

Abstract
We used Affymetrix oligonucleotide microarrays to compare gene expression profiles of the metastatic parental breast cancer cell line MDA-MB-435 (435) and the non-metastatic daughter cell line created by the stable expression of the BReast cancer Metastasis Suppressor 1 (BRMS1) gene in 435 cells, MDA-MB-435-BRMS1 (435/BRMS1). Analysis of microarray data provided insight into some of the potential mechanisms by which BRMS1 inhibits tumor formation at secondary sites. Furthermore, due to the importance of the microenvironment, we also examined gene expression under different growth conditions (i.e., plus or minus serum), however, expression of 565 genes was significantly (adjusted p-value <0.05) altered regardless of in vitro growth conditions. BRMS1 expression significantly increased multiple major histocompatability complex (MHC) genes and significantly decreased expression of several genes associated with protein localization and secretion. The pattern of gene expression associated with BRMS1 expression suggests that metastasis suppression may be mediated by enhanced immune recognition, altered transport, and/or secretion of metastasis-associated proteins.
Keywords: Affymetrix, breast cancer, BRMS1, metastasis, microarray, MHC
Introduction
Breast cancer metastasis suppressor 1 (BRMS1) is one of a growing family of genes termed metastasis suppressors, that block development of metastasis without preventing orthotopic tumor growth [1-3]. For the most part, the mechanisms by which metastasis suppressors inhibit are not well understood. BRMS1 induces several apparently unrelated phenotypic changes – restoration of homotypic gap junctional intercellular communications [4, 5], inhibition of NFκB signaling [6], abrogation of phosphoinositide signaling [7].
The breadth of BRMS1-induced cellular changes are, in part, explained by its role as a component of core mSin3a:histone deacetylase complexes and interaction with the retinoblastoma tumor suppressor [8-10]. Thus, BRMS1 is positioned within the cell to regulate expression of a plethora of genes, yet histone deacetylase inhibitors (and by inference, the acetylases and deacetylases themselves) regulate only approximately 2% of known genes [11]. The objective of the current study was to compare gene expression patterns in BRMS1-expressing vs. non-expressing human breast carcinoma cells. We hypothesized that insights regarding BRMS1-associated changes, in particular, and perhaps cancer metastasis in general, would be gained by using unbiased genome-wide expression analyses. Our data shows that BRMS1 expression significantly altered genes involved in: (1) immune response; (2) protein transport and localization; and, (3) Golgi structure and function.
Materials and Methods
Cell Culture and Harvest for RNA isolation
Cells were cultured in Corning™ T-175 flasks (Corning catalog no. 431080, Fisher Scientific, Santa Clara, CA) in HyQ DMEM high glucose/SF-12 medium (HyClone, Logan, UT) supplemented with 5% fetal bovine serum (FBS) (Atlanta Biologicals, Lawrenceville, GA), 1% nonessential amino acids, 1.0 mM sodium pyruvate and maintained at 37°C with 5% CO2 in a humidified atmosphere. Cells were passaged using 0.125% trypsin for the parental 435 cells or 2.0 mM EDTA for the daughter 435/BRMS1 cells in Ca2+/Mg2+-free Dulbecco's phosphate buffered saline.
After attachment cells were washed with 1× phosphate buffered saline (PBS). Medium was then replaced for one hour with either fresh complete media containing 10% FBS or complete serum-free medium. After one hour medium was removed and cells were harvested by washing once with 1× PBS, followed by addition of 10 mL of Trizol® (Invitrogen, Carlsbad, CA). Trizol® was incubated with cells for 3-5 minutes at room temperature before thorough rinsing of flask bottom and removal into a 15 mL conical tube. Samples were then frozen at −80°C at least overnight prior to isolation of total RNA. Samples were thawed at room temperature for 60 minutes. RNA isolation was done by following the Invitrogen protocol except centrifugation steps were done at 3,200 × g for 30 minutes or 10 minutes at 2 to 8°C in place the “long” or “short” centrifugation steps in the protocol.
Microarray Hybridizations and Analyses
Total RNA was submitted to the Utah State University Affymetrix Core Facility for labeling and hybridization. Samples were assessed for quantity on a NanoDrop ND 1000 spectrophotometer and quality using an Agilent (Santa Clara, CA) Bioanalyzer 2100. Samples were labeled using the Affymetrix GeneChip® One-Cycle Target Labeling and Control Kit (part # 900493) as described in the Affymetrix Eukaryotic RNA One-cycle cDNA Synthesis and Labeling manual using an MJ Research DNA Engine Tetrad 2 thermocycler® (Bio-Rad Laboratories, Hercules, CA). Hybridization was performed using a GeneChip Hybridization Oven 640® (Affymetrix, Santa Clara, CA). Washing and staining was performed using an Affymetrix GeneChip Fluidics Station 650® and chips were scanned using an Affymetrix GeneChip Scanner 3000® and images interpreted and monitored using GCOS version 1.2.
Briefly, single- then double-stranded cDNA was synthesized using a T7-oligo primer followed by sample purification. Synthesis of biotin-labeled cRNA proceeded using clean double-stranded cDNA and the Affymetrix IVT labeling kit. Labeled cRNA was purified, quantified and fragmented prior to addition to pre-warmed U133 +2.0 chips. Following overnight hybridization, chips were washed and subjected to stain solution, then washed to remove excess stain. After scanning, images were assessed for quality using default metric settings. Both raw and quality controlled data were received for each sample.
Data was analyzed by standard methods using R [12] and Bioconductor [13] (open source program; http://www.bioconductor.org) for all statistical analyses. The eight Affymetrix Gene Chip array raw data files were normalized using the robust multi-array analysis (RMA) procedure in Bioconductor's affy package [14], which includes background correction, quantile normalization, and probe set summary. Using Bioconductor's limma package [15], a linear model was then fit to the normalized data and contrasts tested for a cell line-media interaction and cell line and media main effects. The top 750 significant cell line effect probe sets were selected to generate a working gene list, which corresponded to an FDR-adjusted p-value cutoff of p < 0.05 and B (log odds) cutoff of 0.
The working list of significant probe sets was used as the basis for performing a functional over-representation analysis using mappings to the Gene Ontology (GO) [16]. The working list of significant probe sets was first converted to a list of unique Entrez Gene [17] identifiers. Probe sets were then matched to Entrez Gene entries, and the genes were mapped to their representative GO identifiers, as well as all connected GO ancestors. Using a hypergeometric p-value, each GO identifier was evaluated for over-representation in the list of significant genes. Due to the nature of the hypergeometric calculation and its testing of many dependent hypotheses, interpretation of this p-value is not straightforward. In addition, GO identifiers with few annotated genes tend to show artificially small p-values. We therefore used the developer's suggested criteria and focused our attention on GO identifiers having a small p-value (p < 0.05) and at least 10 member genes as significantly over-represented [18].
The expression patterns of genes found in selected over-represented GO terms were plotted on heat maps with the gplots package [19] using Euclidean hierarchical clustering on the genes. Replicates and multiple probe sets when present for a given gene were averaged for display purposes.
qRT-PCR
Total RNA was thawed on ice and aliquots were diluted to provide standards at 100, 50, 10, 0.1 and 0.05 nanograms (ng) final concentration. The hypoxanthine guanine phosphoribosyl transferase (HPRT) gene was used as the standard. Samples were probed for relative copy number using a start concentration of 10 ng of total RNA. Samples were prepared using TaqMan primers and probe sets (GeneScript, Inc., Scotch Plains, NJ) and the Qiagen (Valencia, CA) QuantiTect Probe RT-PCR kit according to the directions. Samples were processed and recorded using a M.J. Research Opticon 2 system. Data analyses were performed using Opticon 2 software.
RESULTS
The experimental and analytical approaches used in this study have been outlined in a flow chart (appendix, Fig 1). RNA was isolated from 435 and 435/BRMS1 cells cultured for one hour under serum-containing or serum-free growth conditions, quantified and hybridized to Affymetrix human U133 plus 2.0 chips. Normalization and analysis of the raw data files from the microarray analyses was performed (see Materials and Methods) and a linear model was then fit to the normalized data and contrasts tested for cell line or media effects. No media effects were identified (effects specific to the presence or absence of serum), but some gene chip probe sets were found to have a significant (adj. p-value<0.05) cell line effect. The top 750 significant cell line effect probe sets were selected, and the redundant probe sets for specific genes were removed to produce a working gene list, which corresponded to an FDR-adjusted p-value cutoff of p < 0.05 and B (log odds) cutoff of 0.
The differentially expressed gene list generated using this approach reveals patterns or groups of genes whose expression is changed as a result of BRMS1 expression in the 435 cell line. From our differentially expressed gene list the 40 genes whose expression showed the greatest fold change induction were compiled (Table 1). This list contains genes in descending order by fold change that have been implicated in the immune response, signal transduction, metabolism, transcription, cytoskeleton, Golgi apparatus, and transport. The 40 genes whose expression showed the greatest fold change repression are shown in Table 2. These genes have been associated with transport, cell adhesion, signal transduction, metabolism, cell proliferation, cytoskeleton, and extracellular matrix.
Table 1.
The top forty induced genes in 435/BRMS cells relative to 435 cells.
| Accession no.a | Gene symbol | Function | Signal Log Ratiob |
FDR adj. P-valuec |
|---|---|---|---|---|
| NM_006169 | NNMT | metabolism | 6.163206 | 0.000737 |
| NM_003785 | PAGE1 | immune response | 5.383853 | 0.000035 |
| NM_152997 | C4orf7 | unknown | 5.344053 | 0.007440 |
| NM_019111 | HLA-DRA | immune response | 5.268353 | 0.000562 |
| NM_015444 | RIS1 | cell cycle, proliferation | 4.663992 | 0.000663 |
| NM_002122, 020056 | HLA-DQA1/A2 | immune response | 4.639360 | 0.000035 |
| NM_002124 | HLA-DRB1 | immune response | 4.247076 | 0.000182 |
| NM_000584 | IL8 | immune response | 4.117897 | 0.002229 |
| NM_002123 | HLA-DQB1 | immune response | 4.110985 | 0.000035 |
| NM_001056, 176825 | SULT1C1 | metabolism | 4.110093 | 0.001317 |
| NM_004114, 033642 | FGF13 | signal transduction | 4.041216 | 0.000035 |
| NM_032307 | C9orf64 | unknown | 4.011214 | 0.000796 |
| NM_001734, 201442 | C1S | immune response | 3.767461 | 0.003596 |
| NM_181607 | KRTAP19-1 | cytoskeleton | 3.721054 | 0.000803 |
| NM_000200, 032866 | HTN3/CGNL1 | immune response | 3.639734 | 0.000796 |
| NM_002159 | HTN1 | immune response | 3.432327 | 0.017445 |
| NM_002124 | HLA-DRB5 | immune response | 3.393892 | 0.000071 |
| NM_021983 | HLA-DRB4 | immune response | 3.367954 | 0.000075 |
| NM_033554 | HLA-DPA1 | immune response | 3.144453 | 0.000508 |
| NM_004616 | TSPAN8 | signal transduction | 3.139513 | 0.002499 |
| NM_005708 | GPC6 | unknown | 3.103186 | 0.001502 |
| NM_004355 | CD74 | immune response, protein localization | 3.033860 | 0.002536 |
| NM_017863 | CXorf48 | DNA binding | 2.988708 | 0.000326 |
| NM_001452 | FOXF2 | transcription | 2.956652 | 0.000237 |
| NM_030812 | LOC81569/ACTL8 | cytoskeleton | 2.920189 | 0.001461 |
| NM_002571 | PAEP | transport | 2.886798 | 0.011394 |
| NM_001187 | BAGE | unknown | 2.777546 | 0.016837 |
| NM_001085 | SERPINA3 | immune response | 2.550661 | 0.004990 |
| NM_001445 | FABP6 | cell proliferation inhibitor, metabolism | 2.477626 | 0.026161 |
| NM_002727 | PRG1 | unknown | 2.461429 | 0.001659 |
| NM_012104, 138971-73 | BACE1 | protein modification, Golgi apparatus | 2.427205 | 0.000663 |
| NM_002121 | HLA-DPB1 | immune response | 2.393904 | 0.001461 |
| NM_004163 | RAB27B | signal transduction | 2.297115 | 0.038656 |
| NM_001223, 033292-95 | CASP1 | signal transduction, apoptosis regulation | 2.295373 | 0.017594 |
| NM_021940 | SMAP1 | regulation of GTPase activity | 2.281316 | 0.043648 |
| --------------- | unknown | unknown | 2.274630 | 0.030001 |
| NM_001472, 001474-77, NM_021123 | GAGE 2, 4-7B | cellular defense | 2.259294 | 0.013588 |
| NM_005195 | CEBPD | transcriptional regulation | 2.210539 | 0.003852 |
| NM_003516 | HIST2H2AA | unknown | 2.175758 | 0.009671 |
| NM_017855 | APIN | unknown | 2.173798 | 0.006827 |
GenBank accession number
Signal Log ratio: power value is 2, for example 6.163206 is equal to 26.163206.
For explanation of processing used to obtain FDR-adjusted P-value see materials and methods.
Table 2.
The forty most repressed genes in 435/BRMS cells relative to 435 cells.
| Accession no.a | Gene symbol | Function | Signal Log Ratiob |
FDR adj. P-valuec |
|---|---|---|---|---|
| NM_032525 | TUBB6 | unknown | −3.877230 | 0.005884 |
| NM_000550 | TYRP1 | metabolism | −3.718408 | 0.003087 |
| NM_000615, 181351 | NCAM1 | cell adhesion | −3.224920 | 0.000796 |
| NM_014227 | SLC5A4 | transport - sugar and ions | −3.217907 | 0.010679 |
| NM_005971, 021910 | FXYD3 | transport - ions | −3.019978 | 0.016583 |
| NM_001647 | APOD | transport - lipid | −2.853765 | 0.014191 |
| NM_00100133, 016229 | CYB5R2 | transport - electron | −2.790324 | 0.001659 |
| NM_006499, 201543-45 | LGALS8 | extracellular sugar binding | −2.782567 | 0.001461 |
| NM_002615 | SERPINF1 | cell proliferation | −2.606028 | 0.005884 |
| NM_00609 | ATP8A1 | transport - aminophospholipid | −2.563899 | 0.000818 |
| NM_005944, 001004196 | CD200 | unknown | −2.436624 | 0.012907 |
| NM_006877 | GMPR | metabolism | −2.384433 | 0.000663 |
| NM_017694 | FLJ20160 | unknown | −2.334477 | 0.012502 |
| XM_372574 | LOC390595 | unknown | −2.315783 | 0.017422 |
| NM_007078 | LDB3 | cytoskeleton, protein binding | −2.312725 | 0.016837 |
| NM_000212 | ITGB3 | cell adhesion, signal transduction | −2.248644 | 0.007499 |
| --------------- | LOC401022 | unknown | −2.228267 | 0.002154 |
| NM_005761 | PLXNC1 | cell adhesion | −2.226256 | 0.036549 |
| NM_005328 | HAS2 | ECM component metabolism | −2.152467 | 0.015516 |
| NM_152309 | PIK3AP1 | lipid binding | −2.150887 | 0.005745 |
| --------------- | LOC283824 | unknown | −2.124596 | 0.013738 |
| NM_024505 | NOX5 | cell proliferation, cytokine secretion | −2.094189 | 0.030001 |
| NM_007085 | FSTL1 | extracellular heparin binding | −2.086846 | 0.001659 |
| NM_053042 | KIAA1729 | nucleic acid binding | −2.073675 | 0.013882 |
| NM_152270 | FLJ34922 | unknown | −1.977802 | 0.026823 |
| NM_203463 | LASS6 | cell proliferation, metabolism | −1.948670 | 0.001461 |
| NM_014654 | SDC3 | cytoskeletal protein binding | −1.928120 | 0.016836 |
| NM_001002034 | LOC150368 | unknown | −1.914849 | 0.000631 |
| NM_012228 | MSRB2 | transcription factor | −1.907740 | 0.019284 |
| NM_014782, 177949 | ARMCX2 | binding | −1.905816 | 0.010707 |
| --------------- | KIAA1295 | unknown | −1.900952 | 0.007121 |
| NM_000070, 024344, 173087-90, 212464-65, 212467 | ||||
| CAPN3 | signal transduction, cell adhesion | −1.866039 | 0.026170 | |
| NM_006236 | POU3F3 | transcription factor | −1.829551 | 0.002536 |
| NM_005380, 182744 | NBL1 | apoptosis | −1.829396 | 0.000799 |
| NM_000935, 182943 | PLOD2 | protein modification | −1.828234 | 0.001461 |
| NM_003500 | ACOX2 | metabolism | −1.793333 | 0.031810 |
| NM_015150 | RAFTLIN | unknown | −1.776159 | 0.015190 |
| NM_005160 | ADRBK2 | signal transduction | −1.763304 | 0.015429 |
| NM_003812 | ADAM23 | cell adhesion | −1.751657 | 0.001461 |
| NM_000660 | TGFB1 | cell proliferation, signal transduction | −1.751519 | 0.013738 |
GenBank accession number
Signal Log ratio: power value is 2, for example −3.877230 is equal to 2−3.877230.
For explanation of processing used to obtain FDR-adjusted P-value see materials and d. methods.
Our gene lists provide some insight into the effect of BRMS1 expression, but information in this format fails to uncover the overall biological relevance of the transcriptional changes that lead to a block in metastasis resulting from BRMS1 expression. A statistically sound method for summarizing significant gene lists into the biological processes they represent has been developed by Gentleman [18]. This technique, known as over-representation analysis, was applied to the data.
The working list of significant probe sets was used as the basis for performing a functional over-representation analysis using mappings to Gene Ontology (GO). This technique helps to highlight the overall processes that our list of significant genes represents, and offers an effective method to reveal the biological relevance of transcriptional changes which may not be apparent examining the list on a gene-by-gene basis.
The results of functional over-representation analysis are shown as directed acyclic graphs (DAG) with each graph node representing a GO term. GO terms found to be significantly over-represented were colored according to the collective trend for induction or repression among the genes at that GO term. Red indicates that genes at that node show collective mean induction in 435/BRMS1 cells, while blue indicates collective mean repression. Nodes not colored are not significantly over-represented, but are included to maintain connectivity. Significantly over-represented GO terms within the biological process (BP) GO category are shown (Fig. 1, A). Some of the most highly over-represented BP GO terms include immune response (Fig. 1, B) and protein localization (Fig. 1, C). For the cellular component (CC) GO category (data not shown), the most notable over-representation was found at the Golgi apparatus.
Figure 1.
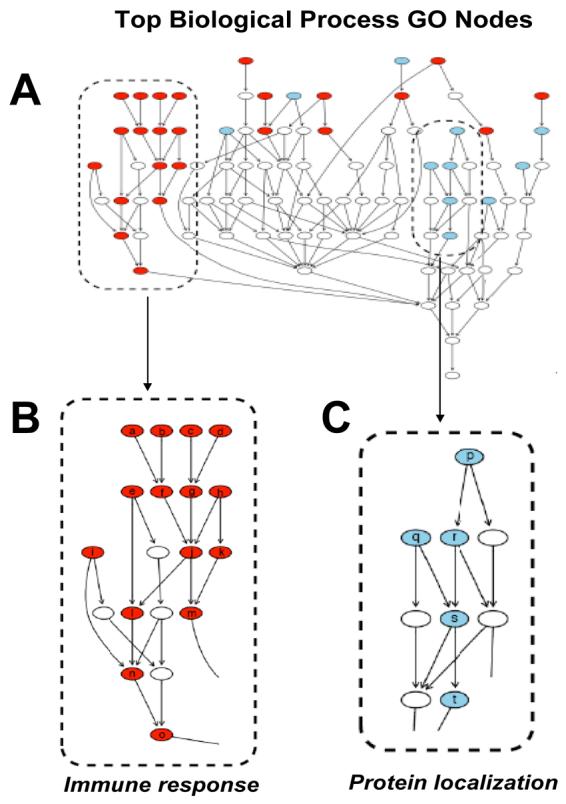
Directed acyclic graph of Gene Ontology (GO) terms over-represented by genes within the data set. Directed acyclic graph (DAG) of all biological process (BP) GO terms which met cut-off criteria (see materials and methods) with enlarged view of terms associated with immune response and protein localization. For all graphs red indicates BP GO terms that were generally induced in 435/BRMS1 cells while blue indicates BP GO terms generally repressed in 435/BRMS1 cells relative to 435 cells. Nodes represent general ancestor BP GO terms at the bottom of the graph proceeding upward to more specific child BP GO terms.
To elucidate which nodes contribute to over-representation within the general GO term of immune response an expansion of the DAG of BP GO terms with both ancestor and child terms is shown in more detail (Fig. 2, A). The function depicted in the expansion proceeds from general in the bottom nodes to very specific in the top nodes (Fig 2, A). To visualize expression of individual genes expanded heat maps were created for specific BP GO terms (appendix Fig. 2, 3, 4). Figure 2 B depicts a combination figure of DAG representation for individual genes which fall within child nodes of the “immune response” node (j in Fig. 2, A) combined with the heat map expression intensities for each.
Figure 2.
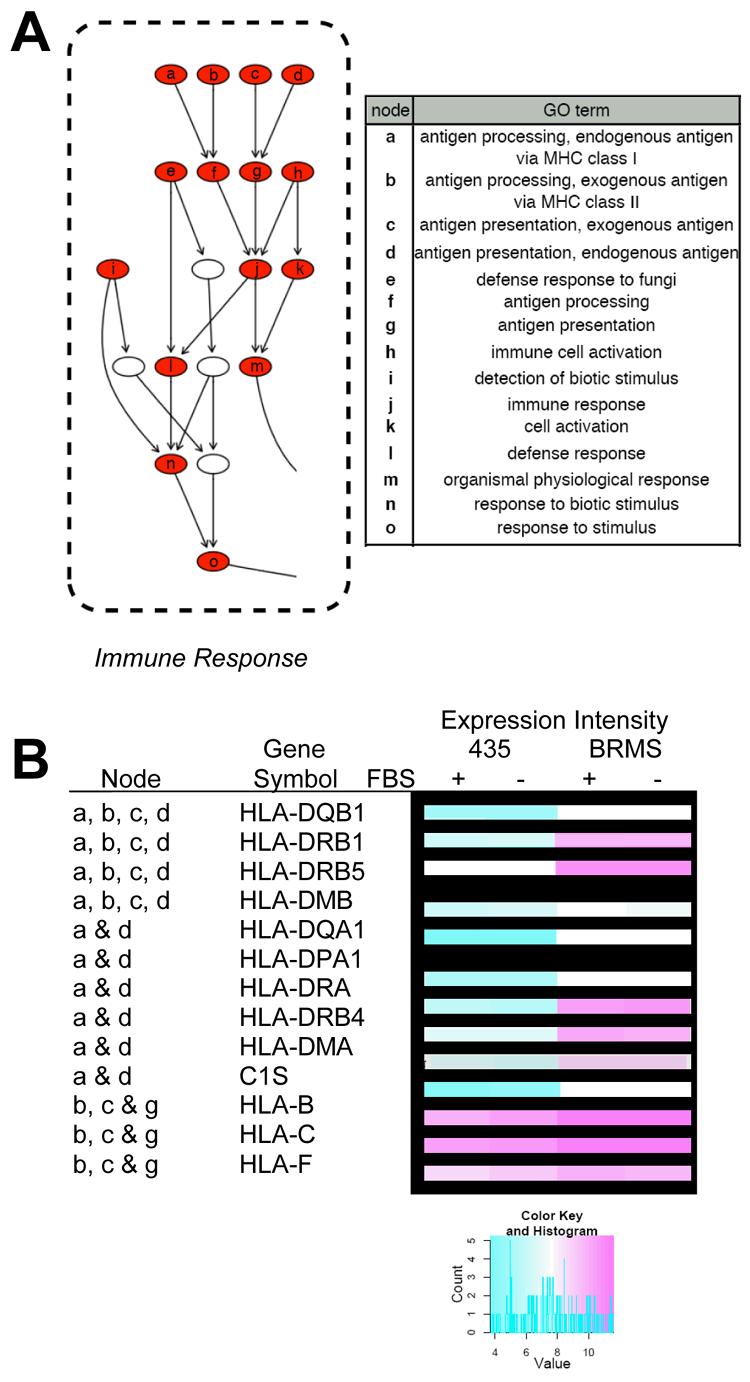
Immune response-associated Gene Ontology (GO) terms and related genes from the data set. A) GO terms are presented from the directed acyclic graph (DAG) of biological process (BP) GO terms which met the cut-off criteria (see materials and methods) with table identifying over-represented BP GO terms. Red nodes indicate BP GO terms which were generally induced in 435/BRMS1 cells relative to 435 cells. Nodes represent general ancestor BP GO terms at the bottom of the graph proceeding upward to more specific child BP GO terms. B) Several genes identified in some of the represented BP GO terms and their expression (as represented in a heat map), are shown in the table.
The full heat map of the expression of the genes within the BP GO term immune response is shown (appendix Fig. 2). A more detailed list of genes significantly (adj. p-value <0.05) induced within the immune response BP GO term is shown in tabular from in the appendix (Table 1, appendix). Of the genes most highly induced in 435/BRMS1 cells are several encoding major histocompatibility complex (MHC), class I and II proteins, including: HLA-DQB1, HLA-DRB1, HLA-DRB5, HLA-DMB, HLA-DQA1, HLA-DPA1, HLA-DRA, HLA-DRB4, HLA-DMA, C1S, HLA-B, HLA-C, and HLA-F.
Our analyses also uncovered changes in the expression of genes associated with protein localization/transport (Fig. 2, A and B). Significant (adj. p-value <0.05) genes were found to be associated with the BP GO term protein localization, as well as child GO terms intracellular protein transport, protein secretion, protein transport, and establishment of protein localization (Fig. 2, A). A more detailed expression profile for individual genes identified in the protein localization BP GO term is shown as a node delineated heat map in Figure 4 (B) and in the full heat map (appendix Fig. 3).
For the protein localization GO term, expression of several genes is repressed coincident with BRMS1 expression including: RBP1, RAB1A, APBA2BP, TIMM22, DNAJC1, RAB8B, SNX1, SSR3, SNX5, and RAB27A. Genes whose expression is induced as a result of BRMS1 expression include: SEC23IP, SEC3L1, SYTL2, COG1, NUP98, COPZ2, CD74 (2), AP1S2, A2M, SCG2, FYB, AP1G2, TGFB1, TLK1, GNAS, and PEX13.
The genes significantly (adj. p-value <0.05) altered in 435/BRMS1 cells identified by over-representational analysis to be involved in the Golgi apparatus GO node under Cellular Component are shown as a heat map (appendix Fig. 4). Those whose expression is significantly repressed coincident with BRMS1 expression include: D4ST1, PIK4CA, ATP7A, ST6GALNAC2, APBA2BP, RAB1A, B3GNT1, LARGE, GOLGA8A, AP1G2, ST6GALT1, SNX1, and B4GALT1. Genes whose expression is significantly induced as a result of BRMS1 expression include: COG1, SEC23IP, PSENEN, AP1S2, COPZ2, BACE1, BICD1, VAMP4, IL15, and HS3ST3A1.
In an effort to test the validity of the microarray data that we obtained, we used quantitative RT-PCR as a second technique to examine changes in gene expression. Five genes from our differentially expressed gene list were selected at random for confirmation using real time quantitative PCR (qRTPCR). TaqMan primers and probes were obtained (GeneScript or Applied Biosystems, Inc.) for BRMS1, Endothelin converting enzyme 2 (Ece2), Ectonucleotide pyrophosphatase phosphodiesterase 2 (Enpp2), Abl-interacting protein 2(Abi2) and Caspase 1 (Casp1). qRT-PCR was performed using a one-step protocol. Under our experimental conditions only amplification of Ece2 produced signal below our lowest control making a reasonable assessment of its expression level in either cell type inconclusive. All other products from amplification (Fig 5, appendix) clearly corroborated our analyses of the microarray data.
Discussion
Metastasis is a complex, multi-step event involving cellular processes such as protein transport, cytoskeletal rearrangements and signal transduction. The metastatic process is sequential and inhibition of any step precludes completion of subsequent steps. Re-expression of BRMS1 prevents metastasis but the step(s) at which BRMS1 exerts its effects are still not completely understood. Preliminary data suggest that BRMS1 controls as multiple steps, including outgrowth at the secondary site (P.A. Phadka and D. Welch, unpublished).
While the amino acid sequence of BRMS1 provided minimal clues regarding function, identification of proteins with which BRMS1 interacts led to the understanding that BRMS1 is involved in transcriptional machinery. Specifically, the BRMS1 protein is a component of multiple mSIN3a:histone deacetylase complexes. Because these complexes regulate chromatin structure, they have the potential to influence gene expression throughout the genome. To better understand BRMS1 function, in particular, its role in regulating gene expression, a genome-wide unbiased expression study was initiated.
Using Affymetrix platforms coupled with robust informatic analyses, we found that BRMS1 re-expression selectively changed expression of certain categories of genes, especially MHC Class I and II and molecules involved in protein transport and secretion. The patterns of gene expression change provide insights into the steps of metastasis influenced by BRMS1 expression.
The most dramatically over-represented gene ontology designation was host immune system, which is a complicated compilation of molecules and events involved in immune response (Table 1, Fig. 2; appendix Table 1, Fig. 2). In response to BRMS1 expression, the expression of several immune response genes was altered including: MHC class I, MHC class II, interleukins, interferon-induced, interferon-induced protein with tetratricopeptide repeats, CD antigens and several with roles in processing or transport of antigens.
Previous studies have shown that some MHC class I genes are not expressed in certain cancers, which may alter the immune response [20-22]. Altered expression of MHC, class I and II molecules has been described in metastatic lesions from breast tumors [23-25] and breast cancer lines [26]. Notably, it has been established that the expression of MHC class I molecules on cancer cells has an important role in the cytotoxic T-lymphocyte (CTL)-mediated immune response [27], and up-regulation of MHC class I genes could provide opportunities for CTL's to recognize and destroy cancer cells [28]. However, a cause and effect relationship has not been clearly established. On a related note, the tumor suppressor pRb appears to be required for up-regulation of some MHC, class II genes [29, 30]. Therefore, some similarity may exist in the mechanism of action of tumor suppressors and metastasis suppressors.
MHC, class II classical molecules, HLA-DPA, -DPB, -DQA, -DQB, -DRA and – DRB, are normally expressed only on professional Antigen Presenting Cells (APC'S) and a few other types of cells. These molecules present peptides derived from proteins degraded in endosomes. Earlier studies suggest that changes in expression of MHC, class II molecules may influence tumor cell evasion of immune surveillance favoring metastatic disease [31, 32]. MHC class II genes were found repressed in highly metastatic cells [30]. Induction of MHC class I and II genes may be one mechanism by which 435/BRMS1 cells are kept at low populations, by triggering an immune response that eliminates them in lung and bone.
In addition to immune response gene expression changes, GO analyses revealed two other over-represented GO categories, the BP GO term protein localization and CC GO term Golgi apparatus. Reciprocally, one is dependent on the other—the structure of the Golgi apparatus is dependent upon protein localization, and appropriate protein localization is dependent upon the Golgi apparatus for transportation.
The secretory pathway of eukaryotic cells is a highly complex bidirectional system which includes the endoplasmic reticulum, the cis-, medial-, and trans-Golgi, the late endosome, the lysosome/vacuole, and the plasma membrane. Proteins transported in this elaborate system are first synthesized at the endoplasmic reticulum (ER) membrane where they remain in the membrane or are injected into the lumen of the ER. Proteins packaged into lipid vesicles are then transported in an anterograde direction to more distal sites in the pathway via a series of vesicle budding, docking and fusion events. Alternatively, proteins can be retained in secretory compartments or retrieved to a more proximal compartment via a retrograde trafficking process.
The processes of lipid vesicle fission (breaking off the donor membrane), transport, and fusion (with the acceptor membrane) are complex and involve a number of membrane associated and transmembrane proteins. Uncovered in this study were genes representing membrane associated proteins such as vesicle-associated membrane protein 4 (Vamp4), coatamer protein complex-subunit zeta 2 (COPZ2), sec3L1, sec23IP, adaptor-related protein complex subunits (AP1G2 and AP1S2), as well as those genes encoding proteins known to cycle from the cytosol to a membrane (vesicle, Golgi, endosome, or plasma membrane).
Other gene products known to have a role in regulation of vesicle-mediated protein trafficking that are found localized to membranes or in the cytosol include the small monomeric GTPases Rab1a, Rab8b, and Rab27a which have been implicated in vesicle formation, uncoating and docking [33-35]. Notably, Rab27a and the synaptotagmin-like 2 (SYTL2) protein are involved in melanosome transport [36]. The sorting nexins (SNX1 and SNX5) which are known to bind phosphoinositides (via PX domains) associate with the retromer complex and regulate protein transport at the endosome [37, 38]. Also a regulator of vesicle-mediated protein trafficking and phosphoinositide metabolism is the PtdIns 4-kinase (PIK4CA) with a role in protein transport from the Golgi to the plasma membrane [39, 40]. Altogether, the data strongly suggest that the expression of BRMS1 alters exocytic protein trafficking, with changes in genes that encode proteins associated with the ER, Golgi, and endosomes. Importantly, changes in proteins associated with Golgi function would have profound affects on protein transport.
Several genes whose protein products have important roles in protein and lipid modification within the Golgi compartments were also altered. Those genes include glycosyltransferases (e.g. LARGE), galactosyltransferase (B4GALT1), N-acetylglucosaminyltransferase (B3gnt1), sialyltransferase (ST6GALNAC2), and sulfotransferase (D4ST1). The decrease in expression of these genes may reflect an overall reduction in post-translational protein modification prior to transport, or be specific to a subset of proteins and lipids.
Two other cellular attributes that seem to be altered in the BRMS1-expressing cells are the cytoskeletal-related genes and cell-adhesion related genes. Several genes whose expression is changed very likely affect how cell-to-cell and cell-to-extracellular matrix interactions occur. Notably, we also observed significant (adj. p-value < 0.05) gene expression changes in connexin 32 (full data set on web site) that was previously implicated in metastasis [5] and associated with BRMS1 expression.
Altogether, what do these results tell us about the mechanism by which metastasis can be suppressed? The inherently simple design of this study was to express a single metastasis suppressor gene, BRMS1, in order to gain insight into potential transcriptional changes that enable switching a highly metastatic cell line to a non-metastatic cell line. The results strongly suggest that BRMS1 suppression of metastasis in part is accomplished by altering the host immune response, intracellular protein transport and Golgi structure and function. In addition to these potentially coordinated effects, changes in expression of numerous genes involved in signal transduction, cell adhesion, cytoskeletal architecture, cell proliferation and metabolism (see summary gene list on web site www.biosystems.usu.edu/ …. were observed in BRMS1-expressing cells. The breadth of changes is consistent with the observed biology.
Many of the changes observed in gene expression could potentiate altering of the microenvironment of micrometastases via changes in protein transport, secretion, or extracellular matrix affects. Alternatively, observed changes may alter how the non-metastatic 435/BRMS1 cells are able to respond to the stimuli encountered in the microenvironment. That is, the non-metastatic cells cannot respond to growth factor mediated signaling, alter their cytoskeleton or that their cell adhesion properties are altered compared to the metastatic cells. While this study provides processes and genes that should be evaluated, follow-up studies carefully examining cause and effect of individual genes or sets of genes and how these modulate metastasis are necessary.
Figure 3.
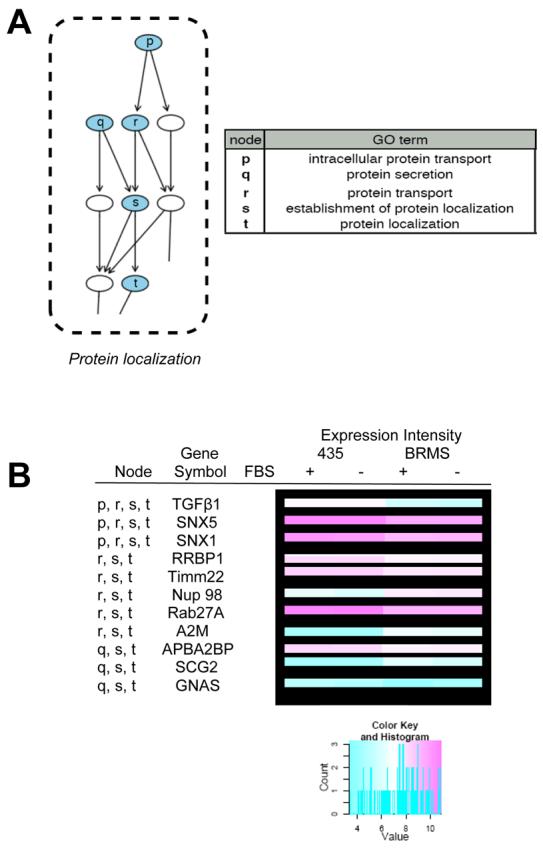
Protein localization-associated Gene Ontology (GO) terms and related genes from the data set. A) GO terms are presented from the directed acyclic graph (DAG) of biological process (BP) GO terms which met the cut-off criteria (see materials and methods) with table identifying over-represented BP GO terms. Blue nodes indicate BP GO terms which were generally repressed in 435/BRMS1 cells relative to 435 cells. Nodes represent general ancestor BP GO terms at the bottom of the graph proceeding upward to more specific child BP GO terms. B) Several genes identified in some of the represented BP GO terms and their expression (as represented in a heat map), are shown in the table.
Acknowledgements
The authors wish to acknowledge the following organization for funding: USDA/CSREES Special Research Grant # WY 2004/2005-06084. We also wish to thank Ning Lin Yin, Ryan Black and Katie Grover of the USU Affymetrix Core Laboratory. We thank Graham Casey (Cleveland Clinic Foundation) and Kent Hunter (N.I.H.) for input. This is Utah Agricultural Experiment Station paper 7864.
Abbreviations
- 435-MDA-MB
435 cells
- ABI
Abl-interactor protein
- BP
biological process
- BRMS
breast cancer metastasis suppressor
- CC
cellular component
- DIAG
diacylglycerol
- Ece
endothelin converting enzyme
- ENPP
ectonucleotide pyrophosphatase phosphodiesterase
- FBS
fetal bovine serum
- GO
gene ontology
- HPRT
hypoxanthine guanine ribosyltransferase
- NK
natural killer cell
- PBS
phosphate buffered saline
- PKC
protein kinase C
- qRT-PCR
quantitative real time polymerase chain reaction
- RNA
ribonucleic acid
- USU
Utah State University
- 435/BRMS1-MDA-MB
435/BRMS1 cells
- ANOVA
analysis of variance
- BRCA
breast cancer gene
- CASP1
caspase 1
- DAG
directed acyclic graph
- DMEM
Dulbecco's modified eagle's medium
- EDTA
ethylene diamine tetraacetic acid
- ER
endoplasmic reticulum
- FDR
false discovery rate
- Her
human epidermal growth factor receptor
- MHC
major histocompatibility complex
- NF-κ-B
nuclear factor-kappa in B-cells
- PDGF
platelet derived growth factor
- pRb
retinoblastoma protein
- RMA
robust multi-array analysis
- Th
Helper T cells
Appendix
Figure 1.

A flow chart of experimental and analytical approaches.
Appendix
Figure 2.
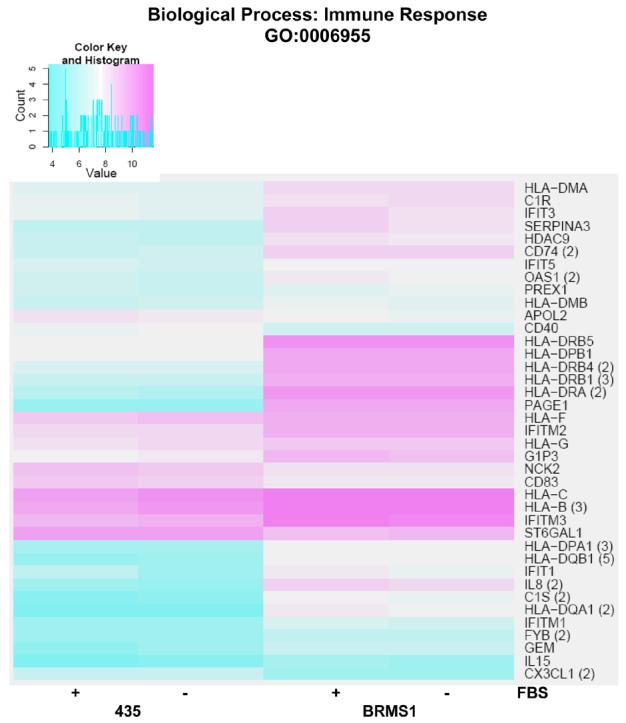
Heat map of significant genes within the biological process (BP) Gene Ontology (GO) term immune response. Color key is shown at top left and represents the log(2) expression intensity for the average of the probe sets for the specified gene determined to be significantly (adj. p-value <0.05) altered in our data. Cyan color indicates lower relative expression and magenta indicates higher relative expression. If more than one probe set for a given gene was determined to be significant, they were averaged together prior to heat mapping and the number of probe sets is indicated in parentheses to the right of the gene symbol on the map. The presence or absence of fetal bovine serum (FBS) in the samples from each cell line is indicated at the bottom of the map.
Appendix
Figure 3.
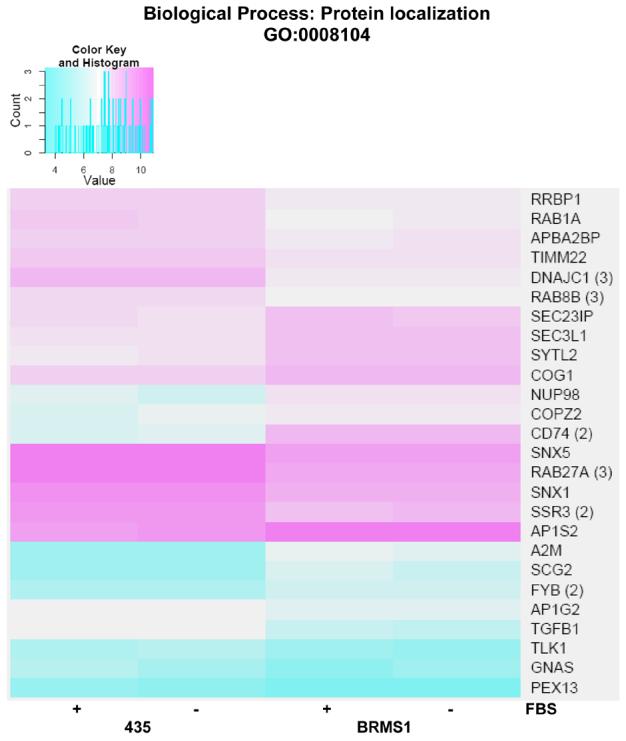
Heat map of significant genes within the biological process (BP) Gene Ontology (GO) term protein localization. Color key is shown at top left and represents the log(2) expression intensity for the average of the probe sets for the specified gene determined to be significantly altered in our data. Cyan color indicates lower relative expression and magenta indicates higher relative expression. If more than one probe set for a given gene was determined to be significant, they were averaged together prior to heat mapping and the number of probe sets is indicated in parentheses to the right of the gene symbol on the map. The presence or absence of fetal bovine serum (FBS) in the samples from each cell line is indicated at the bottom of the map.
Appendix
Figure 4.
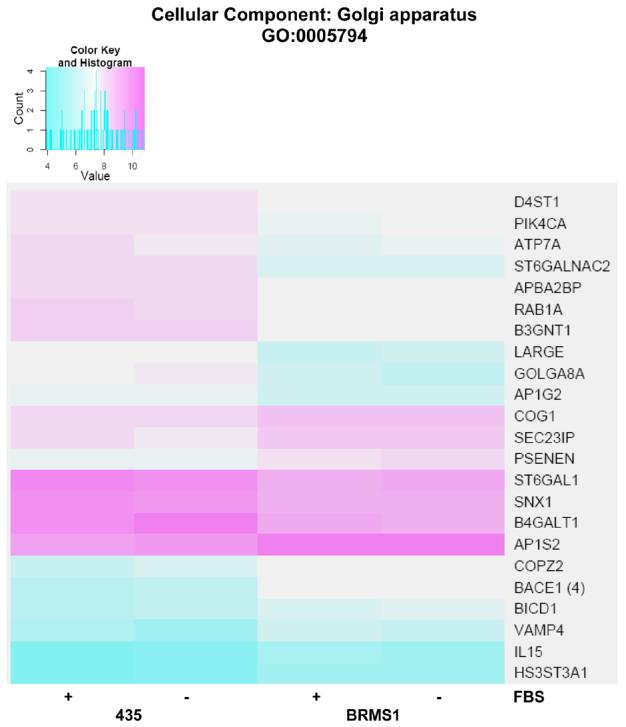
Heat map of significant genes within the cellular component (CC) Gene Ontology (GO) term Golgi apparatus. Color key is shown at top left and represents the log(2) expression intensity for the average of the probe sets for the specified gene determined to be significantly altered in our data. Cyan color indicates lower relative expression and magenta indicates higher relative expression. If more than one probe set for a given gene was determined to be significant, they were averaged together prior to heat mapping and the number of probe sets is indicated in parentheses to the right of the gene symbol on the map. The presence or absence of fetal bovine serum (FBS) in the samples from each cell line is indicated at the bottom of the map.
Appendix
Figure 5.
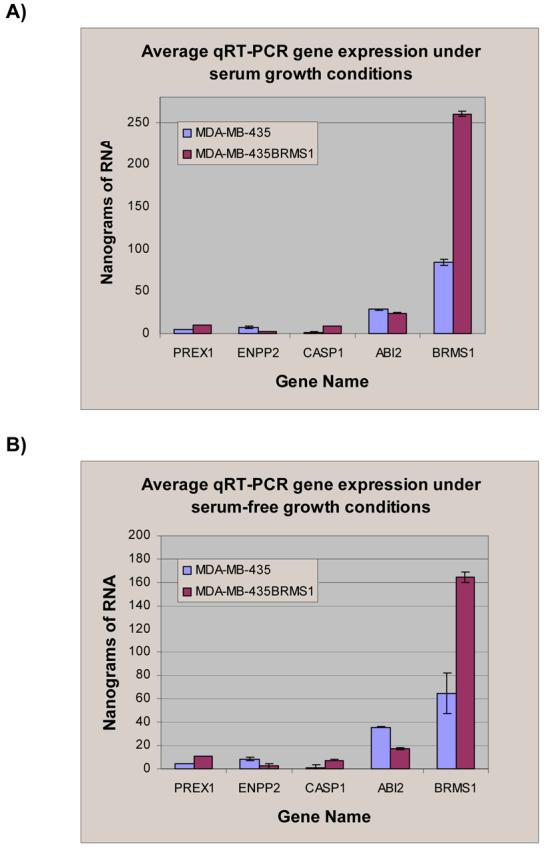
Panel A: Quantitative real-time polymerase chain reaction analysis of select gene expression in nanograms of RNA from 435 and 435/BRMS1 cells grown in serum containing media. Standard deviation of each of the three replicates is shown. Panel B: Quantitative real-time polymerase chain reaction analysis of select gene expression with standard deviation in nanograms of RNA from 435 and 435/BRMS1 cells grown in serum-free media. Standard deviation of each of the three replicates is shown. Gene expression was determined relative to standards from both cell lines using the hypoxanthine guanine phosphoribosyl transferase gene.
Appendix Table 1
Genes involved in immune response induced in 435/BRMS cells relative to 435 cells.
| Accession no.a | Symbol | Signal Log Ratiob |
FDR adj P-Valuec |
|---|---|---|---|
| NM_003785 | PAGE1 | 5.383853 | 0.000035 |
| NM_019111 | HLA-DRA | 5.268353 | 0.000562 |
| NM_002122, 020056 | HLA-DQA1/A2 | 4.639360 | 0.000035 |
| NM_002124 | HLA-DRB1 | 4.247076 | 0.000182 |
| NM_000584 | IL8 | 4.117897 | 0.002229 |
| NM_002123 | HLA-DQB1 | 4.110985 | 0.000035 |
| NM_001734, 201442 | C1S | 3.767461 | 0.003596 |
| NM_002159 | HTN1 | 3.432327 | 0.017445 |
| NM_002124 | HLA-DRB5 | 3.393892 | 0.000071 |
| NM_021983 | HLA-DRB4 | 3.367954 | 0.000075 |
| NM_033554 | HLA-DPA1 | 3.144453 | 0.000508 |
| NM_004355 | CD74 | 3.033860 | 0.002536 |
| NM_001187 | BAGE | 2.777546 | 0.016837 |
| NM_001085 | SERPINA3 | 2.550661 | 0.004990 |
| NM_002121 | HLA-DPB1 | 2.393904 | 0.001461 |
| NM_001472, 001474-77, 021123 | GAGE 2, 4-7B | 2.259294 | 0.013588 |
| NM_001001887, 001548 | IFIT1 | 2.146046 | 0.028883 |
| NM_014707, 058176-77, 178423, 178425 | HDAC9 | 2.109489 | 0.006050 |
| --------------- | HLA-DRB6 | 1.974294 | 0.002037 |
| NM_005261, 181702 | GEM | 1.758017 | 0.005575 |
| NM_002038, 022872, 022873 | G1P3 | 1.682761 | 0.032661 |
| NM_021034 | IFITM3 | 1.576183 | 0.017422 |
| NM_003641 | IFITM1 | 1.554440 | 0.016836 |
| NM_001712 | CEACAM1 | 1.443276 | 0.016670 |
| NM_006120 | HLA-DMA | 1.441918 | 0.015915 |
| NM_006435 | IFITM2 | 1.434174 | 0.013460 |
| NM_002534, 016816 | OAS1 | 1.432204 | 0.016408 |
| NM_001011544, 005366 | MAGEA11 | 1.348985 | 0.046765 |
| NM_005514 | HLA-B | 1.324459 | 0.014377 |
| NM_001733 | C1R | 1.299634 | 0.022136 |
| NM_001549 | IFIT3 | 1.283534 | 0.035789 |
| NM_001465, 199335 | FYB | 1.235783 | 0.023193 |
| NM_012420 | IFIT5 | 1.125754 | 0.038872 |
| --------------- | GAGE3 | 1.082578 | 0.029565 |
| NM_000585, 172174, 172175 | IL15 | 0.966510 | 0.016706 |
| NM_002118 | HLA-DMB | 0.929749 | 0.044733 |
| NM_020820 | PREX1 | 0.898315 | 0.030792 |
| NM_006417 | IFI44 | 0.894730 | 0.033646 |
| 'XM_496804 | HCG22 | 0.876532 | 0.042341 |
| NM_002117 | HLA-C | 0.850252 | 0.043950 |
| NM_018950 | HLA-F | 0.759131 | 0.041014 |
GenBank accession number
Signal Log ratio: power value is 2, for example 5.383853 is equal to 25.383853.
For explanation of processing used to obtain FDR-adjusted P-value see materials and methods.
Appendix Table 2
Genes involved in protein localization/secretion altered in 435/BRMS cells relative to 435 cells.
| Accession no.a | Symbol | Signal Log Ratiob |
FDR adj P-Valuec |
|---|---|---|---|
| Induced | |||
| NM_004355 | CD74 | 3.033860 | 0.002536 |
| NM_000014 | A2M | 2.081898 | 0.004244 |
| NM_003469 | SCG2 | 1.564269 | 0.002536 |
| NM_005387, 016320, 139131-32 | NUP98 | 1.463201 | 0.045012 |
| NM_001465, 199335 | FYB | 1.235783 | 0.023193 |
| NM_012219 | MRAS | 1.165204 | 0.012502 |
| NM_032379, 032943, 206927-30 | SYTL2 | 1.057945 | 0.005790 |
| NM_016429 | COPZ2 | 1.041162 | 0.027706 |
| NM_003916 | AP1S2 | 0.997023 | 0.022876 |
| NM_018261, 178237 | SEC3L1 | 0.924925 | 0.040666 |
| NM_006946 | SPTBN2 | 0.805178 | 0.031828 |
| NM_018714 | COG1 | 0.774169 | 0.041457 |
| NM_007190 | SEC23IP | 0.662779 | 0.041697 |
| Repressed | |||
| NM_003917, 080545 | AP1G2 | −0.593089 | 0.044733 |
| NM_031231, 031232 | APBA2BP | −0.624727 | 0.032046 |
| NM_013337 | TIMM22 | −0.680658 | 0.033443 |
| NM_012290 | TLK1 | −0.697219 | 0.029565 |
| NM_006912 | RIT1 | −0.776814 | 0.029936 |
| NM_004587 | RRBP1 | −0.819226 | 0.036365 |
| NM_002618 | PEX13 | −0.847441 | 0.016837 |
| NM_004161 | RAB1A | −0.869007 | 0.041363 |
| NM_003099, 148955, 152826 | SNX1 | −0.897803 | 0.024230 |
| NM_014426, 152227 | SNX5 | −1.002329 | 0.007902 |
| NM_016530 | RAB8B | −1.003628 | 0.021477 |
| NM_007107 | SSR3 | −1.230001 | 0.022588 |
| NM_022365 | DNAJC1 | −1.634016 | 0.000907 |
GenBank accession number
Signal Log ratio: power value is 2, for example 3.033860 is equal to 23.033860.
For explanation of processing used to obtain FDR-adjusted P-value see materials and methods.
Appendix Table 3
Genes involved in Golgi apparatus altered in 435/BRMS cells relative to 435 cells.
| Accession no.a | Symbol | Signal Log Ratiob |
FDR adj P-Valuec |
|---|---|---|---|
| Induced | |||
| NM_012104, 138971-73 | BACE1 | 2.427205 | 0.000663 |
| NM_003762, 201994 | VAMP4 | 1.090870 | 0.033237 |
| NM_172341 | PSENEN | 1.074803 | 0.013460 |
| NM_016429 | COPZ2 | 1.041162 | 0.027706 |
| NM_006042 | HS3ST3A1 | 1.016080 | 0.013820 |
| NM_003916 | AP1S2 | 0.997023 | 0.022876 |
| NM_000585, 172174-75 | IL15 | 0.966510 | 0.016706 |
| NM_001003398, 001714 | BICD1 | 0.916492 | 0.032477 |
| NM_018714 | COG1 | 0.774169 | 0.041457 |
| NM_007190 | SEC23IP | 0.662779 | 0.041697 |
| Repressed | |||
| NM_003917, 080545 | AP1G2 | −0.593089 | 0.044733 |
| NM_031231-32 | APBA2BP | −0.624727 | 0.032046 |
| NM_130468 | D4ST1 | −0.673337 | 0.039947 |
| NM_002650, 058004 | PIK4CA | −0.701046 | 0.039884 |
| NM_003032, 173216-17 | ST6GAL1 | −0.867223 | 0.022725 |
| NM_004161 | RAB1A | −0.869007 | 0.041363 |
| NM_003099, 148955, 152826 | SNX1 | −0.897803 | 0.024230 |
| NM_000052 | ATP7A | −0.924147 | 0.034246 |
| NM_001497 | B4GALT1 | −0.988029 | 0.026170 |
| NM_006577, 033252 | B3GNT1 | −1.029937 | 0.036966 |
| NM_003574, 194434 | VAPA | −1.108895 | 0.013738 |
| NM_004737, 133642 | LARGE | −1.144102 | 0.026161 |
| NM_015003, 181076-77 | GOLGA8A | −1.551530 | 0.039884 |
| NM_006456 | ST6GALNAC2 | −1.565736 | 0.014191 |
GenBank accession number
Signal Log ratio: power value is 2, for example 2.427205 is equal to 22.427205.
For explanation of processing used to obtain FDR-adjusted P-value see materials and methods.
References
- 1.Shevde LA, Welch DR. Metastasis suppressor pathways--an evolving paradigm. Cancer Lett. 2003;198:1–20. doi: 10.1016/s0304-3835(03)00304-5. [DOI] [PubMed] [Google Scholar]
- 2.Steeg PS. Metastasis suppressors alter the signal transduction of cancer cells. Nat Rev Cancer. 2003;3:55–63. doi: 10.1038/nrc967. [DOI] [PubMed] [Google Scholar]
- 3.Rinker-Schaeffer CW, O'Keefe JP, Welch DR, Theodorescu D. Metastasis suppressor proteins: discovery, molecular mechanisms, and clinical application. Clin Cancer Res. 2006;12:3882–3889. doi: 10.1158/1078-0432.CCR-06-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Samant RS, Seraj MJ, Saunders MM, Sakamaki TS, Shevde LA, Harms JF, Leonard TO, Goldberg SF, Budgeon L, Meehan WJ, Winter CR, Christensen ND, Verderame MF, Donahue HJ, Welch DR. Analysis of mechanisms underlying BRMS1 suppression of metastasis. Clin Exp Metastasis. 2000;18:683–693. doi: 10.1023/a:1013124725690. [DOI] [PubMed] [Google Scholar]
- 5.Saunders MM, Seraj MJ, Li Z, Zhou Z, Winter CR, Welch DR, Donahue HJ. Breast cancer metastatic potential correlates with a breakdown in homospecific and heterospecific gap junctional intercellular communication. Cancer Res. 2001;61:1765–1767. [PubMed] [Google Scholar]
- 6.Cicek M, Fukuyama R, Welch DR, Sizemore N, Casey G. Breast cancer metastasis suppressor 1 inhibits gene expression by targeting nuclear factor-kappaB activity. Cancer Res. 2005;65:3586–3595. doi: 10.1158/0008-5472.CAN-04-3139. [DOI] [PubMed] [Google Scholar]
- 7.DeWald DB, Torabinejad J, Samant RS, Johnston D, Erin N, Shope JC, Xie Y, Welch DR. Metastasis suppression by breast cancer metastasis suppressor 1 involves reduction of phosphoinositide signaling in MDA-MB-435 breast carcinoma cells. Cancer Res. 2005;65:713–717. [PubMed] [Google Scholar]
- 8.Meehan WJ, Samant RS, Hopper JE, Carrozza MJ, Shevde LA, Workman JL, Eckert KA, Verderame MF, Welch DR. Breast cancer metastasis suppressor 1 (BRMS1) forms complexes with retinoblastoma-binding protein 1 (RBP1) and the mSin3 histone deacetylase complex and represses transcription. J Biol Chem. 2004;279:1562–1569. doi: 10.1074/jbc.M307969200. [DOI] [PubMed] [Google Scholar]
- 9.Ohta S, Lai EW, Pang AL, Brouwers FM, Chan WY, Eisenhofer G, de Krijger R, Ksinantova L, Breza J, Blazicek P, Kvetnansky R, Wesley RA, Pacak K. Downregulation of metastasis suppressor genes in malignant pheochromocytoma. Int J Cancer. 2005;114:139–143. doi: 10.1002/ijc.20670. [DOI] [PubMed] [Google Scholar]
- 10.Samant RS, Debies MT, Hurst DR, Moore BP, Shevde LA, Welch DR. Suppression of murine mammary carcinoma metastasis by the murine ortholog of breast cancer metastasis suppressor 1 (Brms1) Cancer Lett. 2006;235:260–265. doi: 10.1016/j.canlet.2005.04.032. [DOI] [PubMed] [Google Scholar]
- 11.Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1:194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 12.Team RDC. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2006. [Google Scholar]
- 13.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Irizarry RAGL, Bolstad BM, Miller C, Astrand M, Cope LM, Gentleman R, Gentry J, Halling C, Huber W, MacDonald J, Rubinstein BIP, Workman C, Zhang J. affy: Methods for Affymetrix Oligonucleotide Arrays. R package; 2005. [Google Scholar]
- 15.GK S. Limma: linear models for microarray data. In: Gentleman VC R, Dudoit R S, Irizarry WH, editors. Bioinformatics and Computational Biology Solutions using R and Bioconductor. Springer; New York: 2005. pp. 397–420. [Google Scholar]
- 16.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maglott D, Ostell J, Pruitt KD, Tatusova T. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 2005;33:D54–58. doi: 10.1093/nar/gki031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gentleman R. Using GO for statistical analyses. In: Antoch J, editor. Proceedings in Computational Statistics. Physica; Verlag: 2004. pp. 171–180. [Google Scholar]
- 19.Lumley BBaT. Warnes G, editor. gplots: Various R programming tools for plotting data. 2006 [Google Scholar]
- 20.Solana R, Romero J, Alonso C, Pena J. MHC class I antigen expression is inversely related with tumor malignancy and ras oncogene product (p21ras) levels in human breast tumors. Invasion Metastasis. 1992;12:210–217. [PubMed] [Google Scholar]
- 21.Goepel JR, Rees RC, Rogers K, Stoddard CJ, Thomas WE, Shepherd L. Loss of monomorphic and polymorphic HLA antigens in metastatic breast and colon carcinoma. Br J Cancer. 1991;64:880–883. doi: 10.1038/bjc.1991.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palmisano GL, Pistillo MP, Fardin P, Capanni P, Nicolo G, Salvi S, Spina B, Pasciucco G, Ferrara GB. Analysis of HLA-G expression in breast cancer tissues. Hum Immunol. 2002;63:969–976. doi: 10.1016/s0198-8859(02)00642-0. [DOI] [PubMed] [Google Scholar]
- 23.Dadmarz R, Sgagias MK, Rosenberg SA, Schwartzentruber DJ. CD4+ T lymphocytes infiltrating human breast cancer recognise autologous tumor in an MHC-class-II restricted fashion. Cancer Immunol Immunother. 1995;40:1–9. doi: 10.1007/BF01517229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garrido F, Ruiz-Cabello F. MHC expression on human tumors--its relevance for local tumor growth and metastasis. Semin Cancer Biol. 1991;2:3–10. [PubMed] [Google Scholar]
- 25.Maiorana A, Cesinaro AM, Fano RA, Collina G. Expression of MHC class I and class II antigens in primary breast carcinomas and synchronous nodal metastases. Clin Exp Metastasis. 1995;13:43–48. doi: 10.1007/BF00144017. [DOI] [PubMed] [Google Scholar]
- 26.Sotiriadou R, Perez SA, Gritzapis AD, Sotiropoulou PA, Echner H, Heinzel S, Mamalaki A, Pawelec G, Voelter W, Baxevanis CN, Papamichail M. Peptide HER2(776-788) represents a naturally processed broad MHC class II-restricted T cell epitope. Br J Cancer. 2001;85:1527–1534. doi: 10.1054/bjoc.2001.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Foss FM. Immunologic mechanisms of antitumor activity. Semin Oncol. 2002;29:5–11. doi: 10.1053/sonc.2002.33076. [DOI] [PubMed] [Google Scholar]
- 28.Remedi MM, Bonacci G, Vides MA, Donadio AC. Immune control of tumors by antigen presentation improvement. Tumour Biol. 2003;24:228–235. doi: 10.1159/000076137. [DOI] [PubMed] [Google Scholar]
- 29.Fan P, Wang S, Liu X, Zhen L, Wu Z. Major histocompatibility complex class II antigen and costimulatory molecule expression on the surface of breast cancer cells. Zhonghua Zhong Liu Za Zhi. 2002;24:327–330. [PubMed] [Google Scholar]
- 30.Shi B, Vinyals A, Alia P, Broceno C, Chen F, Adrover M, Gelpi C, Price JE, Fabra A. Differential expression of MHC class II molecules in highly metastatic breast cancer cells is mediated by the regulation of the CIITA transcription Implication of CIITA in tumor and metastasis development. Int J Biochem Cell Biol. 2006;38:544–562. doi: 10.1016/j.biocel.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 31.Gorelik E, Kim M, Duty L, Henion T, Galili U. Control of metastatic properties of BL6 melanoma cells by H-2Kb gene: immunological and nonimmunological mechanisms. Clin Exp Metastasis. 1993;11:439–452. doi: 10.1007/BF00054935. [DOI] [PubMed] [Google Scholar]
- 32.Walter W, Lingnau K, Schmitt E, Loos M, Maeurer MJ. MHC class II antigen presentation pathway in murine tumours: tumour evasion from immunosurveillance? Br J Cancer. 2000;83:1192–1201. doi: 10.1054/bjoc.2000.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jordens I, Marsman M, Kuijl C, Neefjes J. Rab proteins, connecting transport and vesicle fusion. Traffic. 2005;6:1070–1077. doi: 10.1111/j.1600-0854.2005.00336.x. [DOI] [PubMed] [Google Scholar]
- 34.Grosshans BL, Ortiz D, Novick P. Rabs and their effectors: achieving specificity in membrane traffic. Proc Natl Acad Sci U S A. 2006;103:11821–11827. doi: 10.1073/pnas.0601617103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sannerud R, Marie M, Nizak C, Dale HA, Pernet-Gallay K, Perez F, Goud B, Saraste J. Rab1 defines a novel pathway connecting the pre-Golgi intermediate compartment with the cell periphery. Mol Biol Cell. 2006;17:1514–1526. doi: 10.1091/mbc.E05-08-0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuroda TS, Fukuda M, Ariga H, Mikoshiba K. The Slp homology domain of synaptotagmin-like proteins 1-4 and Slac2 functions as a novel Rab27A binding domain. J Biol Chem. 2002;277:9212–9218. doi: 10.1074/jbc.M112414200. [DOI] [PubMed] [Google Scholar]
- 37.Liu H, Liu ZQ, Chen CX, Magill S, Jiang Y, Liu YJ. Inhibitory regulation of EGF receptor degradation by sorting nexin 5. Biochem Biophys Res Commun. 2006;342:537–546. doi: 10.1016/j.bbrc.2006.01.179. [DOI] [PubMed] [Google Scholar]
- 38.Merino-Trigo A, Kerr MC, Houghton F, Lindberg A, Mitchell C, Teasdale RD, Gleeson PA. Sorting nexin 5 is localized to a subdomain of the early endosomes and is recruited to the plasma membrane following EGF stimulation. J Cell Sci. 2004;117:6413–6424. doi: 10.1242/jcs.01561. [DOI] [PubMed] [Google Scholar]
- 39.Bruns JR, Ellis MA, Jeromin A, Weisz OA. Multiple roles for phosphatidylinositol 4-kinase in biosynthetic transport in polarized Madin-Darby canine kidney cells. J Biol Chem. 2002;277:2012–2018. doi: 10.1074/jbc.M108571200. [DOI] [PubMed] [Google Scholar]
- 40.Wang YJ, Wang J, Sun HQ, Martinez M, Sun YX, Macia E, Kirchhausen T, Albanesi JP, Roth MG, Yin HL. Phosphatidylinositol 4 phosphate regulates targeting of clathrin adaptor AP-1 complexes to the Golgi. Cell. 2003;114:299–310. doi: 10.1016/s0092-8674(03)00603-2. [DOI] [PubMed] [Google Scholar]