

Abstract
Gomesin is a potent antimicrobial peptide (AMP) isolated from hemocytes of the spider Acanthoscurria gomesiana. The present study aimed at determining whether gomesin exerted antitumor activity in vitro and in vivo. Topical treatment of subcutaneous murine melanoma with gomesin incorporated in a cream base significantly delayed tumor growth. A direct cytotoxicity of gomesin in murine melanoma B16F10-Nex2 cells and several human tumor cell lineages was observed in vitro, with IC50 values below 5 µM. The β-hairpin structure of gomesin with disulfide bridges seemed essential for optimal activity. d-Gomesin was equally active. A membrane-permeabilizing activity was suggested, as gomesin bound to the cell membrane and cytoplasmic lactate dehydrogenase was detected extracellularly. At doses causing partial growth of tumor cells, gomesin allowed internalization of macromolecules (immunoglobulins), which increased the cytotoxic effect. The in vivo antitumor effect of gomesin might also involve a cytotoxic effect on endothelial cells because cultured human endothelial cells were killed in vitro at a similar concentration range. This effect represents a novel and potential use for gomesin as a topical agent against unsuccessfully treated intradermal and epithelial skin cancers. To our knowledge, this is the first report on the successful topical use of AMPs in cancer treatment.
Introduction
Cancer chemotherapy has several pitfalls. The majority of drugs is active against rapidly proliferating cancer cells, whereas slow-growing tumors or dormant cells respond poorly to these agents [1]. Cancer cells frequently develop multidrug resistance, which greatly reduces the arsenal of chemotherapeutic drugs [2,3]. Antiangiogenic drugs, which reduce or abrogate the blood supply in solid tumors, are promising new therapeutic agents [4]. Emerging evidence, however, shows that tumors can also develop resistance to angiogenesis inhibitors [5].
Antimicrobial peptides (AMPs) are natural-source drugs that show a potential use as anticancer agents [6]. AMPs, mostly cationic and amphipathic molecules, are expressed in a variety of species (e.g., insects, fish, amphibians, and mammals) and can directly eliminate a broad range of Gram-negative and Gram-positive bacteria, fungi, enveloped viruses, and protozoa [7]. These molecules are grouped in different classes according to their structural characteristics [8].
Some AMPs exhibit in vitro direct cytotoxic activity against cancer cells. Cathelicidins (BMAP-28 and hCAP-18), cecropins, melittin, magainin 2, defensins, lactoferricin, and tachyplesin were cytotoxic to human leukemia, lymphoma, breast, lung, ovarian, cervical, and oral squamous carcinoma cells [6]. Rabbit and human α-defensins isolated from granulocytes killed human and murine tumor cell lines in vitro [9]. α-Defensins inhibited angiogenesis [10] and lactoferricin B killed several murine tumor cells in vitro and showed in vivo activity [11–13]. It has not been possible, however, to predict an antitumor activity based on the peptide structure.
Gomesin (Gm) is a cationic AMP isolated from hemocytes of the unchallenged Brazilian spider Acanthoscurria gomesiana [14]. It contains 18 amino acid residues (ZCRRLCYKQRCVTYCRGRNH2) and carries two posttranslational modifications, the N-terminal pyroglutamic acid (Z) and the C-terminal amidated arginine residue. The hairpin-like two-stranded antiparallel β-sheet structure is maintained by two internal disulfide bridges formed by four cysteine residues, Cys2–15 and Cys6–11, which stabilize a rigid conformation together with six hydrogen bonds in the central part of the molecule as well as at each end of the β-sheet [15]. The peptide is highly amphipathic, with a hydrophobic face formed by residues Leu5, Tyr7, Val12, and Tyr14, and three hydrophilic regions containing positively charged and polar amino acids located at the N-terminus (Arg3 and Arg4), at the C-terminus (Arg16 and Arg18), and within the noncanonical β-turn (Lys8, Gln9, and Arg10) [16].
Gomesin exerts a strong microbicidal activity against Gram-positive and Gram-negative bacteria, filamentous fungi, yeast and parasites, such as Leishmania amazonensis, Plasmodium falciparum, and Plasmodium berghei [14,17].
In the present work, we investigated the direct cytotoxic activity of Gm on murine and human tumor cells, and examined the possible in vivo use of this peptide in the treatment of subcutaneous murine melanoma B16F10-Nex2.
Materials and Methods
Peptide Synthesis
Gomesin and all structural derivatives were synthesized using the classic solid-phase methodology on a 4-methylbenzhydrylamine-resin [15]. Structures and molecular weights of all peptides are depicted on Table 1.
Table 1.
Primary Structures and Molecular Mass of Gm and Derived Peptides.
| Peptide | Sequence | Molecular Mass |
| Gomesin (Gm) |  |
2270.7 |
| d-Gomesin (d-Gm) | 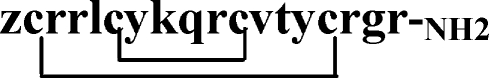 |
2270.7 |
| [Ser2,6,11,15]-Gm | ZSRRLSYKQRSVTYSRGR-nh2 | 2210.5 |
| [Ser2,15]-Gm | 2240.6 | |
| [Ser5]-Gm | 2244.6 | |
| [Ser12]-Gm | 2258.7 | |
| [Ser5,12]-Gm | 2232.6 |
Underlined peptides: Cysteine-to-serine substitutions.
Bold lowercase letters correspond to d-amino acids.
Bold serine units: Serine substitutions of originally hydrophobic residues.
Disulfide bonds are represented by continuous lines connecting cysteine residues.
Tumor Cell Lines and Cell Culture
The murine melanoma cell line B16F10 was originally obtained from the Ludwig Institute for Cancer Research (São Paulo, Brazil). The melanotic B16F10-Nex2 subline, isolated at the Experimental Oncology Unit, is characterized by low immunogenicity and moderate virulence. Human breast adenocarcinoma (SKBr3), colon adenocarcinoma (LS180), and cervical cancer (HeLa) cell lines were obtained from the Ludwig Institute for Cancer Research. Human melanoma cell lines (SKMel 19 and A2058) were provided by Dr. Alan N. Houghton ofMemorial Sloan Kettering Cancer Center, NY. All cells were cultured at 37°C in a humidified atmosphere containing 5% CO2, in RPMI-1640 medium (Invitrogen, Carlsbad, CA) supplemented with 10 mM N-2-hydroxyethylpiperazine-N2 ethanesulfonic acid (Hepes) (Sigma, St. Louis, MO), 24 mM sodium bicarbonate (Sigma), 40 mg/l gentamycin (Schering-Plough, São Paulo, Brazil), pH 7.2, and 10% fetal calf serum (Invitrogen).
Antibodies
Murine anti-α-tubulin (IgG, clone DM1A) was from Sigma. Murine anti-pan-histones antibodies were from Boehringer Mannheim (Germany). Anti-Gm is a polyclonal rabbit antibody [18]. Monoclonal antibody (mAb) A4M is a histone H1-reacting IgM raised against B16F10-Nex2 melanoma cells.
B16F10-Nex2 Nuclear Extract and Chemiluminescent Immunoblot Analysis with mAb A4M
Approximately 200 µl of cell pellet (5 x 107 B16F10-Nex2 cells) was diluted in five volumes of buffer A (10 mM Hepes, 1.5 mM MgCl2, 10 mM KCl, and 0.5 mM DTT) and incubated on ice for 10 minutes. After centrifugation, the original pellet was resuspended in two volumes of buffer A. Tumor cells were lysed in a Potter homogenizer and centrifuged for 20 minutes at 25,000g. The pellet was resuspended in 3 ml of buffer B [20 mM Hepes, 25% (v/v) glycerol, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM PMSF, and 0.5 mM DTT] and was homogenized again. The suspension was softly agitated 30 minutes on ice and centrifuged for 30 minutes at 25,000g. The supernatant was dialysed in 50 volumes of buffer C [20 mM Hepes, 20% (v/v) glycerol, 0.1 M KCl, 0.2 mM EDTA, 0.5 mM PMSF, and 0.5 mM DTT] for 12 hours at 4°C. After centrifugation for 20 minutes at 25,000g, the supernatant was collected and used for immunoblot analysis with mAb A4M. Purified commercial H1 histone from calf thymus (Type III SS; Sigma) was used as control. Melanoma nuclear extract and commercial histones were eletrophoretically separated in 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and transferred to a nitrocellulose membrane. After blocking, membranes were incubated with mAb A4M or murine anti.pan-histone antibody (Boehringer Mannheim) for 12 hours at 4°C under agitation, following incubation with biotinylated total murine anti-IgG or anti-IgM (Sigma) and streptavidin-peroxidase (Sigma), at 37°C, for 1 hour each. Membranes were revealed with enhanced chemiluminescence (Amersham GE, São Paulo, Brazil).
In Vitro Cytotoxic Activity
Gomesin and derivatives were diluted in supplemented RPMI medium and incubated with 5 x 103 B16F10-Nex2 or 104 human tumor cells in 96-well plates; cells were plated 24 hours before treatment. After incubation, viable cells were counted in a Neubauer chamber (Electron Microscopy Sciences, Hatfield, PA) using Trypan blue. To analyze the combined effect of Gm and antibodies, B16F10-Nex2 cells were treated with 2 µM Gm and mAb A4M. Cell viability was measured after 12 hours of incubation. Human umbilical vein endothelial cells (HUVECs), 104 cells plated as described, were treated with Gm and cell viability was then analyzed after 16 hours. All experiments were performed in triplicate.
Flow Cytometry
B16F10-Nex2 cells (106 cells/100 µl) were incubated for 12 hours with 2 µM Gm and 100 µg/ml mAb A4M. As positive permeabilization control, cells were treated with 0.5% saponin and 1% paraformaldehyde in phosphate-buffered saline (PBS), pH 7.2, for 20 minutes, and with mAbs for 12 hours, diluted in the same solution. Cells were incubated sequentially for 1 hour with biotin-conjugated murine anti-IgM (Sigma) at 20 µg/ml and streptavidin-fluorescein isothiocyanate (FITC) (Pharmigen BD Biosciences, San Jose, CA) at 10 µg/ml, both diluted in PBS, protected from light. All steps were performed at 4°C, preceded by washings with PBS. Cells were fixed with PBS-formaldehyde 2% (Merck S.A., São Paulo, Brazil) in a final volume of 500 µl. The reaction was analyzed in a FACS-Calibur equipment (Becton-Dickinson, Franklin Lakes, NJ), using CellQuest software.
Analysis of Morphologic Alterations by Light Microscopy
B16F10-Nex2 cells (104 cells/1 ml) were cultivated in 24-well plates for 24 hours and incubated for 2 hours with 15 µM Gm or with 2 µM Gm associated with 100 µg/ml mAb A4M. Cell morphology was monitored every 5 minutes using a microscope (model IX70; Olympus, Center Valley, PA) at a magnification of x40. Images were analyzed using a software (MetaMorph; Molecular Devices Co., Sunnyvale, CA).
Lactate Dehydrogenase Assay
B16F10-Nex2 melanoma cells (105) were incubated in 24-well plates with Gm, in RPMI without serum. Supernatants (100 µl) were then collected and assayed for the presence of released lactate dehydrogenase (LDH) after adding 120 µl of PBS containing 0.7 mM NADH and 4.7 mM pyruvate. The decrease in A340 after 1 minute of incubation was measured. The initial A340 of NADH added was taken as 100% or 1. Lower values indicated consumptions relative to 100%. Positive control consisted of a Triton X-100 (10%) B16F10-Nex2 cell lysate. All experiments were performed in triplicate.
Confocal Microscopy
B16F10-Nex2 cells (2 x 103) were cultivated in round glass coverslips (13 mm) for 24 hours [19], following incubation with 5 µM Gm for 10 minutes at 37°C, and fixation with 3.7% paraformaldehyde for 15 minutes at room temperature. Cells were then incubated in 150 mM NaCl (Merck), 50 mM Tris (Gibco Invitrogen, Carlsbad, CA), 0.25% BSA (Sigma), and 0.5% Tween 20 (Sigma), pH 7.2 to 7.4, for 1 hour at room temperature, with mouse antibodies against α-tubulin (1:100 ascite dilution) for 12 hours at 4°C, and finally with 4 µg/ml FITC-conjugated goat anti-mouse antibody (Molecular Probes Invitrogen, Carlsbad, CA). Alternatively, cells were incubated with rabbit polyclonal anti-Gm antibodies, 1:200 diluted, and revealed with FITC-conjugated goat anti-rabbit antibody (5 µg/ml) (ICN Biochemicals Inc., Irvine, CA). Staining of actin filaments and nuclei were performed with 0.3 µg/ml phalloidin-rhodamine conjugate (Invitrogen) and 50 µg/ml DAPI (Invitrogen), respectively, for 1 hour at room temperature. The coverslips were mounted using a mounting medium (Vectashield; Vector Laboratories, Burlingame, CA) to reduce bleaching and were examined by a laser scanning fluorescence confocal microscope (MRC 1024/UV System; Bio-Rad, Hercules, CA) equipped with a transmitted light detector for Nomarski differential interference contrast. The images were obtained with a 40x 1.2 NA/water immersion PlanApo objective (Carl Zeiss MicroImaging, Inc., Thornwood, NY); Kalman averaging at least 20 frames using a 2-mm iris (pinhole) [20].
Extracellular Acidification Rate
B16F10-Nex2 cells (3 x 105 cells/ml) were added into 3.0-µm-pore cups (Transwells; Corning Costar, Cambridge, MA) held on 12-well plates 12 hours before the experiment. The extracellular acidification rate of treated and untreated cells was determined using a bioassay (Cytosensor Microphysiometer; Molecular Devices, Sunny Valley, CA). The capsules containing the adherent cells were allowed to equilibrate in low buffered RPMI containing 1% BSA for 20 minutes at 37°C, when the acidification rates were normalized to 100% before drug addition. Cells were perfused for 400 minutes with 200 µM cisplatin or 10 µM Gm. During the flow-off periods, protons released from the drug-treated cells accumulated in the sensor chamber, and the H+ decay profile was quantified [21].
Topical Treatment of Subcutaneous Murine Melanoma
Seven- to eight-week-old male C57BL/6 mice from Centro de Desenvolvimento de Modelos Experimentais, UNIFESP (average weight of 32–34 g each), had the right flank chemically depilated with a hair removal cream (Veet; Reckitt Benckiser, São Paulo, Brazil) and, 24 hours later were injected subcutaneously with 105 B16F10-Nex2 tumor cells. Gomesin (200 µg/100 µl water) was incorporated in 1 g of anionic, oil-in-water cream (final concentration, 0.02% w/w of Gm). Topical treatment started when subcutaneous tumor volumes reached 4 to 10 mm3 and consisted of spreading 20 mg of cream with a small brush over the tumor external surface, three times a week for 4 weeks or until the tumor volume reached a maximum of 3000 to 3300 mm3, and then the animals were sacrificed. The maximum tumor size accepted was calculated based on the weight of the male mice used, i.e., 32 to 34 g. The control group was treated with a waterincorporated cream. Tumor volumes were measured three times a week, before treatment, using the formula V = 0.52 x D12 x D2, where D1 and D2 are short and long tumor diameters, respectively. Animal manipulation was conducted according with the guidelines of the Animal Ethics Committee of Federal University of São Paulo (UNIFESP), protocol no. 316/06.
Statistical Analysis
Two-tailed Student' t test was used for statistical analysis of in vitro experiments. In the case of data presentation using percent values (as in Figure 2), the original numbers were used for statistic comparison with the control. The plots of treated living animals below the maximum allowed tumor size were analyzed statistically by the log rank test compared with the control group. The IC50 value and 95% confidence intervals were calculated using GraphPad InStat3 software (GraphPad Software Inc., San Diego, CA).
Figure 2.

The antimicrobial peptide gomesin showed in vitro cytotoxic effect on B16F10-Nex2 murine tumor cells, in a dose-, time-, and structure-dependent fashion. (A) Tumor cells were incubated for 12 hours with 20 µM gomesin (Gm), its d-enantiomer (d-Gm), monocyclic {[Ser2,15]-Gm}, linear {[Ser2,6,11,15]-Gm}, forms, and with 10 µM analogues with amino acid substitutions disrupting the hydrophobic face of the peptide, [Ser5]-Gm, [Ser12]-Gm, and [Ser5,12]-Gm. (B) Cells were incubated with different concentrations of Gm (1, 5, 10, and 20 µM) for 15 minutes up to 5 hours. Cells were alternatively incubated with Gm for 5 hours, washed to eliminate the peptide, and incubated with fresh medium for 24 hours (5/24 h). Cell viability was determined by counting cells in presence of Trypan blue. Data are a representative experiment of a triplicate set. Bars represent means and standard deviations (SD). *P < .05 and #P < .01 relative to the control.
Results
Local Treatment with a Gomesin-Containing Cream Significantly Increased the Survival Time of B16F10-Nex2-Challenged Mice with Tumors Showing Delayed Growth
Animals with established subcutaneous tumors (4–10 mm3) were treated topically three times a week, with individual doses of 4 µg of Gm in 20 mg of cream. Strikingly, a significant delay in tumor development was observed in treated animals (Figure 1A). Treated animals also had significantly increased survival times as compared with controls, with mice being sacrificed when tumors reached the maximum size (3000 mm3) allowed (P < .01; Figure 1B).
Figure 1.

Topical treatment with a gomesin-containing cream delayed subcutaneous tumor development and increased the time of living animals with tumors below the maximum allowed size. B16F10-Nex2 murine melanoma cells (105 per animal) were injected subcutaneously into the hair-free flank of C57BL/6 male mice. Animals were treated topically with a Gm-containing cream (white circles, n = 10) or a control cream (black circles, n = 5), as described in Materials and Methods. (A) Individual tumor volumes. (B) Treated and control mice bearing tumors bellow 3000 mm3 after different times of tumor implantation. Log rank test, P < .01.
Gomesin Reduced Viability of Murine and Human Tumor Cell Lines, and also of a Human Endothelial Cell Lineage (HUVEC) In Vitro, but Showed Reduced Cytotoxic at Erythrocytes and Macrophages
The antitumor local effect of gomesin in vivo could be due to Gm direct cytotoxicity in tumor cells. In vitro, after 12 hours of incubation with 20 µM Gm, B16F10-Nex2 melanoma cells lost viability (Figure 2A). The IC50 value for these tumor cells was 3.58 µM (Table 2). The enantiomer d-gomesin (d-Gm), which was synthesized employing d-amino acids and containing both disulfide bonds, was also highly cytotoxic implying that chiral recognition is not required for antitumor activity. In contrast, derivatives where cysteine residues had been substituted by serine, resulting in molecules with one (monocyclic [Ser2,15]-Gm) or none (linear [Ser2,6,11,15]-Gm) disulfide bond showed a much reduced tumor cell cytotoxicity, 70% and 30%, respectively (Figure 2A).
Table 2.
Estimated IC50 and 95% Confidence Intervals (CIs) of Gomesin Cytotoxic Effect in Murine and Human Cell Lines.
| Tumor and Endothelial Cells | IC50 (95% CI), µM |
| B16F10-Nex2 | 3.58 (2.76–4.41) |
| SKBr3 | 2.87 (1.30–4.45) |
| LS180 | 4.78 (2.82–6.75) |
| A2058 | 1.36 (0.29–3.02) |
| HeLa | 8.13 (6.00–10.25) |
| SKMel 19 | 2.35 (1.69–3.00) |
| HUVEC | 5.30 (2.21–8.46) |
Disruption of the hydrophobic face (amino acid residues Leu5, Tyr7, Val12, and Tyr14) by serine substitution of Leu5, Val12, or both residues, completely abolished Gm cytotoxicity (Figure 2A). Substitution of a single hydrophobic residue was sufficient to completely abrogate peptide activity.
The cytotoxic effect of Gm was time- and dose-dependent (Figure 2B) and was not reversed by washing, as cells incubated with Gm (10 and 20 µM) for 5 hours, and subsequently with complete medium in the absence of the peptide did not resume growth (Figure 2B).
Gomesin was also cytotoxic to human tumor cell lineages in vitro, the most sensitive being human melanoma A2058, with IC50 value of 1.36 µM. A human endothelial cell lineage (i.e., HUVEC) was also sensitive in vitro to Gm, with IC50 value of 5.3 µM (Table 2).
Gomesin Bound to and Permeabilized Tumor Cell Membrane
Anti-Gm rabbit antibody staining showed that Gm accumulates very rapidly on the tumor cell membrane, forming clusters (Figure 3B). A preimmune rabbit serum did not react with Gm-treated tumor cells (data not shown).
Figure 3.

Gomesin accumulated on the cell membrane and permeabilized B16F10-Nex2 tumor cells. Cells were cultivated on round glass coverslips, treated with 5 µM Gm for 10 minutes, fixed, and incubated with anti-Gm polyclonal monospecific antibody (B), or murine antitubulin antibody (C), both revealed with FITC-conjugated secondary antibodies (green). The fluorescence was analyzed by confocal microscopy as described in Materials and Methods. Red, phalloidin-rhodamine; blue, DAPI staining. (A) Single optical section through control cells treated with phalloidin-rhodamine, DAPI, and antitubulin in the absence of Gm; (B, C) maximum pixel value projections of serial optical sections. Scale bars, 20 µm.
Dose-dependent permeabilization of Gm-treated B16F10-Nex2 cells was shown by the extracellular release of cytoplasmic LDH (Figure 4). Membrane-permeabilized tumor cells internalized IgMs and the fluorescent staining of tubulin by anti-α-tubulin was possible only after Gm treatment (Figure 3C). An irrelevant IgG2a, used as an isotype control antibody, did not stain the tumor cell (data not shown).
Figure 4.

Gomesin treatment of B16F10-Nex2 cells in vitro induced leakage of cytoplasmic LDH. Cells were incubated with 5, 10, or 20 µM Gm for 5, 15, and 30 minutes, and the activity of LDH in the culture supernatants was quantified by NADH consumption, as described in Materials and Methods. Control, untreated cells; Triton, cells treated with 10% of Triton X-100 for maximal lysis of tumor cells. Data are a representative experiment of a triplicate set. Bars represent means and SD. *P < .01 compared to respective control.
A mAb noncytotoxic to whole melanoma cells was then tested against Gm-treated B16F10-Nex2 cells in vitro. MAb A4M, recognizing nuclear histone H1 (Figure 5A), was internalized in B16F10-Nex2 cells (Figure 5B) and showed additive cytotoxic activity with Gm in vitro (Figure 5C).
Figure 5.

Immunoglobulins (IgM) were detected in the cytoplasm of B16F10-Nex2 murine melanoma cells permeabilized with a low dose of gomesin. (A) Chemiluminescent immunoblot analysis showing mAb A4M reactivity with histone 1. NE B16, nuclear extract of B16F10-Nex2 cells; H1 Sigma, commercially purified calf thymus histone from Sigma. Lane 1, 25 µg/ml mAb A4M; Lane 2, 25 µg/ml commercial anti-pan histone antibody; Lane 3, 25 µg/ml an irrelevant mAb. (B) Cells were incubated with a low dose of Gm (2 µM) or with 0.5% saponin and 100 µg/ml mAb A4M for 12 hours. Samples were incubated with FITC-conjugated secondary antibodies and cytoplasmic fluorescence was quantified by FACS. None, untreated cells; Ig-FITC, cells treated with secondary antibodies in the absence of mAbs; FL1-H, fluorescence intensity. (C) Cells were incubated with different concentrations of A4M mAb (25, 50, 80, and 100 µg/ml) in presence (+Gm) or absence (-Gm) of 2 µM Gm for 12 hours. Viable cells were counted in the presence of Trypan blue. None, untreated cells; Gomesin, cells treated with 2 µM Gm. Data are a representative experiment of a triplicate set. Bars represent means and SD.
Gm-treated B16F10-Nex2 cells showed early morphologic alterations. Treatment with 15 µM Gm for 15 minutes increased the granularity in 30% of the cell population and, after 30 minutes, 80% of cells were heavily granulated and shrunk (Figure 6D). No detachment from the substrate was observed. The additive effect of Gm and mAb A4M was detected with 2 µg of Gm, 5 minutes after coincubation with both reagents. This low dose of Gm alone did not affect cell morphology even after 120 minutes (Figure 6B). Round cells were observed suggesting detachment from the extracellular matrix and, after 10 minutes, 100% of the cell population assumed round forms (Figure 6E) and had increased cytoplasmic granularity after 15 minutes (Figure F).
Figure 6.

Morphologic alterations of B16F10-Nex2 cells induced by gomesin and the association of gomesin and mAb A4M analyzed by light microscopy. Cells were either (A) left untreated or treated with (B) 2 µM Gm for 120 minutes, (C) 100 µg/ml mAb A4M for 120 minutes, (D) 15 µM Gm for 30 minutes, and 2 µ Gm associated with 100 µg/ml mAb A4M for either (E) 10 or (F) 15 minutes. A picture was taken every 5 minutes, and those at representative times are depicted (original magnification, x40).
Early Effects of Gomesin on Tumor Cell Respiratory Metabolism
The extracellular acidification rate was measured after B16F10-Nex2 cell treatment with Gm and cisplatin, a control apoptotic drug. Using Cytosensor Microphysiometer, cisplatin at 200 µM slowly reduced the external proton concentration, reaching values 85% lower than the baseline values only after 400 minutes of incubation (Figure 7). Gomesin (10 µM) caused 40% reduction in the extracellular acidification rate after 200 minutes of incubation, keeping it steady until 400 minutes.
Figure 7.

Extracellular acidification response of B16F10-Nex2 cells to gomesin and cisplatin. Cells were added to Transwell cups and placed on the Cytosensor Microphysiometer. After 20 minutes of equilibration in low buffered RPMI-1% BSA, gomesin (10 µM) and cisplatin (200 µM), diluted in the same medium, were added at zero time and maintained during the whole experiment (400 minutes). The cells were monitored for acidification rate every 2 minutes, in relation to untreated control cells.
Discussion
Although numerous AMPs have been discovered in the last years, only a few were tested as in vivo drugs against tumor cells [6]. Human melanoma subcutaneously injected in nude mice was treated with local injections of d-magainin [22] and lactoferricin B-inhibited neuroblastoma xenografts [13].
Antimicrobial peptides have been successfully used for treatment of superficial infections, as topical drugs. In thermally injured and Pseudomonas aeruginosa-infected rat skin, protegrin decreased bacterial counts after topical application or intradermal injection [23].
Topical treatment of melanoma is, sometimes, a preferred alternative. Some patients develop extensive, confluent cutaneous metastases near the primary nodular melanoma, and these lesions are unsuitable for surgical excision or radiotherapy. Some topical treatments have tentatively being used, but only partial responses were obtained with 5-aminolevulinic acid photodynamic therapy [24], imiquimod [25,26], dinitrochlorobenzene [27-29], and diphencyprone [30].
Our results show that topical treatment with Gm, an AMP structurally related to androctonin and protegrin [14,15], significantly delayed subcutaneous murine melanoma development and increased the number of living treated animals with tumors below the allowed maximal size limit. Apparently, the antitumor activity of gomesin was due to a direct effect on tumor cells. Gomesin was cytotoxic in vitro to murine and human tumor cells (melanoma, breast, and colon carcinoma) at doses lower than 5 µM, except for human HeLa cells, which showed an IC50 value of 8.13 µM.
Gomesin was also cytotoxic to HUVECs in vitro with IC50 in the same range as for tumor cells, suggesting that in vivo both tumor and endothelial cells of the microvascular core might be affected by the peptide.
Repeated topical applications of Gm did not affect the peripheral healthy skin of peptide-treated mice. Silva et al. [14] previously have shown a minimal hemolytic activity of gomesin in vitro, and we also observed the complete absence of cytotoxicity of the peptide in thioglycollate-elicited peritoneal murine macrophages in vitro (data not shown). Gomesin did not stimulate the production of nitric oxide in these macrophages, thus differing from human β-defensin 2 [31]. Seemingly normal cells may be resistant to gomesin unlike tumor cells and cultured endothelial cells.
Gm tumor cell cytotoxicity depended on the β-hairpin configuration with at least one disulfide bridge for keeping a significant activity, whereas both bridges are required for high serum stability and optimal activity [16,32]. Apparently, in AMPs with β-hairpin structure, amphiphilicity, charge, and shape are more important to antimicrobial activity than the presence of specific amino acids [33]. Electrostatic and hydrophobic interactions are important for the antimicrobial activity of Gm [32,34,35]. Gomesin apparently did not require a specific surface receptor to reduce tumor cell viability, because d-Gm was as cytotoxic as the native peptide. The same independence of chirality was observed for the antimicrobial effect of Gm [14] and protegrin [33].
Treatment of tumor cells with Gm did not induce apoptosis. After 12 hours of incubation, cellular DNA was not degraded (data not shown). In a dose-dependent manner, gomesin induced morphologic cell alterations with increased granularity, loss of cytoplasmic content by membrane permeabilization, and partial collapse of the proton gradient.
Gm at low concentrations could facilitate the penetration of drugs inside tumor cells, thus potentially reducing toxic doses. Otherwise, Gm could allow molecules that are not directly cytotoxic to cells with intact membranes, act to eliminate these cells after peptide treatment.
There is no data up to now concerning the precise mode of action of Gm; however, the high structural similarities with protegrin [15] suggest that a similar mode of action through formation of pores may also occur [15,36]. As the peptide concentrates at the cell membrane, clusters can be seen suggesting the formation of pore structures.
The putative pore formation by Gm caused the release of LDH from melanoma cells and inhibited the respiration-dependent proton gradient in treated cells. After Gm treatment, tubulin filaments were stained by specific IgG, and this effect did not occur in the absence of the peptide. Moreover, treatment of tumor cells with a low dose of Gm allowed internalization of mAb A4M that reacted with histone H1. The enhanced cytotoxicity observed by the association of Gm and mAb A4M was observed at relatively high concentrations of the antibody (80–100 µg/ml).
Most importantly, our results describe a novel and important use for Gm as a topical drug against intradermal and intraepithelial cancers. To our knowledge, this is the first report describing the successful topical use of AMPs in cancer treatment.
Acknowledgments
We are indebted to Ludwig Institute, São Paulo Branch, and to Alan N. Houghton from the Memorial Sloan Kettering Cancer Center, NY, USA, for providing tumor cell lineages used in this study.
Abbreviations
- AMP
antimicrobial peptide
- FITC
fluorescein isothiocyanate
- Gm
gomesin
- HUVEC
human umbilical vein endothelial cell
- LDH
lactate dehydrogenase
- mAb
monoclonal antibody
Footnotes
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo and Brazilian National Research Council (CNPq). L. R. T., E. G. R., R. A. M., L. N., A. M., and S. D. are recipients of fellowships from CNPq.
References
- 1.Naumov GN, Townson JL, MacDonald IC, Wilson SM, Bramwell VH, Groom AC, Chambers AF. Ineffectiveness of doxorubicin treatment on solitary dormant mammary carcinoma cells or late-developing metastases. Breast Cancer Res Treat. 2003;82:199–206. doi: 10.1023/B:BREA.0000004377.12288.3c. [DOI] [PubMed] [Google Scholar]
- 2.Gottesman MM. Mechanisms of cancer drug resistance. Ann Rev Med. 2002;53:615–627. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 3.Gottesman MM, Ling V. The molecular basis of multidrug resistance in cancer: the early years of P-glycoprotein research. FEBS Lett. 2006;580:998–1009. doi: 10.1016/j.febslet.2005.12.060. [DOI] [PubMed] [Google Scholar]
- 4.Folkman J. Angiogenesis. Ann Rev Med. 2006;57:1–18. doi: 10.1146/annurev.med.57.121304.131306. [DOI] [PubMed] [Google Scholar]
- 5.Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438:967–974. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- 6.Mader JS, Hoskin DW. Cationic antimicrobial peptides as novel cytotoxic agents for cancer treatment. Expert Opin Investig Drugs. 2006;15:933–946. doi: 10.1517/13543784.15.8.933. [DOI] [PubMed] [Google Scholar]
- 7.Bulet P, Stocklin R, Menin L. Anti-microbial peptides: from invertebrates to vertebrates. Immunol Rev. 2004;198:169–184. doi: 10.1111/j.0105-2896.2004.0124.x. [DOI] [PubMed] [Google Scholar]
- 8.Yount NY, Bayer AS, Xiong YQ, Yeaman MR. Advances in antimicrobial peptide immunobiology. Biopolymers. 2006;84:435–458. doi: 10.1002/bip.20543. [DOI] [PubMed] [Google Scholar]
- 9.Lichtenstein A, Ganz T, Selsted ME, Lehrer RI. In vitro tumor-cell cytolysis mediated by peptide defensins of human and rabbit granulocytes. Blood. 1986;68:1407–1410. [PubMed] [Google Scholar]
- 10.Chavakis T, Cines DB, Rhee JS, Liang OD, Schubert U, Hammes HP, Higazi AA, Nawroth PP, Preissner KT, Bdeir K. Regulation of neovascularization by human neutrophil peptides (alpha-defensins): a link between inflammation and angiogenesis. FASEB J. 2004;18:1306–1308. doi: 10.1096/fj.03-1009fje. [DOI] [PubMed] [Google Scholar]
- 11.Yoo YC, Watanabe S, Watanabe R, Hata K, Shimazaki K, Azuma I. Bovine lactoferrin and lactoferricin, a peptide derived from bovine lactoferrin, inhibit tumor metastasis in mice. Jpn J Cancer Res. 1997;88:184–190. doi: 10.1111/j.1349-7006.1997.tb00364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eliassen LT, Berge G, Sveinbjornsson B, Svendsen JS, Vorland LH, Rekdal O. Evidence for a direct antitumor mechanism of action of bovine lactoferricin. Anticancer Res. 2002;22:2703–2710. [PubMed] [Google Scholar]
- 13.Eliassen LT, Berge G, Leknessund A, Wikman M, Lindin I, Lokke C, Ponthan F, Johnsen JI, Sveinbjornsson B, Kogner P, et al. The antimicrobial peptide, lactoferricin B, is cytotoxic to neuroblastoma cells in vitro and inhibits xenograft growth in vivo. Int J Cancer. 2006;119:493–500. doi: 10.1002/ijc.21886. [DOI] [PubMed] [Google Scholar]
- 14.Silva PI, Daffre S, Bulet P. Isolation and characterization of gomesin, an 18-residue cysteine-rich defense peptide from the spider Acanthoscurria gomesiana hemocytes with sequence similarities to horseshoe crab antimicrobial peptides of the tachyplesin family. J Biol Chem. 2000;275:33464–33470. doi: 10.1074/jbc.M001491200. [DOI] [PubMed] [Google Scholar]
- 15.Mandard N, Bulet P, Caille A, Daffre S, Vovelle F. The solution structure of gomesin, an antimicrobial cysteine-rich peptide from the spider. Eur J Biochem. 2002;269:1190–1198. doi: 10.1046/j.0014-2956.2002.02760.x. [DOI] [PubMed] [Google Scholar]
- 16.Fazio MA, Oliveira VX, Bulet P, Miranda MTM, Daffre S, Miranda A. Structure-activity relationship studies of gomesin: importance of the disulfide bridges for conformation, bioactivities, and serum stability. Biopolymers. 2006;84:205–218. doi: 10.1002/bip.20396. [DOI] [PubMed] [Google Scholar]
- 17.Moreira CK, Rodrigues FG, Ghosh A, Varotti FD, Miranda A, Daffre S, Jacobs-Lorena M, Moreira LA. Effect of the antimicrobial peptide gomesin against different life stages of Plasmodium spp. Exp Parasitol. 2007;116:346–353. doi: 10.1016/j.exppara.2007.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lorenzini DM, Fukuzawa AH, da Silva PI, Machado-Santelli G, Bijovsky AT, Daffre S. Molecular cloning, expression analysis and cellular localization of gomesin, an anti-microbial peptide from hemocytes of the spider, Acanthoscurria gomesiana. Insect Biochem Mol Biol. 2003;33:1011–1016. doi: 10.1016/s0965-1748(03)00115-2. [DOI] [PubMed] [Google Scholar]
- 19.Dobroff AS, Rodrigues EG, Moraes JZ, Travassos LR. Protective, anti-tumor monoclonal antibody recognizes a conformational epitope similar to melibiose at the surface of invasive murine melanoma cells. Hybrid Hybridomics. 2002;21:321–331. doi: 10.1089/153685902761022661. [DOI] [PubMed] [Google Scholar]
- 20.Barros HC, Verbisck NV, DaSilva S, Araguth MF, Mortara RA. Distribution of epitopes of Trypanosoma cruzi amastigotes during the intracellular life cycle within mammalian cells. J Eukaryot Microbiol. 1997;44:332–344. doi: 10.1111/j.1550-7408.1997.tb05675.x. [DOI] [PubMed] [Google Scholar]
- 21.Rodrigues EG, Silva LS, Fausto DM, Hayashi MS, Dreher S, Santos EL, Pesquero JB, Travassos LR, Caires ACF. Cyclopalladated compounds as chemotherapeutic agents: antitumor activity against a murine melanoma cell line. Int J Cancer. 2003;107:498–504. doi: 10.1002/ijc.11434. [DOI] [PubMed] [Google Scholar]
- 22.Soballe PW, Maloy WL, Myrga ML, Jacob LS, Herlyn M. Experimental local therapy of human melanoma with lytic magainin peptides. Int J Cancer. 1995;60:280–284. doi: 10.1002/ijc.2910600225. [DOI] [PubMed] [Google Scholar]
- 23.Steinstraesser L, Klein RD, Aminlari A, Fan MH, Khilanani V, Remick DG, Su GL, Wang SC. Protegrin-1 enhances bacterial killing in thermally injured skin. Crit Care Med. 2001;29:1431–1437. doi: 10.1097/00003246-200107000-00022. [DOI] [PubMed] [Google Scholar]
- 24.Wolf P, Rieger E, Kerl H. Topical photodynamic therapy with endogenous porphyrins after application of 5-aminolevulinic acid. An alternative treatment modality for solar keratoses, superficial squamous cell carcinomas, and basal cell carcinomas? J Am Acad Dermatol. 1993;28:17–21. doi: 10.1016/0190-9622(93)70002-b. [DOI] [PubMed] [Google Scholar]
- 25.Steinmann A, Funk JO, Schuler G, von den DP. Topical imiquimod treatment of a cutaneous melanoma metastasis. J Am Acad Dermatol. 2000;43:555–556. [PubMed] [Google Scholar]
- 26.Hesling C, D'Incan M, Mansard S, Franck F, Corbin-Duval A, Chevenet C, Dechelotte P, Madelmont JC, Veyre A, Souteyrand P, et al. In vivo and in situ modulation of the expression of genes involved in metastasis and angiogenesis in a patient treated with topical imiquimod for melanoma skin metastases. Br J Dermatol. 2004;150:761–767. doi: 10.1111/j.0007-0963.2004.05898.x. [DOI] [PubMed] [Google Scholar]
- 27.Malek-Mansour S. Remission of melanoma with D.N.C.B. treatment. Lancet. 1973;2:503–504. doi: 10.1016/s0140-6736(73)92103-x. [DOI] [PubMed] [Google Scholar]
- 28.Illig L, Paul E, Bodeker RH. Epifocal dinitrochlorobenzene therapy in malignant melanoma (experience during the last eight years) Anticancer Res. 1984;4:293–298. [PubMed] [Google Scholar]
- 29.von Nida J, Quirk C. Successful treatment of in-transit melanoma metastases using topical 2–4 dinitrochlorobenzene. Australas J Dermatol. 2003;44:277–280. doi: 10.1046/j.1440-0960.2003.00009.x. [DOI] [PubMed] [Google Scholar]
- 30.Damian DL, Thompson JF. Treatment of extensive cutaneous metastatic melanoma with topical diphencyprone. J Am Acad Dermatol. 2007;56:869–871. doi: 10.1016/j.jaad.2006.12.018. [DOI] [PubMed] [Google Scholar]
- 31.Biragyn A, Ruffini PA, Leifer CA, Klyushnenkova E, Shakhov A, Chertov O, Shirakawa AK, Farber JM, Segal DM, Oppenheim JJ, et al. Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science. 2002;298:1025–1029. doi: 10.1126/science.1075565. [DOI] [PubMed] [Google Scholar]
- 32.Moraes LG, Fazio MA, Vieira RF, Nakaie CR, Miranda MT, Schreier S, Daffre S, Miranda A. Conformational and functional studies of gomesin analogues by CD, EPR and fluorescence spectroscopies. Biochim Biophys Acta. 2007;1768:52–58. doi: 10.1016/j.bbamem.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 33.Chen J, Falla TJ, Liu H, Hurst MA, Fujii CA, Mosca DA, Embree JR, Loury DJ, Radel PA, Cheng CC, et al. Development of protegrins for the treatment and prevention of oral mucositis: structure-activity relationships of synthetic protegrin analogues. Biopolymers. 2000;55:88–98. doi: 10.1002/1097-0282(2000)55:1<88::AID-BIP80>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 34.Miranda A, Fázio MA, Miranda MTM, Daffre S, Lamas WT. Alanine series of the antimicrobial peptide gomesin: a structure-activity relationship study. In: Flegel M, Fridkin M, Gilon C, Slaninova J, editors. Peptides. Prague: Kenes International; 2004. pp. 493–494. [Google Scholar]
- 35.Fazio MA, Jouvensal L, Vovelle F, Bulet P, Miranda MT, Daffre S, Miranda A. Biological and structural characterization of new linear gomesin analogues with improved therapeutic indices. Biopolymers. 2006;88:386–400. doi: 10.1002/bip.20660. [DOI] [PubMed] [Google Scholar]
- 36.Mani R, Cady SD, Tang M, Waring AJ, Lehrer RI, Hong M. Membrane-dependent oligomeric structure and pore formation of a beta-hairpin antimicrobial peptide in lipid bilayers from solid-state NMR. Proc Natl Acad Sci USA. 2006;103:16242–16247. doi: 10.1073/pnas.0605079103. [DOI] [PMC free article] [PubMed] [Google Scholar]