti-apoptotic effect of Dex, but did not completely eliminate its effects. In neutrophils, the combined regulation of Mcl-1L and A1 by Dex may account for the full effect on the rate of apoptosis. A Dex-induced decrease in Bak mRNA was also detected by Madsen-Bouterse et al, but they were unable to detect a change in Bak protein expression in cultured neutrophils. However, Bak protein changes were detected in animals treated with Dex [42]. Although this decrease in Bak expression may be important for the full effects of Dex in vivo, this data does not suggest that changes in Bak are necessary for the Dex-delayed apoptosis that is observed in vitro.
Our findings that Dex exerts its effects through alterations in Bcl-2 family expression are supported by a large number of others. However, it has also become clear that the effect of Dex on Bcl-2 family mRNA and protein is unique among various cell types. In lung epithelial cells, Dex increases expression of the human inhibitor of apoptosis (h-IAP) protein, but does not alter expression of any other Bcl-2 family members [4]. In hepatocytes, Dex prevents death receptor-mediated apoptosis by up-regulating cellular FLICE inhibitory protein (cFLIP) expression, but Dex does not affect the expression of Bcl-2, Bcl-xL, or Mcl-1L [44]. In fibrosarcoma development, Dex inhibits apoptosis by increasing Bcl-xL mRNA and protein expression [45]. In contrast, Dex protects ovarian follicular cells from apoptosis by increasing the rate of Bcl-2 translation, while having no effect on Bcl-2 transcriptional rates [46]. These varied responses to glucocorticoids by different cell types point to the importance of carefully investigating the exact mechanism of action by Dex in each cell type of interest.
The effects of glucocorticoids are generally anti-inflammatory. Glucocorticoids induce apoptosis in inflammatory cells such as lymphocytes and macrophages; while they prevent apoptosis in lung epithelial cells and other tissue resident cells. This produces a model in which glucocorticoids decrease the life span of inflammation-inducing cells, while protecting the surrounding tissue. Consistent with the anti-inflammatory properties of glucocorticoids, Dex induces apoptosis in eosinophils. Paradoxically, Dex delays apoptosis in neutrophils. Data presented here broaden the understanding of how Dex regulates granulocyte apoptosis through alterations in Mcl-1L degradation. Further understanding of these processes may shed light on the anti-inflammatory properties of glucocorticoids. In addition, understanding the mechanisms regulating apoptosis that cause an opposite reaction to Dex in granulocytes may allow therapeutic targets to be identified that will enable selective induction of neutrophil or eosinophil apoptosis.
Acknowledgments
We thank Bridget Fitzgerald for technical assistance. This work was supported by National Institutes of Health grant RO1 HL64226 (DAB).
Abbreviations
- Dex
Dexamethasone
- Cyt c
cytochrome c
- CHX
cycloheximide
- BH
Bcl-2 homology
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Luft FC. From the life of a neutrophil. J Mol Med. 2000;78:301–302. doi: 10.1007/s001090000127. [DOI] [PubMed] [Google Scholar]
- 2.Weller PF, Lim K, Wan HC, Dvorak AM, Wong DT, Cruikshank WW, Kornfeld H, Center DM. Role of the eosinophil in allergic reactions. Eur Respir J Suppl. 1996;22:109s–115s. [PubMed] [Google Scholar]
- 3.Barnes PJ, Pedersen S, Busse WW. Efficacy and safety of inhaled corticosteroids. Am J Respir Crit Care Med. 1998;157:S1–S53. doi: 10.1164/ajrccm.157.3.157315. [DOI] [PubMed] [Google Scholar]
- 4.Wen LP, Madani K, Fahrni JA, Duncan SR, Rosen GD. Dexamethasone inhibits lung epithelial cell apoptosis induced by IFN-γ and Fas. Am J Physiol. 1997;273:L921–L929. doi: 10.1152/ajplung.1997.273.5.L921. [DOI] [PubMed] [Google Scholar]
- 5.Liu YQ, Cousin JM, Hughes J, Van Damme J, Seckl JR, Haslett C, Dransfield I, Savill J, Rossi AG. Glucocorticoids promote nonphlogistic phagocytosis of apoptotic leukocytes. J Immunol. 1999;162:3639–3646. [PubMed] [Google Scholar]
- 6.Stern M, Meagher L, Savill J, Haslett C. Apoptosis in human eosinophils. Programmed cell-death in the eosinophil leads to phagocytosis by macrophages and is modulated by IL-5. J Immunol. 1992;148:3543–3549. [PubMed] [Google Scholar]
- 7.Savill JS, Wyllie AH, Henson JE, Walport MJ, Henson PM, Haslett C. Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J Clin Invest. 1989;83:865–875. doi: 10.1172/JCI113970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Savill J. Apoptosis in resolution of inflammation. J Leukoc Biol. 1997;61:375–380. doi: 10.1002/jlb.61.4.375. [DOI] [PubMed] [Google Scholar]
- 9.Sendo F, Tsuchida H, Takeda Y, Gon S, Takei H, Kato T, Hachiya O, Watanabe H. Regulation of neutrophil apoptosis--its biological significance in inflammation and the immune response. Hum Cell. 1996;9:215–222. [PubMed] [Google Scholar]
- 10.Yamaguchi Y, Suda T, Ohta S, Tominaga K, Miura Y, Kasahara T. Analysis of the survival of mature human eosinophils - Interleukin-5 prevents apoptosis in mature human eosinophils. Blood. 1991;78:2542–2547. [PubMed] [Google Scholar]
- 11.Meagher LC, Cousin JM, Seckl JR, Haslett C. Opposing effects of glucocorticoids on the rate of apoptosis in neutrophilic and eosinophilic granulocytes. J Immunol. 1996;156:4422–4428. [PubMed] [Google Scholar]
- 12.Cox G. Glucocorticoid treatment inhibits apoptosis in human neutrophils. J Immunol. 1995;154:4719–4725. [PubMed] [Google Scholar]
- 13.Liles WC, Dale DC, Klebanoff SJ. Glucocorticoids inhibit apoptosis of human neutrophils. Blood. 1995;86:3181–3188. [PubMed] [Google Scholar]
- 14.Peachman KK, Lyles DS, Bass DA. Mitochondria in eosinophils: functional role in apoptosis but not in respiration. Proc Natl Acad Sci USA. 2001;98:1717–1722. doi: 10.1073/pnas.98.4.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maianski NA, Geissler J, Srinivasula SM, Alnemri ES, Roos D, Kuijpers TW. Functional characterization of mitochondria in neutrophils: a role restricted to apoptosis. Cell Death Differ. 2004;11:143–153. doi: 10.1038/sj.cdd.4401320. [DOI] [PubMed] [Google Scholar]
- 16.Narita M, Shimizu S, Ito T, Chittenden T, Lutz RJ, Matsuda H, Tsujimoto Y. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc Natl Acad Sci USA. 1998;95:14681–14686. doi: 10.1073/pnas.95.25.14681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shimizu S, Eguchi Y, Kamiike W, Funahashi Y, Mignon A, Lacronique V, Matsuda H, Tsujimoto Y. Bcl-2 prevents apoptotic mitochondrial dysfunction by regulating proton flux. Proc Natl Acad Sci USA. 1998;95:1455–1459. doi: 10.1073/pnas.95.4.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 19.Del Poeta G, Venditti A, Del Principe MI, Maurillo L, Buccisano F, Tamburini A, Cox MC, Franchi A, Bruno A, Mazzone C, Panetta P, Suppo G, Masi M, Amadori S. Amount of spontaneous apoptosis detected by Bax/Bcl-2 ratio predicts outcome in acute myeloid leukemia (AML) Blood. 2003;101:2125–2131. doi: 10.1182/blood-2002-06-1714. [DOI] [PubMed] [Google Scholar]
- 20.Ohta K, Iwai K, Kasahara Y, Taniguchi N, Krajewski S, Reed JC, Miyawaki T. Immunoblot analysis of cellular expression of Bcl-2 family proteins, Bcl-2, Bax, Bcl-X, and Mcl-1, in human peripheral blood and lymphoid tissues. Int Immunol. 1995;7:1817–1825. doi: 10.1093/intimm/7.11.1817. [DOI] [PubMed] [Google Scholar]
- 21.Yang T, Buchan HL, Townsend KJ, Craig RW. MCL-1, a member of the BCL-2 family, is induced rapidly in response to signals for cell differentiation or death, but not to signals for cell proliferation. J Cell Physiol. 1996;166:523–536. doi: 10.1002/(SICI)1097-4652(199603)166:3<523::AID-JCP7>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 22.Bae J, Leo CP, Hsu SY, Hsueh AJW. MCL-1S, a splicing variant of the antiapoptotic BCL-2 family member MCL-1, encodes a proapoptotic protein possessing only the BH3 domain. J Biol Chem. 2000;275:25255–25261. doi: 10.1074/jbc.M909826199. [DOI] [PubMed] [Google Scholar]
- 23.Bingle CD, Craig RW, Swales BM, Singleton V, Zhou P, Whyte MKB. Exon skipping in Mcl-1 results in a Bcl-2 homology domain 3 only gene product that promotes cell death. J Biol Chem. 2000;275:22136–22146. doi: 10.1074/jbc.M909572199. [DOI] [PubMed] [Google Scholar]
- 24.Boyum A. Separation of White Blood Cells. Nature. 1964;204:793–794. doi: 10.1038/204793a0. [DOI] [PubMed] [Google Scholar]
- 25.Bass DA, Parce JW, Dechatelet LR, Szejda P, Seeds MC, Thomas M. Flow cytometric studies of oxidative product formation by neutrophils: a graded response to membrane stimuation. J Immunol. 1983;130:1910–1917. [PubMed] [Google Scholar]
- 26.Hansel TT, Devries IJM, Iff T, Rihs S, Wandzilak M, Betz S, Blaser K, Walker C. An improved immunomagnetic procedure for the isolation of highly purified human blood eosinophils. J Immunol Methods. 1991;145:105–110. doi: 10.1016/0022-1759(91)90315-7. [DOI] [PubMed] [Google Scholar]
- 27.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 28.Leuenroth SJ, Grutkoski PS, Ayala A, Simms HH. The loss of Mcl-1 expression in human polymorphonuclear leukocytes promotes apoptosis. J Leukoc Biol. 2000;68:158–166. [PubMed] [Google Scholar]
- 29.Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, Youle RJ. Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol. 1997;139:1281–1292. doi: 10.1083/jcb.139.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moulding DA, Quayle JA, Hart CA, Edwards SW. Mcl-1 expression in human neutrophils: regulation by cytokines and correlation with cell survival. Blood. 1998;92:2495–2502. [PubMed] [Google Scholar]
- 31.Goulding NJ. Corticosteroids - A case of mistaken identify? Br J Rheumatol. 1998;37:477–480. doi: 10.1093/rheumatology/37.5.477. [DOI] [PubMed] [Google Scholar]
- 32.Derouet M, Thomas L, Cross A, Moots RJ, Edwards SW. GM-CSF Signalling and Proteasome Inhibition Delay Neutrophil Apoptosis by Increasing the Stability of Mcl-1. J Biol Chem. 2004;279:26915–26921. doi: 10.1074/jbc.M313875200. [DOI] [PubMed] [Google Scholar]
- 33.Kroemer G, Zamzami N, Susin SA. Mitochondrial control of apoptosis. Immunol Today. 1997;18:44–51. doi: 10.1016/s0167-5699(97)80014-x. [DOI] [PubMed] [Google Scholar]
- 34.Hutcheson J, Scatizzi JC, Bickel E, Brown NJ, Bouillet P, Strasser A, Perlman H. Combined loss of proapoptotic genes Bak or Bax with Bim synergizes to cause defects in hematopoiesis and in thymocyte apoptosis. J Exp Med. 2005;201:1949–1960. doi: 10.1084/jem.20041484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moulding DA, Akgul C, Derouet M, White MRH, Edwards SW. Bcl-2 family expression in human neutrophils during delayed and accelerated apoptosis. J Leukoc Biol. 2001;70:783–792. [PubMed] [Google Scholar]
- 36.Kato T, Kutsuna H, Oshitani N, Kitagawa S. Cyclic AMP delays neutrophil apoptosis via stabilization of Mcl-1. FEBS Lett. 2006;580:4582–4586. doi: 10.1016/j.febslet.2006.07.034. [DOI] [PubMed] [Google Scholar]
- 37.Zhong Q, Gao W, Du F, Wang X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell. 2005;121:1085–1095. doi: 10.1016/j.cell.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 38.Beato M, Chalepakis G, Schauer M, Slater EP. DNA regulatory elements for steroid hormones. J Steroid Biochem. 1989;32:737–747. doi: 10.1016/0022-4731(89)90521-9. [DOI] [PubMed] [Google Scholar]
- 39.Schaaf MJ, Cidlowski JA. Molecular mechanisms of glucocorticoid action and resistance. J Steroid Biochem Mol Biol. 2002;83:37–48. doi: 10.1016/s0960-0760(02)00263-7. [DOI] [PubMed] [Google Scholar]
- 40.Sato T, Hanada M, Bodrug S, Irie SJ, Iwama N, Boise LH, Thompson CB, Golemis E, Fong L, Wang HG, Reed JC. Interactions among members of the Bcl-2 protein family analyzed with a yeast 2-hybrid system. Proc Natl Acad Sci USA. 1994;91:9238–9242. doi: 10.1073/pnas.91.20.9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gardai SJ, Hildeman DA, Frankel SK, Whitlock BB, Frasch SC, Borregaard N, Marrack P, Bratton DL, Henson PM. Phosphorylation of Bax Ser184 by Akt regulates its activity and apoptosis in neutrophils. J Biol Chem. 2004;279:21085–21095. doi: 10.1074/jbc.M400063200. [DOI] [PubMed] [Google Scholar]
- 42.Madsen-Bouterse SA, Rosa GJ, Burton JL. Glucocorticoid modulation of Bcl-2 family members A1 and Bak during delayed spontaneous apoptosis of bovine blood neutrophils. Endocrinology. 2006;147:3826–3834. doi: 10.1210/en.2006-0142. [DOI] [PubMed] [Google Scholar]
- 43.Francois S, El Benna J, Dang PM, Pedruzzi E, Gougerot-Pocidalo MA, Elbim C. Inhibition of neutrophil apoptosis by TLR agonists in whole blood: involvement of the Phosphoinositide 3-Kinase/Akt and NF-κB signaling pathways, leading to increased levels of Mcl-1, A1, and phosphorylated Bad. J Immunol. 2005;174:3633–3642. doi: 10.4049/jimmunol.174.6.3633. [DOI] [PubMed] [Google Scholar]
- 44.Oh HY, Namkoong S, Lee SJ, Por E, Kim CK, Billiar TR, Han JA, Ha KS, Chung HT, Kwon YG, Lee H, Kim YM. Dexamethasone protects primary cultured hepatocytes from death receptor-mediated apoptosis by upregulation of cFLIP. Cell Death Differ. 2006;13:512–523. doi: 10.1038/sj.cdd.4401771. [DOI] [PubMed] [Google Scholar]
- 45.Gascoyne DM, Kypta RM, Vivanco MM. Glucocorticoids inhibit apoptosis during Fibrosarcoma development by transcriptionally activating Bcl-xL. J Biol Chem. 2003;278:18022–18029. doi: 10.1074/jbc.M301812200. [DOI] [PubMed] [Google Scholar]
- 46.Sasson R, Amsterdam A. Pleiotropic anti-apoptotic activity of glucocorticoids in ovarian follicular cells. Biochem Pharmacol. 2003;66:1393–1401. doi: 10.1016/s0006-2952(03)00489-1. [DOI] [PubMed] [Google Scholar]