

Abstract
The flavoprotein 2-aminobenzoyl-CoA monooxygenase/reductase from the eubacterium Azoarcus evansii catalyzes the dearomatization of 2-aminobenzoyl-CoA. The reaction consists in an O2-dependent monooxygenation at the benzene position 5, which is followed immediately by an NADH-dependent hydrogenation of the intermediate at the same catalytic locus. The reaction was studied by 1H, 2H, and 13C NMR spectroscopy of the products. The main product was characterized as 5-oxo-2-aminocyclohex-1-ene-1-carboxyl-CoA by two-dimensional NMR spectroscopy. Thus, [5-2H]2-aminobenzoyl-CoA was converted into [6-2H]5-oxo-2-aminocyclohex-1-ene-1-carboxyl-CoA, indicating a 5 → 6 shift of the [5-2H] label. Label from NAD2H was transferred to the 3 position of the cyclic eneamine, whereas label from solvent D2O was incorporated into the 4 and the 6 positions of 5-oxo-2-aminocyclohex-1-ene-1-carboxyl-CoA. The labeling pattern is compatible with the monooxygenation proceeding via what is formally an NIH shift, yielding 5-oxo-2-aminocyclohex-1,3-diene-1-carboxyl-CoA as a protein-bound intermediate. It is suggested that this shift in flavin-dependent monooxygenation may have general validity.
Flavin monooxygenases catalyze the insertion of one atom of dioxygen into a variety of substrates (1). The overall reaction involves three, chemically distinct steps (a–c).
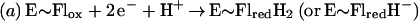 |
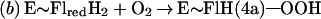 |
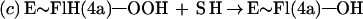 |
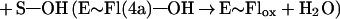 |
Step a is the generation of a reduced flavin (E∼FlredH2), the form that activates dioxygen in step b to form a covalent flavin hydroperoxide [E∼FlH(4a)—OOH]. In step c, the oxygen atom is inserted into a substrate C–H bond (S—H), yielding S—OH.
The flavin-hydroperoxide formed in step (b) is a flavin C(4a) adduct (1). Bruice’s laboratory has contributed prominently to the understanding of its formation from O2 (2). In the present work we address the chemical mechanism of step (c). At its basis is the reactivity of the flavin-4a-hydroperoxide, a multifaceted functional group that can behave in mechanistically divergent ways [e.g., Baeyer–Villiger-type reactions (3) or generation of excited states in bacterial luciferase (4)]. In the case of true monooxygenases the distal oxygen is assumed to react electrophilically with the substrate (1). This is supported by linear free energy-type correlations, which indicate that the flavin C(4a) group behaves as an acid (5, 6). Studies with fluorinated phenols (7) and theoretical calculations on the reactivity of the peroxide (8) also are consistent with this type of mechanism. However, experimental evidence allowing the unequivocal identification of a specific mechanistic variant for the monooxygenation step has not yet emerged.
Flavin-dependent monooxygenases react specifically with aromatic substrates “activated” by the presence of electron-rich substituents (—O−, —NH2, —SH) (1). Pteridine–iron-type catalysts such as in phenylalanine hydroxylase hydroxylate “nonactivated” aromatic substrates (9). These and heme-type catalysts (P450 hydroxylases) work by the so-called “NIH-shift” mechanism (10, 11). In this, the hydrogen atom at the aromatic position to be hydroxylated migrates to an adjacent one in the final product. Whether or not this shift occurs via arene oxide intermediates is still disputed (12).
With this background we have studied the properties of the flavoprotein 2-aminobenzoyl-CoA monooxygenase/reductase (ACMR) from the eubacterium Azoarcus evansii. The enzyme is a dimer comprising two 85-kDa chains and containing one FAD cofactor per subunit, i.e., per polypeptide chain (13). However, the two chains combine asymmetrically to form two centers, each containing one FAD molecule. These centers have markedly different properties, in that the flavins carry out two “opposite” types of chemical reactions while acting in sequence on the same substrate (14, 15). First, the CoA conjugate of anthranilic acid (AB-CoA) is monooxygenated by the “ACM” functionality (Fig. 1A) at position 5, i.e., para to the amino group. Subsequently, the reductase functionality of the enzyme (“ACR”) hydrogenates this intermediate to yield the nonaromatic cyclohexene product AOB-CoA (the CoA conjugate of 2-amino-5-oxocyclohex-1-ene-1-carboxylate) (Fig. 1B). The transfer of the hydride via FAD occurs stereoselectively from the pro-R site of NADH (C.H. and S.G., unpublished data).
Figure 1.

Reactions catalyzed by ACMR. The monooxygenase functionality (ACM) first is reduced by NADH and subsequently reacts with dioxygen (in the presence of the substrate AB-CoA) probably to form a flavin 4a-hydroperoxide (not shown). This then inserts oxygen into AB-CoA to form the assumed 5-oxo-2-aminocyclohexadiene intermediate (A). The latter is hydrogenated rapidly by the reductase functionality (ACR) to yield the more stable AOB-CoA (B). At limiting NADH concentrations, the intermediate spontaneously rearranges to the aromatic 5-OH-AB-CoA (C). Reaction B thus constitutes an efficient trapping of the intermediate.
This system partitions (14, 15): at low concentrations of NADH, path B becomes inefficient; this leads, via rearomatization of the intermediate shown, to production of the probably nonphysiological, aromatic side product 5-hydroxy-2-aminobenzoyl-CoA via path C (14, 15) (Fig. 1). The salient peculiarity of ACMR is thus that it efficiently traps the assumed 5-oxo-cyclohexadieneamine intermediate (Fig. 1 Center), the likely primary product of oxygen insertion.
Based on this we reasoned that this would be an optimal system for the study of the mechanism of oxygen insertion via elucidation of the structure of the final products. We thus have investigated the mode of insertion/retention of label originating from [4R-2H]NAD2H, [5-2H]AB-CoA, [ring-13C6]AB-CoA, and 1H2O/2H2O during the reaction catalyzed by ACMR. The results allow an unambiguous mechanistic conclusion, which is discussed in the general context of flavin-dependent monooxygenation.
EXPERIMENTAL PROCEDURES
Growth of Organisms, Enzyme Purification, and Assays.
ACMR was isolated from Azoarcus evansii (earlier called Pseudomonas sp. KB740) (16) grown aerobically at 28°C. Minimal medium, anthranilate, was used as the single carbon source (17). Enzyme purification was done according to refs. 14 and 18, and enzyme activity was done as described in refs. 13 and 14.
Preparation of Labeled Substrates.
Sources were 2-aminobenzoic acid (Aldrich); [ring-13C6]2-aminobenzoate (Deutero, Germany); and [5-2H]2-aminobenzoate (gift from B. Langkau; ref. 14). AB-CoA thioesters were prepared enzymatically. A typical reaction mixture contained 10 mmol Tris⋅HCl (pH 8.4), 200 μmol MgCl2, 50 μmol 2-aminobenzoic acid, 100 μmol CoA (Waldhoff, Germany), and 100 μmol ATP in a total volume of 100 ml. The reaction was started by the addition of AB-CoA synthetase [from A. evansii (19)] and was incubated at 37°C for 2 h. It then was acidified, and AB-CoA was purified as described earlier (14). [4R-2H]NADH was synthesized as will be described elsewhere (C.H. and S.G., unpublished data).
Preparation of AOB-CoA.
A mixture containing 500 μmol KPi (pH 7.8), 100 μmol NADH, 10 μmol AB-CoA, and 5.6 units ACMR in a volume of 100 ml was incubated aerobically at 37°C. After 20 min the enzyme was inactivated by the addition of 100 μl of 0.1 M methylmethanethiosulfonate (in H2O). The mixture then was applied to a Waters Sep Pak C18-column (35 ml, 10 g). The column was washed with 40 ml of 5 mM KPi buffer (pH 7.8) and developed with 50% methanol in 5 mM KPi buffer (pH 7.8). AOB-CoA-containing fractions (strong absorption at 320 nm) were pooled and lyophilized. Further purification of AOB-CoA was achieved by preparative HPLC (C18, 250 × 20 mm; Waters). Development was achieved by using 4.5 mM KPi (pH 7.8), containing 86% acetonitrile and 9.5% methanol. The flow rate was 5 ml/min. AOB-CoA retention time was 25 min. Fractions containing AOB-CoA were lyophilized and desalted. Storage occurred at −20°C.
NMR Spectroscopy.
One- and two-dimensional 1H, 2H, and 13C NMR spectra were recorded at 7°C in 19 mM KPi, pH 7.6, containing 10% D2O (no D2O in the case of 2H NMR spectra). The instrument used was a Bruker DRX 500 spectrometer with four frequency channels, pulsed-field Z-gradient accessory, a lock-switch unit, and ASPECT station. Setup and data processing were done according to standard Bruker software (xwinnmr; Bruker, Billerica, MA).
RESULTS
For the preparation of labeled products, (labeled) AB-CoA was incubated with ACMR in the presence of O2 and (labeled) NADH, and product formation was analyzed by HPLC. The time of incubation and extent of conversion were optimized for the different conditions because the presence of deuterium label affects both the rates of product formation and its stability. The reaction mixture was subjected to preparative HPLC to yield 1–8 mg of purified products, which then were analyzed by 1H, 2H, and 13C NMR spectroscopy (Table 1).
Table 1.
NMR data of 2-amino-5-oxocylohex-1-ene-1-carboxyl-CoA obtained in 19 mM phosphate buffer (pH 7.6) containing 10% D2O
| Position | Chemical shifts, ppm
|
Coupling constants, Hz
|
Isotope shifts¶, ppb
|
Correlation experiments
|
|||||
|---|---|---|---|---|---|---|---|---|---|
| 1H* | 13C† | JCC‡ | JCP§ | JHH§ | Δ1H(2H) | Δ13C(2H) | TOCSY | HMBC | |
| COSCoA | 190.42 | e1, 6 | |||||||
| 1 | 96.94 | 68.6(2), 46.9(6), 2.9 | 67(6) | 6, 3 | |||||
| 2 | 162.59 | 68.4(1), 40.6(3), 1.8 | 25(6) | 6, 3, 4 | |||||
| 3 | 2.55 (t) | 31.46 | 40.6(2), 31.6(4), 2.0 | 6.8 | 4 | 4 | |||
| 4 | 2.42 (t) | 38.61 | 39.6(5), 31.7(3), 10.0(6), 2.9 | 6.8 | 88(3) | 3 | 3.6(w) | ||
| 5 | 218.14 | 39.5(4), 37.8(6), 3.5 | 52(6) | 6, 3, 4 | |||||
| 6 | 3.01 (s) | 40.65 | 46.9(1), 37.7(5), 10.3(4), 1.8 | 22(6) | |||||
| a2 | 8.11 (s) | 155.38 | |||||||
| a4 | 151.85 | a8, a2, 1′(w) | |||||||
| a5 | 121.11 | a8, NH2-a10(w), a2(w) | |||||||
| a6 | 158.13 | a2 | |||||||
| a8 | 8.47(s) | 143.32 | 1′ | ||||||
| NH2-a10 | 6.85 (s) | ||||||||
| 1′ | 6.07 (d) | 89.21 | 7.4 | 2′, 3′, 4′ | 2′, 3′ | ||||
| 2′ | 4.76 (m) | 77.36 | 3.3 | ND | 3′, 4′, 1′ | 1′ | |||
| 3′ | 4.71 (m) | 76.49 | 5.3 | ND | 4′, 2′, 1′ | 4′, 5′ | |||
| 4′ | 4.52 (m) | 86.76 | ND | ND | 5′, 3′, 2′, 1′ | 3′, 5′, 1′(w) | |||
| 5′ | 4.18 (m) | 68.57 | 5.3 | ND | 4′ | 4′(w), 3′(w) | |||
| e1 | 2.84 (m) | 29.86 | ND | c2, NH-c3 | |||||
| e2 | 3.23 (m) | 42.29 | ND | e1, NH-e3 | NH-e3, e1(w) | ||||
| NH-e3 | 8.24 (t) | 5.7 | e2, e1 | ||||||
| p1 | 176.73 | NH-c3, p2, c2(w), p3(w) | |||||||
| p2 | 2.36 (t) | 38.21 | 6.5 | p3, NH-p4 | p3(w) | ||||
| p3 | 3.38 (m) | 38.35 | p2, NH-p4 | p2, NH-p4 | |||||
| NH-p4 | 8.10 (t) | 5.9 | p3, p2 | ||||||
| p5 | 177.69 | NH-p4, p6, p3(w) | |||||||
| p6 | 3.99 (s) | 76.70 | p8, p9, p10, p10′ | ||||||
| p7 | 41.09 | 8.2 | p8, p9, p6, p10(w), p10′(w) | ||||||
| p8 | 0.70 (s) | 20.76 | p6, p9, p10(w), p10′(w) | ||||||
| p9 | 0.85 (s) | 23.76 | p8, p6, p10, p10′ | ||||||
| p10 | 3.49 (dd) | 74.63 | 5.5 | 9.5, 4.7 | p10′ | p8, p9, p6 | |||
| p10′ | 3.80 (dd) | 9.5, 4.7 | p10 | p8, p9, p6 | |||||
ND, not determined; w, weak cross-peak intensity.
Referenced to external trimethylsilylpropanesulfonate; signal multiplicities are indicated in parentheses (s, singlet; d, doublet; t, triplet; m, multiplet; dd, double doublet).
Referenced to external trimethylsilylpropanesulfonate.
13C13C coupling constants as determined from 13C NMR signals of a sample obtained from [ring-13C6]AB-CoA; coupling partners as determined from INADEQUATE experiments are indicated in parentheses.
Obtained from one-dimensional spectra.
Obtained from one-dimensional 13C NMR data of deuterated isotopomers. The position of deuteration is specified in parentheses.
To increase the sensitivity and selectivity of the NMR experiments, we used [ring-13C6]2-aminobenzoyl-CoA as substrate. 1H and 13C NMR chemical shifts of the product, as well as 1H1H and 1H13C correlation patterns from two-dimensional total correlation spectroscopy (TOCSY) and heteronuclear multiple bond correlation (HMBC) experiments, are summarized in Table 1. The 13C NMR spectrum of the sample obtained from the 13C-labeled precursor was dominated by six double-doublets (Fig. 2; coupling constants in the range from 30 to 70 Hz), indicating simultaneous 13C13C coupling to two directly bonded, adjacent 13C atoms. Thus, the connectivity of the ring carbon atoms in the benzoyl substrate is retained in the enzymatic product. The detected fine splitting of the signals is explained as a result of 13C13C coupling via two or three bonds.
Figure 2.

1H decoupled 13C NMR signals of purified AOB-CoA from reaction of ACMR with [ring-13C6]AB-CoA. 13C coupling patterns are indicated above the signals. Coupled carbon atoms to the respective index signal are indicated as analyzed from two-dimensional INADEQUATE experiments. For numbering, see Fig. 4 legend.
The 13C chemical shifts at 96.9 and 162.6 ppm are compatible with the presence of a CC double-bond in the product. More specifically, they are typical for C-2 and C-1 of an eneamine moiety, respectively (20). The signal at 218.1 ppm suggests the presence of a keto function. The 1H NMR signals of the carbocyclic moiety display a singlet at 3.01 ppm [C(6)H2] and two triplets at 2.55 and 2.42 ppm (CH2s at C-3 and C-4, respectively) (Fig. 3A).
Figure 3.

Partial NMR spectra of purified AOB-CoA obtained from ACMR-catalyzed conversion of AB-CoA with NADH and O2; variation of labeling patterns. (A) 13C decoupled 1H NMR spectrum of AOB-CoA obtained from conversions of [ring-13C6]AB-CoA with NADH in H2O (buffer). (B) 1H NMR spectrum of AOB-CoA from [5-2H]AB-CoA and NADH in H2O. (C) 2H decoupled 1H NMR spectrum of sample described in B. (D) 2H decoupled 1H NMR spectrum of AOB-CoA from AB-CoA and [4R-2H]NAD2H in H2O. (E) 2H decoupled 1H NMR spectrum of AOB-CoA from AB-CoA and NADH in 2H2O. 1H coupling patterns are indicated above the signals. Signals resulting from 2H labeled isotopomers of AOB-CoA are indicated. For numbering, see Fig. 4 legend.
The carbon connectivity of the 13C-labeled ring moiety was established by two-dimensional incredible natural abundance double-quantum coherence transfer experiment (INADEQUATE) and HMBC experiments and by a detailed analysis of 13C13C coupling constants (Table 1). INADEQUATE, HMBC, heteronuclear multiple quantum coherence, and TOCSY experiments give no less than 22 different homo- and heterocorrelations between 1H and 13C atoms of the cyclic nucleus, establishing the structure as AOB-CoA (Table 1 and Fig. 4). This structure has been proposed previously based on the UV/visible spectra and the chemical properties of the enzyme product (15, 21). Importantly, and as reported earlier, aerobic incubation of purified, nonaromatic products with ACMR in the absence of NADH leads to formation of aromatic 5-OH-AB-CoA (Fig. 6) (15). This demonstrates that, in the species investigated, the eneamine C(2) nitrogen has not been hydrolyzed. The compound is stable for several days at 7°C in KPi, pH 7.6. At higher temperatures and at different pH values, degradation yielding unidentified products was observed.
Figure 4.

Structure and carbon numbering of AOB-CoA (2-amino-5-oxocyclohex-1-ene-1-carboxyl-CoA).
Figure 6.

Pattern of label incorporation and transfer during the reactions catalyzed by ACMR. ACM and ACR are the monooxygenase and the reductase functionalities of ACMR. ■H, label originally present at position C(5) of the substrate AB-CoA; *H, label originating from [4R-2H]NAD2H; and ●H, label originating from solvent 2H2O. The dashed arrows indicate the steps involving reversal of hydrogenation and that of aromatization of the intermediate, which occur under limiting NADH concentrations (15, 21). For details of the first step catalyzed by the ACM functionality, see Fig. 7 legend.
To trace the origin of the hydrogen atoms in AOB-CoA, and to elucidate the mechanism of formation of the latter, ACMR was incubated with the labeled substrates: (a) [5-2H]AB-CoA (>85% 2H enrichment) and (b) [4R-2H]NAD2H (>90% 2H enrichment) and, further, with (c) unlabeled substrates in 2H2O buffer (99.9% 2H enrichment). The products were isolated, purified by HPLC, and analyzed by 1H and 2H NMR spectroscopy.
The 1H NMR spectrum of AOB-CoA obtained from [5-2H]2-aminobenzoyl-CoA (Fig. 3B) is identical to the spectrum of the nondeuterated sample (Fig. 3A) except for the signal of the hydrogens at C-6. As shown in Fig. 3B, the intensity of the singlet at 3.01 ppm is decreased approximately 3-fold as compared with unlabeled sample. On the other hand, an up-field-shifted satellite signal is observed that displays smaller line widths under 2H decoupling (Fig. 3C). The up-field shift of 22 ppb as well as the line-shape modulation by 2H decoupling are consistent with formation of [6-2H]AOB-CoA. In line with this, we observed a 2H NMR signal at 3 ppm, confirming the presence of the 2H atom at position 6 (Fig. 5A). From the relative intensities of the 1H NMR signals, a ratio of 1:1.4 was estimated for unlabeled AOB-CoA/[6-2H]AOB-CoA.
Figure 5.

Partial 2H NMR spectra of AOB-CoA. (A) Spectra obtained from [5-2H]AB-CoA (>85% 2H enrichment) and NADH in H2O. (B) Spectra from AB-CoA and [4R-2H]NAD2H (>90% 2H enrichment) in H2O. (C) Spectra from AB-CoA and NADH in 2H2O (99.9% 2H enrichment).
The 2H-decoupled 1H NMR spectrum of AOB-CoA obtained from the experiment with [4R-2H]NAD2H is shown in Fig. 3D. Notably, the signal at 2.42 ppm (hydrogen atoms at C-4) is a doublet, indicating scalar coupling to only one hydrogen atom at the adjacent C-3. Moreover, the intensity of the triplet signal at 2.55 ppm (hydrogens at C-3) also reflects the presence of only one 1H. The 2H NMR spectrum of the sample shows a signal at the chemical shift of H-3 (2.5 ppm) again compatible with deuteration at C-3 (Fig. 5B). In summary, it is evident that the deuterium atom from [4R-2H]NAD2H is transferred specifically to C-3 of AOB-CoA without significant loss by exchange with solvent protons.
The 2H-decoupled 1H NMR spectrum of AOB-CoA obtained from incubation in 2H2O is shown in Fig. 3E. The doublet signature of the signal at 2.55 ppm (hydrogens at C-3) is compatible with the presence of a single 1H at C-4. The relative intensities in the 1H NMR spectrum as well as the signal at 2.4 ppm in the 2H NMR spectrum (Fig. 5C) give additional evidence for incorporation of 2H at C-4 of AOB-CoA. Moreover, an up-field-shifted satellite signal for H-6 and a 2H-NMR signal at 3.0 ppm correspond to a partial deuteration at C-6. This is explained by unspecific hydrogen exchange because of keto-enol tautomerization during incubation in deuterated buffer resulting in [4,6-2H2]AOB-CoA. It should be pointed out that incorporation of solvent-borne label at position C-4 of isolated AOB-CoA does not occur by chemical exchange via keto/enol tautomerization subsequent to its formation. To establish this point, the half-lives of 1H → 2H exchange with solvent-borne 2H were estimated by 1H NMR for the positions C(4)—H2 and C(6)—H2 as 23 and 2 h at 37°C and as 300 and 25 h at 7°C, respectively (conditions: 19 mM KPi, pH 7.6, in 2H2O). Thus, chemical exchange at C(4)—H is much slower than at C(6)—H. Consequently, the finding of partial exchange at C(6)—H (Figs. 3E and 5C) provides the necessary control for the much slower exchange at C(4)—H.
DISCUSSION
The structure of AOB-CoA proposed earlier (21) (Fig. 4) is unambiguously confirmed in this work. The paths and the kinetic features leading to its formation have been dissected earlier (14, 15). They involve a monooxygenation and a hydrogenation catalyzed by two distinct functionalities, each containing a FAD cofactor. This structure provides the basis for the interpretation of the reactions catalyzed by ACMR, in particular, of the step(s) involving insertion of oxygen into the aromatic substrate. Fig. 6 summarizes all reactions involved as well as the positions and modes of introduction and migration of hydrogen label in AOB-CoA. First, we address the fate of the 2H label at C-5 of AB-CoA, shown in Figs. 6 and 7, mechanistically the most important feature: the results are unequivocal in showing migration of the label to position 6 of the cyclic nucleus. This occurs either concomitantly or, more probably, immediately after the primary step of oxygen addition. This can proceed by nucleophilic displacement at the distal oxygen of the flavin hydroperoxide catalyzed by the ACM functionality to yield (A) as shown in Fig. 7. The hydride shift then can ensue from (A) in a concerted process, as formulated originally for the famed NIH-shift mechanism, to yield the intermediate (B) directly (Fig. 7).
Figure 7.

Possible structures of the first intermediates formed upon reaction of the flavin-4a-hydroperoxide with the aromatic moiety of AB-CoA and mode of decay. A is formed by nucleophilic attack at the distal oxygen of the flavin hydroperoxide. It is shown to rearrange via shift of the C(5)—H as hydride to position C(6), and to yield B. This formulation is analogous to an NIH shift.
Alternate modes of label transfer to yield (B) can be excluded: abstraction of the activated C(5)—H as H+ would yield a carbanionic species (not shown), which, in turn, is equivalent to the aromatic product 5-OH-AB-CoA (Fig. 6). Formation of the latter would be irreversible, thus precluding incorporation of the label at position C(6). It is interesting that, within the limit of detection of 10–20% for 1H, migration occurs by 1,2-shift toward C-6 and not toward C-4. The occurrence of epoxides, which could be formed either directly or by cyclization of (A) at positions C-4 or C-6 (Fig. 7), cannot be substantiated or excluded by the present data. Whether or not such subspecies occur, the conclusion is inescapable that the monooxygenation process occurs by a migration of hydrogen to the adjacent carbon, most probably via a hydride shift, i.e., by what can be described as an NIH-shift mechanism. This variant has been considered at various occasions, e.g., by Strickland et al. (22), who did not find label migration with melilotate hydroxylase. As suggested by these authors, this failure could be due to the incorporation of oxygen into an ortho position of the activating group, which leads to obligatory loss of label. In a general context, and to our knowledge, there are no experimental data pertaining to flavin-dependent monooxygenases that would contradict an NIH-shift mechanism.
The success of the present study is due to the fact that the enzyme ACR functionality is set up to trap the intermediate, unsaturated eneamine-ketone by hydrogenating it via a Michael-like addition of probably a hydride, as shown in Fig. 6. Interestingly, at low concentrations of the trapping agents, i.e., when the availability of reduced flavin on the ACR moiety is low because of limiting NADH concentrations, trapping becomes inefficient and the eneamine intermediate rearomatizes to 5-OH-AB-CoA, probably by loss of one C(6)—H (Figs. 1C and 6). The labeling pattern, i.e., insertion of deuterium originating from [4R-2H]NAD2H (Fig. 6) into position C(3), is in full agreement with the proposed mechanism. It should be noted that there is no or only negligible loss of label in spite of the fact that the same first is transferred to the flavin position N(5), where it theoretically could exchange with solvent. Absence of exchange of label in flavin-dependent processes, however, has been documented amply in flavin enzymes, the case of d-amino acid oxidase being typical (23), and is taken as reflecting hindered accessibility of solvent to the active center. Incorporation of solvent-borne label (Fig. 6) into position C(4), in turn, is compatible with formulation of the enol form resulting from hydride addition to C(3), as shown. It is intriguing that nature has devised such an efficient trapping. The metabolism of aromatic substrates funnels these toward nonaromatic and acyclic species. This drive to dearomatize substrates probably is the selective force for this unusual event.
Furthermore, the point should be stressed that incubation of purified AOB-CoA with ACMR aerobically and in the absence of NADH leads to the aromatic 5-OH-AB-CoA. This is assumed to proceed by dehydrogenation (i.e., via reversal of the hydrogenation step; Fig. 6) (15, 21). This is a further important argument for the structural identity of AOB-CoA and, in particular, for the absence of hydrolysis of the eneamine under the experimental conditions used.
A survey of flavin-dependent monooxygenases acting on aromatic substrates reveals many mechanistic features in common with ACMR, suggesting the same basic mechanism. The results of analysis of product distribution in the reaction catalyzed by para-hydroxybenzoate hydroxylase by using various fluorinated substrate analogs also is in accordance with an NIH-type reaction (8). We thus propose that an NIH-shift-type mechanism may be of general relevance for flavin-mediated monooxygenations of aromatic compounds.
Acknowledgments
We thank A. Werner and F. Wendling for help in the preparation of the manuscript. This work was supported by grants from the Deutsche Forschungsgemeinschaft (Gh 2/6-2 and Schwerpunkt Neuartige Reaktionen und Katalysemechanismen bei anaeroben Mikroorganismen), the Fonds der Chemischen Industrie, and European Community Grant ERBCHRXCT930166.
ABBREVIATIONS
- ACMR
2-aminobenzoyl-CoA monooxygenase/reductase
- ACM
monooxygenase activity of ACMR, or enzyme lacking the reductase activity
- ACR
reductase activity of ACMR
- AB-CoA
2-aminobenzoyl-CoA
- AOB-CoA
CoA conjugate of 2-amino-5-oxocyclohex-1-ene-1-carboxylate
- INADEQUATE
incredible natural abundance double quantum coherence transfer experiment
- TOCSY
total correlation spectroscopy
- HMBC
heteronuclear multiple bond coherence
References
- 1.van Berkel W J H, Müller F. In: Chemistry and Biochemistry of Flavoenzymes. Müller F, editor. II. Boca Raton, FL: CRC; 1991. [Google Scholar]
- 2.Bruice T C. Israel J Chem. 1984;24:54–61. [Google Scholar]
- 3.Latham J, Walsh C. Ann N Y Acad Sci. 1986;471:208–216. doi: 10.1111/j.1749-6632.1986.tb48036.x. [DOI] [PubMed] [Google Scholar]
- 4.Eckstein J W, Hastings J W, Ghisla S. Biochemistry. 1993;32:404–411. doi: 10.1021/bi00053a004. [DOI] [PubMed] [Google Scholar]
- 5.Ortiz-Maldonado M, Ballou D P, Massey V. In: Flavins and Flavoproteins. Stevenson K J, Massey V, Williams C H Jr, editors. Calgary: Univ. of Calgary Press; 1997. pp. 323–326. [Google Scholar]
- 6.Bruice T C, Noar J B, Ball S S, Venkataram U V. J Amer Chem Soc. 1983;105:2452–2463. [Google Scholar]
- 7.Peelen S, Rietjens I M C M, van Berkel W J H, van Workum W A T, Vervoort J. Eur J Biochem. 1993;218:345–353. doi: 10.1111/j.1432-1033.1993.tb18383.x. [DOI] [PubMed] [Google Scholar]
- 8.Peelen S, Rietjens I M C M, Boersma M G, Vervoort J. Eur J Biochem. 1995;227:284–291. doi: 10.1111/j.1432-1033.1995.tb20386.x. [DOI] [PubMed] [Google Scholar]
- 9.Hufton S E, Jennings I G, Cotton R G H. Biochem J. 1995;311:353–366. doi: 10.1042/bj3110353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guroff G, Daly J W, Jerina D M, Renson J, Witkop B, Udenfriend S. Science. 1967;157:1524–1530. doi: 10.1126/science.157.3796.1524. [DOI] [PubMed] [Google Scholar]
- 11.Rahimtula A D, O’Brien P J, Seifried H E, Jerina D M. Eur J Biochem. 1978;89:133–141. doi: 10.1111/j.1432-1033.1978.tb20905.x. [DOI] [PubMed] [Google Scholar]
- 12.Vannelli T, Hooper A B. Biochemistry. 1995;34:11743–11749. doi: 10.1021/bi00037a011. [DOI] [PubMed] [Google Scholar]
- 13.Buder R, Fuchs G. Eur J Biochem. 1989;185:629–635. doi: 10.1111/j.1432-1033.1989.tb15159.x. [DOI] [PubMed] [Google Scholar]
- 14.Langkau B, Vock P, Massey V, Fuchs G, Ghisla S. Eur J Biochem. 1995;230:676–685. [PubMed] [Google Scholar]
- 15.Langkau B, Ghisla S. Eur J Biochem. 1995;230:686–697. doi: 10.1111/j.1432-1033.1995.0686h.x. [DOI] [PubMed] [Google Scholar]
- 16.Anders H J, Kaetzke A, Kämpfer P, Ludwig W, Fuchs G. Int J Syst Bacteriol. 1995;45:327–633. doi: 10.1099/00207713-45-2-327. [DOI] [PubMed] [Google Scholar]
- 17.Braun K, Gibson D T. Appl Environ Microbiol. 1984;48:102–107. doi: 10.1128/aem.48.1.102-107.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buder R, Ziegler K, Fuchs G, Langkau B, Ghisla S. Eur J Biochem. 1989;185:637–643. doi: 10.1111/j.1432-1033.1989.tb15160.x. [DOI] [PubMed] [Google Scholar]
- 19.Altenschmidt U, Oswald B, Fuchs G. J Bacteriol. 1991;173:5494–5501. doi: 10.1128/jb.173.17.5494-5501.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tourwé D, Van Binst G, De Graaf S A G, Pandit U K. Org Magn Reson. 1975;7:433–441. [Google Scholar]
- 21.Langkau B, Ghisla S, Buder R, Ziegler K, Fuchs G. Eur J Biochem. 1990;191:365–371. doi: 10.1111/j.1432-1033.1990.tb19131.x. [DOI] [PubMed] [Google Scholar]
- 22.Strickland S, Schopfer L M, Massey V. Biochemistry. 1975;14:2230–2235. doi: 10.1021/bi00681a029. [DOI] [PubMed] [Google Scholar]
- 23.Walsh C T, Schonbrunn A, Abeles R H. J Biol Chem. 1971;246:6855–6866. [PubMed] [Google Scholar]