

Abstract
Expression of vascular endothelial growth factor (VEGF), an endothelial cell-specific mitogen and a potent angiogenic factor, is upregulated by a variety of factors including hypoxia, growth factors and hormones. In the adrenal cortex, regulation of VEGF expression by the pituitary hormone ACTH ensures the maintenance of the organ vasculature. We have previously shown that ACTH evokes a rapid and transient increase in VEGF mRNA levels in primary adrenocortical cells through transcription-independent mechanisms. We further demonstrated that the zinc finger RNA-binding protein Tis11b destabilizes VEGF mRNA through its 3′-untranslated region (3′-UTR) and that Tis11b is involved in the decay phase of ACTH-induced VEGF mRNA expression. In the present study, we attempted to determine the mechanisms underlying ACTH-elicited increase in VEGF mRNA levels in adrenocortical cells. We show that ACTH triggers an increase in the levels of the mRNA-stabilizing protein HuR in the cytoplasm and a concomitant decrease in the levels of HuR in the nucleus. This process is accompanied by an increased association of HuR with the nucleocytoplasmic shuttling protein pp32, indicating that ACTH induces HuR translocation from the nuclear to the cytoplasmic compartment. Leptomycin B, a specific inhibitor of CRM1-dependent nuclear export of pp32, significantly reduced ACTH-induced VEGF mRNA levels. Furthermore, RNAi-mediated depletion of HuR in adrenocortical cells abrogated ACTH-induced VEGF mRNA expression. Finally, we show that Tis11b and HuR exert antagonistic effects on VEGF 3′-UTR in vitro. Although both proteins could bind simultaneously on VEGF 3′-UTR, Tis11b markedly decreases HuR-binding to this RNA sequence. Altogether, these results suggest that the RNA-stabilizing protein HuR is instrumental to ACTH-induced expression of VEGF mRNA and that the nuclear export of HuR is a rate-limiting step in this process. HuR appears to transiently stabilize VEGF transcripts following ACTH stimulation of adrenocortical cells, and Tis11b appears to subsequently trigger their degradation.
Keywords: 3' Untranslated Regions; Active Transport, Cell Nucleus; Adrenal Cortex; cytology; drug effects; Adrenocorticotropic Hormone; pharmacology; Animals; Antigens, Surface; genetics; metabolism; Base Sequence; Cattle; Cells, Cultured; DNA-Binding Proteins; genetics; metabolism; Fatty Acids, Unsaturated; pharmacology; Gene Expression Regulation; drug effects; RNA Interference; RNA Stability; drug effects; RNA, Messenger; genetics; metabolism; RNA-Binding Proteins; genetics; metabolism; Retroviridae Proteins; metabolism; Tetradecanoylphorbol Acetate; pharmacology; Vascular Endothelial Growth Factor A; genetics
Keywords: VEGF, Tis11b, BRF1, ZFP36L1, HuR, mRNA stability, Adrenal Cortex, ACTH
INTRODUCTION
Vascular endothelial growth factor (VEGF) is the major angiogenic cytokine contributing to the regulation of physiological and pathological angiogenesis (1, 2). Its expression levels in mammalian cells are very tightly regulated. In fact, a 50% reduction of VEGF expression during embryogenesis, as observed in VEGF +/− heterozygous mice, results in lethality (3, 4). Reciprocally, overexpression of VEGF in adult tissues leads to the formation of angiomas and perturbs organ development (5). VEGF expression is regulated by a number of environmental factors including hypoxia, growth factors and several hormones (1, 6). With most of these factors, different levels of regulation are observed. First, VEGF transcription is activated by the binding of a number of transcription factors onto specific response elements located in the promoter of the VEGF gene. These include HIF-1α and AP-1 for the hypoxic response (7, 8), Sp1 and AP-2 for the growth factor response (9, 10). Second, VEGF mRNA stability is regulated in response to some of these effectors through the binding of stabilizing and destabilizing proteins to AU-rich elements (AREs) located in the 3′-untranslated region (3′-UTR) of VEGF mRNA (11–14). Third, translation of VEGF mRNA into protein is also a controlled mechanism implying several alternative initiation codons (15). The choice of the preferential initiation codon is modified under hypoxic conditions (16) or under oncogenic transformation (17). These different levels of regulation may cooperate to amplify the effects of such or such regulator. For example, hypoxia-induced stimulation of VEGF expression results from both HIF-1α-mediated stimulation of transcription (7) and HuR-mediated stabilization of VEGF mRNA (11).
HuR is a ubiquitously expressed member of the ELAV (embryonic-lethal abnormal visual in Drosophila melanogaster) family of RNA binding proteins, which also comprises the neuron-specific proteins HuD, HuC and Hel-N1 (18, 19). The protein products of all four genes bind with high affinity and specificity to AREs in a variety of mRNAs, such as those encoding VEGF, c-fos, c-jun, IL-3, TNFα, and GM-CSF, and are believed to increase mRNA stability, mRNA translation or both (20, 21). While the precise mechanisms regulating HuR function in mRNA stabilisation are poorly understood, increasing evidence indicates that HuR function is intimately linked to its subcellular localization (22–24). Indeed, HuR contains three classical RNA-binding domains [RNA Recognition Motifs (RRM)]: the first two (RRM1 and RRM2) have been implicated in ARE recognition whereas the third (RRM3) has been suggested to bind the poly A tail of targets mRNA (25). The region located between RRM2 and RRM3, known as the hinge region, is essential for HuR subcellular localization. This region contains a shuttling domain, HNS (for HuR Nucleocytoplasmic Sequence) which is involved in the interaction of HuR with the acidic phosphoproteins ligands, pp32, SETα/β and APRIL (26). Like HuR, these proteins are primarily nucleoplasmic but shuttle between the nucleus and the cytoplasm. pp32 and APRIL contain domains homologous to nuclear export signals (NES) known to interact with CRM1 (Chromosomal region maintenance 1), the nuclear export receptor for the HIV-1 Rev protein. It has been shown recently that pp32 and APRIL mediate CRM1-dependent export of HuR in response to heat shock (27).
We have previously reported that adrenocorticotropin (ACTH) rapidly and transiently stimulates VEGF expression by primary cultures of bovine adrenocortical cells (28). Interestingly, we observed that this increase in VEGF mRNA was transcription-independent as it was still observed in the presence of DRB, a potent transcription inhibitor (28). We then characterized Tis11b (TPA-inducible-sequence 11b) as a zinc finger RNA-destabilizing protein whose synthesis is also induced by ACTH, although slightly later than that of VEGF (29). We hypothesized that Tis11b might play a role in the control of VEGF mRNA stability. Indeed, we recently characterized the interaction between Tis11b and VEGF mRNA 3′-UTR, and identified a 75 bp sequence in this 3′-UTR, containing two AU-rich elements, as the Tis11b binding-element (TBE) (30). We could show that Tis11b binding to TBE induces destabilization of VEGF mRNA, resulting in a reduction of VEGF mRNA half-life from 130 min down to 60 min. In the present work, we attempted to identify the mechanism by which ACTH induces VEGF mRNA. We identified HuR as a potential regulator and could establish that ACTH induces a rapid increase of HuR levels in the cytoplasm and a concomitant decrease of HuR levels in the nucleus. Moreover, co-immunoprecipitations experiments revealed that ACTH induces a molecular complex between the nucleocytoplasmic shuttling protein pp32 and HuR in the cytoplasm, suggesting that pp32 is involved in HuR accumulation in this compartment. Finally, as the Tis11b- and HuR-binding sites are quite close to each other on the VEGF mRNA 3′-UTR, we wondered whether these two proteins might antagonize each other in the control of VEGF mRNA stability.
RESULTS
Regulation of VEGF, Tis11b and HuR mRNAs expression by ACTH
We have previously shown that ACTH rapidly and transiently induces the expression of VEGF in primary cultures of adrenocortical cells (28). Moreover, we recently observed that Tis11b, through its mRNA destabilizing-activity, is involved in the decay phase of ACTH-elicited VEGF mRNA levels (30). However, the mechanisms involved in the induction phase of VEGF mRNA remained to be determined. As this induction was still observed in the presence of the transcription inhibitor DRB (28), we postulated that stabilization of VEGF mRNA might be involved in this process. Since the ELAV family members HuC, HuD and Hel-N1 were reported as neural-specific RNA-stabilizing proteins, we focused on HuR, the ubiquitously expressed member of ELAV proteins. We first examined the possible involvement of HuR in ACTH-induced increase of VEGF mRNA. Fig. 1A illustrates an RT-PCR analysis of VEGF, Tis11b and HuR mRNA levels in adrenocortical cells stimulated with 10 nM ACTH for various periods of time ranging from 0 to 5 h. As previously reported (29), VEGF mRNA levels peaked after 2–3 h whereas the hormone induced the expression of Tis11b between 3 and 5 h of treatment (Fig. 1A and 1B). In contrast, HuR mRNA levels were unchanged over the period of ACTH stimulation (Fig. 1A and 1B). Northern blot analysis of HuR mRNA levels in ACTH-treated cells confirmed that they were very stable for up to 24 h of hormone treatment (maximal value reaching 137 ± 40% of mRNA levels measured in non-treated cells, n=3, data not shown).
Figure 1. Effect of ACTH on VEGF, Tis11b and HuR mRNAs expression.
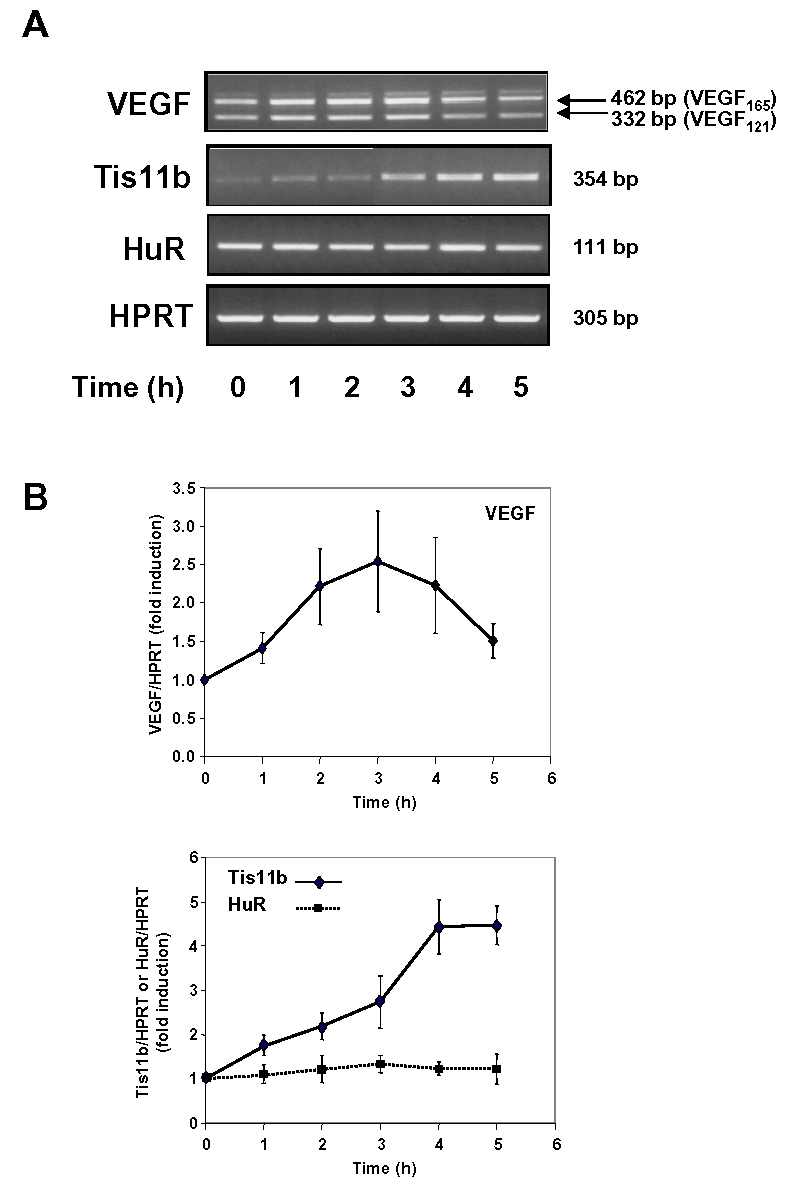
A, Representative ethidium bromide staining of VEGF, Tis11b and HuR mRNAs amplified by RT-PCR. Primary cultures of BAC cells were treated with 10 nM ACTH for the indicated periods of time. VEGF, Tis11b and HuR mRNA levels were then analyzed by RT-PCR as described in Materials and Methods. B, Quantitation of VEGF, Tis11b and HuR mRNAs levels of independent experiments (n=3 to 5). mRNA level values were normalized to HPRT mRNA levels and are expressed as fold induction over control values at time 0 (unstimulated cells).
ACTH does not affect total HuR protein content of BAC cells
The above results prompted us to examine whether ACTH could exert a post-transcriptional effect by acting on HuR mRNA translation. As shown in Fig. 2A and 2B, following exposure of BAC cells to 10 nM ACTH, no significant changes were observed in HuR protein content of whole cell extracts. In contrast, ACTH induced a progressive increase in Tis11b protein content (Fig. 2A and 2B). This increase was apparent as early as 1 h after stimulation and peaked after 5–6 h. Tis11b protein expression remained slightly elevated 24 h after ACTH treatment (data not shown).
Figure 2. Effect of ACTH on Tis11b and HuR protein levels in total cell extracts and subcellular fractions.
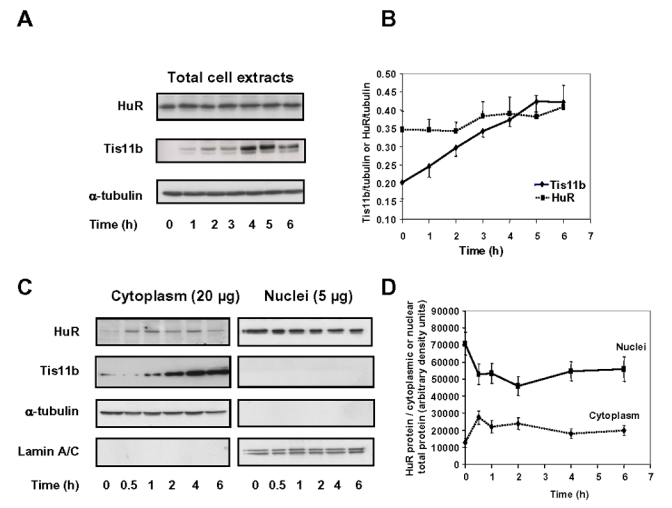
A, BAC cells were treated with 10 nM ACTH for the indicated periods of time. Tis11b and HuR protein levels of whole cell extracts (10 μg) were analysed by Western blot as outlined in Materials and Methods. The Western blot was subsequently probed with an anti-α-tubulin monoclonal antibody to assess equal loading of samples. B, Quantitation of HuR and Tis11b protein levels of total cell extracts in three independent experiments. Protein level values were normalized to α-tubulin protein levels. C, BAC cells were treated with 10 nM ACTH as indicated in (A). Nuclear (5 μg) and cytoplasmic (20 μg) fractions were prepared as described in Materials and Methods and subjected to Western blot analysis to monitor Tis11b and HuR expression. The same membranes were sequentially probed with antibodies recognizing cytoplasm- and nucleus-specific proteins (α-tubulin and lamin A/C, respectively) to assess the quality of the fractionation process and to check for equal protein loading. D, Cytoplasmic and nuclear HuR protein levels were normalized to α-tubulin and lamin protein levels respectively and are expressed as a fraction of total cytoplasmic or total nuclear protein content (n=2).
ACTH increases cytoplasmic levels of HuR
HuR appears predominantly located in the nucleus within all cell types examined so far (24). Moreover, HuR-induced stabilization of its mRNA targets is associated with an increase in cytoplasmic levels of HuR (24). To assess the subcellular distribution of HuR in BAC cells as well as the potential effect of ACTH on HuR localization, BAC cells were incubated with 10 nM ACTH for various periods of time ranging from 0 to 6 h, then subfractionated into nuclear and cytoplasmic fractions. As shown in Fig. 2C, HuR was weakly present in the cytoplasm (20 μg of cy toplasmic extracts) but was abundant in the nucleus (5 μg of nuclear extracts) of unstimulated cells. Quantitation of cytoplasmic and nuclear HuR revealed that ACTH treatment led to a 2 to 3-fold increase of cytoplasmic HuR as early as 30 min after stimulation and a concomitant decrease in nuclear HuR (Fig. 2D). Cytoplasmic HuR levels remained elevated for up to 3 h then decreased by 4 h of stimulation. In unstimulated cells, HuR was estimated to be about 5 to 6-fold more abundant in the nucleus. Hybridization of the same membranes with antibodies recognizing cytoplasm- and nucleus-specific proteins (α-tubulin and lamin A/C, respectively) allowed us to verify that nuclear proteins did not leak into the cytoplasmic fractions during cell fractionation (Fig. 2C). To determine the subcellular localization of Tis11b in BAC cells, the same blots were probed with affinity-purified polyclonal anti-Tis11b antibodies (30). As shown on Fig. 2C, Tis11b was exclusively detected in the cytoplasmic fractions. Upon prolonged exposure of the membrane (15 min), a very weak Tis11b signal could be detected in the nuclear fractions (data not shown). Exposure of BAC cells to ACTH induced a robust increase in Tis11b protein levels in the cytoplasm (Fig. 2C), which was correlated to the induction of Tis11b that we observed in whole cell extracts (Fig. 2A).
Altogether, these results indicate that ACTH treatment of BAC cells leads to a rapid increase of HuR protein content in the cytoplasm with a concomitant decrease of HuR protein content in the nucleus and to a delayed increase of Tis11b protein in the cytoplasm.
HuR interacts with the nucleocytoplasmic shuttling protein pp32 in BAC cells
In light of the observation that increases in cytoplasmic HuR are largely due to the CRM1-dependent export of nuclear HuR which involves complexes between HuR and the nucleocytoplasmic shuttling proteins pp32 and/or APRIL (26, 27), we further tested whether these proteins might contribute to ACTH-induced accumulation of HuR in the cytoplasm using co-immunoprecipitation experiments. As we could not detect APRIL in whole BAC cell extracts (data not shown), we focused on pp32. Cytoplasmic and nuclear BAC extracts were exposed to anti-pp32 antibody. Probing the immunoprecipitates with anti-HuR antibody revealed that pp32 is associated with HuR in the cytoplasm and that ACTH treatment increases this association (Fig 3A and 3B). The same blot was probed with anti-pp32 antibody to check for pp32 immunoprecipitation. In the nucleus, HuR-pp32 complexes are less abundant as compared to the cytoplasm and are not significantly altered by hormone treatment (Fig 3A and 3B). These results suggest that ACTH-induced increase in cytoplasmic HuR levels involves pp32-mediated HuR delocalization from the nucleus to the cytoplasm of BAC cells.
Figure 3. Co-immunoprecipitation of HuR from BAC cytoplasmic and nuclear extracts using anti-pp32 antibodies.
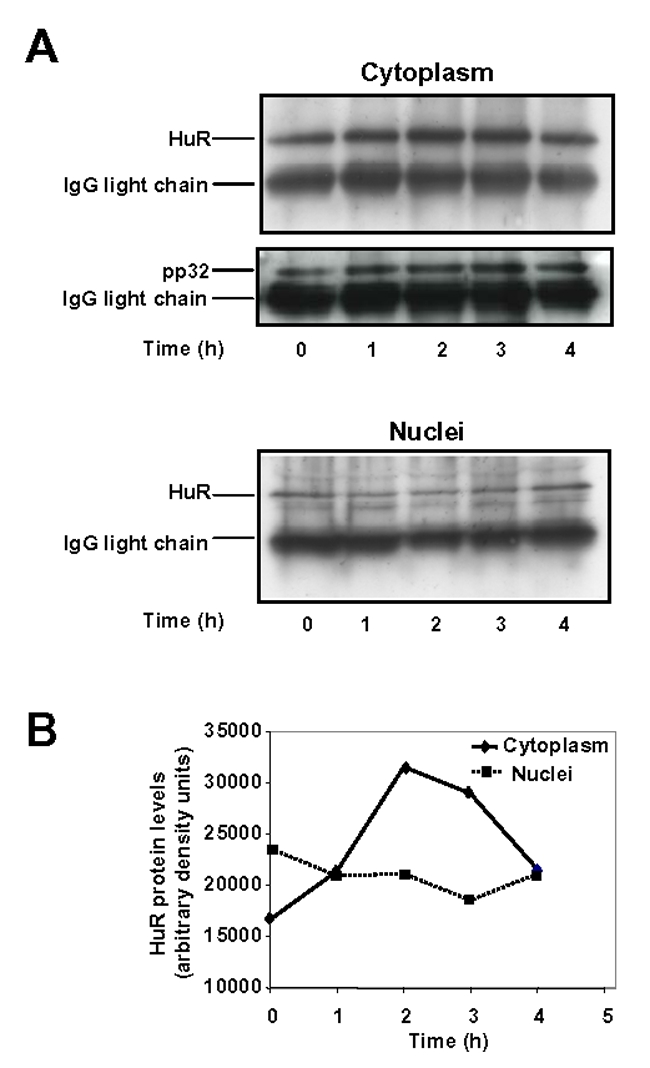
A, BAC cells were treated with 10 nM ACTH for the indicated periods of time. Cytoplasmic (250 μg protein) and nuclear fractions (70 μg protein) were immunoprecipitated with anti-human pp32 antibodies as mentioned in Materials and Methods. Precipitates were electrophoresed on a 12% denaturing gel, transferred to PVDF membrane, and probed with HuR or pp32 polyclonal antibodies. B, Quantitation of HuR levels in cytoplasmic and nuclear extracts. HuR protein levels were normalized to the IgG light chain bands.
Leptomycin B prevents ACTH-induced expression of VEGF mRNA
CRM1-dependent nucleocytoplasmic shuttling of NES-containing proteins such as pp32 has been shown to be impaired by leptomycin B (LMB), a compound which covalently modifies a critical cystein residue in CRM1 and thereby prevents CRM1-pp32 interaction via NES (31). Moreover, CRM1 has been shown to be instrumental in the nuclear export of HuR (26, 27). Our observation that ACTH treatment stimulates the formation of HuR-pp32 complexes in the cytoplasm prompted us to examine the effect of LMB on ACTH-induced increase in VEGF mRNA levels. BAC cells were stimulated with ACTH in the absence or in the presence of leptomycin B (LMB). As shown in Fig. 4A, the hormone-induced increase of HuR in the cytoplasm was abolished at all time points of stimulation in the presence of LMB (5 ng/ml).
Figure 4. Effect of Leptomycin B (LMB) on ACTH-induced increase in VEGF mRNA levels.
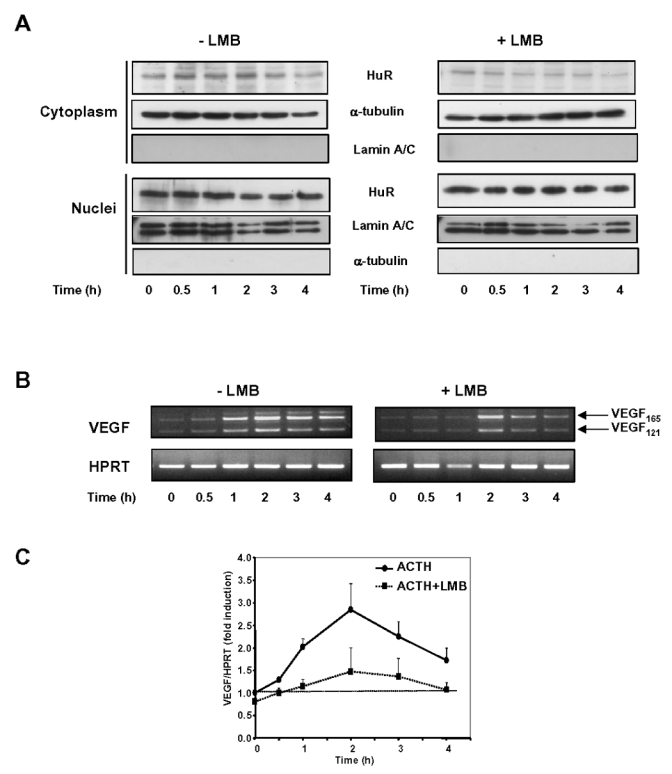
A, BAC cells were treated with 10 nM ACTH for the indicated periods of time, in the presence or in the absence of Leptomycin B (5 ng/ml). When used, LMB was added 15 min before ACTH treatment. Cytoplasmic (30 μg) and nuclear fractions (5 μg) were subjected to Western blot analysis of HuR. B, Representative ethidium bromide staining of VEGF mRNA levels amplified by RT-PCR in BAC cells stimulated with ACTH in the presence or in the absence of LMB (5 ng/ml). C, Quantitation of VEGF mRNA levels in BAC cells stimulated with ACTH in the presence or in the absence of LMB, expressed as fold induction of mRNA levels at time 0 (unstimulated cells). Each point is the mean value from two separate experiments.
RT-PCR analysis of VEGF mRNA levels revealed that co-treatment of BAC cells with ACTH and LMB prevented the increase in VEGF mRNA expression elicited by ACTH (Fig. 4B). Quantitation of two independent experiments showed that after 2 h of ACTH treatment, VEGF mRNA levels were induced up to 290 ± 60% of control levels and that, in the presence of LMB, maximal induction reached only 147 ± 60% of control levels. LMB also had a discrete effect by itself on the basal expression of VEGF mRNA.
HuR knockdown inhibits ACTH-induced increase in VEGF mRNA levels
To provide further evidence of the involvement of HuR in the regulation of VEGF mRNA expression by ACTH, we used RNA interference to more selectively target HuR. Using a specific siRNA targeting exon 2 in HuR gene, we showed by both RT-PCR and Western blotting that this siRNA was effective in decreasing HuR mRNA and protein levels in BAC cells after 48 h of treatment (Fig. 5A and 5B, time point 0). Quantitation of HuR mRNA level in three independent experiments revealed that HuR gene expression was knocked down by 70 to 85% (data not shown). In these experiments, HuR protein levels were barely detectable upon prolonged exposure of the Western blot (data not shown). BAC cells pre-treated either in the presence of a negative control siRNA or in the presence of HuR siRNA were further stimulated with 10 nM ACTH and VEGF mRNA levels were analyzed by RT-PCR. In this particular experiment, cells pre-treated with negative control siRNA displayed a 2.5-fold increase in VEGF mRNA expression after 4 h of exposure to ACTH (Fig. 5A and 5C). In contrast, HuR siRNA impaired ACTH-elicited up-regulation of VEGF mRNA levels (Fig. 5A and 5C). Moreover, silencing HuR expression led to a substantial reduction in basal VEGF mRNA expression down to 39 ± 19% of controls (n=3, data not shown).
Figure 5. Effect of HuR repression by RNAi on ACTH-induced increase in VEGF mRNA levels.
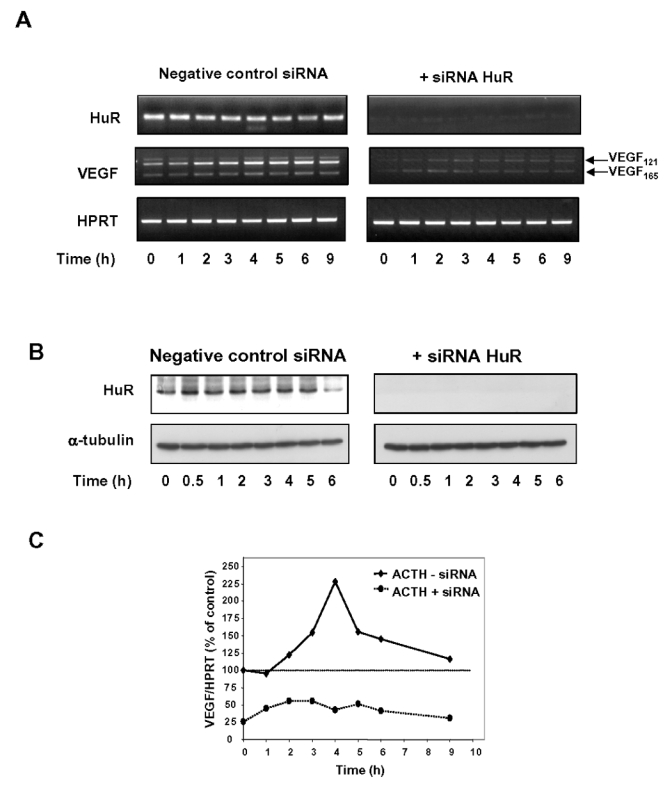
A, BAC cells were transfected either with HuR specific siRNA or a negative control siRNA as described in Materials and Methods. Forty-eight hours later, culture medium was changed and cells were treated for the indicated periods of time with or without 10 nM ACTH. At each time point of stimulation, total RNA was isolated and RT-PCR analysis was performed to determine HuR, VEGF or HPRT mRNA expression levels. B, Western blot analysis of HuR protein levels in whole-cell extracts (10 μg), showing that HuR siRNA was effective in knocking down HuR protein levels. In this particular experiment, blots for HuR were exposed for 2 min. Despite prolonged exposure of the membrane (15 min), HuR was barely detectable in protein extracts derived from HuR siRNA-treated cells. C, Quantitation of the RT-PCR experiment represented in (A), in which HuR repression was evaluated to 85 %. Results obtained from three independent experiments revealed that ACTH-induced increase in VEGF mRNA levels was altered to a lesser extent when HuR repression was about 70% (data not shown).
Antagonistic effects of Tis11b and HuR on VEGF mRNA
A previous study using co-transfections of Tis11b (pCMV-Tis11 b) and firefly luciferase cDNA cloned upstream of VEGF mRNA 3′-UTR (Luc-V3′ construct) allowed us to demonstrate that VEGF mRNA 3′-UTR confers a Tis11 b-mediated decrease in reporter gene activity, which was closely related to a decrease in luciferase transcript stability (30). Furthermore, similar experiments using co-transfections of HuR and Luc-V3′ construct revealed that HuR-induced increase in luciferase activity reflects an increase in luciferase transcript stability (32). Because HuR and Tis11b appear to have antagonistic activity on VEGF mRNA and since the Tis11b binding site on VEGF mRNA 3′-UTR is very close to a recently described HuR binding site (32), we investigated whether these two proteins could compete with each other to regulate VEGF mRNA expression. We first determined the effect of expressing each protein alone on luciferase activity. COS7 cells were transiently transfected with increasing doses of either pCMV-Tis11b or pCMV-HuR plasmids and a fixed concentration of Luc-V3′. Fig. 6A shows that luciferase activity was decreased in a dose-dependent manner by Tis11b, a statistically significant decrease being observed with a plasmid amount as low as 0.5 ng (62.5 ± 7.9%, of controls, p<0.01, n=3). In contrast, HuR expression led to an increase in the reporter gene activity over a narrow window of the cotransfected HuR plasmid (Fig. 6B). A statistically significant increase of luciferase activity was observed with 0.1 ng of pCMV-HuR (156.8 ± 13.8% of controls, p<0.001, n=3, Fig. 6A). Experiments were then performed to determine the competitive effects of HuR and Tis11b co-expression on VEGF mRNA 3′-UTR. As shown in Fig. 6C, the luciferase activity recorded following co-transfection of 1 ng of pCMV-Tis11b and 0.1 ng of pCMV-HuR was significantly lower than the luciferase activity obtained with 0.1 ng of pCMV-HuR alone (81.8 ± 2.3% as compared to 152.2 ± 6.3% of controls, respectively, p<0.001, n=3). This inhibitory effect of Tis11b on HuR-induced luciferase activity was observed with a concentration of pCMV-Tis11b as low as 0.2 ng. Conversely, the luciferase activity measured following co-transfection of 1 ng of pCMV-Tis11b and 0.1 ng of pCMV-HuR was significantly higher than the one measured with pCMV-Tis11b alone (81.8 ± 2.3% as compared to 48.6 ± 2.1% of controls, respectively, p<0.01, n=3). Western blot analysis of cell extracts from these transfection experiments revealed that COS7 cells constitutively express HuR protein but not Tis11b protein (Fig. 6C, lower panel). As expected, transfection of either 0.05 ng or 0.1 ng of pCMV-HuR plasmid led to an increased expression of HuR. By contrast, Tis11b expression was barely detectable with 0.2 ng of pCMV-Tis11b plasmid but clearly detected after transfection of 1 ng of plasmid.
Figure 6. Antagonistic effects of Tis11b and HuR in the regulation of VEGF mRNA stability.
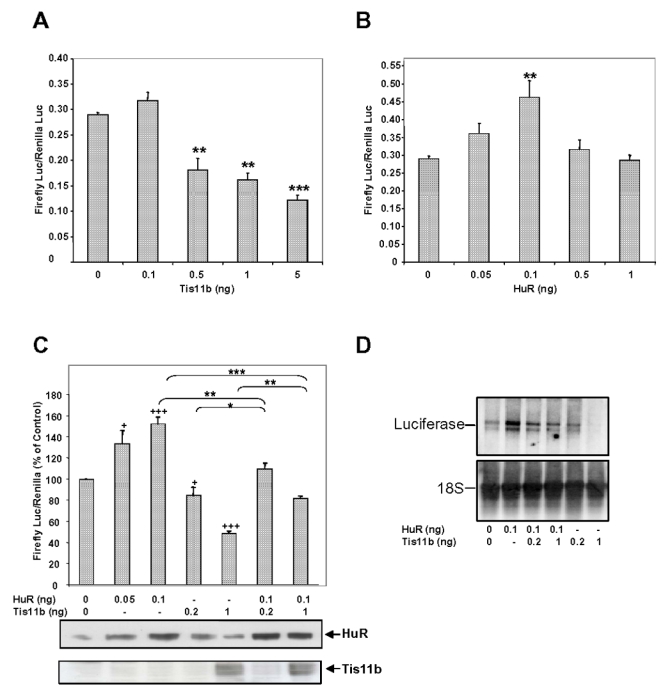
COS7 cells were transfected as outlined in Materials and Methods. The pLuc-V3′ construct contains the full-length 3′-UTR of the rat VEGF mRNA (2201 bp) (30). A, Dose-dependent effect of Tis11b on pLuc-V3′ reporter gene activity. Results are expressed as relative light units of firefly luciferase activity over relative light units of renilla luciferase activity. B, Dose-dependent effect of HuR on pLuc-V3′ reporter gene activity. C, Effect of Tis11b and HuR co-expression on pLuc-V3′ reporter gene activity. The dose giving the maximal effect of HuR on luciferase activity (0.1 ng) was used with 0.2 or 1 ng of Tis11 b to perform competition studies. Transfections were performed in triplicate and values are means ± S.E from three independent experiments. +, +++, significantly different from control (0 ng of pCMV-Tis11b or pCMV-HuR) with p<0.05 and p<0.001, respectively. There was a statistically significant decrease in luciferase activity for (HuR 0.1 ng + Tis11b 1 ng) compared to HuR 0.1 ng (***, p<0.001), as well as a statistically significant increase in luciferase activity for (HuR 0.1 ng + Tis11b 1 ng) compared to Tis11b 1 ng (**, p<0.01). There was a statistically significant decrease in luciferase activity for (HuR 0.1 ng + Tis11b 0.2 ng) compared to HuR 0.1 ng (**, p<0.01), as well as a statistically significant increase in luciferase activity for (HuR 0.1 ng + Tis11b 0.2 ng) compared to Tis11b 0.2 ng (**, p<0.05). In the lower panel, COS7 cell extracts (10 μg) were immunoblotted using anti-HuR or anti-Tis11 b antibodies to check for HuR and Tis11 b protein expression in transfected cells. HuR and Tis11b are indicated with arrows. In this cell line, Tis11b is indetectable at basal levels. D, Effect of Tis11b and HuR co-expression on pLuc-V3′ reporter gene mRNA levels. COS7 cell total RNA (20 μg) were analyzed by Northern blot as indicated in Materials and Methods.
We next investigated the effect of HuR and Tis11b co-expression on Luc-V3′ mRNA levels (Fig. 6D). Northern blot analysis of COS7 total RNA revealed that transfection of 0.1 ng of pCMV-HuR increased Luc-V3′ mRNA levels similarly to the reporter gene activity. Co-transfection of 0.2 and 1 ng of pCMV-Tis11b with 0.1 ng of pCMV-HuR abolished HuR-induced increase in Luc-V3′ mRNA. Finally, Luc-V3′ mRNA level was decreased in a dose-dependent manner by Tis11b. Altogether, these results suggest that Tis11b and HuR could antagonize each other to regulate VEGF mRNA expression through its 3′-UTR.
Tis11b and HuR compete for binding to the 3′UTR of VEGF mRNA
Because Tis11b and HuR appeared to antagonize each other in the regulation of VEGF mRNA expression, experiments were performed to determine whether these proteins could compete with each other for the binding to VEGF 3′-UTR. Tis11b-expressing bacterial extract was mixed with increasing amounts of purified GST-HuR fusion protein and purified GST-HuR was mixed with increasing amounts of Tis11b-expressing bacterial extract. These mixtures were exposed to UV light in cross-linking assays with VEGF 3′-UTR RNA probe. As shown in Fig. 7B, a covalent ribonucleoprotein complex with an apparent molecular weight of 38 kDa was detected when Tis11b-expressing bacterial extract was incubated with VEGF 3′-UTR (lane 2). This complex was not observed in the presence of control bacterial extracts (lane 1). On the other hand, a covalent ribonucleoprotein complex with an apparent molecular weight of 64 kDa was detected when GST-HuR fusion protein was incubated with VEGF 3′-UTR (lane 6). When 1 μg of GST-HuR was add ed to 2 μg of Tis11b, both complexes of 38 and 64 kDa were detected, indicating that Tis11b and HuR could bind simultaneously to the VEGF 3′-UTR RNA probe (lane 3). Increasing the amount of GST-HuR in the presence of 2 μg of Tis11b resulted in a slight decrease in the intensity of Tis11b-RNA complex and a corresponding increase in HuR-RNA complex (lanes 4 and 5). Increasing the amount of Tis11b in the presence of 1 μg of HuR result ed in a marked decrease in the intensity of HuR-RNA complex (lanes 7–9). Unexpectedly, no increase in Tis11b-RNA complex was observed, suggesting that optimal Tis11b binding to VEGF 3′-UTR requires additional factors which remain to be identified. Indeed, we could observe an increase of Tis11b-binding to VEGF 3′-UTR over a narrow window of Tis11b doses ranging from 1 to 3 μg, followed by a decrease of Tis11b-binding to VEGF 3′-UTR at higher doses (data not shown). Quantitation of 2–4 independent experiments shows that increasing doses of Tis11b markedly decreased HuR binding to VEGF 3′-UTR (to 23.2 ± 9.5% of HuR binding in the absence of Tis11 b, Fig 7C, n=3).
Figure 7. Binding of HuR and Tis11 b to VEGF 3′-UTR RNA.
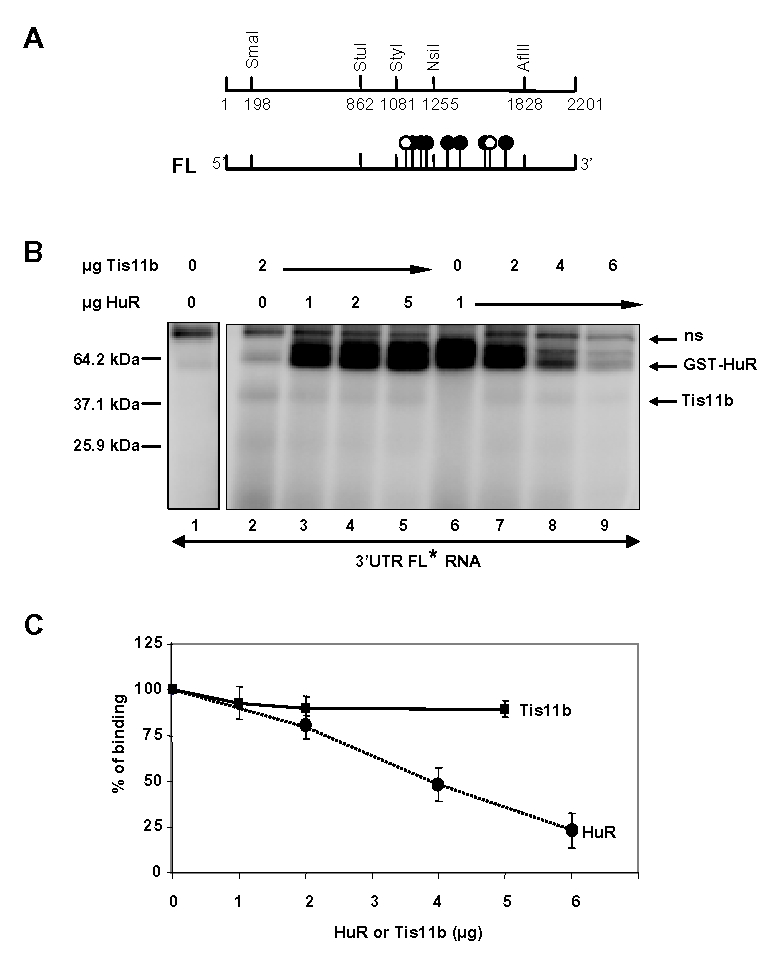
A, Restriction map of the 2201 bp-long 3′-UTR of VEGF mRNA. Tis11b binding element (TBE) is located between nucleotides 1161 and 1235 (30). The 40-bp functional HuR binding site is located between nucleotides 1285 and 1325 (32). Flags with white circles represent the nonameric ARE motifs UUAUUUA(A/U)(A/U) and those with black circles represent the pentameric motif AUUUA. B, VEGF full length 3′UTR RNA probe was mixed either with bacterial cell extracts containing Tis11b (2 μg) and increasing doses of purified GST-HuR (0, 1, 2, or 5 μg) or purified GST-HuR (1μg) and increasing doses of Tis11b (0, 2, 4, or 6 μg). The reaction mixtures were treated with UV radiation and were analyzed by electrophoresis as outlined in Materials and Methods. The positions of migration of the HuR and Tis11 b RNA-protein complexes are indicated with arrows, (ns) represents a non-specific band observed with the control bacterial extract. C, Quantitation of Tis11 b binding to VEGF 3′-UTR in the presence of increasing doses of HuR (squares) and of HuR binding to VEGF 3′-UTR in the presence of increasing doses of Tis11b (circles) in 2 to 4 independent experiments. 100 % represents either the binding of Tis11b in the absence of HuR or the binding of HuR in the absence of Tis11 b.
DISCUSSION
Vascular endothelial growth factor VEGF has been shown in mammals to be a key mediator of angiogenesis in such diverse physiological and pathological processes as embryogenesis, female oestrus cycle, diabetic retinopathy and tumor development (1). The expression of the VEGF gene is controlled at many levels including transcription (10), mRNA stability through the binding of regulatory proteins to the 3′ untranslated region (33) and mRNA translation via IRES sequences located in the 5′ untranslated region (15). Post-transcriptional regulation of VEGF mRNA is a key control point in VEGF expression under hypoxic conditions. Indeed, hypoxia-mediated increase of VEGF mRNA levels is in a large part due to an increase in VEGF mRNA half-life following its stabilization by the RNA-binding protein HuR (11).
Transcriptional regulation of VEGF expression by hormones in endocrine tissues, including endometrium, ovaries and adrenal cortex, has been extensively studied (34–39). In contrast, the possible involvement of post-transcriptional regulation in hormonally-regulated expression of VEGF has received much less attention to date. We have previously shown that the trophic hormone ACTH triggers a rapid and transient increase in VEGF mRNA levels in adrenocortical cells via transcription-independent mechanisms (28). We could further demonstrate that the decay phase of ACTH-induced VEGF mRNA levels involves the RNA-destabilizing protein Tis11b (30). In the current study, we aimed at investigating the molecular mechanisms of ACTH-elicited increase in VEGF mRNA levels. Four major conclusions can be drawn from the present work: (1) ACTH induces a rapid delocalization of the RNA-stabilizing protein HuR from the nucleus to the cytoplasm in adrenocortical cells without affecting total cellular HuR mRNA and protein levels. (2) Blocking nuclear export of NES-bearing proteins by leptomycin B impairs ACTH-induced expression of VEGF mRNA. (3) Silencing the expression of HuR by siRNA markedly inhibits ACTH-mediated induction of VEGF mRNA levels. (4) Tis11b and HuR exert an antagonistic action on VEGF mRNA in vitro. First, our finding that the levels of cytoplasmic and nuclear HuR are potently influenced by the trophic hormone ACTH provides a new insight into the regulation of HuR in adrenocortical cells, suggesting a connection between the ability of HuR to stabilize ACTH-induced labile mRNAs and the subcellular targeting of these mRNAs. Various other stimuli can shift the nucleo-cytoplasmic distribution of HuR. Cytoplasmic localization of HuR is associated with conditions of cellular stress including heat shock (40), UV irradiation (41), and amino acid starvation (42), as well as stimulation with LPS in the case of macrophages (43). HuR can bind to AU-rich regions in c-fos mRNA, a CU-rich region in c-jun mRNA and a U-rich domain in c-myc and VEGF mRNAs (11, 20, 44, 45), all of these genes being early-response genes to ACTH in the adrenal cortex (28, 46). Our observation that ACTH induces an increase in cytoplasmic levels of HuR suggests that HuR may play a role in stabilizing these labile mRNAs. Our results showing that (i) HuR co-immunoprecipitates with the nucleocytoplasmic shuttling protein pp32 in the cytoplasm of ACTH-stimulated cells and (ii) ACTH increases transiently the association of HuR with pp32 in the cytoplasm argue that the hormone triggers nuclear export of HuR to the cytoplasm. Interestingly, there is accumulating evidence that the nuclear-cytoplasmic localization of HuR is modulated by signal transduction pathways (47–51). ACTH is known to increase intracellular cAMP levels in cultured adrenocortical cells. We have previously shown that raising endogenous cAMP levels by forskolin was as potent as ACTH in eliciting an increase of VEGF mRNA (28). In the present study, stimulation of BAC cells with ACTH in the presence of H89 (10 μM), a potent and specific inhibitor of the protein kinase A, completely abolished the hormone-induced increase of HuR in the cytoplasm (data not shown). Moreover, stimulation of BAC cells with forskoline (10 μM) led to a substantial in crease of HuR levels in the cytoplasm (up to 165 % of HuR protein content of non-stimulated cells after 1–2 h of stimulation, data not shown). These observations suggest that cAMP and protein kinase A are involved in the hormone-induced delocalization of HuR to the cytoplasm. The potential targets of protein kinase A remain however to be determined in future studies. Since pp32 is a phosphoprotein (26), it is tempting to speculate that pp32 shuttling or its interaction with HuR could be regulated by phosphorylation. This hypothesis is worth testing since we failed to identify phosphorylated forms of HuR in response to ACTH (data not shown). Second, we observed that leptomycin B (LMB), a specific inhibitor of the nuclear export receptor CRM1 significantly impaired ACTH-induced increase in VEGF mRNA. Although it is possible that LMB inhibits other components of the nucleocytoplasmic trafficking machinery, we hypothesize that ACTH-induced association of pp32 and HuR most likely confers CRM1-dependent export on HuR. Therefore, disruption of the nuclear export of HuR protein partners by LMB might lead to retention of HuR in the nucleus, and thereby might impair ACTH-induced increase in VEGF mRNA levels. Indeed, we observed that LMB prevented the ACTH-mediated increase of HuR in the cytoplasm of BAC cells as well as the hormone-induced increase in VEGF mRNA levels, thus indicating that HuR export to the cytoplasm is instrumental in ACTH-induced VEGF mRNA.
Because BAC cells already have a high level of HuR, defining the functions of HuR in the cellular response to ACTH using the overexpression approach could lead to effects that might not reflect the true function of the endogenous protein. To clearly establish the role of HuR in ACTH-induced VEGF mRNA, we disrupted the expression of endogenous HuR in BAC cells using RNA interference (RNAi). Interestingly, RNAi-mediated depletion of HuR leads to a complete inhibition of hormone-induced expression of VEGF, indicating that HuR is a limiting factor in the induction phase of VEGF mRNAs by ACTH. HuR expression was significantly reduced (by 80 to 85 %) 48 h before ACTH treatment. In our RNAi experiments in which HuR expression was reduced by only 70%, inhibition of ACTH-elicited VEGF mRNA levels was not complete (data not shown), suggesting that low levels of HuR were sufficient to stabilize VEGF mRNA. These results, together with those we reported on the involvement of the mRNA-destabilizing protein Tis11b in the decay phase of ACTH-elicited VEGF mRNA levels, indicate that ACTH controls VEGF expression in adrenocortical cells mainly by post-transcriptional mechanisms. The pivotal role of HuR in the stabilisation of VEGF mRNA by hypoxia has been extensively studied (52). To our knowledge, this work is the first to report a hormonal up-regulation of VEGF mRNA expression which is mediated by the RNA-stabilizing protein HuR.
Fourth, concomitant overexpression of Tis11b and HuR revealed that Tis11b completely abrogated HuR-induced luciferase activity on a heterologous transcript consisting of luciferase cDNA cloned upstream of VEGF mRNA 3′-UTR (Luc-V3′). Our results showing that HuR increases luciferase activity derived from Luc-V3′ in vitro are similar to those reported by Goldberg-Cohen et al (32). Using a similar reporter construct, these authors have shown further that the increase in reporter activity was related to HuR binding to a 40-bp RNA element (nucleotides 1285–1325) within VEGF mRNA 3′-UTR, which confers increased stability to the heterologous transcript. Tis11 b and HuR bind to distinct but very close RNA elements on VEGF mRNA 3′-UTR (nucleotides 1161–1235 (30) and 1285–1325 (32) for Tis11b and HuR, respectively). Our UV cross-linking experiments indicate that Tis11b and HuR can simultaneously as well as individually bind to VEGF 3′-UTR RNA probe. However, Tis11b potently prevents HuR binding to VEGF 3′-UTR, a finding which is consistent with the inhibitory effect of Tis11b on HuR-mediated increase in reporter gene activity and mRNA level (co-transfection experiments, Fig. 6C and 6D). For as yet unidentified reasons, the decrease in HuR-binding to VEGF 3′-UTR in the presence of increasing doses of Tis11b was not paralleled by an increase in Tis11b binding. Similar results showing that HuR and tristetraprolin (Tis11, TTP), the most studied member of the Tis11 protein family, can bind simultaneously and competitively to GM-CSF 3′-UTR have been reported by Raghavan et al (53). More recently, Ashish et al provided evidence that HuR and the mRNA-destabilizing factor AUF1 can bind target transcripts on both distinct, non-overlapping sites, and on common sites in a competitive fashion (54). They propose that the fate of the mRNA target depends on HuR and AUF1 abundance, the target RNA sequence and the subcellular compartment investigated. Experiments aiming at dissecting the molecular mechanisms governing the binding of Tis11b and HuR to specific or common sites on VEGF 3′-UTR are under way.
At the functional level, following stimulation of adrenocortical cells by ACTH, HuR which predominates in the nucleus may shuttle to the cytoplasm where it may stabilize VEGF mRNA allowing it to be translated. Subsequently, Tis11 b expression is induced and the cytoplasmic level of Tis11 b increases. The relative levels and binding affinities of HuR and Tis11b for their specific sequences in VEGF mRNA 3′-UTR may determine the fate of VEGF transcripts, with HuR predominance promoting VEGF mRNA stabilisation and Tis11b predominance promoting VEGF mRNA degradation. At some point (between 4 and 6 h of stimulation by ACTH, according to our data), Tis11b may predominate and facilitate VEGF mRNA degradation. This model provides a mechanism by which VEGF gene could be transiently expressed in BAC cells.
In conclusion, this work reports for the first time that ACTH triggers a rapid nuclear export of HuR into the cytoplasm of adrenocortical cells followed by an induction of Tis11 b protein synthesis and cytoplasmic accumulation, two processes that appear to be responsible for the transient stimulation of VEGF mRNA and protein accumulation. In an in vivo model of adrenal cortex tissue regression triggered by the suppression of pituitary ACTH secretion, we recently observed that adrenocortical VEGF expression is dramatically reduced during this process, resulting in massive disorganization and regression of the capillary network (55). It will therefore be of great interest to establish the respective contributions of HuR and Tis11b to the in vivo regulation of adrenal vasculature by ACTH in future studies.
MATERIALS AND METHODS
Reagents
Synthetic ACTH1–24 was purchased from Neosystem (Strasbourg, France). Culture media and sera were from Invitrogen (Cergy Pontoise, France). All chemicals were obtained from Sigma (St Louis, MO) and were of the highest purity grade available.
Bovine adrenocortical cell (BAC) culture and treatments
Bovine adrenal glands were obtained from a local slaughterhouse. Zona fasciculata-reticularis cells were prepared by enzymatic dispersion with trypsin and primary cultures were established as described in detail elsewhere (56). BAC cells were kept at 37 C in Ham’s F12 medium supplemented with 10% horse serum, 2.5% fetal calf serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 20 μg/ml gentamycin, under 5% CO2 - 95% air atmosphere. On day 4, cells cultured in 10-cm petri dishes (3 × 106 cells/dish) were stimulated with 10 nM ACTH for the indicated periods of time before subcellular fractionation or processing for total RNA isolation as described hereafter. In experiments designed to silence HuR expression (RNAi), BAC cells were transfected on day 2 of culture.
Preparation of subcellular fractions
Total cell extracts, as well as nuclear and cytoplasmic fractions were prepared using the PARIS protein and RNA isolation kit (Ambion, Austin, TX) according to the manufacturer’s instructions. A protease inhibitor cocktail (10 μg/ml of leupeptin, 1 μg/ml of aprotinin, 1 μg/ml of pepstatin and 25 μg/ml of AEBSF) was added to the provided buffers. Briefly, 10-cm petri dishes (3 × 106 BAC cells/dish) were lysed on ice in 400 μl of Cell Disruption buffer and collected with a rubber spatula. A portion of the total cell lysate was used for RNA isolation (300 μl) and the remainder was kept for protein analysis (100 μl).
For nuclear and cytoplasmic lysate preparation, BAC cells (3 × 106 cells/10-cm petri dish) were trypsinized, washed in PBS then suspended in 400 μl of Cell Fractionation Buffer. Following a 15 min-incubation on ice, nuclear and cytoplasmic fractions were separated by centrifugation (5 min at 500 × g). The nuclear pellet was lysed in 400 μl of Cell Disruption buffer and kept 10 min on ice to ensure complete disruption before processing the sample for protein analysis.
Protein concentration was determined using a Micro BCA protein Assay Kit (Pierce, Rockford, IL).
RNA isolation and Reverse Transcription-Polymerase Chain Reaction
BAC cell total RNA was extracted using the RNAgents kit (Promega Corp., Charbonnieres, France) according to the manufacturer’s instructions. This system consistently yields 50–80 μl total RNA/3 × 10 6 cells. For RT-PCR analysis of Tis11b, VEGF, HuR or HPRT gene expression, 1 μl of total RNA were reverse transcribed with Superscript II reverse transcriptase (Invitrogen, Cergy Pontoise, France) and PCR amplified using Taq polymerase (QBiogen, Illkirch, France). Amplification of VEGF mRNA isoforms was performed using the primers and amplification conditions described by Gaillard et al (28). The size of the expected amplified fragments was 462-bp and 332-bp for the two major VEGF transcripts, VEGF165 and VEGF121, respectively. The primers for PCR of Tis11b were as follows: 5′-CGAAGAAAACGGTGCCTGTAAG-3′ and 5′-AGTAGGTGAGCCCAAGAGGTCATC-3′. This primer pair sequence amplifies a 354-bp fragment. The amplification conditions were as follows: 94° C for 5 min followed by 25 amplification cycles, each consisting of 94°C for 1 min, 55°C for 1 min, 72°C for 1 min, and 72°C for 5 min for final extension. The primers for HPRT amplification were as follows: 5′-GCCATCACATTGTAGCCCTCT-3′ and 5′-TGCGACCTTGACCATCTTTGG-3′. This primer pair sequence amplifies a 305-bp fragment. The amplification conditions were as follows: 94° C for 5 min followed by 25 amplification cycles, each consisting of 94°C for 1 min, 55°C for 1 min, 72°C for 1 min, and 72°C for 5 min for final extension. The primers for HuR amplification were as follows: 5′-ATGACCCAGGATGAGTTACGAAGC-3′ and 5′-GTTCACAAAGCCATAGCCCAAG-3′. This primer pair sequence amplifies a 111-bp fragment. The amplification conditions were as follows: 94° C for 5 min followed by 25 amplification cycles, each consisting of 94°C for 1 min, 52°C for 1 min, 72°C for 1 min, and 72°C for 5 min for final extension. PCR products were analyzed on 2% agarose ethidium bromide-containing gels and quantitated using ImageQuant software (Fluorlmager SI, Vistra Fluorescence). Preliminary experiments were done to select the starting amounts of RNA so that there was a linear relationship between the amount of input RNA and the optical density of the HPRT band on ethidium bromide-stained gel.
SDS-Polyacrylamide Gel Electrophoresis
SDS-polyacrylamide gel electrophoresis was performed according to Laemmli (57). Total proteins or subcellular fraction extracts (5–20 μg/lane) were solubilized in sample buffer (60 mM Tris-HCI, pH 6.8, 2% SDS, 5% β-mercaptoethanol, 10% glycerol, 0.01% bromophenol blue), boiled for 5 min and loaded onto a 12% SDS-PAGE minigel (Mini Protean II System, BioRad). Electrophoresis was performed at 150 V for 1 h.
Western blot analysis
SDS-PAGE-resolved proteins were electrophoretically transferred onto a PVDF membrane according to Towbin et al. (58). Following transfer, the membrane was incubated in a blocking buffer (PBS buffer containing 0.1% Tween 20 and 5% non-fat dry milk) for 1 h at room temperature. The blots were probed sequentially with antibodies to a peptide fragment (amino acids 49-63) of Tis11b protein (1:500, CovalAb, Lyon, France), anti-human HuR (1:1000, Santa Cruz Biotechnology, Santa Cruz, CA), monoclonal anti-α-tubulin (1:200000, a generous gift from Dr D. Job, CS-U366 INSERM, CEA-Grenoble, France), and anti-lamin A/C (1:15000, a generous gift from Dr. Deloulme, TS-EMI 01-04 INSERM, CEA-Grenoble, France) for 2 h in PBS containing 0.1% Tween. The membrane was thoroughly washed with the same buffer (3×10 min), then incubated for 1 hour with either horseradish peroxidase (HRP)-labelled goat anti-rabbit IgG (immunodetection of Tis11 b and HuR) or HRP-labelled goat anti-mouse IgG (immunodetection of tubulin), or HRP-labelled goat anti-guinea pig IgG (immunodetection of lamin A/C). The PVDF sheet was washed as above and the antigen-antibody complex revealed by Enhanced Chemiluminescence, using the Western blotting detection kit from Amersham Biosciences (Buckinghamshire, England) and BioMax Kodak films (Sigma, St Louis, MO).
Immunoprecipitation assay
BAC cells cultured in 10-cm petri dishes (3 × 106 cells/dish) were stimulated with 10 nM ACTH for the indicated periods of time. Subcellular fractions (cytoplasm and nuclei) were prepared as indicated previously. Lysates was cleared by centrifugation for 5 min at 12,000 × g at 4 C. The anti-human pp32 (I1PP2A) polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA) was added at a concentration of 2 μg/ml to whole supernatants (about 250 μg protein for cytoplasm and 70 μg protein for nuclei), which were then gently rocked for 2 h at 4 C, before being incubated for 30 min with Protein A/G Sepharose beads. Immunoprecipitates were pelleted, washed four times with RIPA buffer, analyzed by SDS-PAGE and transferred to a PVDF membrane. Blots were probed with anti-HuR or anti-pp32 antibodies which were used at 1:1,000.
RNA interference
Expression of HuR (ELAVL1, GenBank accession no NM_001419) was inhibited by transfection of a pre-designed siRNA duplex targeted to exon 2 of human HuR gene (Ambion, Austin, TX). siRNA sense and antisense sequences were 5′-GGAUGAGUUACGAAGCCUGtt-3′ and 5′-CAGGCUUCGUAACUCAUCCtt-3′, respectively. One day after plating, adrenocortical cells were transfected with 10 nM of either HuR siRNA duplex or negative control siRNA (Ambion), using siPORT lipid reagent (Ambion). Typically, cells were analyzed for the loss of HuR mRNA and protein expression 48 hours after transfection using RT-PCR and immunoblotting. At this time point, culture medium was changed and cells were treated for the indicated periods of time with or without 10 nM of ACTH.
Transient Transfections and Dual Luciferase Activity Assay
COS7 cells were grown in DMEM supplemented with 10% fetal calf serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 25 μg/ml gentamycin, and transfected in 12 well-plates using lipofectamine (Invitrogen, Cergy-Pontoise, France) according to the manufacturer’s recommendations. Plasmid pLuc-V3′ contains the firefly luciferase cDNA cloned upstream of the rat VEGF 3′ UTR and downstream of the thymidine kinase (TK) promoter (30). Plasmid pCMV-murine HuR was provided by Dr. Jonathan LaMarre (University of Guelph, Ontario, Canada). pCMV-Tis11b plasmid was described previously (30). pRL-TK encoding renilla luciferase was obtained from Promega Corp. (Charbonnières, France).
Various amounts of either pCMV-Tis11b, pCMV-HuR or both (0.05–1 ng) were co-transfected with 500 ng pLuc-V3′, 25 ng of pRL-TK, and pUC19 up to a total of 700 ng plasmid DNA into 1.5 × 105 cells. Renilla and firefly luciferase activities were measured sequentially 48 h after transfection with the Dual-Luciferase reporter assay system (Promega Corp.) on a LUMAT LB 9507 luminometer (EGG-Berthold, Bad, Wildbad, Germany). Results are expressed as relative light units of firefly luciferase activity over relative light units of renilla luciferase activity to compensate for variations in transfection efficiency. Each transfection condition was performed in triplicate.
Northern hybridization
COS7 cells total RNA was extracted using the RNAgents kit (Promega,) according to the instructions of the manufacturer. 15–20 μg RNA were size-fractionated on a 1% formaldehyde agarose gel, vacuum-transferred onto Hybond-N+ membranes (Amersham) and fixed by UV cross-linking. Northern blots were pre-hybridized in Rapid Hybridization Buffer (Amersham) at 65 C for 30 min. [α32P]dCTP labeled luciferase cDNA probe (2 × 106 cpm/ng DNA, Rediprime random primer labeling kit, Amersham) was then added and the incubation was continued for 2 h at 65 C. Blots were washed for 5 min and 15 min successively at room temperature in 2 × saline sodium citrate (SSC), 0.1% SDS, and then for 15 min in 1 × SSC, 0.1% SDS. The final wash was performed at 65 C for 15 min in 0.5 × SSC, 0.1% SDS. RNA-cDNA hybrids were visualized on phosphor screen (Molecular Dynamics) after a 12- to 24-h exposure period. Blots were stripped and reprobed with 18s cDNA probe to assess RNA loading.
Recombinant protein expression and purification
Tis11b recombinant protein was produced as reported in (30). GST-HuR fusion protein was produced using E. Coli BL21 strain transformed with pGEX-5X2-HuR plasmid (provided by Dr JA Steitz, Yale University school of Medicine, New Haven, USA). Bacteria were grown at 37°C to an A600 nm of 0.6 in 2YT medium (16 g/l tryptone, 10 g/l yeast extract, 5 g/l NaCl) containing 100 μg/ml of ampicillin. The GST-HuR fusion protein was induced with 0.1 mM IPTG for 2 hours, and purified using MicroSpin GST Purification Module (Amersham Biosciences) according to the manufacturer’s instructions. Briefly, cells were harvested by centrifugation (2,500 × g at 4 C for 5 min). The pellet was resuspended with ice-cold PBS containing 0.1 mg/ml lysozyme and lysed by repeated freeze/thawing (10 times). Clarified lysate was mixed for 10 min with Glutathione Sepharose 4B Microspin column. After two washes with PBS, GST-HuR fusion protein was eluted with 10 mM reduced glutathione. Purity of GST-HuR protein was examined by Coomassie blue staining following SDS-PAGE analysis.
RNA-protein UV cross-linking assay
[32P]-rUTP labeled and unlabeled riboprobes were synthesized in vitro using pSp64 plasmid containing the entire VEGF 3′-UTR and the Riboprobe SP6 in vitro Transcription System (Promega). Integrity of RNA transcripts was visualized on 1% agarose ethidium bromide-containing gel. 1 × 106 cpm of RNA transcripts were incubated for 20 min at room temperature with 1 to 6 μg of either Tis11b-containing bacterial extract or GST-HuR fusion protein, in 10 mM HEPES pH 7.6, 3 mM MgCl2, 40 mM KCl, 5% glycerol, 0,5% NP40 and 2 mM DTT. Yeast tRNA (50 ng/μl) and heparin (2 μg/μl were then added for 10 min. Mixtures were exposed to UV light for 30 min on ice. 100 U of RNase T1 (Invitrogen) were then added for 20 min and RNA-protein complexes were analyzed by 12% SDS-PAGE and autoradiography.
Statistical analysis
Results are expressed as means ± S.E. The mean values were compared by ANOVA using Fisher’s test. A value of p<0.05 was considered as statistically significant. Quantitation of immunoblots and autoradiograms was performed using a Molecular Imager FX and Quantity One software (Biorad).
Acknowledgments
This work was supported by the Institut National de la Santé et de la Recherche Médicale (INSERM, EMI 01-05), the Commissariat à I’Energie Atomique (DSV/DRDC), the Cancéropole Rhône-Alpes and the Fondation de France (grant 2004009572). We are indebted to the Ligue Nationale Contre le Cancer and the Fondation de France for their financial support to NC.
We thank Dr. Jonathan LaMarre (Department of Biomedical Sciences, Ontario Veterinary College, University of Guelph, Canada) and Dr JA Steitz (Yale University school of Medicine, New Haven, USA) for their generous gift of pCMV-HuR and pGEX-HuR plasmids, respectively. We also thank Dr. Didier Job and Dr. Jean-Christophe Deloulme for providing us the anti-tubulin and anti-lamin A/C antibodies, respectively.
References
- 1.Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev. 2004;25:581–611. doi: 10.1210/er.2003-0027. [DOI] [PubMed] [Google Scholar]
- 2.Ferrara N. Role of vascular endothelial growth factor in physiologic and pathologic angiogenesis: therapeutic implications. Semin Oncol. 2002;29:10–14. doi: 10.1053/sonc.2002.37264. [DOI] [PubMed] [Google Scholar]
- 3.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 4.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O’Shea KS, Powell-Braxton L, Hillan KJ, Moore MW. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 5.Dor Y, Djonov V, Abramovitch R, Itin A, Fishman Gl, Carmeliet P, Goelman G, Keshet E. Conditional switching of VEGF provides new insights into adult neovascularization and pro-angiogenic therapy. Embo J. 2002;21:1939–1947. doi: 10.1093/emboj/21.8.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 7.Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levy AP, Levy NS, IIiopoulos O, Jiang C, Kaplin WG, Jr, Goldberg MA. Regulation of vascular endothelial growth factor by hypoxia and its modulation by the von Hippel-Lindau tumor suppressor gene. Kidney Int. 1997;51:575–578. doi: 10.1038/ki.1997.82. [DOI] [PubMed] [Google Scholar]
- 9.Milanini J, Vinals F, Pouyssegur J, Pages G. p42/p44 MAP kinase module plays a key role in the transcriptional regulation of the vascular endothelial growth factor gene in fibroblasts. J Biol Chem. 1998;273:18165–18172. doi: 10.1074/jbc.273.29.18165. [DOI] [PubMed] [Google Scholar]
- 10.Pages G, Pouyssegur J. Transcriptional regulation of the Vascular Endothelial Growth Factor gene: a concert of activating factors. Cardiovasc Res. 2005;65:564–573. doi: 10.1016/j.cardiores.2004.09.032. [DOI] [PubMed] [Google Scholar]
- 11.Levy NS, Chung S, Furneaux H, Levy AP. Hypoxic stabilization of vascular endothelial growth factor mRNA by the RNA-binding protein HuR. J Biol Chem. 1998;273:6417–6423. doi: 10.1074/jbc.273.11.6417. [DOI] [PubMed] [Google Scholar]
- 12.Claffey KP, Shih SC, Mullen A, Dziennis S, Cusick JL, Abrams KR, Lee SW, Detmar M. Identification of a human VPF/VEGF 3′ untranslated region mediating hypoxia-induced mRNA stability. Mol Biol Cell. 1998;9:469–481. doi: 10.1091/mbc.9.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shih SC, Mullen A, Abrams K, Mukhopadhyay D, Claffey KP. Role of protein kinase C isoforms in phorbol ester-induced vascular endothelial growth factor expression in human glioblastoma cells. J Biol Chem. 1999;274:15407–15414. doi: 10.1074/jbc.274.22.15407. [DOI] [PubMed] [Google Scholar]
- 14.Onesto C, Berra E, Grepin R, Pages G. Poly(A)-binding protein-interacting protein 2, a strong regulator of vascular endothelial growth factor mRNA. J Biol Chem. 2004;279:34217–34226. doi: 10.1074/jbc.M400219200. [DOI] [PubMed] [Google Scholar]
- 15.Huez I, Creancier L, Audigier S, Gensac MC, Prats AC, Prats H. Two independent internal ribosome entry sites are involved in translation initiation of vascular endothelial growth factor mRNA. Mol Cell Biol. 1998;18:6178–6190. doi: 10.1128/mcb.18.11.6178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stein I, Itin A, Einat P, Skaliter R, Grossman Z, Keshet E. Translation of vascular endothelial growth factor mRNA by internal ribosome entry: implications for translation under hypoxia. Mol Cell Biol. 1998;18:3112–3119. doi: 10.1128/mcb.18.6.3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mezquita P, Parghi SS, Brandvold KA, Ruddell A. Myc regulates VEGF production in B cells by stimulating initiation of VEGF mRNA translation. Oncogene. 2005;24:889–901. doi: 10.1038/sj.onc.1208251. [DOI] [PubMed] [Google Scholar]
- 18.Good PJ. A conserved family of elav-like genes in vertebrates. Proc Natl Acad Sci U S A. 1995;92:4557–4561. doi: 10.1073/pnas.92.10.4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma WJ, Cheng S, Campbell C, Wright A, Furneaux H. Cloning and characterization of HuR, a ubiquitously expressed Elav-like protein. J Biol Chem. 1996;271:8144–8151. doi: 10.1074/jbc.271.14.8144. [DOI] [PubMed] [Google Scholar]
- 20.Peng SS, Chen CY, Xu N, Shyu AB. RNA stabilization by the AU-rich element binding protein, HuR, an ELAV protein. Embo J. 1998;17:3461–3470. doi: 10.1093/emboj/17.12.3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fan XC, Steitz JA. Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. Embo J. 1998;17:3448–3460. doi: 10.1093/emboj/17.12.3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Atasoy U, Watson J, Patel D, Keene JD. ELAV protein HuA (HuR) can redistribute between nucleus and cytoplasm and is upregulated during serum stimulation and T cell activation. J Cell Sci. 1998;111:3145–3156. doi: 10.1242/jcs.111.21.3145. [DOI] [PubMed] [Google Scholar]
- 23.Fan XC, Steitz JA. HNS, a nuclear-cytoplasmic shuttling sequence in HuR. Proc Natl Acad Sci U S A. 1998;95:15293–15298. doi: 10.1073/pnas.95.26.15293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brennan CM, Steitz JA. HuR and mRNA stability. Cell Mol Life Sci. 2001;58:266–277. doi: 10.1007/PL00000854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma WJ, Chung S, Furneaux H. The Elav-like proteins bind to AU-rich elements and to the poly(A) tail of mRNA. Nucleic Acids Res. 1997;25:3564–3569. doi: 10.1093/nar/25.18.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brennan CM, Gallouzi IE, Steitz JA. Protein ligands to HuR modulate its interaction with target mRNAs in vivo. J Cell Biol. 2000;151:1–14. doi: 10.1083/jcb.151.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gallouzi IE, Brennan CM, Steitz JA. Protein ligands mediate the CRM1-dependent export of HuR in response to heat shock. Rna. 2001;7:1348–1361. doi: 10.1017/s1355838201016089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gaillard I, Keramidas M, Liakos P, Vilgrain I, Feige JJ, Vittet D. ACTH-regulated expression of vascular endothelial growth factor in the adult bovine adrenal cortex: a possible role in the maintenance of the microvasculature. J Cell Physiol. 2000;185:226–234. doi: 10.1002/1097-4652(200011)185:2<226::AID-JCP7>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 29.Chinn AM, Ciais D, Bailly S, Chambaz E, LaMarre J, Feige JJ. Identification of two novel ACTH-responsive genes encoding manganese-dependent superoxide dismutase (SOD2) and the zinc finger protein TIS11b [tetradecanoyl phorbol acetate (TPA)-inducible sequence 11b] Mol Endocrinol. 2002;16:1417–1427. doi: 10.1210/mend.16.6.0844. [DOI] [PubMed] [Google Scholar]
- 30.Ciais D, Cherradi N, Bailly S, Grenier E, Berra E, Pouyssegur J, Lamarre J, Feige JJ. Destabilization of vascular endothelial growth factor mRNA by the zinc-finger protein TIS11b. Oncogene. 2004;23:8673–8680. doi: 10.1038/sj.onc.1207939. [DOI] [PubMed] [Google Scholar]
- 31.Kudo N, Matsumori N, Taoka H, Fujiwara D, Schreiner EP, Wolff B, Yoshida M, Horinouchi S. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc Natl Acad Sci U S A. 1999;96:9112–9117. doi: 10.1073/pnas.96.16.9112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldberg-Cohen I, Furneauxb H, Levy AP. A 40-bp RNA element that mediates stabilization of vascular endothelial growth factor mRNA by HuR. J Biol Chem. 2002;277:13635–13640. doi: 10.1074/jbc.M108703200. [DOI] [PubMed] [Google Scholar]
- 33.Levy AP, Levy NS, Goldberg MA. Post-transcriptional regulation of vascular endothelial growth factor by hypoxia. J Biol Chem. 1996;271:2746–2753. doi: 10.1074/jbc.271.5.2746. [DOI] [PubMed] [Google Scholar]
- 34.Ancelin M, Buteau-Lozano H, Meduri G, Osborne-Pellegrin M, Sordello S, Plouet J, Perrot-Applanat M. A dynamic shift of VEGF isoforms with a transient and selective progesterone-induced expression of VEGF189 regulates angiogenesis and vascular permeability in human uterus. Proc Natl Acad Sci U S A. 2002;99:6023–6028. doi: 10.1073/pnas.082110999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lebovic DI, Shifren JL, Ryan IP, Mueller MD, Korn AP, Darney PD, Taylor RN. Ovarian steroid and cytokine modulation of human endometrial angiogenesis. Hum Reprod. 2000;15(Suppl 3):67–77. doi: 10.1093/humrep/15.suppl_3.67. [DOI] [PubMed] [Google Scholar]
- 36.Mueller MD, Vigne JL, Minchenko A, Lebovic DI, Leitman DC, Taylor RN. Regulation of vascular endothelial growth factor (VEGF) gene transcription by estrogen receptors alpha and beta. Proc Natl Acad Sci U S A. 2000;97:10972–10977. doi: 10.1073/pnas.200377097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mueller MD, Vigne JL, Pritts EA, Chao V, Dreher E, Taylor RN. Progestins activate vascular endothelial growth factor gene transcription in endometrial adenocarcinoma cells. Fertil Steril. 2003;79:386–392. doi: 10.1016/s0015-0282(02)04577-6. [DOI] [PubMed] [Google Scholar]
- 38.Laitinen M, Ristimaki A, Honkasalo M, Narko K, Paavonen K, Ritvos O. Differential hormonal regulation of vascular endothelial growth factors VEGF, VEGF-B, and VEGF-C messenger ribonucleic acid levels in cultured human granulosa-luteal cells. Endocrinology. 1997;138:4748–4756. doi: 10.1210/endo.138.11.5500. [DOI] [PubMed] [Google Scholar]
- 39.Buteau-Lozano H, Ancelin M, Lardeux B, Milanini J, Perrot-Applanat M. Transcriptional regulation of vascular endothelial growth factor by estradiol and tamoxifen in breast cancer cells: a complex interplay between estrogen receptors alpha and beta. Cancer Res. 2002;62:4977–4984. [PubMed] [Google Scholar]
- 40.Gallouzi IE, Brennan CM, Stenberg MG, Swanson MS, Eversole A, Maizels N, Steitz JA. HuR binding to cytoplasmic mRNA is perturbed by heat shock. Proc Natl Acad Sci U S A. 2000;97:3073–3078. doi: 10.1073/pnas.97.7.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang W, Furneaux H, Cheng H, Caldwell MC, Mutter D, Liu Y, Holbrook N, Gorospe M. HuR regulates p21 mRNA stabilization by UV light. Mol Cell Biol. 2000;20:760–769. doi: 10.1128/mcb.20.3.760-769.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yaman I, Fernandez J, Sarkar B, Schneider RJ, Snider MD, Nagy LE, Hatzoglou M. Nutritional control of mRNA stability is mediated by a conserved AU-rich element that binds the cytoplasmic shuttling protein HuR. J Biol Chem. 2002;277:41539–41546. doi: 10.1074/jbc.M204850200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dean JL, Wait R, Mahtani KR, Sully G, Clark AR, Saklatvala J. The 3′ untranslated region of tumor necrosis factor alpha mRNA is a target of the mRNA-stabilizing factor HuR. Mol Cell Biol. 2001;21:721–730. doi: 10.1128/MCB.21.3.721-730.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen CY, Xu N, Shyu AB. Highly selective actions of HuR in antagonizing AU-rich element-mediated mRNA destabilization. Mol Cell Biol. 2002;22:7268–7278. doi: 10.1128/MCB.22.20.7268-7278.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lafon I, Carballes F, Brewer G, Poiret M, Morello D. Developmental expression of AUF1 and HuR, two c-myc mRNA binding proteins. Oncogene. 1998;16:3413–3421. doi: 10.1038/sj.onc.1201895. [DOI] [PubMed] [Google Scholar]
- 46.Viard I, Hall SH, Jaillard C, Berthelon MC, Saez JM. Regulation of c-fos, c-jun and jun-B messenger ribonucleic acids by angiotensin-II and corticotropin in ovine and bovine adrenocortical cells. Endocrinology. 1992;130:1193–1200. doi: 10.1210/endo.130.3.1311231. [DOI] [PubMed] [Google Scholar]
- 47.Dean JL, Brook M, Clark AR, Saklatvala J. p38 mitogen-activated protein kinase regulates cyclooxygenase-2 mRNA stability and transcription in lipopolysaccharide-treated human monocytes. J Biol Chem. 1999;274:264–269. doi: 10.1074/jbc.274.1.264. [DOI] [PubMed] [Google Scholar]
- 48.Ming XF, Kaiser M, Moroni C. c-jun N-terminal kinase is involved in AUUUA-mediated interleukin-3 mRNA turnover in mast cells. Embo J. 1998;17:6039–6048. doi: 10.1093/emboj/17.20.6039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Winzen R, Kracht M, Ritter B, Wilhelm A, Chen CY, Shyu AB, Muller M, Gaestel M, Resch K, Holtmann H. The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. Embo J. 1999;18:4969–4980. doi: 10.1093/emboj/18.18.4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang W, Fan J, Yang X, Furer-Galban S, Lopez de Silanes I, von Kobbe C, Guo J, Georas SN, Foufelle F, Hardie DG, Carling D, Gorospe M. AMP-activated kinase regulates cytoplasmic HuR. Mol Cell Biol. 2002;22:3425–3436. doi: 10.1128/MCB.22.10.3425-3436.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lindstein T, June CH, Ledbetter JA, Stella G, Thompson CB. Regulation of lymphokine messenger RNA stability by a surface-mediated T cell activation pathway. Science. 1989;244:339–343. doi: 10.1126/science.2540528. [DOI] [PubMed] [Google Scholar]
- 52.Levy AP. Hypoxic regulation of VEGF mRNA stability by RNA-binding proteins. Trends Cardiovasc Med. 1998;8:246–250. doi: 10.1016/s1050-1738(98)00020-6. [DOI] [PubMed] [Google Scholar]
- 53.Raghavan A, Robison RL, McNabb J, Miller CR, Williams DA, Bohjanen PR. HuA and tristetraprolin are induced following T cell activation and display distinct but overlapping RNA binding specificities. J Biol Chem. 2001;276:47958–47965. doi: 10.1074/jbc.M109511200. [DOI] [PubMed] [Google Scholar]
- 54.Lai A, Mazan-Mamczarz K, Kawai T, Yang X, Martindale JL, Gorospe M. Concurrent versus individual binding of HuR and AUF1 to common labile target mRNAs. Embo J. 2004;23:3092–3102. doi: 10.1038/sj.emboj.7600305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thomas M, Keramidas M, Monchaux E, Feige JJ. Dual hormonal regulation of endocrine tissue mass and vasculature by adrenocorticotropin in the adrenal cortex. Endocrinology. 2004;145:4320–4329. doi: 10.1210/en.2004-0179. [DOI] [PubMed] [Google Scholar]
- 56.Duperray A, Chambaz EM. Effect of prostaglandin E1 and ACTH on proliferation and steroidogenic activity of bovine adrenocortical cells in primary culture. J Steroid Biochem. 1980;13:1359–1364. doi: 10.1016/0022-4731(80)90098-9. [DOI] [PubMed] [Google Scholar]
- 57.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 58.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]