

Abstract
The ability of peroxynitrite to modify amino acid residues in glutamine synthetase (GS) and BSA is greatly influenced by pH and CO2. At physiological concentrations of CO2 (1.3 mM), the generation of carbonyl groups (0.2–0.4 equivalents/subunit) is little affected by pH over the range of 7.2–9.0, but, in the absence of CO2, carbonyl formation increases (from 0.1- 1.2 equivalents/subunit) as the pH is raised from 7.2 to 10.5. This increase is attributable, in part but not entirely, to the increase in peroxynitrite (PN) stability with increasing pH. Of several amino acid polymers tested, only those containing lysine residues yielded carbonyl derivatives. In contrast, the nitration of tyrosine residues of both GS and BSA at pH 7.5 almost completely depends on the presence of CO2. However, the pH profiles of tyrosine nitration in GS and BSA are not the same. With both proteins, nitration decreases ≈65% with increasing pH over the range of 7.2–8.4, but, then in the case of GS only, there is a 3.4-fold increase in the level of nitration over the range pH 8.4–8.8. The oxidation of methionine residues in both proteins and in the tripeptide Ala-Met-Ala was inhibited by CO2 at both high and low pH values. These results emphasize the importance of controlling the pH and CO2 concentrations in studies involving PN and indicate that PN is not likely to contribute appreciably to carbonyl formation or oxidation of methionine residues of proteins at physiological pH and CO2 concentrations.
With the discovery that peroxynitrite (PN) is generated in vivo by reaction of nitric oxide with superoxide anion, and the demonstration that PN can modify a number of biological molecules, including proteins, lipids, and nucleic acids, considerable attention has been given to the role of PN in oxidative cellular damage. Among other changes, PN has been shown to promote the nitration of tyrosine residues (1, 2) and the oxidation of methionine (3–7) and cysteine (8–11) residues of proteins.
Because various kinds of reactive oxygen species have been shown to convert some amino acid residues of proteins to carbonyl derivatives, the level of protein carbonyl groups has been widely accepted as a reliable marker of reactive oxygen species-promoted tissue damage. Studies in this laboratory (B.S.B., R.L.L., and E.R.S., unpublished work) and results of others (12) demonstrated that exposure of proteins to PN leads to only marginal increases in the carbonyl content. In contrast, it was reported (13) that high yields of protein carbonyls (8–9 mol/mol) were obtained when preparations of Ca-ATPase from the sarcoplasmic reticulum were treated with PN. Because Ca-ATPase preparations likely contain microsomal lipids, it is possible that lipid peroxidation contributes to the formation of carbonyl groups in the studies with this enzyme. In view of the fact that CO2 reacts rapidly with PN to form a derivative with greatly enhanced ability to nitrate tyrosine residues of proteins (14–17) and relatively little ability to oxidize methionine residues compared with PN alone (6, 7), the possibility that CO2 affects also the ability of PN to generate carbonyl groups in proteins must be considered. Moreover, because stock solutions of PN preparations are maintained in strongly alkaline solutions, substantial changes in pH of reaction mixtures will occur in the absence of adequate buffering capacity when, as is necessary, the reactions are initiated by addition of PN.
The present work was undertaken to define more clearly the effects of pH, CO2, and unsaturated fatty acids on the PN-dependent modification of proteins. Such information is fundamental to assessment of the role of PN in oxidative damage associated with aging, oxidative stress, and various pathologies. Because much information is already available on the modification of BSA and glutamine synthetase (GS) by PN and other forms of reactive oxygen species, these proteins were used for the present studies.
MATERIALS AND METHODS
Chemicals.
PN was prepared by two methods: the reaction of ozone with sodium nitrite (18) and the reaction of isoamylnitrite with H2O2 (19). MnO2 was used to remove residual H2O2 in the latter method. All experiments were performed with both PN preparations to ensure lack of H2O2 involvement. Lack of H2O2 involvement also was confirmed by showing that addition of H2O2 neither stimulated nor inhibited reaction yields. PN was quantitated at 302 nm (ɛ302 nm = 1,670 M−1⋅cm−1). Bacterial GS (ɛ278 nm = 31,000 M−1⋅cm−1) was isolated as described (20). BSA (ɛ278 nm = 41,000 M−1⋅cm−1), 2,4-dinitrophenylhydrazine, and egg yolk lecithin were from Sigma.
Carbonyl Determination.
Protein carbonyls were quantitated by spectrophotometric measurement of their 2,4-dinitrophenylhydrazone derivatives (ɛ370 nm = 22,000 M−1⋅cm−1) as described by Levine et al. (21). Because nitrotyrosine also absorbs at 370 nm, its spectral contribution was determined in samples not treated with 2,4-dinitrophenylhydrazine, and this value was subtracted from samples treated with 2,4-dinitrophenylhydrazine.
Lipid Peroxidation.
Linoleic acid hydroperoxides were synthesized from soybean lipoxygenase, and the 13-hydroperoxide was purified by HPLC (22). Liposomes were made from lecithin by sonication. In brief, lecithin was dissolved in CHCl3 to yield 100 mg/ml. For 1.0 ml of liposomes, 0.2 ml of the CHCl3 preparation (20 mg) was evaporated under nitrogen, and 1.0 ml of nitrogen-purged water was added. The anaerobic solution then was sonicated as described (23). Thiobarbituric acid-reactive material was determined spectrophotometrically at 535 nm as described by Buege and Aust (24).
Peroxynitrite Decomposition.
The time-dependent losses of PN at pH 8.6 and 9.5 that occurred during the incubation of PN with GS were calculated values, based on the experimentally determined rate constants for PN at pH 8.6 (kobs = 0.0375 s−1) and at pH 9.5 (kobs = 0.0123 s−1), under our experimental conditions. For these calculations, the fraction of PN consumed because of its reaction with GS was neglected because it represents a very minor amount compared with the loss attributable to spontaneous decomposition of PN. This follows from the fact that the amount of PN added (3,000 nmol) was an order of magnitude greater than the sum of all amino acid residues (200 nmol) of GS that disappeared, based on amino acid analysis, during the incubation period.
Buffers.
Reaction mixtures (1.0 ml) containing the calculated amount of KHCO3 needed to obtain the desired pH after equilibration with 5% CO2/95% N2 (25) were placed in 12- × 75-mm test tubes fitted with rubber sleeve stoppers (Kontes). The mixtures were purged with the CO2/N2 gas mixture, which was delivered into the reaction mixture by means of an 18-gauge hyperdermic needle inserted through the stopper. A second 18-gauge needle inserted well above the reaction mixture served as an exhaust vent. Complete equilibration at the higher pH values required purging for 30 min. After purging, the reactions were initiated by adding PN to the mixture by means of a gas-tight syringe, the gassing was continued for 5 min, and then samples were taken for assay of protein modification. It is impracticable to study the effect of physiological concentrations of CO2 at pH values >8.8 because of an unfavorable CO2-HCO3− equilibrium.
A similar test tube assembly was used to obtain CO2-free reaction systems. Mixtures containing 0.3 M K-PO4 at the desired pH were purged for 15 min with 100% N2, before adding the PN as described above.
Results of initial experiments on the generation of carbonyl groups in GS by PN were highly variable because of the fact that the buffering capacity of the reaction mixtures was insufficient to prevent significant changes in pH when aliquots of the highly alkaline PN stock solution were added. Therefore, when necessary, an amount of HCl needed to neutralize an equivalent amount of base in the aliquot of PN solution used was added to the reaction mixtures just before adding the PN. In all cases, the reported pH values refer to the pH measured after the reaction was over.
RESULTS
Direct Comparison of the Effects of pH and CO2 on PN-Dependent Modification of Amino Acid Residues of GS.
PN reacts rapidly with CO2 to form an adduct that, based on the physiological concentration of CO2 (1.3 mM) and the rate constant, has been proposed to be the major intermediate in all physiological PN-dependent reactions (14–17). Results of earlier studies at pH 7.4 demonstrated that nitration of tyrosine residues of GS almost completely depends on the presence of CO2 whereas the oxidation of methionine residues is almost completely prevented by physiological concentrations of CO2 (6, 7). To investigate the possibility that the effect of CO2 is influenced by pH, we compared directly the PN-dependent modification of GS under anaerobic conditions at both pH 7.5 and 8.6 in the presence of CO2 (bicarbonate buffers) and in the absence of CO2 (phosphate buffers), which were prepared as described in Materials and Methods. As shown in Fig. 1, the level of methionine oxidation (methionine sulfoxide formation) in the absence of CO2 was slightly higher at pH 7.5 than at pH 8.6; however, at both pH values, the oxidation was almost completely prevented by physiological concentrations of CO2.
Figure 1.

Effect of CO2 on modification of GS by PN at pH 7.5 and pH 8.6. Reaction mixtures (1.0 ml) containing 1.0 mg of GS and 3.0 mM PN were incubated in the presence (bicarbonate buffer) or absence (0.3 M K-PO4 buffer) of 1.3 mM CO2. After 5 min at room temperature, the reaction mixtures were assayed for carbonyl, nitrotyrosine, and methionine sulfoxide content. The buffers were prepared as described in Materials and Methods.
A very different pattern was observed for the nitration of tyrosine residues in GS. At pH 7.5, nitration was almost completely dependent on the presence of CO2. At pH 8.6, CO2 had little or no effect on nitration, and the extent of nitration was only about half that observed at pH 7.5 in the presence of CO2 (Fig. 1). This suggests (i) that nitrating species other than OONOCO2− may contribute to the nitration of tyrosine residues at the higher pH values or (ii) that, at the higher pH, proteins undergo conformational changes that expose more tyrosine residues to nitration. Finally, although CO2 is without effect on the generation of carbonyl groups at pH 7.5, it inhibits significantly the formation of carbonyl groups at pH 8.6 (Fig. 1).
pH Profiles of Carbonyl Formation, Tyrosine Nitration, and Methionine Oxidation.
Carbonyl formation in the absence of CO2. The results, summarized in Fig. 2, show that, in the absence of CO2, the carbonyl content of GS increases exponentially, from a value of 0.1 to a value of 1.2 mol/mol subunit, as a function of increasing pH over the range of pH 7.2–10.25. This increase in carbonyls is probably attributable in part to an increase in PN stability because there is a parallel pH-dependent increase in the half-life of PN (Fig. 2). However, from a comparison of the time courses of carbonyl formation and the decomposition of PN at pH 8.6 and 9.5 (as described in Materials and Methods), it is evident that factors other than PN stability are also involved. Thus, as shown in Fig. 3, at pH 8.6, carbonyl formation reaches a maximum value of ≈0.3 mol/mol subunit after 20 s when only 50% of the added PN had been consumed. In contrast, at pH 9.5, the yield of carbonyl groups was 0.45 mol/mol subunit after 20 s when only 20% of the PN had been consumed and continued to increase to a value of 0.6 mol/mol after 40 s. But at both pH values, the added PN was not totally consumed during the 40 s of incubation.
Figure 2.

Comparison of the time-dependent changes in carbonyl group formation in GS and in the levels of PN during incubation at pH 8.6 and 9.5. Reaction mixtures (1.0 ml) containing 1.0 mg GS, 3.0 mM PN, and 0.3M K-PO4 buffer at either pH 8.6 (closed triangles) or pH 9.5 (closed circles) were incubated at room temperature. At the times indicated, the levels of protein carbonyl groups were determined, and the levels of PN were calculated from the measured rate constants for the decomposition of PN at pH 8.6 (open triangles) and at pH 9.5 (open circles) as described in Materials and Methods.
Figure 3.

Effect of pH on PN stability and carbonyl formation in GS in the absence of CO2. Reaction mixtures (1.0 ml) contained 3 mM PN, 1.0 mg GS, and 0.3 M K-PO4 buffer at the specified pH values. After 20 min at room temperature, protein carbonyls (circles) and the half-life of PN (triangles) were determined as described in Materials and Methods.
Carbonyl formation in the presence of CO2.
In contrast to the results summarized in Fig. 2, carbonyl group formation in the presence of physiological concentrations of CO2 is almost independent of variations in pH over the range of 7.0–9.0 (Fig. 4). With both GS and BSA, only small amounts of carbonyl groups (0.1–0.4 mol/mol subunit) were formed in the presence of CO2.
Figure 4.

Effect of pH at physiological concentrations of CO2 on the PN-dependent formation of carbonyls in GS and BSA. Reaction mixtures (1.0 ml) contained 1.0 mg GS (open circles) or 1.0 mg BSA (closed circles) and amounts of KHCO3 (15–700 mM) as needed to obtain the indicated pH values in the presence of 1.3 mM CO2. After purging for 30 min with a 5% CO2/95% N2 gas mixture, 3.0 mM PN was added, the samples were incubated at room temperature for 5 min and then was assayed for carbonyl content as described in Materials and Methods.
Tyrosine nitration.
In the presence of CO2, the nitration of GS and BSA exhibit very different pH profiles. As illustrated in Fig. 5, tyrosine nitration in BSA decreases from a value of ≈3 mol/mol of subunit to a limiting value of ≈2 mol/mol as the pH is raised from 7. 3 to 8.8. Likewise, the nitration of tyrosine residues in GS decreases (from 2.3 to 1.0) as the pH is increased from 7.3 to 8.2. But, in contrast to BSA, further increases in pH from 8.2 to 8.8 leads to a dramatic increase in nitration, to a value of 4.5 mol/mol. It is evident from these results that the effect of pH on the nitration of tyrosine residues of proteins is complex and unpredictable. Because the number of tyrosine residues in GS and BSA are almost the same (17 and 19/subunit, respectively), the large difference in nitration behavior at high pH values cannot be explained by differences in tyrosine content or by pH-dependent changes in the nitrating species involved. This raises the possibility that, at high pH values, GS, but not BSA, undergoes changes in protein conformation that lead to exposure of more tyrosine residues to nitration or to deprotonation of other residues in the protein that affect the ability of tyrosine residues to be nitrated.
Figure 5.

Comparison of the pH-dependence of tyrosine nitration of GS and BSA in the presence of physiological concentrations of CO2. Reaction mixtures (1.0 ml) containing 3.0 mM PN and 1.0 mg of BSA (A) or 1.0 mg of GS (B) and bicarbonate concentrations needed to obtain the indicated pH values were prepared and incubated as described in Fig. 4. Then, the levels of nitrotyrosine were determined as described in Materials and Methods.
Oxidation of methionine in the tripeptide Ala-Met-Ala.
To investigate the possibility that the effect of CO2 on the oxidation of methionine residues of proteins might be related to the protein structure, we examined the effect of CO2 on oxidation of the peptide ala-met-ala. Here again, we see that the PN-dependent oxidation of the Met residue is strongly inhibited by CO2 (Fig. 6) but is relatively independent of pH.
Figure 6.

Effect of pH and CO2 on the PN-dependent oxidation of methionine residues of the tripeptide Ala-Met-Ala. Reaction mixtures (1.0 ml) containing 0.075 mg of the tripeptide (A-M-A) and 3.0 mM PN were incubated in both the absence (open circles) or presence (closed circles) of CO2: i.e., in 0.3 M K-PO4 buffer or in bicarbonate buffer prepared as described in Fig. 4. After incubation for 5 min at room temperature, the samples were analyzed for methionine sulfoxide content as described in Materials and Methods.
Identification of Amino Acids That Are Susceptible to Oxidation to Carbonyl Derivatives.
Measurements of the amino acid content in protein hydrolysates of a protein are subject to an error of 5%; therefore, it is not possible to detect the loss of only one residue of any amino acid that is present in amounts >20 residues per mole of protein. Accordingly, because at alkaline pH only 0.25–1.5 carbonyl groups per mole were formed, it was not possible from amino acid analysis alone to identify which amino acids are the targets for the PN-dependent generation of carbonyl derivatives. Therefore, a series of homo- and heteroamino acid polymers of the more likely candidates were examined for their ability to form carbonyl derivatives on PN treatment. Specifically, reaction mixtures containing 3 mM PN, 0.3 M K-PO4 (pH 8.6), and 1 mg/ml of either poly Glu, poly Pro, poly His, Poly Arg, poly Lys, and poly Lys-Arg were incubated for 20 min at 37°C and then were assayed for carbonyl content. Poly Arg-Lys and poly Lys were the only polymers that yielded detectable amounts of carbonyl groups (data not shown). Therefore, lysine residues are likely candidates for the generation of carbonyl groups by PN-treatment at an alkaline pH.
Effect of Transition Metals and H2O2.
Addition of 0.1 mM concentrations of either FeCl3, CuSO4, or H2O2 to the PN reaction mixtures at pH values varying from 6.6 to 10.5 had no significant effect on the generation of protein carbonyl groups (data not shown).
Effect of PN Degradation Products.
To investigate the possibility that breakdown products of PN, such as nitrate or nitrite, contribute to the generation of carbonyl groups, two experiments were performed. First, after incubation with PN in K-PO4 buffer at pH 9.8, the sample was passed through a column of Sephadex G-25 (Amersham Pharmacia) to remove nitrates and nitrites and any other PN degradation products, and then the protein samples were analyzed for carbonyl content. There was no significant difference between the carbonyl content of GS measured before and after removal of the PN degradation products by gel filtration. In a second experiment, GS was added 5 min after the addition of PN (long enough for total destruction of PN), and then the mixtures were incubated for an additional 20 min. No protein carbonyls were detected in these “reverse addition” experiments.
Effect of Lipid Peroxidation.
Viner et al. (13) demonstrated that PN can induce appreciable carbonyl formation (4–7 mol/mol subunit) in the sarcoplasmic reticulum Ca-ATPase at physiological pH. This finding is contrary to the above results and data of earlier studies (B.S.B., R.L.L. and E.R.S., unpublished work) showing that barely detectable amounts of carbonyl groups are generated on treatment of either GS or BSA at the physiological pH of 7.4. Because the Ca-ATPase preparations likely contain some sarcoplasmic membrane-derived lipids, and because PN is known to promote lipid peroxidation (26, 27), we investigated the possibility that lipids and or lipid peroxides might enhance the PN-mediated generation of protein carbonyl groups. In confirmation of the results of Radi et al. (26), we showed that the PN-dependent oxidation of phosphatidylcholine liposomes leads to formation of thiobarbituric acid-reactive material, presumably malondialdehyde. Thus, addition of 0.1 mM PN to reaction mixtures containing 2 mg/ml egg yolk phosphatidylcholine in 0.3 M K-PO4 (pH 7.4) led to the formation of 0.5 mM equivalents of thiobarbituric acid reactive products; increasing the concentration of PN to higher values, up to 0.8 mM, led to further increases in the level of thiobarbituric acid reactive substances to a value of 0.8 mM.
To examine the effect of lipid peroxidation on formation of protein carbonyl groups, reaction mixtures containing BSA (1 mg/ml), 0.3 M K-PO4 buffer (pH 7.4), 1.5 mM PN, and various amounts of phosphatidylcholine were incubated for 20 min at 37°C and then were assayed for carbonyl groups (21). Supplementation of the reaction mixture with phosphatidylcholine did not stimulate carbonyl formation. In fact, as the concentration of phosphatidylcholine was increased from 0 to 8.0 mg/ml, there was a progressive slight decrease in the amount of carbonyl groups formed from a value of 1.06 to 0.87 mol/mol, respectively.
Lipid peroxidation could play a role in PN-dependent protein carbonyl formation through the generation of lipid hydroperoxides. We examined the ability of linoleic-13-hydroperoxide to convert BSA to carbonyl derivatives in the presence of either Cu2+ or Fe3+, both in the presence or absence of reducing agent (ascorbate). For comparison, similar experiments were performed with H2O2. The results, summarized in Fig. 7, show that (i) lipid hydroperoxides in the presence of either copper or iron promote the formation of carbonyl groups in BSA, (ii) linoleic-13-hydroperoxide is more effective than H2O2 in the metal-catalyzed formation of BSA carbonyl groups, (iii) Cu2+ is more effective than Fe3+ in the generation of carbonyl groups by either H2O2 or linoleic-13-hydroperoxide, and (iv) ascorbate enhances the production of carbonyl groups in all combinations studied.
Figure 7.

Lipid hydroperoxide-dependent carbonyl formation. Reaction mixtures (1.0 ml) contained 1.0 mg BSA in 0.2 M K-PO4 buffer (pH 7.4) and the following additions as indicated in the figure: 10 mM ascorbate, 0.1 mM CuSO4, 0.1 mM FeCl2, 1.0 mM H2O2, and 1.0 mM linoleic-13-hydroperoxide. After 4 hr of incubation at room temperature, protein carbonyls were measured as described in Materials and Methods.
DISCUSSION
It is evident from the data presented here that only small amounts (0.1–0.4 mol per mol subunit) of carbonyl groups are formed when GS or BSA are treated with PN at physiological pH and CO2 concentrations (Fig. 4). However, in the absence of CO2, the yield of carbonyl groups in GS increased exponentially as a function of increasing pH over the range of 7.0 to 10.5 (Fig. 2). Although the yield of carbonyl groups correlates well with the pH-dependent increase in PN stability, it is evident from a comparison of the time courses of carbonyl formation at pH 8.6 and 9.5 that PN stability alone cannot fully account for the pH effect. Among other possibilities, it is suggested that the pH-dependent increase in carbonyl formation also might reflect changes in protein conformation that expose functional groups of amino acid residues to PN attack or to the deprotonation of some amino acid residues, making them more susceptible to oxidation by PN. If, in fact, lysine residues of proteins are targets for PN conversion to carbonyl groups, as is suggested by the studies with amino acid polymers, then the pH-dependent increase in carbonyl formation might reflect the deprotonation of the ɛ-amino groups of lysine residues in proteins over the pH range of 9.8–10.4. Because of insensitivity of the amino acid analysis of protein hydrolysates and the fact that only small amounts of carbonyl groups are formed by the PN-treatment, it was not possible to demonstrate directly that the generation of carbonyl derivatives is linked to the modification of lysine residues.
In confirmation of earlier reports (6, 7), we found that nitration of tyrosine residues in GS at pH 7.5 is almost completely dependent on the presence of CO2 (Fig. 1). When reactions are carried out in bicarbonate buffers at physiological CO2 concentrations (1.3 mM), the nitration of tyrosine in both GS and BSA decreased with increasing pH over the range of 7.3 to 8.2. However, with further increases in pH from 8.2 to 8.8, there was an enormous increase in the nitration of GS but no additional increase in BSA (Fig. 5). To the contrary, it was recently reported that treatment of BSA with PN in the presence of 0.06 mM CO2 lead to a 4-fold increase in the level of nitrotyrosine with increasing pH over the range of 5–9. There is no obvious explanation for this discrepancy. The experimental conditions of the two studies are very different. As noted in Materials and Methods, our studies were carried out in a closed system in which the pH was fixed by equilibration of 5% CO2–95% N2 gas mixtures with an amount of bicarbonate needed to yield the desired pH, and 1.3 mM dissolved CO2.; the studies of Gow et al. (28) were carried out in an open system containing 100 mM PBS and amounts of bicarbonate calculated to yield 0.06 mM CO2. Significantly, the solution concentrations of both CO2 and HCO3− were orders of magnitude higher in our studies. In our studies, the concentrations of bicarbonate needed to obtain equilibrium mixtures containing a physiological concentration of CO2 (1.3 mM) varied from 12 to 700 mM as the pH was raised from 7.3 to 8.8. Even so, it is not evident why the pH-dependence profiles of tyrosine nitration for GS and BSA are so different in the higher pH range because the experimental conditions were identical and the number of tyrosine residues present per milligram of protein are almost the same for BSA and GS. As noted earlier, the difference in nitration at the higher pH values might reflect differences in the effects of pH and/or ionic strength on protein conformation. We showed previously that the tyrosine residues in GS, which were nitrated at pH 7.4, were generally those that were solvent-exposed in the crystal structure (7). In contrast to tyrosine nitration, the PN-dependent oxidation of methionine residues in GS and in the tripeptide ala-met-ala is almost independent of pH and is strongly inhibited by physiological concentrations of CO2 (Figs. 1 and 6).
Attempts to explain why treatment of the sarcoplasmic Ca-ATPase with PN leads to formation of high levels of carbonyl groups (13) were unsuccessful. Treatment of GS with PN in the presence of egg white phosphatidylcholine had little or no effect on the production of protein carbonyls. However, the fact that lipid hydroperoxides are more effective than H2O2 in mediating the transition-metal-catalyzed generation of carbonyl groups in GS suggests that lipids might contribute to carbonyl generation under some conditions.
Furthermore, under our experimental conditions, PN is known to undergo the reactions depicted in Scheme 1, where X, Y, and Z represent “caged radical pairs” for [ȮH, ṄO2] and [Ȯ2−, ṄO] or [Ȯ−, ṄO2] and [ĊO3−, ṄO2], respectively. The rate constants for the decomposition of PNH and PN− to form NO3− + H+ and NO2− + O2 has been reported (29) to be 0.66 s−1 at 20°C and 7.5 × 10−6 s−1, respectively, at pH 13. The rate and equilibrium constants for generation of ȮH in reaction (1), Ȯ2− in reaction (2), Ȯ− in reaction (2′), and ĊO3− in reaction (3), from their respective parent compounds, have been shown to be 0.5 s−1 and 10−10 M (29), 1.7 × 10−2 s−1 and 3 × 10−12 M (27), 3 × 10−6 s−1 and 1 × 10−15 M (27), and 1.5 × 106 s−1 and 3 × 10−3 M (30), respectively. The second-order rate constant for the formation of peroxynitrite-CO2 adduct (PNCO2−) has been reported to be 3 × 104 M−1⋅s−1 (14). From the kinetic and equilibrium constants, it is clear that, in the presence of 1.3 mM CO2, the predominant radicals will be ĊO3− and ṄO2. The ĊO3− will react with the aromatic ring of tyrosine to form a tyrosyl radical, which will react with ṄO2 to form a nitrated tyrosine. However, ĊO3− is not an effective oxidizing agent for generating methionine sulfoxide and carbonyl derivatives of amino acid residues. In the absence of CO2, one obtains a relatively small quantity of ȮH, Ȯ−, and Ȯ2−, known oxygen-containing free radicals that are capable of oxidizing methionine and generating protein carbonyl derivatives (31). At alkaline pH, and in the absence of CO2, the anionic PN− is stable, and, thus, it could supply relatively low levels of the free radicals produced in reactions (1), (2), and (2′) (Scheme 1) for reaction with proteins over a much longer time interval. Thus, the observed protein modifications by PN are consistent with free radical-mediated reactions. 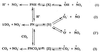
In view of the fact that barely detectable amounts of carbonyl groups are formed when proteins are treated with PN at pH 7.4 at physiological CO2 concentrations, it seems unlikely that PN contributes significantly to the increase in protein carbonyl derivatives, as occurs during aging and various age-related diseases. This suggests that the presence of high levels of protein carbonyls in tissues may serve as a measure of oxidative damage caused by forms of reactive species other than PN.
ABBREVIATIONS
- PN
peroxynitrite
- GS
Escherichia coli glutamine synthetase
References
- 1.Beckman J S, Ischiropoulos H, Zhu L, Van der Woerd M, Smith C, Chen J, Harrison J, Martin J C, Tsai M. Arch Biochem Biophys. 1992;298:438–445. doi: 10.1016/0003-9861(92)90432-v. [DOI] [PubMed] [Google Scholar]
- 2.Kong S K, Yim M B, Stadtman E R, Chock P B. Proc Natl Acad Sci USA. 1996;93:3377–3382. doi: 10.1073/pnas.93.8.3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pryor W A, Jin X, Squadrito G L. Proc Natl Acad Sci USA. 1994;91:11173–11177. doi: 10.1073/pnas.91.23.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pryor W A, Squadrito G L. Am J Physiol. 1995;268:L699–L722. doi: 10.1152/ajplung.1995.268.5.L699. [DOI] [PubMed] [Google Scholar]
- 5.Jensen J L, Miller B L, Zang X P, Hug G L, Schoneich C. J Am Chem Soc. 1997;119:4749–4757. [Google Scholar]
- 6.Berlett B S, Friguet B, Yim M B, Chock P B, Stadtman E R. Proc Natl Acad Sci USA. 1996;93:1776–1780. doi: 10.1073/pnas.93.5.1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berlett B S, Levine R L, Stadtman E R. Proc Natl Acad Sci USA. 1998;95:2784–2789. doi: 10.1073/pnas.95.6.2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karoui H, Hogg N, Frejaville C, Tordo P, Kalyanaraman B. J Biol Chem. 1996;271:6000–6009. doi: 10.1074/jbc.271.11.6000. [DOI] [PubMed] [Google Scholar]
- 9.Mohr S, Stamler J S, Brune B. FEBS Lett. 1994;348:223–227. doi: 10.1016/0014-5793(94)00596-6. [DOI] [PubMed] [Google Scholar]
- 10.Gatti R M, Radi R, Agusto O. FEBS Lett. 1994;348:287–290. doi: 10.1016/0014-5793(94)00625-3. [DOI] [PubMed] [Google Scholar]
- 11.Kato Y, Kawakski S, Aoki T, Itakura K, Osawa T. Biochem Biophys Res Commun. 1997;234:82–84. doi: 10.1006/bbrc.1997.6587. [DOI] [PubMed] [Google Scholar]
- 12.Ischiropoulos H, Al-Medi A B. FEBS Lett. 1995;364:279–282. doi: 10.1016/0014-5793(95)00307-u. [DOI] [PubMed] [Google Scholar]
- 13.Vinor R L, Huhmer A F R, Biglow D J, Schoneich C. Free Radical Res. 1996;24:243–259. doi: 10.3109/10715769609088022. [DOI] [PubMed] [Google Scholar]
- 14.Lymar S V, Hurst K. J Am Chem Soc. 1995;117:8867–8868. [Google Scholar]
- 15.Uppu R M, Squadrito G L, Pryor W A. Arch Biochem Biophys. 1996;327:335–343. doi: 10.1006/abbi.1996.0131. [DOI] [PubMed] [Google Scholar]
- 16.Denicola A, Freeman B A, Trujillo M, Radi R. Arch Biochem Biophys. 1996;333:49–56. doi: 10.1006/abbi.1996.0363. [DOI] [PubMed] [Google Scholar]
- 17.Lymar S V, Jiang Q, Hurst J K. Biochemistry. 1996;35:7855–7861. doi: 10.1021/bi960331h. [DOI] [PubMed] [Google Scholar]
- 18.Pryor W A, Cueto R, Jin X, Koppenol W H, Ngu-Schwemlein M, Squadrito G L, Uppu P L, Uppu R M. Free Radical Biol Med. 1995;18:75–83. doi: 10.1016/0891-5849(94)00105-s. [DOI] [PubMed] [Google Scholar]
- 19.Uppu R M, Pryor W A. Anal Biochem. 1996;236:242–249. [PubMed] [Google Scholar]
- 20.Miller R E, Shelton E, Stadtman E R. Arch Biochem Biophys. 1974;163:155–171. doi: 10.1016/0003-9861(74)90465-2. [DOI] [PubMed] [Google Scholar]
- 21.Levine R L, Garland D, Oliver C N, Amici A, Climent I, Lenz A, Ahn B W, Shaltiel S, Stadtman E R. Methods Enzymol. 1990;186:464–478. doi: 10.1016/0076-6879(90)86141-h. [DOI] [PubMed] [Google Scholar]
- 22.Nikaloev V, Reddanna P, Whelan J, Hildenbrandt G, Reddy C C. Biochem Biophys Res Commun. 1990;170:491–496. doi: 10.1016/0006-291x(90)92118-j. [DOI] [PubMed] [Google Scholar]
- 23.Pederson T C, Buege J A, Aust S D. J Biol Chem. 1973;248:7134–7141. [PubMed] [Google Scholar]
- 24.Buege J A, Aust S D. Methods Enzymol. 1978;51:302–310. doi: 10.1016/s0076-6879(78)52032-6. [DOI] [PubMed] [Google Scholar]
- 25.Umbreit W W. In: Manometric Techniques. Umbreit W W, Burris R H, Stauffer J W, editors. Minneapolis: Burgess; 1957. pp. 18–27. [Google Scholar]
- 26.Radi R, Beckman J S, Bush K M, Freeman B A. Arch Biochem Biophys. 1991;288:481–487. doi: 10.1016/0003-9861(91)90224-7. [DOI] [PubMed] [Google Scholar]
- 27.Rubbo H, Freeman B A. Methods Enzymol. 1996;269:385–394. doi: 10.1016/s0076-6879(96)69039-9. [DOI] [PubMed] [Google Scholar]
- 28.Gow A, Duran D, Thom S R, Ischiropoulos H. Arch Biochem Biophys. 1996;333:42–48. doi: 10.1006/abbi.1996.0362. [DOI] [PubMed] [Google Scholar]
- 29.Merenyi G, Lind J, Goldstein S, Czapski G. Chem Res Toxicol. 1998;11:712–713. doi: 10.1021/tx980043h. [DOI] [PubMed] [Google Scholar]
- 30.Merenyi G, Lind J. Chem Res Toxicol. 1997;10:1216–1220. doi: 10.1021/tx970101j. [DOI] [PubMed] [Google Scholar]
- 31.Berlett B S, Stadtman E R. J Biol Chem. 1997;272:20313–20316. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]