

Abstract
Objective
We have previously reported the ability of a mesenchymal stem cell (MSC)-based serum-free culture system to expand human cord blood (CB) hematopoietic stem cells (HSC) along the myeloid pathway and simultaneously generate a CD7+CD34- population. In this study, we investigated the ability of the CD7+CD34- population to differentiate into natural killer and dendritic cells.
Materials and Methods
CB CD34+ cells were expanded over a MSC layer in serum-free medium supplemented with SCF, bFGF, LIF, and FL for 2 weeks. Cultured cells were harvested and CD7+CD34-Lin- cells sorted and plated for two additional weeks in either natural killer (NK)- or dendritic cell (DC)-inductive medium.
Results
culture of CD34+ cells for the first 2 weeks in this system resulted in expansion of the stem cell pool and the myeloid component of the graft, and also produced a 58 fold-increase in the CD7+CD34- cell population. When sorted CD7+CD34-Lin- cells were induced towards a NK phenotype, further expansion was observed during this time in culture, and differentiation was confirmed by cytotoxic activity and by flow cytometry, with cells displaying CD16 and CD56 in the absence of CD3. The generation of DC cells in culture was also verified by observing both the characteristic dendritic morphology and the dendritic phenotypes HLA-DRbrightCD123brightCD11c- and HLA-DRbrightCD11c+.
Conclusion
These results demonstrate the ability of an ex-vivo culture system to drive the expansion of human CB HSCs while promoting the immune maturation of the graft and the generation of DC and NK cells that could then be utilized for adoptive cancer cellular immunotherapy.
Keywords: Cord Blood, Expansion, Cell therapy, Transplantation, Natural Killer Cells, Dendritic Cells
Introduction
The use of umbilical cord blood (UCB) as a source of hematopoietic stem cells (HSC) for allogeneic stem cell transplantation has increased over the last 18 years to become a standard and safe alternative to bone marrow transplantation [1-3]. This procedure has extended the boundaries of HSC transplantation to patients who otherwise would not be suitable for this therapy [4-6] and offers considerable rational and clinical advantages over other allogeneic hematopoietic stem cell sources. [2, 3, 7]. Some of the advantages of using CB as a source of HSC are due to the prompt availability of frozen UCB units, the decreased risk of transmission of viruses, and the lower incidence and severity of acute graft-versus-host disease (GvHD). In addition, the relative absence of mature immune effector cells in cord blood permits the use of grafts with 1 to 2 HLA mismatches, increasing the probability of finding a compatible donor, especially amongst under-represented minorities [8]. Nevertheless, UCB transplantation has also been associated with some inherent limitations such as a greater risk of graft failure due to the inadequate number of stem/progenitor cells available per unit, as well as delayed immune and hematologic reconstitution, because of the higher prevalence of developmentally immature cells [9-14]. Furthermore, in the case of cancer relapse, CB transplantation shows a major disadvantage, since donor lymphocytes and/or dendritic cells are not available for subsequent immunotherapy. Thus, the development of methods to overcome these limitations would greatly improve the utility of CB as an alternative source of HSC for transplantation and would allow its widespread use in the clinic. We have previously reported that human MSC are able to effectively support the ex vivo expansion / maintenance of human UCB HSCs using a serum-free culture system [15]. Some of the major advantages of this culture system include the use of pre-established human allogeneic stromal layers, thus overcoming the limitations of using xenogeneic and/or transformed and immortalized human stromal cell lines, this culture system’s serum-free conditions, and its ability to expand CB cells along the myeloid pathway, while simultaneously generating a population expressing a marker of early lymphopoiesis, CD7+. Since several different investigators had reported the ability to generate NK, B, DCs and T cells [10, 16-22] from a CD7+CD34+ cell population from fresh CB and BM, in the present studies, we examined whether the CD7+CD34negative population obtained after CB expansion over MSC layers, in serum-free medium, also had lymphocytic differentiative potential. Specifically, we investigated the ability of the CD7+ population generated in our culture system to give rise to two of the major constituents of the immune system, mature NK cells and dendritic cells (DCs) that could ultimately be used in cellular immunotherapy.
Materials and Methods
Cord Blood cell isolation
CB samples were obtained from the Pediatric Stem Cell Transplant Program at Duke University Medical Center (Durham, NC, USA) after maternal donor consent. Samples were collected into sterile bags containing citrate-phosphate dextrose anticoagulant and diluted 1:3 in Iscove’s Modified Dulbecco’s Medium (IMDM; Gibco Laboratories, Grand Island, NY, USA) before separation of mononuclear cells (MNC). Low-density MNC were separated on a Ficoll-Histopaque density gradient centrifugation (1.077 g/mL; Sigma, St. Louis, MO, USA) and washed twice in IMDM. CBMNC from each donor were enriched for CD34+cells using the Direct CD34 Progenitor Isolation Kit (Miltenyi Biotec Inc. Auburn, CA, USA).
Human bone marrow Stro-1+ stroma layer cell cultures
Heparinized human bone marrow was obtained from healthy donors after informed consent. Low-density bone marrow mononuclear cells (BMMNC) were separated on a Ficoll-Histopaque density gradient centrifugation (1.077 g/mL; Sigma, St. Louis, MO, USA). For each donor, Stro-1+ cells were isolated magnetically. Briefly, BMMNC were incubated for 30 min at 4°C with Stro-1 antibody (R&D Systems, Minneapolis, MN), washed, incubated with Rat-Anti-mouse IgM beads for 15 min, and separated using a MiniMacs column (Miltenyi Biotec Inc. Auburn, CA, USA). Cells were cultured in gelatin-coated T25 flasks with Mesenchymal Stem Cell Basal Medium (MSCBM®; Poietics™, Cambrex Bioscience, Baltimore, MD, USA) supplemented with MSCGM SingleQuot® Kit. Stroma layers were cultured to confluence and then γ-irradiated with a 137Cs source as previously described [15, 23]
Ex vivo expansion of CD34+
CD34+cells were cultured in QBSF-60 serum-free medium with L-Glutamine (Quality Biological, Inc, Gaithersburg, MD, USA) supplemented with 100 ng/ml stem cell factor (SCF), 10 ng/ml leukemia inhibitor factor (LIF), 5 ng/ml basic fibroblast growth factor (bFGF) and 100 ng/ml Flt-3 ligand (FL) (all cytokines from PeproTech Inc., Rocky Hill, NJ, USA), on irradiated Stro-1+ stromal layers, at 37°C in a 5% (v/v) CO2 incubator. Every 3 days, half of the medium was replaced with fresh medium and half the cultures were harvested for the following analyses: cell count, viability using trypan blue stain 0.4% solution (Gibco Laboratories, Grand Island, NY, USA), and phenotype by flow cytometry.
Differentiation assays
NK Cells
Ex-vivo expanded CD34+ cells (as described above) were harvested at day 12 of culture and CD7+CD2-CD3-CD5-CD16-CD56-CD34- cells were sorted on a FACSVantage (Becton-Dickinson Immunocytometry Systems (BDIS), San Jose, CA, USA) and cultured in HAM’S F12 Medium (Gibco Laboratories, Grand Island, NY, USA) supplemented with 5 μM β-mercaptoethanol (βME) (Sigma, St. Louis, MO, USA), insulin, transferrin and selenium (ITS) (Sigma, St. Louis, MO, USA), 10ng/ml of SCF, interleukin-7 (IL-7), IL-15, FL and 1000 U/ml IL-2 (all cytokines from PeproTech Inc., Rocky Hill, NJ, USA), seeded on irradiated Stro-1+ stromal layers. Every 3 days half of the medium was changed and the cells counted and analyzed by flow cytometry (for the presence of CD3, CD16 and CD56). After 2 weeks in culture, cytotoxic assays (CytoTox96® Non Radioactive Cytototoxic Assay; Promega, Madison, WI, USA) were performed to demonstrate the cytolytic activity of this cell population.
Dendritic cells
CD34+ cells ex-vivo expanded for 12 days were harvested and sorted for CD7+ CD2-CD3-CD5-CD14-CD16-CD56-CD34- cells, on a FACSVantage. Sorted cells were then cultured in the absence of stroma in RPMI-1640 (Gibco Laboratories, Grand Island, NY, USA) supplemented with 5 μM β–mercaptoethanol (Sigma, St. Louis, MO, USA), 200U/ml of interleukin-4 (IL-4), 0.2U/ml of Granulocyte/Macrophage Colony-Stimulating Factor (GM-CSF) and 10 ng/ml FL (all cytokines from PeproTech Inc., Rocky Hill, NJ, USA). Every 3 days, cells were harvested and analyzed for proliferation, morphology, and phenotype (presence of CD1a, CD11c, CD83 and CD123); half of the medium was replaced with fresh supplemented medium. Morphological analysis was conducted after cytocentifugation onto slides and staining with Wright Giemsa.
Flow Cytometric Analysis
The phenotype of fresh and cultured cells was assessed by flow cytometry. Briefly, harvested cells were incubated with fluorescent monoclonal antibodies against CD1a, CD3, CD7, CD11c, CD16, CD34, CD56, CD83, CD123 and HLA-DR (Becton-Dickinson Immunocytometry Systems (BDIS), San Jose, CA, USA) for 15 minutes at room temperature. The cells were then washed in phosphate-buffered saline (PBS) 1% sodium azide (Sigma, St. Louis, MO, USA) and fixed with 4% paraformaldehyde. Isotype controls (Simultest control γ1/γ 1 and Simultest control γ 1/γ 2a) were included in every experiment to evaluate the unspecific binding. Samples were analyzed using a FACScan (Becton-Dickinson Immunocytometry Systems (BDIS), San Jose, CA, USA) with CellQuest analysis software (Becton Dickinson).
Cytotoxic assays
Cytototoxic activity of cultured cells was determined with a colorimetric assay that measures lactate dehydrogenase (LDH), a stable cytosolic enzyme that is released upon cell lysis. The conversion of a tetrazolium salt (INT) into a red formazan product by the released enzyme is then measured in a coupled enzymatic assay, in a plate reader at visible wavelength. In brief, target cells (T) (NK-sensitive cell line K562) were incubated with effector (E) cells, at different E:T ratios (10:1, 5:1, 2.5:1, 1.25:1) for 4h; after incubation, supernatants were collected and incubated for 30 minutes with the Substrate Mix, the reaction was then stopped by adding 1M acetic acid (Stop solution). Triplicates of each E:T ratio were performed. Spontaneous LDH release was measured by incubating the target cells in the absence of effector cells. Maximum LDH release was determined by adding Lysis solution (0.9 % (v/v) Triton X-100). The amount of LDH released was measured in a plate reader (Bio-Rad Model 3550 – UV) and the percent cytotoxicity was calculated as follows, for each E:T ratio:
Results
CD7+ cells obtained at day 9 in culture are derived from expansion and differentiation of a more primitive CD34+CD7+ cell population and not only the result of expansion of the pre-existing CD34-CD7+ population at day 0.
We have previously reported [15, 23] that CB-derived CD34+ cells could be expanded ex vivo in a MSC-based serum-free culture system containing SCF, FL, LIF and bFGF, differentiating primarily towards a myeloid phenotype, while maintaining a population of cells that expressed CD7, a marker of early lymphopoiesis. In this culture system, total CB CD34+ enriched cells expanded 124-358 fold, CD34+ cells increased by 35 fold, and CD34+CD38- cells by 48 fold by the end of culture. The total fold increase in clonogenic potential was 137.46±2.2 times that of the initial culture [15]. Although it was clear from these studies that a population of cells positive for CD7 was maintained in this culture system, we were unable to determine with certainty whether the population of CD7+ cells obtained after HSC expansion reflected the ability of our culture system to support expansion and differentiation of the more primitive CD34+CD7+ cell pool, or whether these CD7+ cells were derived from the expansion of the small number of pre-existing CD7+CD34- cells present at day 0.
In order to address this question , we started by expanding CB CD34+ cells under culture conditions identical to those previously described [15, 23] and analyzed the kinetics of expansion and differentiation of the CD7+CD34+ and CD7+CD34- cell populations each 3 days. As can be seen in Table I, during the first 3 days of culture, we first observed a significant increase in the CD7+CD34+ population from 4.10 ± 0.95% to 24.1 ± 5.12% (p<0.001), while no significant variation in the numbers of CD7+CD34- cells (12.3 ± 4.51% to 16.4 ± 2.60%) was observed. From day 3 to day 9, a decrease in CD34 expression was seen within the CD7+CD34+ cells. This population decreased from 24.1±5.12% to 9.92±1.70%, to give rise to a population of cells possessing a CD7+CD34- phenotype. In fact, at day 9, CD7+CD34- cells constituted 62.3 ± 6.79% (5.76×106±0.65×106) of the total cells present in culture (Table I), corresponding to a 58-fold increase in the CD7+34- population. The expanded CD7+CD34- obtained in culture can be further differentiated into cell types that can be used for cellular immunotherapy.
Table I.
Flow cytometric analysis of CB-CD34+enriched cells. Relative percentage of CD34 and CD7 cells with time in culture (data are presented mean percentage ± SEM, n=5)
| Day | CD34+ 7-(%) | CD34+7+ (%) | CD7+ 34- (%) |
|---|---|---|---|
| 0 | 79.4 ± 6.81 | 4.10 ± 0.95 | 12.3 ± 4.51 |
| 3 | 71.9 ± 7.60 | 24.1 ± 5.12 | 16.4 ± 2.60 |
| 6 | 51.7 ± 4.48 | 19.5 ± 4.56 | 47.7 ± 3.71 |
| 9 | 30.1 ± 1.27 | 9.92 ± 1.79 | 62.3 ± 6.79 |
In order to investigate whether the CD7+34- population obtained after expansion in culture could be further differentiated into functionally mature NK cells and dendritic cells (DCs) that could ultimately be used in cellular immunotherapy, we sorted respectively, CD7+ CD2,CD3,CD5,CD16,CD34,CD56 negative cells or CD7+CD2-CD3-CD5-CD14-CD16-CD56-CD34- cells, at day 12 of culture and replated these cells in specific media inductive of NK or DC differentiation.
CD7+CD34- obtained in culture are able to differentiate into NK Cells
Cells were sorted from the initial culture system at day 12 based on CD7 positivity and CD2,CD3,CD5,CD16,CD34,CD56 negativity, and plated over new stromal layers and cultured for 12 additional days, in the presence of media inductive of NK cell differentiation, as described in the Materials and Methods section. Every three days cultures were evaluated for cell expansion, and phenotypic analysis; a cytotoxic assay was performed on the last day of culture. Between day 0 and day 3 of culture, a 2-fold increase in total cell numbers was observed, increasing from 1.28×106±0.45×106 cells to 2.84×106±1.42×106 cells (n=5); no further increase was found between day 3 and day 9, with the cell count reaching a plateau at approximately 1.10×106±0.23×106. (Figure 1).
Figure 1.
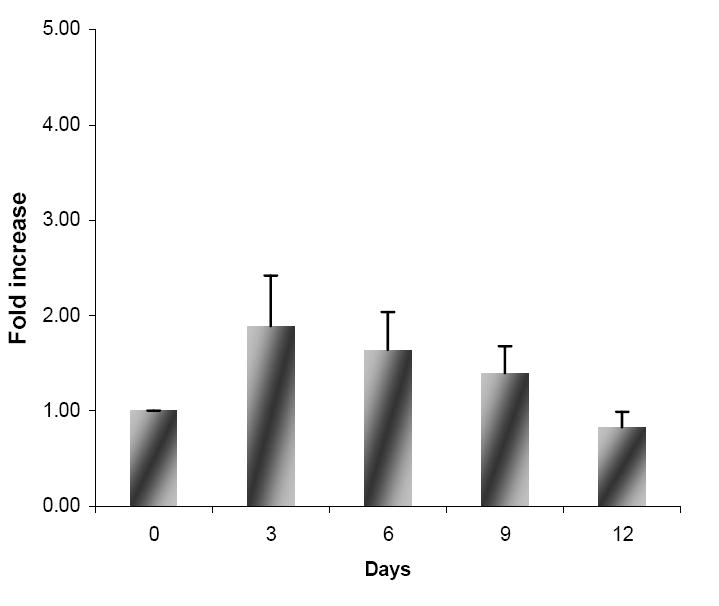
Fold increase of total cell number of viable sorted CD7+CD34-Lin--cells under NK-inductive conditions (data are expressed as mean fold expansion ± SEM (n=5)).
Immunophenotypic characterization showed that the cells acquired a phenotype consistent with that of NK cells as early as day 3 of culture, becoming CD16+CD3- and CD56+CD3-, as shown in Figure 2. On day 3, the percentages of CD16+CD3- cells and CD56+CD3- cells were 19.6 ± 9.31% and 11.9 ± 4.78%, respectively. At day 6, the percentage of cells exhibiting a phenotype of CD16+CD3- was 21.1±9.77, and at day 12, it was 24.8±12.0, while the percentages of CD56+CD3- cells were 24.5 ± 10.8% on day 6 and 25.3 ± 4.18% on day 12 (Table II). Of note is that although the culture conditions we employed were not ideal for T cell differentiation, a small CD3+ population was obtained between day 3 and day 12 in culture, 1.89 ± 0.25 % and 8.75 ± 0.48% respectively (Table II).
Figure 2.

Flow cytometric plots of cultured cells in NK inductive medium with time in culture. The dot plot on the top left represents the isotype control; the top 3 plots show the expression of CD16 and CD3; the bottom 3 plots display the expression of CD56 and CD3. These plots show day 3, 6, and 9 in culture.
Table II.
Flow cytometric analysis of cells cultured under NK inductive conditions in the presence of stroma and SCF, FL, IL-2, IL-7and IL-15 (each time point represents the mean percentage ± SEM, n= 5).
| Day | CD16+CD3- (%) | CD56+CD3- (%) | CD3+ (%) |
|---|---|---|---|
| 0 | 0.00 | 0.00 | 0.00 |
| 3 | 19.6 ± 9.31 | 11.9 ± 4.78 | 1.89 ± 0.25 |
| 6 | 21.1 ± 9.77 | 24.5 ± 10.8 | 3.44 ± 0.23 |
| 9 | 27.0 ± 12.1 | 28.5 ± 6.86 | 7.50 ± 0.73 |
| 12 | 24.8 ± 12.0 | 25.3 ± 4.18 | 8.75 ± 0.48 |
Cytotoxic activity of CD7+CD34--derived NK cells
In order to investigate whether CD7+CD34--derived NK cells were functional, we harvested these cells after 12 days of culture and tested them as effector cells against NK-sensitive targets (K562) as described in the material and methods section. CD7+CD34-–derived NK cells were confirmed to be functional as shown by the cytotoxic activity that increased in an E:T ratio-dependent fashion. As shown in Figure 3 at an E:T ratio of 1.25:1 the cytotoxicity was 2.70 ± 0.47 and increased to 19.7 ± 0.47 at an E:T ratio of 10:1 showing that CD7+CD34--derived NK cells effectively killed K562 targets.
Figure 3.
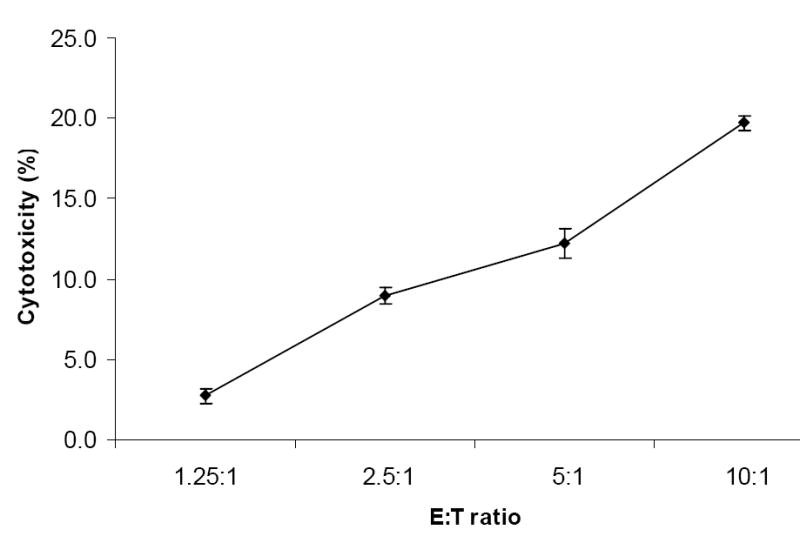
Cytolytic activity of CD7+CD34--derived NK cells. Cytotoxic activity was analyzed by colorimetric assay. Cultured cells were incubated with K562 cells for 4 hours at 37°C, the supernatants were collected and absorbance was read at 490nm (data are presented as mean percentage of triplicates ± SEM, n=3).
Expansion and characterization of CD7+CD34-- derived Dendritic cells
In order to investigate whether CD7+CD2-CD3-CD5-CD14-CD16-CD56-CD34- cells also had the potential to differentiate into dendritic cells, we cultured these cells in the presence of IL-4, GM-CSF and FL and found that a 1.8 fold increase in total cell numbers occurred during the 12 days in culture. These cultures were started with 0.93×106 ± 0.09×106 cells; at day 3, the total number of cells in culture increased to 1.72×106 ± 0.18×106, and at day 9 1.15×106 ± 0.07×106 cells were still present (Figure 4).
Fig 4.
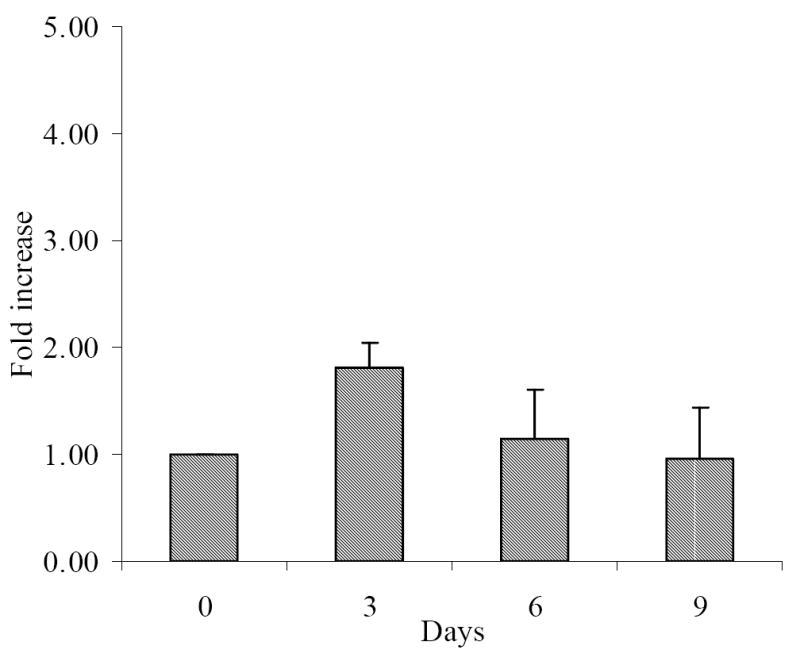
Fold increase of viable cells under dendritic cell inductive conditions after cell sorting (data are presented as mean fold increase ± SEM, n=3)
Differentiation towards a dendritic phenotype was assessed by performing flow cytometry on the cultured cells by gating for lineage negativity (CD3-, CD14-, CD16-, CD19-, CD20- and CD56-), and high expression of HLA-DR. This double gated population was then evaluated for the expression of CD11c and CD123. Using this 3-color assay, we were able to separate two populations: HLA-DRbrightCD123brightCD11c-, indicative of a plasmacytoid dendritic subset and a HLA-DRbrightCD11c+, a myeloid dendritic subset. Differentiation towards a plasmacytoid phenotype was initially seen at day 3 with 4.22 ± 0.94% of the cells displaying a CD123+HLA-DR+ phenotype. This number increased to 18.6±0.75 by day 6 of culture, and by day 9, 38.5 ± 4.22% of cells had differentiated into CD123+HLA-DR+ cells. Differentiation towards a myeloid dendritic subset was achieved more rapidly, with 8.08 ± 0.34% of the cells possessing a CD11c+HLA-DR+ phenotype at day 3. This population increased to 14.3± 1.03% at day 6 of culture, and by day 9, 20.6 ± 1.63% of the cells were CD11c+HLA-DR+ (Table III). Furthermore, these cells exhibited a phenotype associated with maturation of dendritic cells, expression since CD1a and CD83 positive cells increased progressively during the time in culture. By day 9, 10% of the total cells were CD1a and CD83 positive (Table III).
Table III.
Expression of the dendritic cell markers CD1a, CD83, CD11c, CD123 and HLA-DR in CD7+CD34-- derived DC during culture (data are expressed as mean percentage ± SEM, n=3).
| Day | CD123+HLA-DR+ (%) |
CD11c+ HLA-DR+ (%) |
CD1a
(%) |
CD83
(%) |
|---|---|---|---|---|
| 0 | 0.00 | 0.00 | 0.00 | 0.00 |
| 3 | 4.22 ± 0.94 | 8.08 ± 0.34 | 2.90 ± 0.35 | 0.57 ± 0.35 |
| 6 | 18.6 ± 0.74 | 14.3 ± 1.03 | 7.55 ± 2.51 | 7.53 ± 0.63 |
| 9 | 38.5 ± 4.22 | 20.6 ± 1.63 | 9.26 ± 2.98 | 10.7 ± 0.85 |
Despite the typical dendritic cell phenotype determined by flow cytometry, we wished to further confirm the generation of these cells by performing cytospins for evaluation of cell morphology. As can be seen in Fig 5, these cells displayed the typical dendritic cell morphology, characterized by an irregular form, extending long and thin processes in many directions from the cell body [24].
Figure 5.
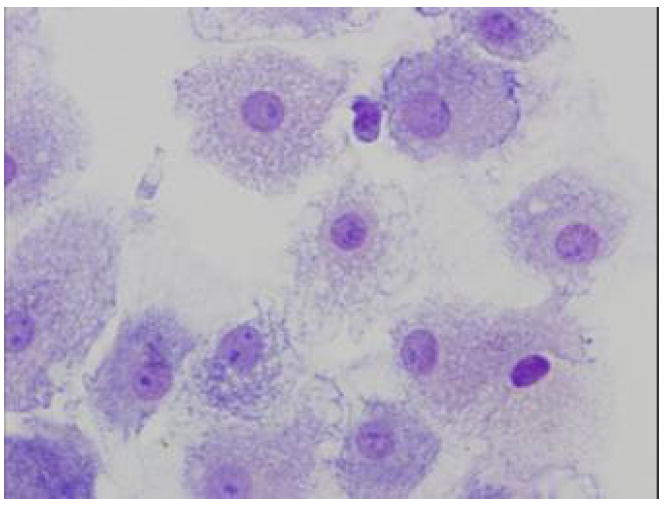
Cytospin preparation from cell cultures in dendritic inductive media, at day 12 showing large cells with a typical dendritic morphology.
Discussion
The use of CB as a viable alternative to marrow and PB transplantation, in adults who lack a matched related or unrelated donor, is limited by the very intrinsic biologic nature of cord blood. The inadequate number of primitive HSC and the predominance of immature immune cells lead to delayed engraftment and abnormal immune reconstitution, potentially resulting in higher mortality post-transplantation than with HSC from adult sources.
Thus, the main focus for CB transplantation continues to be the pediatric patient, since a very high percentage of adults referred for UBC transplantation are ineligible for the procedure based on the recommended cell dose for their given body weight [20].
In order to overcome this shortcoming, several different approaches have been put forward to increase the number of total mononuclear cells available for transplantation into patients [11]. The use of multiple cords has been quite successful, but it remains uncertain whether this approach will provide the patient with a normal and prompt immune reconstitution or if it will be able to minimize the relapse of the primary disease. Another successful approach relies on the ex-vivo expansion of whole CB unit prior to transplantation or the combination of a partially expanded unit with the unmanipulated fraction. However, it seems that most of the available ex-vivo expansion methods have failed to improve engraftment due to expansion of the more mature cells instead of the primitive HSC, alteration in stem cell homing, cell cycling, and even induction of apoptosis. [20]. Also, it has been reported that although ex-vivo expansion of CB does not seem to impair T-cell development, it does appear to decrease dendritic cell differentiation, contributing to altered immune function post-transplantation.
Thus, the development of efficient methods that could allow CB manipulation ex-vivo in a way that promoted expansion of primitive HSC without losing grafting ability and simultaneously allowed maturation of the immune cellular component of the graft would make this already valuable source of HSC available to a wider range of transplant applications. The use of all-trans retinoic acid, epigenic modification, and copper chelation [25-28] are all novel methods that may prove useful to allow expansion of the primitive pool of HSC in CB without inducing engraftment defects.
We have previously reported the development of a stroma-based serum-free culture system [15, 23] in which co-cultivation of CB CD34+ enriched cells with stroma in a cytokine cocktail (SCF, FL, bFGF and LIF) resulted in a 48-fold increase of the CD34+CD38− cell population with a total fold increase in clonogenic potential of 137.46 ± 2.2 at day 24. Kinetic analysis of this culture system showed that the presence of stroma was the primary contributing factor for the observed decrease of cell death [29]. Although the overall differentiative potential of the cells in culture was biased towards the myeloid lineage, we also observed an increase in the percentage of CD7+ cells [15]. Since the CD7+ subpopulation of CD34+CD38- CB cells has been reported as a clonogenic, primitive and progenitor population with NK, B and DC potential [17, 30, 31], we hypothesized that the stroma-based serum-free culture system reported by da Silva et al. would be able to expand primitive HSC while simultaneously leading the expanded cells into the immunologic maturity needed for successful transplantation. Furthermore, since there is no mechanism at this time to use cord blood for adoptive cancer cellular immunotherapy after CB transplantation, in the current work, we investigated the potential of the CD7+CD34- cells obtained during HSC expansion, to give rise to NK and DC populations. We focused our attention on the CD7+CD34- population, since concomitant with the HSC expansion phase, a 58-fold increase was seen in the CD7+CD34- cells, corresponding to 62.3 ± 6.79 % of the total cells within 12 days of culture. In order to evaluate the differentiative potential of the CD7+CD34- cells that were not yet committed towards the NK or DC lineage, CD7+CD34-cells obtained in culture were depleted of mature cells by cell sorting with lineages markers, before culturing under conditions inductive of NK or DC differentiation. At day 9 of culture in the presence of stroma and a cocktail of cytokines known to induce NK differentiation (SCF, IL-2, IL-7, IL-15 and FL), we obtained a population, 28.5 ± 6.86 which were CD56+CD3- cells, a phenotype consistent with that reported for NK cells [32]. In order to demonstrate the functionality of the differentiated cells, the cytolytic activity of this cell population was also evaluated, and proved to be similar to freshly isolated NK cells from cord blood and bone marrow [33], as well as NK cells derived from a CD34+CD7+ population [30]. Although the culture conditions were not ideal to generate T cells, by day 12, 8.75±0.48% of the cells in culture expressed CD3, a marker of T cells.
The dendritic cell population has been the target of several studies because of their relevance in the immune system as potent initiators of adaptive immune response by presenting antigens to T cells and regulating the production of cytokines [16, 34]. In the present study, we were also able to differentiate lineage depleted CD7+CD34- into dendritic cells. After 9 days of culture, we were able to observe two distinct dendritic populations: a plasmacytoid-like (CD123+HLA-DR+; 38.5±4.22%) and a myeloid-like (CD11c+HLA-DR+; 20.6±1.63%) population. Since dendritic cells can also be evaluated for their maturation/activation based on surface markers such as CD83 [35], we also examined the expression of this marker in our cultures. After 9 days in culture, 10.7±0.85% of the cells expressed a phenotype of mature/activated DC (CD83+). Furthermore, morphologic analysis of these cells showed an irregular form with long and thin processes characteristic of dendritic cells. In conclusion, we have developed an in vitro culture system that fulfills the essential criteria for paving the way to the more widespread usage of cord blood for HSC transplantation. This culture system reproducibly drives the ex vivo expansion of human cord blood HSCs while simultaneously promoting the maturation of the immune cellular component of the graft by generating, at earlier time points, a known lympho-progenitor population CD34+ CD7+, and at later time points, considerable numbers of CD34-CD7+ cells that are able to differentiate into NK cells and DC and could thus be used for adoptive cancer cellular immunotherapy after CB transplantation.
Acknowledgments
This work was supported by grants HL70566, HL73737, NAG9-1340 from National Institutes of Health, and NASA. Ana M. Frias is a recipient of a scholarship SFRH/BD/8970/2002 awarded from Fundação para a Ciência e a Tecnologia, Portugal
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ayello J, van de Ven C, Fortino W, et al. Characterization of cord blood natural killer and lymphokine activated killer lymphocytes following ex vivo cellular engineering. Biol Blood Marrow Transplant. 2006;12:608–622. doi: 10.1016/j.bbmt.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 2.Schoemans H, Theunissen K, Maertens J, Boogaerts M, Verfaillie C, Wagner J. Adult umbilical cord blood transplantation: a comprehensive review. Bone marrow transplantation. 2006;38:83–93. doi: 10.1038/sj.bmt.1705403. [DOI] [PubMed] [Google Scholar]
- 3.Rocha V, Gluckman E. Clinical use of umbilical cord blood hematopoietic stem cells. Biol Blood Marrow Transplant. 2006;12:34–41. doi: 10.1016/j.bbmt.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 4.Copelan EA. Hematopoietic stem-cell transplantation. The New England journal of medicine. 2006;354:1813–1826. doi: 10.1056/NEJMra052638. [DOI] [PubMed] [Google Scholar]
- 5.Wagner JE, Kernan NA, Steinbuch M, Broxmeyer HE, Gluckman E. Allogeneic sibling umbilical-cord-blood transplantation in children with malignant and non-malignant disease. Lancet. 1995;346:214–219. doi: 10.1016/s0140-6736(95)91268-1. [DOI] [PubMed] [Google Scholar]
- 6.Broxmeyer HE. Cord blood as an alternative source for stem and progenitor cell transplantation. Current opinion in pediatrics. 1995;7:47–55. doi: 10.1097/00008480-199502000-00010. [DOI] [PubMed] [Google Scholar]
- 7.Rubinstein P. Why cord blood? Human immunology. 2006;67:398–404. doi: 10.1016/j.humimm.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 8.Gluckman E, Rocha V. Donor selection for unrelated cord blood transplants. Current opinion in immunology. 2006;18:565–570. doi: 10.1016/j.coi.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 9.Bracho F, van de Ven C, Areman E, et al. A comparison of ex vivo expanded DCs derived from cord blood and mobilized adult peripheral blood plastic-adherent mononuclear cells: decreased alloreactivity of cord blood DCs. Cytotherapy. 2003;5:349–361. doi: 10.1080/14653240310003017. [DOI] [PubMed] [Google Scholar]
- 10.Satwani P, Ayello J, Ven C, et al. Immaturity of IL-18 gene expression and protein production in cord blood (CB) versus peripheral blood (PB) mononuclear cells and differential effects in natural killer (NK) cell development and function. British journal of haematology. 2005;130:284–292. doi: 10.1111/j.1365-2141.2005.05592.x. [DOI] [PubMed] [Google Scholar]
- 11.Robinson S, Niu T, de Lima M, et al. Ex vivo expansion of umbilical cord blood. Cytotherapy. 2005;7:243–250. doi: 10.1080/14653240510027172. [DOI] [PubMed] [Google Scholar]
- 12.Robinson SN, Ng J, Niu T, et al. Superior ex vivo cord blood expansion following co-culture with bone marrow-derived mesenchymal stem cells. Bone marrow transplantation. 2006;37:359–366. doi: 10.1038/sj.bmt.1705258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Introna M, Franceschetti M, Ciocca A, et al. Rapid and massive expansion of cord blood-derived cytokine-induced killer cells: an innovative proposal for the treatment of leukemia relapse after cord blood transplantation. Bone marrow transplantation. 2006;38:621–627. doi: 10.1038/sj.bmt.1705503. [DOI] [PubMed] [Google Scholar]
- 14.Kim YJ, Stringfield TM, Chen Y, Broxmeyer HE. Modulation of cord blood CD8+ T-cell effector differentiation by TGF-beta1 and 4-1BB costimulation. Blood. 2005;105:274–281. doi: 10.1182/blood-2003-12-4343. [DOI] [PubMed] [Google Scholar]
- 15.da Silva CL, Goncalves R, Crapnell KB, Cabral JM, Zanjani ED, Almeida-Porada G. A human stromal-based serum-free culture system supports the ex vivo expansion/maintenance of bone marrow and cord blood hematopoietic stem/progenitor cells. Exp Hematol. 2005;33:828–835. doi: 10.1016/j.exphem.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 16.Gluckman JC, Canque B. Dendritic Cell and Lymphoid Progenitors in Cord Blood. In: Broxmeyer HA, editor. Cord Blood:Biology, Immunology, Banking, and Clinical Transplantation. 2004. pp. 163–186. [Google Scholar]
- 17.Miller JS, Alley KA, McGlave P. Differentiation of natural killer (NK) cells from human primitive marrow progenitors in a stroma-based long-term culture system: identification of a CD34+7+ NK progenitor. Blood. 1994;83:2594–2601. [PubMed] [Google Scholar]
- 18.Kobari L, Giarratana MC, Gluckman JC, Douay L, Rosenzwajg M. Ex vivo expansion does not alter the capacity of umbilical cord blood CD34+ cells to generate functional T lymphocytes and dendritic cells. Stem cells (Dayton, Ohio) 2006;24:2150–2157. doi: 10.1634/stemcells.2006-0102. [DOI] [PubMed] [Google Scholar]
- 19.Kobari L, Pflumio F, Giarratana M, et al. In vitro and in vivo evidence for the long-term multilineage (myeloid, B, NK, and T) reconstitution capacity of ex vivo expanded human CD34(+) cord blood cells. Experimental hematology. 2000;28:1470–1480. doi: 10.1016/s0301-472x(00)00557-9. [DOI] [PubMed] [Google Scholar]
- 20.Hofmeister CC, Zhang J, Knight KL, Le P, Stiff PJ. Ex vivo expansion of umbilical cord blood stem cells for transplantation: growing knowledge from the hematopoietic niche. Bone marrow transplantation. 2007;39:11–23. doi: 10.1038/sj.bmt.1705538. [DOI] [PubMed] [Google Scholar]
- 21.Galy A, Travis M, Cen D, Chen B. Human T, B, natural killer, and dendritic cells arise from a common bone marrow progenitor cell subset. Immunity. 1995;3:459–473. doi: 10.1016/1074-7613(95)90175-2. [DOI] [PubMed] [Google Scholar]
- 22.Borras FE, Matthews NC, Lowdell MW, Navarrete CV. Identification of both myeloid CD11c+ and lymphoid CD11c- dendritic cell subsets in cord blood. British journal of haematology. 2001;113:925–931. doi: 10.1046/j.1365-2141.2001.02840.x. [DOI] [PubMed] [Google Scholar]
- 23.Goncalves R, Lobato da Silva C, Cabral JM, Zanjani ED, Almeida-Porada G. A Stro-1(+) human universal stromal feeder layer to expand/maintain human bone marrow hematopoietic stem/progenitor cells in a serum-free culture system. Experimental hematology. 2006;34:1353–1359. doi: 10.1016/j.exphem.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 24.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 25.Araki H, Mahmud N, Milhem M, et al. Expansion of human umbilical cord blood SCID-repopulating cells using chromatin-modifying agents. Experimental hematology. 2006;34:140–149. doi: 10.1016/j.exphem.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 26.Peled T, Glukhman E, Hasson N, et al. Chelatable cellular copper modulates differentiation and self-renewal of cord blood-derived hematopoietic progenitor cells. Experimental hematology. 2005;33:1092–1100. doi: 10.1016/j.exphem.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 27.Peled T, Mandel J, Goudsmid RN, et al. Pre-clinical development of cord blood-derived progenitor cell graft expanded ex vivo with cytokines and the polyamine copper chelator tetraethylenepentamine. Cytotherapy. 2004;6:344–355. doi: 10.1080/14653240410004916. [DOI] [PubMed] [Google Scholar]
- 28.Milhem M, Mahmud N, Lavelle D, et al. Modification of hematopoietic stem cell fate by 5aza 2’deoxycytidine and trichostatin A. Blood. 2004;103:4102–4110. doi: 10.1182/blood-2003-07-2431. [DOI] [PubMed] [Google Scholar]
- 29.Goncalves R, da Silva CL, Ferreira BS, et al. Kinetic analysis of the ex vivo expansion of human hematopoietic stem/progenitor cells. Biotechnology letters. 2006;28:335–340. doi: 10.1007/s10529-005-5932-2. [DOI] [PubMed] [Google Scholar]
- 30.Hao QL, Zhu J, Price MA, Payne KJ, Barsky LW, Crooks GM. Identification of a novel, human multilymphoid progenitor in cord blood. Blood. 2001;97:3683. doi: 10.1182/blood.v97.12.3683. [DOI] [PubMed] [Google Scholar]
- 31.Haddad R, Guardiola P, Izac B, et al. Molecular characterization of early human T/NK and B-lymphoid progenitor cells in umbilical cord blood. Blood. 2004;104:3918–3926. doi: 10.1182/blood-2004-05-1845. [DOI] [PubMed] [Google Scholar]
- 32.Robertson MJ, Ritz J. Biology and clinical relevance of human natural killer cells. Blood. 1990;76:2421–2438. [PubMed] [Google Scholar]
- 33.Gardiner CM, Meara AO, Reen DJ. Differential cytotoxicity of cord blood and bone marrow-derived natural killer cells. Blood. 1998;91:207–213. [PubMed] [Google Scholar]
- 34.Navarrete CV, Gómez J, Borras FE. Cord Blood Dendritic Cells. In: Broxmeyer HA, editor. Cord Blood:Biology, Immunology, Banking, and Clinical Transplantation. 2004. [Google Scholar]
- 35.Lechmann M, Berchtold S, Hauber J, Steinkasserer A. CD83 on dendritic cells: more than just a marker for maturation. Trends Immunol. 2002;23:273–275. doi: 10.1016/s1471-4906(02)02214-7. [DOI] [PubMed] [Google Scholar]